

Abstract
Mutations in glucosidase, beta, acid (GBA) are associated with cognitive impairment in Parkinson disease (PD) as well as dementia with Lewy bodies. For both of these diseases, dementia and hallucinations are typically treated with cholinesterase inhibitors and antipsychotics. However, in some lysosomal storage disorders certain antipsychotic medications are poorly tolerated. This study examined cholinesterase inhibitor and antipsychotic use in monoallelic GBA-related PD to explore potential pharmacogenetic relationships. Monoallelic GBA mutation carriers with PD (GBA-PD) with at least two clinic visits (n = 34) were matched for age-of-onset and gender to GBA and leucine-rich repeat kinase 2 (LRRK2) mutation negative idiopathic PD subjects (IPD) (n = 60). Information regarding cholinesterase inhibitor and antipsychotic use as well as impaired cognition (UPDRS Mentation >1) and hallucinations (UPDRS Thought Disorder >1) were obtained. GBA-PD more frequently reported hallucinations (HR = 5.0; p = 0.01) and they were more likely to have cognitive impairment but this was not statistically significant (HR 2.2, p = 0.07). Antipsychotic use was not significantly different between GBA-PD and IPD (HR = 1.9; p = 0.28), but GBA-PD were more likely to have sustained cholinesterase inhibitor use (HR = 3.1; p = 0.008), even after adjustment for cognition and hallucinations. Consistent with reports of worse cognition, GBA-PD patients are more likely to use cholinesterase inhibitors compared to IPD. While there was no difference in antipsychotic use between IPD and GBA-PD, persistent use of quetiapine in GBA-PD suggests that it is tolerated and that a significant interaction is unlikely. Further prospective study in larger samples with more extensive cognitive assessment is warranted to better understand pharmacogenetic relationships in GBA-PD.
Introduction
Gaucher disease (GD) is a rare lysosomal storage disease caused by homozygous or compound heterozygous glucosidase, beta, acid (GBA) mutations. In addition to GD, both biallelic and monoallelic GBA mutations have been linked to Parkinson disease (PD) and dementia with Lewy bodies (DLB): individuals with GD type 1 have an increased risk of PD (Bultron et al. 2010), and heterozygous GBA mutations have emerged as the most important genetic contributor to PD and DLB risk (Sidransky et al. 2009; Nalls et al. 2013). Estimates of the prevalence of heterozygous GBA mutations in PD vary from 3 to 20% depending on the population assessed (Sidransky et al. 2009). Although heterozygous GBA mutations have been considered a risk factor rather than causal for PD, a reported penetrance as high as 30% by age 80 (Anheim et al. 2012; Kumar et al. 2012) suggests autosomal dominant inheritance with reduced penetrance.
While overall clinically similar to idiopathic PD (IPD), PD patients with monoallelic GBA mutations (GBA-PD) as a group have greater cognitive impairment (Sidransky et al. 2009; Neumann et al. 2009; Seto-Salvia et al. 2012; Brockmann et al. 2011; Alcalay et al. 2012). In a multicenter analysis of 5,691 PD subjects, cognitive changes were more prevalent in GBA-PD (Sidransky et al. 2009), and in pathologically confirmed PD with GBA mutations, dementia or cognitive decline was a common finding (Neumann et al. 2009). Another study found that GBA-PD subjects have a 6-fold greater risk of dementia compared to IPD after adjustment for age of onset and duration of disease (Seto-Salvia et al. 2012). Prospective studies with quantitative assessments demonstrate greater cognitive impairment in GBA-PD compared to IPD: GBA-PD subjects had lower Montreal Cognitive Assessment (MoCA) scores (Brockmann et al. 2011) and lower Mini-Mental State Exam scores (Alcalay et al. 2012). As cognitive impairment is associated with psychosis in PD and GBA mutations are associated with DLB (Nalls et al. 2013; Fenelon et al. 2000), it is not surprising that there is a high prevalence of hallucinations in GBA-PD (Neumann et al. 2009). Cognitive impairment is also a prominent feature of PD in patients with biallelic GBA mutations (Saunders-Pullman et al. 2010), but this represents a much smaller group of patients compared to those with monoallelic GBA mutations.
As monoallelic GBA mutations are the most important genetic risk factor for PD and DLB, it is important to understand the pharmacogenetic implications of GBA mutations in these two conditions. In biallelic lysosomal storage disorders, cationic amphiphilic drugs, which include quetiapine and clozapine, may worsen lysosomal function (Anderson and Borlak 2006). As a first step to assess pharmacogenetic implications of GBA-PD, we performed a retrospective study to determine whether GBA-PD is associated with differential medication usage of cholinesterase inhibitors and antipsychotic medications.
Methods
Participants. At the Movement Disorder Center at Beth Israel Medical Center, patients with two or more Ashkenazi Jewish grandparents who met diagnostic criteria for PD were invited to participate in a genetic study of PD (Pankratz et al. 2002). Consent was obtained, blood or saliva samples were collected, and DNA was isolated and screened for the eight common Ashkenazi Jewish GBA mutations (N370S, L444P, 84GG, IVS2+1G→A, V394L, del55bp, D409H, and R496H) (Saunders-Pullman et al. 2010) and the leucine-rich repeat kinase 2 (LRRK2) G2019S mutation (Ozelius et al. 2006). The institutional review board at Beth Israel Medical Center approved the study procedures, and all subjects provided informed consent.
After excluding subjects with symptom onset prior to January 1990, only one clinical visit, carriers of the LRRK2 G2019S mutation, and GBA mutation homozygotes or compound heterozygotes, 34 PD subjects with heterozygous GBA mutations were identified. Each of these was gender-matched and age-of-onset-matched (±2 years) with at least one PD subject without GBA mutations from the same patient population. Controls met the same criteria as cases. Potential subjects with symptom onset prior to 1990 were excluded because quetiapine and donepezil were approved in the USA in 1997 and 1996, respectively. The gene status of subjects was unknown to treating clinicians during the study period; therefore, clinical care and treatment were provided irrespective of genotype.
Data Collection
A retrospective chart review blinded to gene status was performed to assess demographics and UPDRS scores and to determine: (1) the date after which subjects were rated as having cognitive impairment for two or more consecutive visits based on patient and/or caregiver report; (2) whether a cholinesterase inhibitor was prescribed and compliance reported for two or more consecutive visits; (3) the date after which subjects reported hallucinations for two or more consecutive visits; and (4) whether an antipsychotic medication was prescribed and compliance reported for two or more consecutive visits. All clinic visits prior to April 1, 2012 were considered. In order to reduce the likelihood of detecting a single aberrant finding or acute but limited cognitive worsening, cognitive impairment was defined as a United Parkinson Disease Rating Scale (UPDRS) Mentation score (Item I.1) >1 for at least 2 visits encompassing greater than 6 months. Similarly, hallucinations were defined as a UPDRS Thought Disorder (Item I.2) score >1 for greater than 6 months. Cholinesterase inhibitor and antipsychotic medication usage were only recorded if the subject reported medication use for more than 6 months. For each date a subject met an outcome and for the last recorded clinic date, medications and UPDRS scores were recorded. Duration of disease was calculated by determining the time between the date of symptom onset and visit date. A subset of patients also had formal testing with the MoCA. A MoCA score of <26, which was previously identified as the optimal screening cutoff for mild cognitive impairment in PD (Dalrymple-Alford et al. 2010), was used to differentiate mild cognitive impairment and dementia from normal cognition in PD. Subjects with any missing UPDRS items were not included in analyses utilizing motor UPDRS.
Statistical Analysis
Mann–Whitney and Chi-squared or Fisher’s exact tests were used for bivariate comparisons of continuous and categorical variables, respectively. Cox proportional hazard models were applied for the outcomes of: reported cognitive impairment, sustained psychotic symptoms, and usage of cholinesterase inhibitor as well as atypical antipsychotics. The predictor of interest was heterozygous GBA mutation status. Significance of the Cox proportional hazard models was determined using the Wald statistic with a p value set at 0.05. All models met the proportional hazards assumption based on Schoenfeld residuals. Analyses were performed using Stata 11.1 (Statacorp, College Station, TX, USA). Post-hoc assessment was also performed in the subgroup with available MoCA scores.
Results
Demographic characteristics of GBA-PD and age-of-onset and gender-matched IPD subjects are presented in Table 1. As expected, there were no differences in gender distribution (p = 0.76), median age of onset (p = 0.44), disease duration (0.99), and duration of clinical follow-up (p = 0.83). Levodopa equivalent dose at last visit between the GBA-PD and IPD groups also did not differ (p = 0.22). There were also no significant differences between the GBA-PD and IPD groups in the UPDRS III score (p = 0.26) at the last visit.
Table 1.
Demographics, clinical features, and medication use in GBA-PD and IPD
| GBA-PD (n = 34) | IPD (n = 60) | p value | |
|---|---|---|---|
| Women, n (%) | 17 (50.0) | 28 (46.7) | 0.76 |
| Age of onset, years | 57.5 (48, 65) | 59 (51, 66) | 0.44 |
| Time to first evaluation, years | 2.4 (1.0, 5.7) | 2.0 (1.4, 5.0) | 0.90 |
| Duration of disease, years | 9.3 (5.9, 15.1) | 8.4 (7.0, 12.5) | 0.99 |
| Duration of clinical follow-up, years | 5.1 (3.1, 9.2) | 5.4 (3.0, 8.5) | 0.83 |
| Education, n (%) | 0.28 | ||
| High School | 4 (11.8) | 2 (1.8) | |
| Undergraduate | 13 (38.2) | 27 (48.2) | |
| Graduate | 17 (50.0) | 27 (48.2) (n = 56) | |
| UPDRS III at last visit | 20.5 (15, 30) | 16 (13, 26.5) (n = 52) | 0.26 |
| Levodopa equivalents at last visit, mg | 715.8 (400, 1,000) | 598.5 (432.5, 887.5) | 0.22 |
| Dopamine agonist at last visit, n (%) | 12 (35.3) | 26 (43.3) | 0.29 |
| Mentation consistently >1, n (%) | 9 (26.5) | 7 (11.7) | 0.07 |
| Cholinesterase inhibitor >6 months, n (%) | 14 (41.2) | 9 (15.0) | 0.005 |
| Hallucinations >1, n (%) | 17(50.0) | 18 (30.0) | 0.054 |
| Hallucinations >1 for 6 months, n (%) | 9 (26.5) | 4 (6.7) | 0.01 |
| Antipsychotic >6 months, n (%) | 6 (17.7) | 7 (11.7) | 0.54 |
Continuous variables are reported as medians with interquartile ranges. GBA-PD Parkinson disease with heterozygous GBA mutations, IPD idiopathic Parkinson disease, UPDRS Unified Parkinson’s Disease Rating Scale
Of the 34 GBA-PD subjects, 27 had a mild mutation (22 with N370S and 5 with R496H) and 7 had a severe mutation (4 with 84GG, 2 with L444P, and 1 with V394L) (Beutler et al. 2005). Demographic characteristics of the GBA-PD groups with mild and severe mutations are presented in Table 2. While the duration of clinical follow-up time was longer in the mild mutation group compared to the severe mutation group (9.5 vs. 4.6 years, p = 0.02), there were no significant differences in gender, age of onset, disease duration, UPDRS III, or levodopa equivalent dose at the last visit between the two groups.
Table 2.
Demographics, clinical features, and medication use in GBA-PD with severe mutations versus GBA-PD with mild mutations
| GBA-PD with mild mutations (n = 27) | GBA-PD with severe mutations (n = 7) | p value | |
|---|---|---|---|
| Women, n (%) | 11 (40.7) | 6 (85.7%) | 0.09 |
| Age of onset, years | 57 (48, 67) | 58 (45, 62) | 0.61 |
| Time to first evaluation, years | 2.5 (1.0, 6.6) | 1.9 (1.1, 3.4) | 0.80 |
| Duration of disease, years | 7.8 (5.0, 12.8) | 11.6 (9.0, 16.9) | 0.06 |
| Duration of clinical follow-up, years | 4.6 (2.3, 7.2) | 9.5 (5.2, 15.0) | 0.02 |
| UPDRS III at last visit | 20 (15, 32) | 26 (8, 30) | 0.65 |
| Levodopa equivalents at last visit, mg | 675 (400, 1,000) | 733 (575, 1,330.5) | 0.69 |
| Mentation consistently >1, n (%) | 8 (29.6) | 1 (14.3) | 0.64 |
| AcheI >6mo, n (%) | 9 (33.3) | 5 (71.4) | 0.10 |
| Hallucinations >1, n (%) | 11 (40.7) | 6 (85.7) | 0.09 |
| Hallucinations >1 for 6 months, n (%) | 7 (25.9) | 2 (28.6) | 1.0 |
| Antipsychotic >6 months, n (%) | 5 (18.5) | 1 (14.3) | 1.0 |
Continuous variables are reported as medians with interquartile ranges. GBA-PD Parkinson disease with heterozygous GBA mutations, UPDRS Unified Parkinson’s Disease Rating Scale
GBA-PD subjects had a significantly higher risk of reporting hallucinations (HR = 5.0; 95%CI: 1.5, 16.9) compared to IPD subjects. GBA-PD subjects also were more likely to report cognitive impairment, but this was not statistically significant (HR = 2.2; 95%CI: 0.8, 5.8). GBA-PD subjects were maintained on cholinesterase inhibitor more often than IPD subjects (HR = 3.1; 95%CI: 1.3, 7.2) (see Fig. 1). Of these, 19 received donepezil (12 GBA-PD) and 4 received rivastigmine (2 GBA-PD). Neither adjustment for the UPDRS mentation score nor presence of hallucinations at the time subjects met this outcome or at the last visit date altered the finding that GBA-PD subjects were more likely to use cholinesterase inhibitors (HR = 3.2; 95%CI: 1.3, 7.9).
Fig. 1.
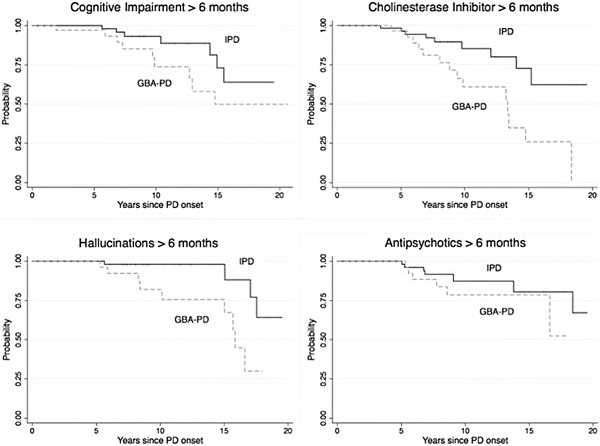
Kaplan–Meier survival curves for cognitive outcomes in GBA-PD vs. IPD
GBA-PD were not significantly more likely be prescribed an antipsychotic for greater than 6 months (HR = 1.9; 95%CI: 0.6, 5.8). Of those treated, 12 received quetiapine (6 GBA-PD) and 1 received clozapine (IPD). While a greater proportion of GBA-PD subjects with severe mutations reported hallucinations and used cholinesterase inhibitors compared to GBA-PD subjects with mild mutations, the differences were not significant (see Fig. 2). As anticipated, for the subset with available MoCA scores, UPDRS mentation scores >1 were more frequently observed in the group with MoCA scores <26 compared to those with scores ≥ 26 (45.5% vs 8%, p = 0.04).
Fig. 2.
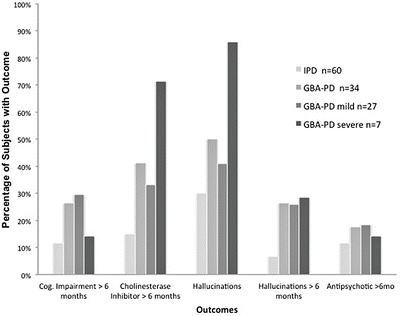
Percentage of IPD and GBA-PD subjects meeting cognitive outcomes. Bar graph representing percentage of subjects in each group that met outcomes of UPDRS mentation score >1 for >6 months, usage of cholinesterase inhibitor >6 months, ever having hallucinations, UPDRS thought disorder score >1 for > 6 months, and usage of antipsychotic medication >6 months
Discussion
Our study found no difference in sustained use of quetiapine and clozapine between GBA-PD and IPD, despite the fact that GBA-PD was more likely to have sustained hallucinations. The greater use of cholinesterase inhibitors in GBA-PD suggests that PD genotype could be associated with differential medication use. The greater cholinesterase inhibitor use and sustained hallucinations in GBA-PD are consistent with previous reports of greater cognitive impairment and hallucinations in this condition (Sidransky et al. 2009; Neumann et al. 2009; Seto-Salvia et al. 2012; Brockmann et al. 2011; Alcalay et al. 2012). While greater cognitive impairment in GBA-PD is likely related to higher cholinesterase use, the association with cholinesterase use was maintained even when adjusted for cognitive score, raising the possibility that additional factors, such as better response, may be present.
In some autosomal recessive metabolic diseases, medications may worsen symptoms. For example, in Tay–Sachs disease, treatment with risperidone, haloperidol, and chlorpromazine may precipitate neurological worsening and should be avoided (Shapiro et al. 2006). This effect is postulated to result from reduction in residual lysosomal function. Because monoallelic GBA mutations may disturb lysosomal dysfunction (Westbroek et al. 2011), there is a parallel concern for GBA-PD. As cationic amphiphilic drugs, both quetiapine and clozapine tend to accumulate in lysosomes and can interfere with lysosomal function and autophagy (Anderson and Borlak 2006). While this study found no difference in quetiapine and clozapine use between GBA-PD and IPD, suggesting tolerability of this medication in GBA-PD, we lack the data to assess whether the response to quetiapine, both its benefit and side effect profile, was the same in both groups.
While our study is limited by its retrospective nature and the increased frequency of cholinesterase use is not a proxy for efficacy, more and persistent use of cholinesterase inhibitors in GBA-PD is consistent with the expected response based on the prominence of Lewy body pathology in GBA-related parkinsonism. Lewy bodies have been reported in both monoallelic GBA-related PD and DLB (Goker-Alpan et al. 2006; Clark et al. 2009; Poulopoulos et al. 2012; Tsuang et al. 2012), as well as in GD1 patients with parkinsonism, who had Lewy bodies in the cortex and hippocampal pyramidal cell layers CA2–CA4 (Wong et al. 2004). The presence of Lewy bodies has been associated with greater reduction in midfrontal choline acetyltransferase compared to Alzheimer’s disease (AD) pathology (Tiraboschi et al. 2000), a rationale for better cholinesterase inhibitor efficacy in dementia associated with PD (PDD) and DLB compared to AD. Increased hallucinations in GBA-PD may also be explained by the predominance of Lewy body pathology. Evidence linking Lewy body pathology to visual hallucinations as well as a greater cortical cholinergic deficit helps to explain the finding that PDD subjects with visual hallucinations showed greater benefit from rivastigmine compared to those without visual hallucinations (Williams and Lees 2005; Harding et al. 2002; Burn et al. 2006).
Consistent with prior studies that demonstrated greater PD risk and earlier PD onset with severe GBA mutations (Gan-Or et al. 2009; Gan-Or et al. 2008; Barrett et al. 2013), among the GBA-PD group, 86% of those with severe mutations reported hallucinations during clinical follow-up compared to 41% of those with mild mutations, suggesting a more malignant phenotype. Comparatively, a pathological series found that the lifetime prevalence of hallucinations in PD was 50% (Williams and Lees 2005).
While this study is retrospective and exploratory, it has the advantages of including a large number of GBA-PD subjects and extended follow-up time. One limitation is that by targeted sequencing of common Ashkenazi Jewish GBA mutations, it is possible, albeit unlikely, that there was misclassification of IPD subjects. It is also possible that genetic mutations linked to PD, besides those in LRRK2 and GBA, were present in the GBA-PD or IPD groups. As this study relied on retrospective clinical information, we did not capture short medication trials, especially those outside routine clinic visits, nor did we formally measure medication compliance. Therefore, we cannot assess whether GBA-PD subjects were less likely to stop cholinesterase inhibitors once they were started. Additionally, as Part I of the UPDRS is determined with input from the patient and caregiver, and the source of information was not systematically recorded, reporting bias may have affected the mentation score. Lastly, clinician practice was not standardized and may have evolved over time; however, this would not be expected to differentially affect our comparison groups as they were both sampled from the same time period. To address the need for standardization in the assessment of neuropsychiatric symptoms, a prospective study evaluating cognitive outcomes and medication effects, including detailed pre- and post-medication assessment of the quality and frequency of psychotic symptoms, cognition with neuropsychological testing, and adverse events, is warranted. Further studies will also be necessary to determine if cholinesterase inhibitor use is increased in GBA-PD secondary to increased dementia, better response to treatment, or a combination of both.
Acknowledgments
The research presented in this manuscript was supported by the Empire State Clinical Research Training Program, the Marcled Foundation, and NIH-NINDS NS073836.
Sources of Support
Empire State Clinical Research Training Program, the Marcled Foundation, and NIH-NINDS NS073836.
Synopsis Sentence
Monoallelic GBA mutation carriers with PD have the same rate of antipsychotic medication use as PD patients without GBA mutations and are more likely to use cholinesterase inhibitors.
Compliance with Ethics Guidelines
Conflict of Interest
Neither Matthew Barrett, Vicki Shanker, Lawrence Severt, Debbie Raymond, Susan Gross, Nicole Schreiber-Agus, Ruth Kornreich, Laurie Ozelius, Susan Bressman, nor Rachel Saunders-Pullman have any disclosures, financial or otherwise, that would represent a conflict of interest with the research presented in this manuscript.
Other Disclosures
Dr. Barrett reports no disclosures. Dr. Shanker reports no disclosures.
Dr. Severt served as a speaker and/or on advisory boards for Teva, Allergan, Merz, Impax, and UCB. Ms. Raymond reports no disclosures. Dr. Gross serves as PI on a research grant sponsored by PerkinElmer Inc. for development of new genetic platforms and as co-PI on an NIH research grant for preterm labour (primary PI Dr. S. Gennaro at Boston College). She received no personal compensation of any kind from either grant. All research funds are to the hospital system. Dr. Schreiber-Agus served as an investigator on a research grant sponsored by PerkinElmer Inc. for development of new genetic platforms. She received no personal compensation of any kind. All research funds were to the hospital system. Dr. Kornreich reports no disclosures. Dr. Ozelius receives salary support from NIH [NS058949, NS037409, NS075881, DC011805] and has received grant support from the Bachmann-Strauss Dystonia and Parkinson Foundation, the Benign Essential Blepharospasm Research Foundation, and The Dystonia Medical Research Foundation. She is a current member of the scientific advisory boards of the National Spasmodic Dysphonia Association, the Benign Essential Blepharospasm Research Foundation, and Tourette Syndrome Association, Inc. Dr. Ozelius receives royalty payments from Athena Diagnostics related to patents. Dr. Bressman serves on the advisory boards of the Michael J. Fox Foundation, the Dystonia Medical Research Foundation, the Bachmann Strauss Dystonia and Parkinson’s Foundation, and the Board of We Move. She has consulted for Bristol Meyer Squibb. She has received research support from the Michael J. Fox Foundation, National Institutes of Health (NIH), and Dystonia Medical Research Foundation. Dr. Bressman received royalty payments from Athena Diagnostics related to patents. Dr. Saunders-Pullman serves on the Scientific Advisory Board of the Dystonia Medical Research Foundation. She receives research support from the NIH (K02 NS073836), the Michael J Fox Foundation for Parkinson’s Research, the Bachmann-Strauss Dystonia and Parkinson’s Foundation, the Marcled Foundation, and the Empire State Clinical Research Training Program.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Animal Rights
No animals were involved in the conduct of this study.
Individual Author Contributions
Study conception and design: Dr. Barrett, Dr. Saunders-Pullman; Conduct of research: Dr. Barrett, Dr. Shanker, Dr. Severt, Ms. Raymond, Dr. Gross, Dr. Schreiber-Agus, Dr. Kornreich, Dr. Ozelius, Dr. Bressman, Dr. Saunders-Pullman. Analysis and interpretation of data: Dr. Barrett, Dr. Bressman, Dr. Saunders-Pullman; Drafting the manuscript or revising it critically for important intellectual content: Dr. Barrett, Dr. Shanker, Dr. Severt, Ms. Raymond, Dr. Gross, Dr. Schreiber-Agus, Dr. Kornreich, Dr. Ozelius, Dr. Bressman, Dr. Saunders-Pullman.
Guarantor
Dr. Barrett
Contributor Information
M. J. Barrett, Email: mjbarrett@virginia.edu
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology. 2012;78:1434–1440. doi: 10.1212/WNL.0b013e318253d54b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N, Borlak J. Drug-induced phospholipidosis. FEBS Lett. 2006;580:5533–5540. doi: 10.1016/j.febslet.2006.08.061. [DOI] [PubMed] [Google Scholar]
- Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78:417–420. doi: 10.1212/WNL.0b013e318245f476. [DOI] [PubMed] [Google Scholar]
- Barrett MJ, Giraldo P, Capablo JL, et al. Greater risk of parkinsonism associated with non-N370S GBA1 mutations. J Inherit Metab Dis. 2013;36:575–580. doi: 10.1007/s10545-012-9527-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cell Mol Dis. 2005;35:355–364. doi: 10.1016/j.bcmd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Brockmann K, Srulijes K, Hauser AK, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77:276–280. doi: 10.1212/WNL.0b013e318225ab77. [DOI] [PubMed] [Google Scholar]
- Bultron G, Kacena K, Pearson D, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33:167–173. doi: 10.1007/s10545-010-9055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord. 2006;21:1899–1907. doi: 10.1002/mds.21077. [DOI] [PubMed] [Google Scholar]
- Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol. 2009;66:578–583. doi: 10.1001/archneurol.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple-Alford JC, MacAskill MR, Nakas CT, et al. The MoCA: Well-suited screen for cognitive impairment in Parkinson disease. Neurology. 2010;75:1717–1725. doi: 10.1212/WNL.0b013e3181fc29c9. [DOI] [PubMed] [Google Scholar]
- Fenelon G, Mahieux F, Huon R, Ziegler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain. 2000;123:733–745. doi: 10.1093/brain/123.4.733. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Giladi N, Rozovski U, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70:2277–2283. doi: 10.1212/01.wnl.0000304039.11891.29. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Giladi N, Orr-Urtreger A. Differential phenotype in Parkinson’s disease patients with severe versus mild GBA mutations. Brain. 2009;132:e125. doi: 10.1093/brain/awp161. [DOI] [PubMed] [Google Scholar]
- Goker-Alpan O, Giasson BI, Eblan MJ, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67:908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Broe GA, Halliday GM. Visual hallucinations in Lewy body disease relate to Lewy bodies in the temporal lobe. Brain. 2002;125:391–403. doi: 10.1093/brain/awf033. [DOI] [PubMed] [Google Scholar]
- Kumar KR, Lohmann K, Klein C. Genetics of Parkinson disease and other movement disorders. Curr Opin Neurol. 2012;25:466–474. doi: 10.1097/WCO.0b013e3283547627. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013;70:727–735. doi: 10.1001/jamaneurol.2013.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132:1783–1794. doi: 10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Nichols WC, Uniacke SK, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet. 2002;71:124–135. doi: 10.1086/341282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord. 2012;27:831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders-Pullman R, Hagenah J, Dhawan V, et al. Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov Disord. 2010;25:1364–1372. doi: 10.1002/mds.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto-Salvia N, Pagonabarraga J, Houlden H, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord. 2012;27:393–399. doi: 10.1002/mds.24045. [DOI] [PubMed] [Google Scholar]
- Shapiro BE, Hatters-Friedman S, Fernandes-Filho JA, Anthony K, Natowicz MR. Late-onset Tay-Sachs disease: adverse effects of medications and implications for treatment. Neurology. 2006;67:875–877. doi: 10.1212/01.wnl.0000233847.72349.b6. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiraboschi P, Hansen LA, Alford M, et al. Cholinergic dysfunction in diseases with Lewy bodies. Neurology. 2000;54:407–411. doi: 10.1212/WNL.54.2.407. [DOI] [PubMed] [Google Scholar]
- Tsuang D, Leverenz JB, Lopez OL, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012;79:1944–1950. doi: 10.1212/WNL.0b013e3182735e9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. 2011;17:485–493. doi: 10.1016/j.molmed.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: a retrospective autopsy study. Lancet Neurol. 2005;4:605–610. doi: 10.1016/S1474-4422(05)70146-0. [DOI] [PubMed] [Google Scholar]
- Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]