

Abstract
MET, the receptor of hepatocyte growth factor, plays important roles in tumorigenesis and drug resistance in numerous cancers including non-small cell lung cancer. As increasing numbers of MET inhibitors are being developed for clinical applications, antibody fragment based immuno-positron emission tomography (immunoPET) has the potential to rapidly quantify in vivo MET expression levels for drug response evaluation and patient stratification for these targeted therapies. Here, fully human single-chain variable fragments (scFvs) isolated from a phage display library were re-formatted into bivalent cys-diabodies (scFv-cys dimers) with affinities to MET ranging from 0.7 nM to 5.1 nM. The candidate with the highest affinity, H2, was radiolabeled with 89Zr for immunoPET studies targeting non-small cell lung cancer xenografts: low MET expressing Hcc827 and the gefitinib-resistant Hcc827-GR6 with 4-fold MET over-expression. ImmunoPET at as early as 4 hours post injection produced high contrast images, and ex vivo biodistribution analysis at 20 hours post injection showed about 2-fold difference in tracer uptake levels between the parental and resistant tumors (p < 0.01). Further immunoPET studies using a larger fragment, the H2 minibody (scFv-CH3 dimer) produced similar results at later time points. Two of the antibody clones (H2 and H5) showed in vitro growth inhibitory effects on MET-dependent gefitinib-resistant cell lines, while no effects were observed on resistant lines lacking MET activation. In conclusion, these fully human antibody fragments inhibit MET-dependent cancer cells and enable rapid immunoPET imaging to assess MET expression levels, showing potential for both therapeutic and diagnostic applications.
Introduction
Since its discovery in the mid-1980s, MET, the receptor of hepatocyte growth factor (HGF), has been found to be very important in embryonic development, cell migration, cell growth, cell survival, epithelial-mesenchymal transition, wound healing and tumorigenesis (1-3). Activation of MET has been found in various cancers, including bladder, breast, cervical, colorectal, gastric, kidney, liver, lung, ovarian and prostate (1). MET amplification has also been found to be an important mechanism for acquired resistance to anti-EGFR therapies in non-small cell lung cancer (4, 5). Because of the important roles of HGF-MET signaling in various cancers, many inhibitors targeting this pathway are currently being developed for clinical applications, including both small-molecule inhibitors and monoclonal antibodies (3). A humanized one-armed anti-MET antibody, onartuzumab (MetMAb), has been evaluated in clinical trials for advanced non-small cell lung cancer in combination with erlotinib. While patients with MET positive tumor benefited from such combination treatment, the MET negative patients actually had worse overall survival when treated with onartuzumab plus erlotinib, compared to with erlotinib plus placebo (6). Such results emphasize the importance to evaluate MET expression level for patient stratification to improve these anti-MET therapies.
Compared to traditional biopsy and immunohistochemistry, antibody based positron emission tomography, or immunoPET, offers a unique opportunity for non-invasive evaluation of the in vivo expression levels of various biomarkers. The whole body information provided by immunoPET scans can help illuminate the heterogeneity of the primary tumor and metastatic lesions, and the evolving molecular status of tumors can be easily monitored via serial immunoPET scans to aid treatment planning and follow-up (7). Previously, anti-MET immunoPET imaging has been successfully demonstrated in preclinical mouse models using the intact monoclonal mouse antibody DN-30 or the humanized one-armed antibody onartuzumab (8, 9). However, these antibodies with full Fc domains require relatively long imaging delays (3 days to 1 week) to clear from the circulation in order to produce high contrast images.
By using smaller antibody fragments with shorter serum half lives, such as diabodies and minibodies (described in greater detail below), immunoPET can be performed at earlier time points with similar or even higher contrast, highly desired for clinical imaging applications (7, 10-12). Compared to an intact antibody (150 kDa) with heavy and light chain variable and constant domains, a single-chain variable fragment (scFv; ∼27 kDa) is a small monovalent fragment consisting of the antibody VH and VL domains linked by a flexible linker. A diabody is related to an scFv, comprised of only the VH and VL domains, but with a shorter linker that induces dimerization, resulting in a bivalent fragment (∼55 kDa). The bivalent minibody fragment is formed by fusion of the scFv to the immunoglobulin CH3 constant domain for dimerization. Their higher molecular weight (∼80 kDa) promotes longer serum persistence, facilitating higher uptake levels in target tissues. Cys-diabodies are modified diabodies with engineered cysteines at their C-termini to allow site-specific conjugation and labeling (13-18). Figure 1 shows sizes and structures of these antibody fragments in comparison with an intact antibody. The availability of engineered antibody fragments allows selection of the optimal format for an imaging probe based on the target and application.
Figure 1.
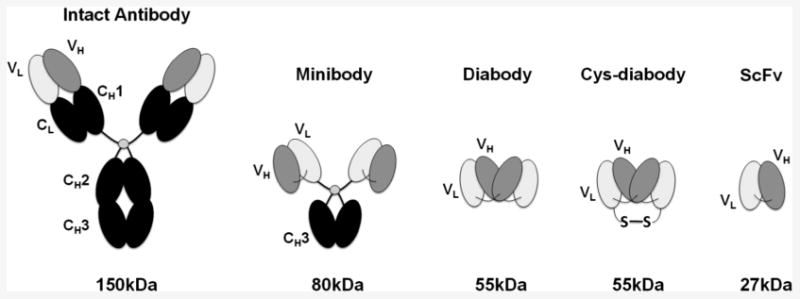
Schematic showing sizes and structures of the intact antibody and different antibody fragments.
For clinical use, fully human antibodies and their respective fragments are preferred due to the lower risk of immunogenicity, and phage display technology offers a reliable source of high affinity fully human antibodies (19-21). With FDA approved Humira (adalimumab) and Benlysta (belimumab), and dozens more in clinical trials (22), phage display technology is now one of the major sources of fully human monoclonal antibodies for therapeutic applications. Furthermore, because the human antibody gene sequences are already in hand, reformatting selected scFvs into diabodies or minibodies is very fast and simple. This approach has been employed to generate several previous antibody fragment imaging probes from phage libraries (16, 23, 24). As immunoPET is playing an increasingly important role in diagnosis, and in the development and implementation of targeted therapies (25, 26), phage display can be an effective path for the rapid generation of antibody fragment-based imaging probes. Instead of the months required to generate and screen conventional hybridomas, isolate the antibody genes, humanize, and conduct affinity maturation, by using phage display technology, fully human antibodies can be identified and reformatted in just a few weeks. This can lead to significantly faster discovery and development of novel antibodies and fragments for various applications.
In this study, we utilized a previously reported fully human scFv phage display library (27) to screen for novel human antibodies recognizing human MET. One of the antibody clones, H2, was reformatted into a cys-diabody and a minibody for MET-specific immunoPET studies in mouse models to evaluate the potential of assessing MET expression levels in vivo. In parallel, the potential biological activity of the human anti-MET antibodies was assessed, since avoidance of agonistic biological activity is desirable in an imaging agent.
Materials and Methods
Details of antibody reformatting, expression and purification, radiolabeling of antibody fragments and the immunoreactivity assay can be found in the supplementary methods.
Cell lines and tumor models
Hcc827 parental cells and the gefitinib resistant Hcc827-GR6 cells (4) were obtained from Dr. Jeffrey A. Engelman's laboratory (Massachusetts General Hospital) in 2012. Hcc827-/+ cells were derived from Hcc827 cells by 15 days of treatment with 50 ng/mL HGF and 1 μM erlotinib (LC Laboratories) (replenished every 3 days). H1975 and H1650 cells were obtained from ATCC (American Type Culture Collection) in 2011. All these cell lines were maintained with RPMI 1640 media (Cellgro) supplemented with 10% FBS (fetal bovine serum, Gemini), 1% sodium pyruvate (Cellgro) and 1% penicillin-streptomycin (Gibco). The Hcc827-GR6 media was also supplemented with 100 nM gefitinib (Santa Cruz biotechnology), and Hcc827-/+ cells were maintained with 1 μM erlotinib. C6 rat glioma cells (from the laboratory of Sanjiv Gambhir in 2002) were maintained in Deficient DME High Glucose media (Irvine Scientific) supplemented with 10% FBS, 1% penicillin-streptomycin and 2 mM L-glutamine. The cell lines were not authenticated by our laboratory. Female SCID mice (Jackson Laboratories) were injected subcutaneously into the left or right shoulders with cells (2 – 4 × 106) in growth media: 50% Matrigel (BD Biosciences). The tumors were allowed to develop for 2 – 4 weeks before imaging. All animal studies were carried out under a protocol approved by the Chancellor's Animal Research Committee of the University of California in Los Angeles.
Phage library selection
A naïve human scFv phage display library from James D. Marks' laboratory (UCSF) (27) was used to perform three rounds of selection against recombinant human MET protein using previously published method (28). Briefly, recombinant MET protein was used to coat an immunotube (Nunc). After washing and blocking, the immunotube was then incubated with 1012 ∼1013 phage in 2 mL 1% milk-PBS for 2 hours at room temperature. The unbound phage were washed off with PBS and 0.05% PBS-Tween, and the bound phage were eluted with 1 mL of 100 mM triethylamine for 8 minutes and neutralized with 0.5 mL of 1 M Tris-HCl (pH = 7.5). The eluted phage were used to infect exponentially growing E. coli TG1, and the infected bacteria were then used to produce phage for the next round of selection. 100 μg of MET protein from R&D Systems (358-MT/CF) was used for the first round. Reduced amounts of MET protein from another supplier (Sino Biological Inc., 10692-H08H) were used for the following 2 rounds.
ELISA
Phage ELISA was performed as previously described (28) to screen for binding clones. Briefly, antibody-displaying phage were expressed from 93 clones from the 3rd round of selection to detect human MET protein immobilized on a 96-well. HRP conjugated anti-M13 antibody (GE Healthcare, Cat#: 27-9421-01) was used to detect the bound phage. 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, Sigma, A9941) was used as the HRP substrate. The 64 clones with the highest positive signals (A405) were subjected to DNA sequencing.
To determine cross reactivity to mouse MET protein, purified anti-MET cys-diabodies were used to coat a ELISA plate (anti-EpCAM cys-diabody or 2% milk-PBS were used for negative controls). 0.1 μg human or mouse MET-hIgG Fc fusion protein (Sino Biological Inc.) was then applied to each well. The captured human or mouse MET-hIgG Fc were then detected with alkaline phosphatase conjugated goat anti-human IgG. Each combination was done in triplicate.
Flow cytometry
For the standard anti-MET flow cytometry, murine anti-human MET antibody (Cell Signaling Technology, #5631) was used for primary incubation and FITC or PE conjugated goat anti-mouse IgG was used for secondary incubation. To determine the copy number of MET protein per cell, quantitative flow was performed using QIFIKIT (Dako, K0078) according to the recommended protocol.
To estimate affinities, Hcc827-GR6 cells were incubated with various concentrations of anti-MET cys-diabodies at 4°C for 2 hours, then cells were quickly washed with flow buffer and incubated on ice with excess mouse anti-MYC IgG for 20 minutes, and washed again before a final incubation on ice with excess PE-conjugated goat anti-mouse IgG. Mean fluorescence intensities (MFI) at different concentrations were calculated using FlowJo software, and the binding curves were analyzed using Graphpad Prism to calculate dissociation constants using the “one site-specific binding” model.
For phage flow cytometry, 30X concentrated phage were incubated with cells in flow buffer (PBS / 1% FBS / 2 mM EDTA) with 4% milk on ice for 1 hour. After washing 3 times with flow buffer, FITC conjugated anti-M13 antibody (Santa Cruz Biotech, sc-53005 FITC) was added to detect the phage.
In vitro MTS assay
Cell growth and inhibition were evaluated by MTS assay using the CellTiter96 AQueous Assay Kit from Promega (Cat# G5421), according to the recommended protocol. 1 × 104 cells were seeded in each well of the 96-well plates and treated with different combination of antibodies and drugs for 3 days before the MTS assay. Each combination of cell line and drug and antibody treatment was evaluated in at least triplicate wells. The results were normalized to the cells treated with media only.
Binding and internalization assay of H2 cys-diabody
MET positive Hcc827-GR6 cells were seeded into a 24-well plate and cultured until confluent. The media was removed and the cells were incubated with ice cold PBS / 1% FBS with excess 89Zr labeled H2 cys-diabody for 1 hour to saturate cell surface MET. Following washing with ice cold PBS-1% FBS three times, the cells were incubated at 37°C in growth media with excess 89Zr labeled cys-diabody. At each time point (0 h, 1 h, 2 h, 4 h, and 24 h), cells were washed with ice cold PBS and the membrane bound cys-diabody was stripped with two 3-min treatment of 1 mL cold stripping buffer (0.1 M glycine, 0.15 M NaCl, pH = 2.5). The 2 mL stripping buffer was combined and considered as the membrane bound fraction. The cells were then lysed with 1 mL lysis buffer (PBS / 10 mM Tris / 0.5% SDS, pH = 7.4) at room temperature for 3 min. Each well was washed one more time with 1 mL lysis buffer and the combined lysis buffer was considered as internalized fraction. The radioactivity of each fraction was measured by gamma counting. The assay was performed in triplicate for each time point.
Small animal PET imaging and ex vivo biodistribution studies
Tumor bearing mice (tumor weight: 58∼467 mg) were injected via tail vein with approximately 13 – 25 μg of 89Zr labeled cys-diabody or minibody in PBS (specific activity: 0.08 - 0.2 MBq/μg). 10-minute microPET acquisitions were performed at 4 and 20 hours post injection for cys-diabody, and 24 and 44 hours post injection for minibody, with a Inveon microPET Scanner (Siemens). The mice were anesthetized with 2% isoflurane during the scanning process. MicroPET images were reconstructed using a filtered backprojection algorithm, and analyzed with AMIDE software. Ex vivo biodistribution studies were performed after the final imaging time point. Organs, tumors and blood were harvested and weighed, and the radioactivities were measured by gamma counting. The percent injected dose per gram of tissue (%ID/g) was calculated after decay correction to evaluate the uptake level in each organ. About half of the Hcc827-GR6 tumors were cystic, and the biodistribution of these tumors was calculated excluding the cystic fluid. The radioactivities of some kidney samples were too high to be accurately measured by gamma counting, so the uptake levels were estimated by image quantitation using previously described method (29).
Pharmacokinetic studies
89Zr labeled H2 cys-diabody (22 μg, 1.15 MBq per mouse) or minibody (29 μg, 1.20 MBq per mouse) was injected into female SCID mice via tail vein. 5 μL of blood samples were taken at different time points (0.5, 1, 2, 4, 6, 12, 24 hours post injection for the cys-diabody and 1, 2, 4, 6, 12, 24, 48, 72 hours post injection for the minibodyand the radioactivities were measured by a gamma counter. The blood curves were analyzed with Graphpad Prism, and fitted with the “one phase decay” model to calculate the terminal half lives.
Statistical analysis
Statistical analysis was performed using Prism (Graphpad.com). Student's t-tests were employed to compare the in vitro MTS assay results, and the tracer uptake levels. Data are presented as mean ± SD.
Results
Anti-MET phage selection and screening
Following 3 rounds of selection on immobilized recombinant MET protein, 93 clones were picked and ranked based on binding to MET protein via phage ELISA. DNA sequencing results of the top 64 clones reveal 19 distinct functional sequences. Figure 2A shows the ELISA results of the 19 representative clones, each demonstrating at least 2-fold increase above background signal. To further confirm binding to the native cell surface antigen, all 19 phage clones were incubated with MET positive Hcc827-GR6 cells and analyzed by flow cytometry (determined by quantitative flow, Hcc827-GR6 cell line has 2.6 × 105 cell surface MET per cell while the parental Hcc827 cell line has 6.7 × 104 per cell). Eight of the clones (A12, C2, E9, F1, F11, G1, H2 and H5) showed good binding profiles in flow analysis. Figure 2B shows the flow results of 2 representative clones. The sequences of these eight clones were analyzed using the abYsis online tool (http://www.bioinf.org.uk/abysis/) to identify the germline genes (based on IMGT definition and classification) and the CDRs. The VH germline genes showed an over-representation of the IGHV3 subgroup, which is consistent with the original Sheets library (27). The eight clones use very different VH CDR3 sequences, with lengths ranging from 6 to 19 residues (Figure 2C).
Figure 2.
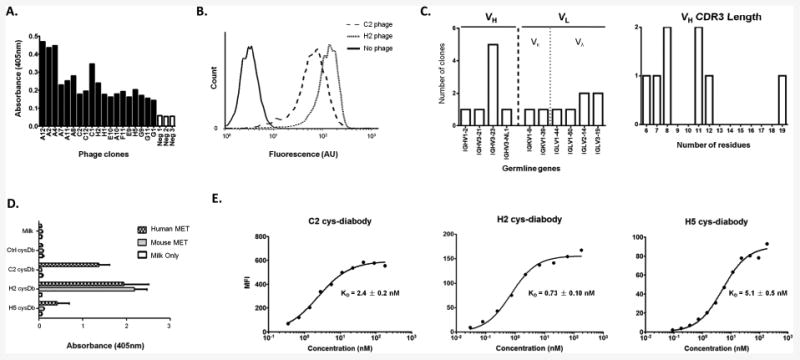
Characterization of anti-MET antibodies. A. Binding of phage clones and controls (helper phage, Neg 1-3) to immobilized antigen (MET) in ELISA. Bound phage were detected with HRP conjugated anti-M13 antibody. B. Binding of two representative phage clones (C2 and H2) to MET-expressing cells by flow cytometry. C. Germline gene usage and the VH CDR3 lengths of the eight clones which bound well to MET positive cells. D. Binding of C2, H2 and H5 cys-diabodies (cysDb) to immobilized human and mouse MET in ELISA. Only the H2 cys-diabody showed cross-reactivity, while C2 and H5 bind to human MET exclusively. Non-specific cysDb (ctrl cysDb) and secondary Ab only were included as controls. E. The affinities of C2, H2 and H5 cys-diabodies were estimated by flow cytometry using MET expressing Hcc827-GR6 cells. The binding curves were fitted in Graphpad Prism to calculate the dissociation constants (KD) using the “one site-specific binding” model.
These clones were reformatted into cys-diabodies by shortening the 15-aa linker to 6-aa linker and incorporation of C-terminal tags (see Materials and Methods). Three of the cys-diabody clones, C2, H2 and H5, were successfully expressed, purified (yields ranging from 1 to 3 mg/L, stable at 4 °C for months) and characterized further. Other clones were dropped because of low expression level and purity.
In vitro characterization of cys-diabodies
The purified C2, H2 and H5 cys-diabodies retained the ability to bind to MET protein. Cross reactivity analysis showed that the H2 cys-diabody clearly bound to both human and mouse MET, while C2 and H5 have no significant cross reactivity to the mouse MET (Figure 2D). Binding curves on Hcc827-GR6 showed H2 cys-diabody has the highest binding affinity (KD = 0.73 ± 0.10 nM), while the other two cys-diabodies also have low nanomolar affinities, as shown in Figure 2E.
Cell growth inhibition assays
To assess off-target toxicity, first, the parental Hcc827 cells were treated with cys-diabodies alone for 3 days in culture media. No significant effects were observed overall, but the H5 cys-diabody appeared to have a small activating effect at very high concentration (180 nM) (Figure 3A), which might also be due to experimental variation. Next, the cys-diabodies were used to treat two gefitinib-resistant cell lines lacking MET amplification, H1650 and H1975, in combination with gefitinib. No significant inhibitory effects were observed on these two cell lines (Figure 3B). For the H1975 cells, although the C2 cysDb treated at 18 nM appeared statistically different compared to the no antibody group, differences at either higher or lower concentrations were not significant, and the mean cell activities were all very similar to the no antibody group. C2 showed a slight activating effect on the H1650 cell line as the cys-diabody concentration was increased..
Figure 3.
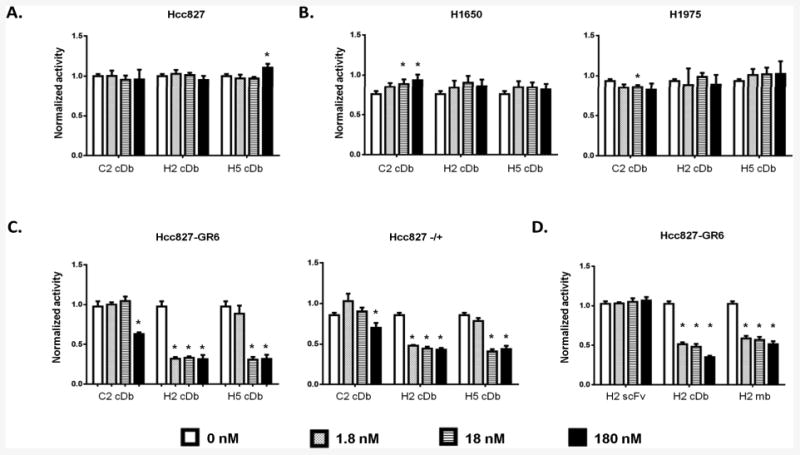
Anti-MET antibody fragments inhibit cell growth of MET dependent cell lines. Different cell lines were treated with combinations of anti-MET antibody fragments (1.8 – 180 nM) and gefitinib (1 μM) for 3 days as indicated. Cell viability was assessed by MTS assay and normalized to untreated cells. A. Hcc827 parental cell line treated with cys-diabodies. B. Two gefitinib-resistant cell lines without MET amplification (H1650 and H1975) treated with the cys-diabodies in combination with gefitinib. C. Two cell lines with amplified MET signal, Hcc827-GR6 and Hcc827-/+ cells treated with cys-diabodies and gefitinib. D. Hcc827-GR6 treated with either H2 cys-diabody, minibody or scFv in combination with gefitinib. (*: p<0.01; cDb: cys-diabody; mb: minibody.)
The anti-MET inhibitory effects were tested by applying these cys-diabodies to two cell lines with amplified MET signaling, Hcc827-GR6 and Hcc827-/+. The Hcc827-GR6 cell line was generated through prolonged selection from the parental Hcc827 treated with gefitinib (4). The Hcc827-/+ cells were obtained by short-term treatment of Hcc827 with HGF and erlotinib (see Materials and Methods). Figure 3C shows the H2 cys-diabody has the strongest inhibitory effect at lowest concentration, while C2 and H5 only showed inhibitory effects at higher concentrations. Because of the high affinity and inhibitory effect of H2 cys-diabody, we further reformatted H2 into a minibody and expressed the H2 minibody in 293F cells. The protein A purified H2 minibody was then tested for inhibitory effects on Hcc827-GR6 in comparison with the cys-diabody and scFv. Similar inhibitory effects were observed for the minibody as shown in Figure 3D.
Internalization and uptake of 89Zr labeled H2 cys-diabody
Following site-specific conjugation of H2 cys-diabody with maleimide-DFO and radiolabeling with 89Zr (Figure 4A), the labeling efficiency was 49%, the radiochemical purity was 95% after column purification, and the specific activity was 0.13 MBq/μg. When MET expressing Hcc827-GR6 cells were cultured with excess radiolabeled H2 cys-diabody, the membrane bound radioactivity decreased in the first 6 hours while the internalized activity increased, giving a constant total cell radioactivity in the first few hours. The membrane radioactivity remained low and the internalized 89Zr activity continued to accumulate over the 24 hour time course (Figure 4B). Such results indicate rapid internalization of the radiolabeled H2 cys-diabody, making it necessary to use residualizing isotope to achieve better imaging results.
Figure 4.
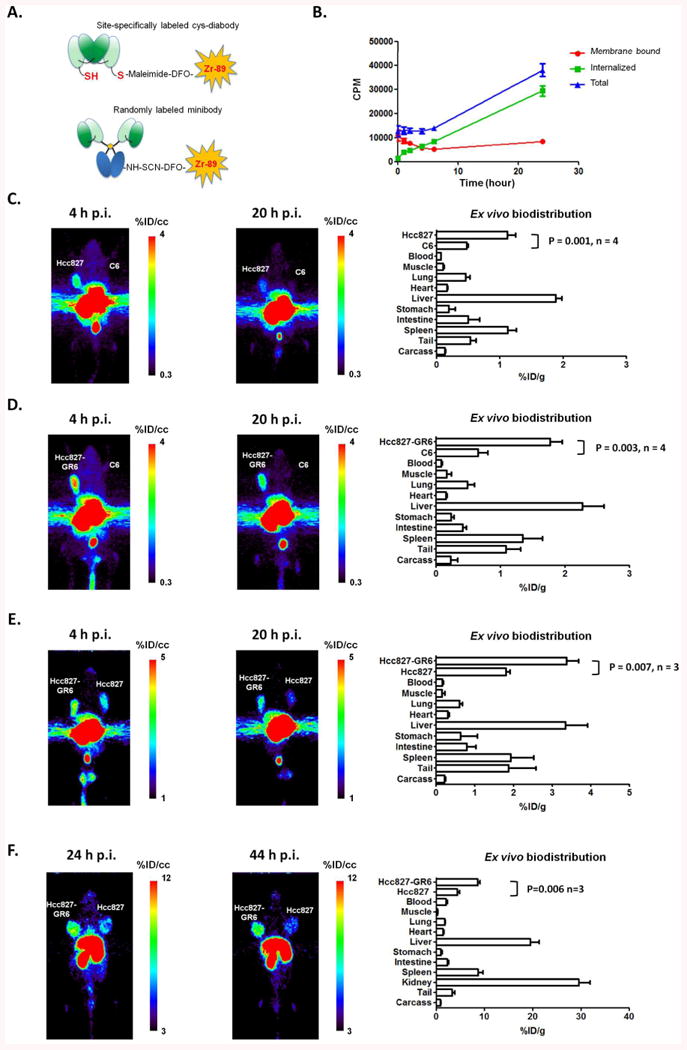
ImmunoPET using 89Zr labeled antibody fragments. A. H2 cys-diabody was site-specifically conjugated with maleimide-DFO, and the minibody was randomly conjugated with SCN-DFO. The fragments were then radiolabeled with 89Zr. B. Binding and internalization of 89Zr-DFO-H2 cys-diabody by Hcc827-GR6 cells. C. 89Zr-DFO-H2 cys-diabody imaging of MET expressing Hcc827 tumor in a SCID mouse with Hcc827 tumor (212mg) on left shoulder and C6 tumor (101mg) on right shoulder. The ex vivo biodistribution analysis was performed at 20 hours post injection (n = 4). D. 89Zr-DFO-H2 cys-diabody imaging of MET expressing Hcc827-GR6 tumor in a SCID mouse bearing Hcc827-GR6 tumor (262mg) and C6 tumor (59mg). The ex vivo biodistribution analysis was performed at 20 hours post injection (n = 4). E. 89Zr-DFO-H2 cys-diabody imaging of MET expressing Hcc827-GR6 tumor (202mg) and Hcc827 tumor (295mg) in a SCID mouse. The ex vivo biodistribution analysis was performed at 20 hours post injection (n = 3). F. 89Zr-DFO-H2 minibody imaging of a SCID mouse with Hcc827-GR6 tumor (270mg) and Hcc827 parental tumor (306mg). The ex vivo biodistribution analysis were performed at 44 hours post injection (n = 3).
In vivo PET imaging and ex vivo biodistribution
89Zr labeled H2 cys-diabody (89Zr-DFO-H2 cys-diabody) was initially used for immunoPET studies in mice bearing MET positive tumors (Hcc827 or Hcc827-GR6) and MET negative C6 tumors. 89Zr-DFO-H2 cys-diabody showed specific uptake in antigen positive tumors (Hcc827, Hcc827-GR6) and high contrast images were obtained at as early as 4 hours post injection. As shown in Figure 4C and 4D, tracer uptake in MET positive tumors was higher than the MET negative C6 tumors in the PET scans at both 4 hours and 20 hours post injection. The ex vivo biodistribution data confirmed these observations. In the Hcc827 vs. C6 group, the MET positive Hcc827 tumors had uptake levels of 1.1 ± 0.1 %ID/g while the negative C6 tumors showed uptake levels of 0.47 ± 0.02 %ID/g (positive tumor : negative tumor = 2.4 ± 0.2; positive tumor : blood = 18 ± 2; positive tumor : muscle = 12 ± 4). In the Hcc827-GR6 vs. C6 group, the Hcc827-GR6 tumors had higher uptake levels of 1.8 ± 0.2 %ID/g, and the negative C6 tumors had uptake levels of 0.65 ± 0.15 %ID/g (positive tumor : negative tumor = 2.9 ± 0.8; positive tumor : blood = 23 ± 5; positive tumor : muscle = 13 ± 7). For better comparison of Hcc827-GR6 and Hcc827 tumors, the high-MET gefitinib resistant tumor and the low-MET parental tumor were imaged in the same mouse as shown in Figure 4E. The images show clear differences between high-MET Hcc827-GR6 tumor and the low-MET Hcc827 parental tumor, confirmed by the ex vivo biodistribution analysis. The high-MET Hcc827-GR6 tumors had uptake levels of 3.4 ± 0.3 %ID/g (Hcc827-GR6 tumor : Hcc827 tumor = 1.9 ± 0.1; Hcc827-GR6 tumor : blood = 22 ± 2; Hcc827-GR6 tumor : muscle = 27 ± 14). Table 1 summarizes the ex vivo biodistribution study of the H2 cys-diabody imaging. The kidney uptakes were not shown in the biodistribution results because the signals were too high to be accurately measured by the gamma counter. However, we estimated the uptake levels to be about 83 – 104 %ID/g (Hcc827 vs. C6 group: 85 ± 1 %ID/g; Hcc827-GR6 vs. C6 group: 99 ± 2 %ID/g; Hcc827-GR6 vs. Hcc827 group: 98 ± 9 %ID/g) by image quantitation using a previously described method (29). The high kidney uptakes are due to the renal clearance of these low molecular weight cys-diabody proteins.
Table 1.
Ex vivo biodistribution of 89Zr-DFO-maleimide-H2 cysDb at 20 hours post injection in female SCID mice bearing MET-positive (Hcc827, Hcc827-GR6) and MET-negative (C6) tumors.
| Hcc827 vs. C6 (n = 4) |
Hcc827-GR6 vs. C6 (n = 4) |
Hcc827-GR6 vs. Hcc827 (n = 3) |
|
|---|---|---|---|
| Immuno-reactivity | 67% | 71% | 78% |
| Injected dose (μg, MBq) | 13 μg, 2.7 MBq | 19 μg, 3.4 MBq | 22 μg, 1.7 MBq |
|
| |||
| Tumor Size (mean ± SD) | |||
|
| |||
| Hcc827 tumor | 183 ± 43 mg | N.A. | 236 ± 182 mg |
|
| |||
| Hcc827-GR6 tumor | N.A. | 221 ± 58 mg | 177 ± 62 mg |
|
| |||
| C6 tumor | 102 ± 45 mg | 128 ± 52 mg | N.A. |
|
| |||
| %ID/g (mean ± SD) | |||
|
| |||
| Hcc827 tumor | 1.1 ± 0.1 | N.A. | 1.8 ± 0.1 |
| Hcc827-GR6 tumor | N.A. | 1.8 ± 0.2 | 3.4 ± 0.3 |
| C6 tumor | 0.47 ± 0.02 | 0.65 ± 0.15 | N.A. |
| Blood | 0.064 ± 0.002 | 0.080 ± 0.012 | 0.16 ± 0.02 |
| Muscle | 0.10 ± 0.02 | 0.16 ± 0.07 | 0.15 ± 0.07 |
| Lung | 0.46 ± 0.07 | 0.48 ± 0.11 | 0.60 ± 0.07 |
| Heart | 0.16 ± 0.01 | 0.15 ± 0.02 | 0.29 ± 0.04 |
| Liver | 1.9 ± 0.1 | 2.3 ± 0.3 | 3.3 ± 0.6 |
| Stomach | 0.19 ± 0.11 | 0.23 ± 0.05 | 0.63 ± 0.43 |
| Intestine | 0.50 ± 0.18 | 0.41 ± 0.06 | 0.79 ± 0.23 |
| Spleen | 1.1 ± 0.1 | 1.3 ± 0.3 | 1.9 ± 0.6 |
| Tail | 0.53 ± 0.09 | 1.1 ± 0.2 | 1.9 ± 0.7 |
| Carcass | 0.13 ± 0.01 | 0.22 ± 0.11 | 0.21 ± 0.03 |
|
| |||
| Ratio (mean ± SD) | |||
|
| |||
| High-MET tumor / Blood | 18 ± 2 | 23 ± 5 | 22 ± 2 |
| High-MET tumor / Muscle | 12 ± 4 | 13 ± 7 | 27 ± 14 |
| High-MET Tu / Low-MET Tu | 2.4 ± 0.2 | 2.9 ± 0.8 | 1.9 ± 0.1 |
To further confirm the specificity of anti-MET targeting, H2 minibody was also evaluated in immunoPET studies. For the minibody imaging, tumor bearing SCID mice were scanned at 24 hours and 44 hours post injection. Figure 4F shows clear differences in uptake levels between the high-MET Hcc827-GR6 tumor and the low-MET Hcc827 tumor, both at 24 hours and 44 hours post injection. Due to the longer serum persistance of the minibody, the ex vivo biodistribution analysis at 44 hours post injection revealed higher uptake levels of 8.6 ± 0.5 %ID/g in Hcc827-GR6 tumors, and the Hcc827 tumors also showed higher uptake level of 4.5 ± 0.4 %ID/g. However, the image contrast was not improved due to higher background signal in the blood (Hcc827-GR6 Tumor : Blood = 4.2 ± 0.5, details shown in Table 2). To further compare the pharmacokinetic properties, radiolabeled H2 cys-diabody and minibody were also injected into tumor-free female SCID mice, and their terminal half lives were estimated to be 24 ± 3 min (n=2) and 10 ± 1 h (n=4) respectively.
Table 2.
Ex vivo biodistribution of 89Zr-DFO-maleimide-H2 cysDb at 44 hours post injection in female SCID mice bearing Hcc827 and Hcc827-GR6 tumors.
| Hcc827-GR6 vs. Hcc827 (n = 3) |
|
|---|---|
|
| |
| Immuno-reactivity | 65% |
| Injected dose (μg, MBq) | 25μg, 0.74MBq |
|
| |
| Tumor Size (mean ± SD) | |
|
| |
| Hcc827 tumor | 321 ± 80 mg |
|
| |
| Hcc827-GR6 tumor | 244 ± 25 mg |
|
| |
| %ID/g (mean ± SD) | |
|
| |
| Hcc827 tumor | 4.5 ± 0.4 |
| Hcc827-GR6 tumor | 8.6 ± 0.5 |
| Blood | 2.1 ± 0.3 |
| Muscle | 0.33 ± 0.3 |
| Lung | 1.8 ± 0.1 |
| Heart | 1.5 ± 0.2 |
| Liver | 20 ± 2 |
| Stomach | 1.0 ± 0.2 |
| Intestine | 2.4 ± 0.2 |
| Spleen | 8.7 ± 1.0 |
| Kidney | 30 ± 2 |
| Tail | 3.4 ± 0.5 |
| Carcass | 0.96 ± 0.07 |
|
| |
| Ratio (mean ± SD) | |
|
| |
| Hcc827-GR6 Tu/Blood | 4.2 ± 0.5 |
| Hcc827-GR6 Tu/Muscle | 26 ± 1 |
| Hcc827-GR6 Tu/Hcc827 Tu | 1.9 ± 0.2 |
Discussion
In this work, we isolated multiple anti-MET antibody clones from a large naïve human scFv phage display library. Three of these clones were successfully reformatted into cys-diabodies for in vitro characterization, and demonstrated high affinities ranging from 0.7 to 5.1 nM. The H2 cys-diabody shows the highest affinity (KD = 0.73 ± 0.10 nM), best anti-MET inhibitory effect (significant inhibitory effects at 1.8 nM on MET dependent cells, p < 0.01), and cross-reacts with murine MET. ImmunoPET studies using 89Zr radiolabeled H2 cys-diabody and minibody were able to distinguish a MET over-expressing gefitinib resistant tumor from the parental tumor at as early as 4 hours post injection. The ex vivo biodistribution analysis also showed significant differences in tracer uptake levels in the MET over-expressing Hcc827-GR6 tumors vs. the parental tumors (p < 0.01). Using the H2 cys-diabody, very high positive tumor-to-blood ratios (18 to 22) were achieved at 20 hours post injection. These results indicate the potential for rapid and even same day imaging for patient stratification.
In this study, we sequenced 64 positive phage clones and found 19 distinct antibody sequences, 11 of which occurred only once, indicating significant diversity of the MET binding phage in the library. Among the solitary isolates, the H2 scFv showed excellent affinity, and reformatted versions were employed as immunoPET agents, producing high contrast images of MET-positive tumor xenografts. It is likely that there are additional good candidates in the phage pool remaining after the third round of selection. During the phage selection process, high affinity candidates are not always the most enriched ones. Individual antibody sequences can vary in the ability to be expressed, or may encode proteins that are toxic or inhibit robust growth of the host bacterium. Rare clones may express antibodies that perform better in vivo due to unique pharmacokinetic properties and stability. Complementary to the more traditional medium-throughput approach we used in this study, high-throughput approaches empowed by deep sequencing, low-cost gene synthesis, and high-throughput protein production technologies can enable researchers to screen higher numbers of candiates. This will make the rare clones more accessible to reasearchers, including clones with unique properties. For example, although anti-tumor effects are desirable for therapeutic applications, it is typically preferable to employ biologically inert binders for strictly imaging applications. The ability to screen and evaluate larger numbers of candidate clones would increase the odds of finding the best fit for specific applications.
While the intent was to isolate antibody fragments for in vivo PET imaging, it was also important to avoid antibodies that may stimulate MET signaling. H2 and H5 cys-diabodies inhibited cell proliferation of two MET-activated cell lines (Hcc827-GR6 and Hcc827-/+), but showed no effects on two other gefitinib-resistant cell lines lacking MET amplification (H1650 and H1975), indicating the inhibitory effects were MET specific. Focusing on the H2 clone alone, the cys-diabody, minibody and scFv formats were further compared. Interestingly, while the bivalent H2 cys-diabody and minibody showed similar inhibitory effects on Hcc827-GR6 cells, the monovalent H2 scFv failed to inhibit the growth of the MET over-expressing cells even at very high concentration (180nM), implying bivalent binding is required for the anti-MET inhibitory effect. Previously, many anti-MET antibodies that have been isolated exhibit a partial agonistic effect, limiting their potential utility as cancer therapeutics. Despite blocking HGF-induced activation, the bivalent engagement of anti-MET antibodies appeared to cause at least partial activation of MET signaling. To address this problem, DN-30 and MetMAb (onartuzumab) were engineered into monovalent fragments, in order to exert their inhibitory effects without activating MET kinase (30-32). Engelman et al showed that Hcc827-GR6 acquired gefitinib resistance by activating ERBB3 through MET over-expression (4). Given the requirement of bivalency, it is unlikely that H2 antibody fragments' inhibitory effects are due to competition with HGF or directly blocking of binding site for other cell surface receptors such as ERBB3. As the internalization assay shows, binding of H2 cys-diabody can cause rapid internalization of cell surface MET, suggesting a mechanism for downregulation of MET signaling similar to that of recently reported non-agonistic bivalent murine antibodies (33). Alternatively, the bivalent binding of H2 antibody fragments may lock MET into a conformation that prohibits downstream signaling. Investigation of the detailed mechanism is ongoing.
Given the fact that H2 cys-diabody is rapidly internalized upon binding to cell surface MET, a residualizing radiolabeling approach using chelated 89Zr was selected, the same as two previous preclinical anti-MET immunoPET studies (8, 9). It is difficult to directly compare among these studies because of the differences in tumor models and mouse strains. However, it is known that radiolabeled diabodies usually have lower tumor uptake levels compared to larger fragments or intact antibodies, but high contrast images can be produced at earlier time points, due to the rapid blood clearance (34). With the high tumor-to-blood ratio (∼20:1) at only 20 hours post injection, these results once more demonstrated the unique advantage of diabodies for rapid and high contrast immunoPET imaging. Thus far, anti-MET imaging studies using similar small antibody fragments in patients have not been described, motivating the current efforts to develop these fully human antibody fragments for in vivo PET imaging. Previously, Santimaria et al. had demonstrated diabody-based imaging of the ED-B fibronectin domain in patients (35). Due to the fast clearance of the diabody fragment, they were able to demonstrate tumor targeting at as early as 6 hours post injection instead of the 4 – 7 day delay typically required when imaging with intact antibodies (7). With the short serum half lives of the H2 antibody fragments, similar rapid imaging in patients can be expected in the future.
In the current cys-diabody imaging studies, some variations were observed between experiments. For example, the cys-diabody imaging of mice bearing dual Hcc827-GR6 and Hcc827 tumors showed higher tumor uptake levels relative to the other experiments. This may reflect the effects of variations in radiolabeling, tail vein injection, or perhaps the ages of the mice and variations in physiology. Despite difference in absolute uptake levels, the tumor-to-blood ratios remain similar among the three groups, suggesting that the variations in uptake levels are likely caused by differences in availability of the radiotracer in the blood.
Compared to cys-diabodies, minibodies have longer serum persistence, allowing higher tracer uptake in the tumors. The imaging and biodistribution results indeed showed improved tumor uptake levels when using H2 minibody, but the tumor-to-blood contrast was less impressive (∼4:1, at 44 hours post injection). Overall, cys-diabody is the preferred choice for rapid immunoPET applications. A further potential advantage of the H2 antibody fragments is their cross reactivity to mouse MET, allowing more representative imaging studies in the presence of endogenous MET expression in normal tissues of the mouse.
In conclusion, the present study demonstrates a successful example of rapid development of fully human antibody fragment based immunoPET probes from a human antibody phage display library. Compared to previous anti-MET immunoPET studies using intact or one-armed antibodies, imaging using the H2 cys-diabody provides high contrast images at early time points, with clear potential for same day imaging. Considering the growing interest in MET-targeted therapies, a non-invasive imaging tool for detecting and assessing MET expression in tumors could be useful for diagnosis and patient selection. Of interest, the H2 antibody fragments also show anti-MET inhibitory effects, making these antibody fragments candidates for potential therapeutic applications as well.
Supplementary Material
Acknowledgments
The authors thank past and present members of the Wu lab for helpful discussions and contributions. We are grateful to Dr. Jeffrey A. Engelman for providing cell lines used in these studies. We thank Waldemar Ladno, Darin Williams, and Dr. David Stout, of the Crump Institute for Molecular Imaging for assistance with the small animal PET scans.
Financial support: Research funding was provided by the Stanford Center for Cancer Nanotechnology Excellence and Translation (CCNE-T; NIH U54 CA151459) (A.M. Wu, K. Li, K. Zettlitz, S. Mumenthaler, P. Mallick). Additional support was provided by an unrestricted gift from the Ralph and Marjorie Crump Foundation (A.M.Wu). Support for the Small Animal Imaging Facility and Flow Cytometry Core was provided by the UCLA Jonsson Comprehensive Cancer Center (NIH CA016042)
Footnotes
Conflict of Interest statement: Anna M. Wu and James D. Marks are shareholders and consultants to ImaginAb, Inc.
References
- 1.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature reviews Molecular cell biology. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 2.Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nature reviews Cancer. 2006;6:637–45. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 3.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nature reviews Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 4.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 5.Zucali PA, Ruiz MG, Giovannetti E, Destro A, Varella-Garcia M, Floor K, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2008;19:1605–12. doi: 10.1093/annonc/mdn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Surati M, Patel P, Peterson A, Salgia R. Role of MetMAb (OA-5D5) in c-MET active lung malignancies. Expert opinion on biological therapy. 2011;11:1655–62. doi: 10.1517/14712598.2011.626762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knowles SM, Wu AM. Advances in immuno-positron emission tomography: antibodies for molecular imaging in oncology. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:3884–92. doi: 10.1200/JCO.2012.42.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perk LR, Stigter-van Walsum M, Visser GW, Kloet RW, Vosjan MJ, Leemans CR, et al. Quantitative PET imaging of Met-expressing human cancer xenografts with 89Zr-labelled monoclonal antibody DN30. Eur J Nucl Med Mol Imaging. 2008;35:1857–67. doi: 10.1007/s00259-008-0774-5. [DOI] [PubMed] [Google Scholar]
- 9.Jagoda EM, Lang L, Bhadrasetty V, Histed S, Williams M, Kramer-Marek G, et al. Immuno-PET of the hepatocyte growth factor receptor Met using the 1-armed antibody onartuzumab. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2012;53:1592–600. doi: 10.2967/jnumed.111.102293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nature reviews Cancer. 2002;2:683–93. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 11.Wu AM. Antibodies and antimatter: the resurgence of immuno-PET. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- 12.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23:1137–46. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Olafsen T, Anderson AL, Wu A, Raubitschek AA, Shively JE. Reduction of kidney uptake in radiometal labeled peptide linkers conjugated to recombinant antibody fragments. Site-specific conjugation of DOTA-peptides to a Cys-diabody. Bioconjugate chemistry. 2002;13:985–95. doi: 10.1021/bc025565u. [DOI] [PubMed] [Google Scholar]
- 14.Sirk SJ, Olafsen T, Barat B, Bauer KB, Wu AM. Site-specific, thiol-mediated conjugation of fluorescent probes to cysteine-modified diabodies targeting CD20 or HER2. Bioconjugate chemistry. 2008;19:2527–34. doi: 10.1021/bc800113v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barat B, Sirk SJ, McCabe KE, Li J, Lepin EJ, Remenyi R, et al. Cys-diabody quantum dot conjugates (immunoQdots) for cancer marker detection. Bioconjugate chemistry. 2009;20:1474–81. doi: 10.1021/bc800421f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCabe KE, Liu B, Marks JD, Tomlinson JS, Wu H, Wu AM. An engineered cysteine-modified diabody for imaging activated leukocyte cell adhesion molecule (ALCAM)-positive tumors. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2012;14:336–47. doi: 10.1007/s11307-011-0500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olafsen T, Sirk SJ, Olma S, Shen CK, Wu AM. ImmunoPET using engineered antibody fragments: fluorine-18 labeled diabodies for same-day imaging. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2012;33:669–77. doi: 10.1007/s13277-012-0365-8. [DOI] [PubMed] [Google Scholar]
- 18.Girgis MD, Federman N, Rochefort MM, McCabe KE, Wu AM, Nagy JO, et al. An engineered anti-CA19-9 cys-diabody for positron emission tomography imaging of pancreatic cancer and targeting of polymerized liposomal nanoparticles. The Journal of surgical research. 2013;185:45–55. doi: 10.1016/j.jss.2013.05.095. [DOI] [PubMed] [Google Scholar]
- 19.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annual review of immunology. 1994;12:433–55. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 20.Marks C, Marks JD. Phage libraries--a new route to clinically useful antibodies. The New England journal of medicine. 1996;335:730–3. doi: 10.1056/NEJM199609053351008. [DOI] [PubMed] [Google Scholar]
- 21.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–16. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 22.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nature reviews Drug discovery. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 23.Robinson MK, Doss M, Shaller C, Narayanan D, Marks JD, Adler LP, et al. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005;65:1471–8. doi: 10.1158/0008-5472.CAN-04-2008. [DOI] [PubMed] [Google Scholar]
- 24.Fu M, Brewer S, Olafsen T, Wu AM, Gordon LK, Said J, et al. Positron emission tomography imaging of endometrial cancer using engineered anti-EMP2 antibody fragments. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2013;15:68–78. doi: 10.1007/s11307-012-0558-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCabe KE, Wu AM. Positive progress in immunoPET--not just a coincidence. Cancer biotherapy & radiopharmaceuticals. 2010;25:253–61. doi: 10.1089/cbr.2010.0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Dongen GA, Vosjan MJ. Immuno-positron emission tomography: shedding light on clinical antibody therapy. Cancer biotherapy & radiopharmaceuticals. 2010;25:375–85. doi: 10.1089/cbr.2010.0812. [DOI] [PubMed] [Google Scholar]
- 27.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, et al. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A. 1998;95:6157–62. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marks JD, Bradbury A. Selection of Human Antibodies from Phage Display Libraries. In: Lo BKC, editor. Antibody engineering : methods and protocols. Totowa, N.J.: Humana Press; 2004. p. 161. [DOI] [PubMed] [Google Scholar]
- 29.Knowles SM, Zettlitz KA, Tavare R, Rochefort MM, Salazar FB, Stout DB, et al. Quantitative immunoPET of prostate cancer xenografts with 89Zr- and 124I-labeled anti-PSCA A11 minibody. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2014;55:452–9. doi: 10.2967/jnumed.113.120873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J Biol Chem. 2010;285:36149–57. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R, et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12:6144–52. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- 32.Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110:E2987–96. doi: 10.1073/pnas.1302725110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenall SA, Gherardi E, Liu Z, Donoghue JF, Vitali AA, Li Q, et al. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PloS one. 2012;7:e34658. doi: 10.1371/journal.pone.0034658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundaresan G, Yazaki PJ, Shively JE, Finn RD, Larson SM, Raubitschek AA, et al. 124I-labeled engineered anti-CEA minibodies and diabodies allow high-contrast, antigen-specific small-animal PET imaging of xenografts in athymic mice. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2003;44:1962–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Santimaria M, Moscatelli G, Viale GL, Giovannoni L, Neri G, Viti F, et al. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin Cancer Res. 2003;9:571–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.