

Abstract
Neuronal activity is controlled by a fine-tuned balance between intrinsic properties and extrinsic synaptic inputs. Moreover, neighbouring astrocytes are now recognized to influence a wide spectrum of neuronal functions. Yet, how these three key factors act in concert to modulate and fine-tune neuronal output is not well understood. Here, we show that in rat hypothalamic magnocellular neurosecretory cells (MNCs), glutamate NMDA receptors (NMDARs) are negatively coupled to the transient, voltage-gated A-type K+ current (IA). We found that activation of NMDARs by extracellular glutamate levels influenced by astrocyte glutamate transporters resulted in a significant inhibition of IA. The NMDAR–IA functional coupling resulted from activation of extrasynaptic NMDARs, was calcium- and protein kinase C-dependent, and involved enhanced steady-state, voltage-dependent inactivation of IA. The NMDAR–IA coupling diminished the latency to the first evoked spike in response to membrane depolarization and increased the total number of evoked action potentials, thus strengthening the neuronal input/output function. Finally, we found a blunted NMDA-mediated inhibition of IA in dehydrated rats. Together, our findings support a novel signalling mechanism that involves a functional coupling between extrasynaptic NMDARs and A-type K+ channels, which is influenced by local astrocytes. We show this signalling complex to play an important role in modulating hypothalamic neuronal excitability, which may contribute to adaptive responses during a sustained osmotic challenge such as dehydration.
Key points
In hypothalamic magnocellular neurosecretory cells, activation of glutamate NMDA receptors leads to inhibition of the transient voltage-gated A-type K+ current (IA), in a Ca2+- and protein kinase C-dependent manner.
The negative NMDAR–IA functional coupling involves activation of extrasynaptic (e)NMDARs. The eNMDAR–IA coupling is engaged by endogenous extracellular glutamate, whose levels are controlled by astrocyte glutamate GLT1 transporters.
The eNMDAR–IA coupling is enhanced during dehydration, a condition in which astrocyte GLT1 efficiency is blunted.
The eNMDAR–IA coupling results in increased neuronal excitability and firing activity in magnocellular neurosecretory neurons.
Taken together these studies support the concept that the eNMDAR–IA coupling is a powerful mechanism by which glutamate increases magnocellular neurosecretory excitability and firing activity.
Introduction
Complex interactions among intrinsic membrane properties and synaptic inputs influence the degree and pattern of neuronal activity in the central nervous system. Moreover, astrocytes are now recognized to influence a wide spectrum of neuronal functions, ranging from fine-tuning of synaptic strength to changes in overall neuronal network output (Araque & Navarrete, 2010; Giaume, 2010). In addition to releasing a variety of neuroactive substances, including ATP and d-serine (Panatier et al. 2006; Gordon et al. 2007) astrocytes affect neuronal function via the expression of selective transporters that regulate ambient neurotransmitter levels (Rothstein et al. 1996). Yet the precise mechanisms by which these three pivotal factors (i.e. intrinsic properties, synaptic mechanisms and astrocytes) act in concert to modulate neuronal output have not been explored in detail.
The hypothalamic magnocellular system constitutes an ideal network model to study this phenomenon. It is a simple, well-characterized system comprising magnocellular neurosecretory vasopressin and oxytocin cells (MNCs) in the supraoptic (SON) and paraventricular (PVN) nuclei. MNC firing activity directly correlates with neurosecretion from the posterior pituitary (Cazalis et al. 1985). The key intrinsic and synaptic properties influencing MNC activity are well characterized.
The A-type potassium current (IA) is a dominant intrinsic, voltage-dependent subthreshold current that determines spike onset and interspike interval during MNC repetitive firing (Bourque, 1988; Luther & Tasker, 2000). Moreover, we showed that downregulation of IA contributes to increased hypothalamic neuronal excitability during hypertension (Sonner et al. 2008, 2011).
Glutamate is the major extrinsic excitatory neurotransmitter in the hypothalamus (van den Pol et al. 1990). Acting primarily on ionotropic NMDA receptors (NMDARs), glutamate not only stimulates overall MNC activity, but also contributes to the adoption of bursting firing, which is critical to optimize the neurosecretory output of this system (Hu & Bourque, 1992; Nissen et al. 1995).
Finally, the magnocellular system possesses a unique and dynamic neuro-glial microenvironment. Under basal conditions, astroglial processes closely enwrap neurons and their synapses. This tight microenvironment enables astrocyte glutamate transporters to efficiently buffer extracellular glutamate, restricting activation of extrasynaptic NMDARs (eNMDARs) (Fleming et al. 2011) and presynaptic metabotropic receptors (Oliet et al. 2001; Boudaba et al. 2003). Physiological challenges including dehydration and lactation evoke a rapid and reversible retraction of astroglial processes (Perlmutter et al. 1984; Oliet et al. 2001) enhancing ambient glutamate levels and actions at these extra/perisynaptic sites (Oliet et al. 2001; Fleming et al. 2011). This dynamic neuro-glial remodelling is necessary for the adoption of bursting firing patterns during conditions of high hormonal demand (Theodosis & Poulain, 1993; Tasker et al. 2012). Thus, given the importance of astrocytes, NMDARs and IA in MNCs, we explored whether these mechanisms acted in an interrelated manner to influence MNC firing activity. Our results show that in addition to inducing a direct excitatory inward current, activation of eNMDARs are negatively coupled, in a Ca2+- and protein kinase C (PKC)-dependent manner, to IA. Moreover, we show that, by modulating ambient glutamate levels, astrocyte GLT1 transporters influence the degree of activation of the NMDAR–IA coupling. Finally, we demonstrate that this functional coupling efficiently influences MNC membrane excitability and firing output.
Methods
Ethical approval
All experimental procedures were in strict compliance with NIH guidelines, and were approved by the Georgia Regents University Institutional Animal Care and Use Committee.
Animals
Male Wistar rats purchased from Harlan Laboratories (Indianapolis, IN, USA) (4–5 weeks old) were housed under standardized conditions (12 h:12 h light–dark cycle, lights on 07.00 h) with food and water available ad libitum. For dehydration, water was removed for 48 h with free access to dry food. In these rats, plasma osmolarity was significantly elevated (328.5 ± 1.8 vs. 308 ± 2.1 mosmol l−1, in dehydrated and euhydrated rats, respectively, P < 0.05).
Hypothalamic slices
Rats were anaesthetized with pentobarbital (50 mg kg–1 i.p.), quickly decapitated and brains dissected out. Coronal slices were cut (250 μm thick) utilizing a vibroslicer (Leica VT1000). An oxygenated ice-cold artificial cerebrospinal fluid (ACSF) was used during slicing (containing in mm): 119 NaCl, 2.5 KCl, 1 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 20 d-glucose, 0.4 ascorbic acid, 2.0 CaCl2 and 2.0 pyruvic acid; pH 7.4; 290–310 mosmol l−1). After sectioning, slices were placed in a holding chamber containing ACSF and kept at room temperature (22°C) until used.
Drugs
Tetrodotoxin (TTX) and (2S,3S,4R)-2-carboxy-4-isopropyl-3-pyrrolidineacetic acid (DHK, dihydrokainic acid) were purchased from Abcam Biochemicals (Cambridge, MA, USA). N-Methyl-d-aspartate (NMDA), bicuculline, MK801, 4-aminopyridine, 4-hydroxyquinoline-2-carboxylic acid (kynurenic acid), 6,7-dinitroquinoxaline-2,3-dione (DNQX) and glycine were purchased from Sigma-Aldrich (St Louis, MO, USA). 1,2-bis(o-Aminophenoxy)ethane- N,N,N′,N′-tetraacetic acid (BAPTA) was purchased from Invitrogen (Grand Island, NY, USA). dl-2-Amino-5-phosphonopentanoic acid (AP5) and chelerythrine chloride were purchased from Tocris Bioscience (Bristol, UK).
Electrophysiological recordings
Slices were placed in a submersion style recording chamber, and bathed with solutions (3.0 ml min−1) that were bubbled continuously with a gas mix of 95% O2–5% CO2, and maintained at near physiological temperature (32°C). Thin-walled (1.5 mm o.d., 1.17 mm i.d.) borosilicate glass (G150TF-3, Warner Instruments, Sarasota, FL, USA) was used to pull patch pipettes (3–6 MΩ) on a horizontal Flaming/Brown micropipette puller (P-97, Sutter Instruments, Novato, CA, USA). Whole-cell patch-clamp recordings from SON neurons were visually made using diffraction interference contrast (DIC) videomicroscopy. Recordings were obtained with a Multiclamp 700A amplifier (Axon Instruments, Union City, CA, USA). The voltage output was digitized at 16-bit resolution, 10 kHz (Digidata1320A, Axon Instruments), and saved on a computer to be analysed offline using pCLAMP9 software (Axon Instruments). Mean series resistance (SR) was 13.4 ± 0.5 MΩ and experiments were discarded in cases in which the series resistance was unstable (changes >20%). In a control set of experiments, SR was compensated for ∼60% and a similar degree of NMDAR-mediated IA inhibition was observed as we report without SR compensation. Moreover, the overall magnitude of the recorded currents was minimally affected by the compensation (less than 20% at the highest voltage tested). Finally, MNCs in SON/PVN are electrotonically compact, displaying very rudimentary dendritic trees (Armstrong & Smith, 1990; Stern & Armstrong, 1998; Luther & Tasker, 2000; Stern, 2001), minimizing potential space clamp problems during recordings. Given this, and the fact that the goal of our study was to report changes in IA current ‘before and after’ a treatment, rather than reporting precise amplitude/kinetic values of these currents, all subsequent studies were performed without SR compensation. The internal solution contained (in mm): 140 potassium gluconate, 0.2 EGTA, 10 Hepes, 10 KCl, 0.9 MgCl2, 4 MgATP, 0.3 NaGTP and 20 phosphocreatine (Na+); pH 7.2–7.3. All recordings were obtained in a low Mg2+ ACSF (10 μm MgSO4) in order to facilitate measurements of NMDA currents (INMDA).
Voltage clamp
Unless otherwise stated, glycine (10 μm) and tetrodotoxin (0.5 μm) were added to the ACSF for voltage-clamp recordings. INMDA was activated either by focal, transient application of NMDA via a picospritzer or via bath application of NMDA. INMDA was also activated by inducing build-up of extracellular glutamate levels following blockade of glial GLT1 transporters with dihydrokainate (DHK, 300 μm; Fleming et al. 2011). Cells not showing an evident INMDA following DHK (n = 2) were not included in the study. INMDA was quantified as the change in the holding current measured as the average of a 2 min segment of steady-state baseline obtained before and after a 5–10 min application of NMDA or DHK. The voltage-dependent properties of activation of IA were assessed before and after NMDA/DHK application (when a new steady-state current was achieved), using conventional protocols (depolarizing steps from −70 to +20 mV in 10 mV increments, 200 ms; Sonner et al. 2008). These protocols evoked both IA and the delayed-rectifier current IKDR. For bath-applied NMDA/DHK, due to the very slow nature of these experiments, we were unable to electronically or pharmacologically isolate these two components. Nonetheless, at the range of membrane potentials tested, IA was clearly differentiated from IKDR based on several characteristic properties including hyperpolarized activation threshold, overall larger magnitude and faster activation kinetics and rapid inactivation (see Fig. 1). During transient NMDAR activation, however, IA was isolated electronically by digital subtraction of currents evoked from two separate protocols, in which preconditioning steps of either −80 mV and −10 mV were used, to fully evoke, and to completely inactivate IA, respectively (Sonner et al. 2008). Since the focus of the present study was on IA, the properties of IKDR were not further studied. To determine the IA half-activation potential, current amplitudes normalized to the maximum peak were plotted as a function of the command step potentials, and fitted with a Boltzmann function. To determine the voltage dependence of inactivation, IA currents were evoked by voltage steps to +10 mV (300 ms duration) from conditioning steps between −120 and −30 mV in 10 mV increments (50 ms duration) before and after application of NMDA/DHK. To determine the half-inactivation potential, current amplitudes normalized to the maximum peak were plotted as a function of the conditioning step (Sonner et al. 2008).
Figure 1. Sustained NMDA receptor activation inhibits IA.
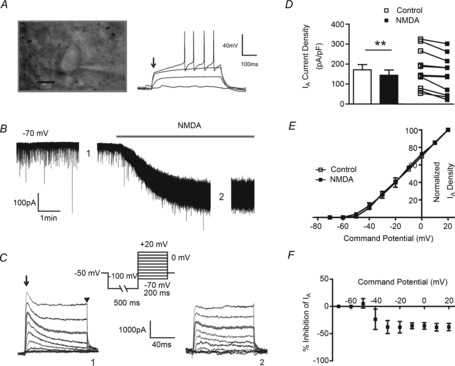
A, DIC image of a patched MNC (left) showing the characteristic transient outward rectification during evoked depolarizations (right, arrow, 50 pA step–1). B, NMDA (30 μm)-induced inward shift in Iholding evoked in the same neuron as in A. C, samples of IA (arrow) and delayed rectifier (IKDR, arrowhead) outward K+ currents activated before and during NMDA application (at time points 1 and 2 in B, respectively). D, mean (left) and before–after (right) plots summarizing the effect of NMDAR activation on IA density evoked at 0 mV before (open symbols) and during (filled symbols) NMDAR activation (n = 12), E, mean plot of the normalized IA density vs. command potential before (open squares) and during (filled squares) NMDA application (n = 9). F, plot of mean percentage IA inhibition in the presence of NMDA vs. the command potential (n = 9). **P < 0.01; paired t test. Scale bar in A, 10 μm.
Current clamp
Action potentials were triggered with depolarizing current steps of varying amplitudes (Δ50 pA) or duration (Δ200 ms) while holding neurons at ∼−80 mV. Plots of number of action potentials as a function of step current amplitude or duration were constructed. The mean time delay to the first spike, and the total number of action potentials triggered were calculated and compared before and after drug applications. In the case of bath-applied NMDA or DHK, neurons were hyperpolarized with an additional DC current injection to maintain neurons at ∼−80 mV before depolarizing steps were applied again. To test for the influence of IA on evoked firing properties, the K+ channel blocker 4-aminopyridine (4-AP) was used. Since 4-AP facilitates the presynaptic release of neurotransmitter (Flores-Hernandez et al. 1994), which could mask the direct effects of 4-AP on intrinsic properties, these experiments were performed in the presence of the GABAA and AMPA receptor blockers bicuculline (20 μm) and DNQX (30 μm), respectively, a condition in which the DHK-mediated current was still present (not shown).
Statistical analysis
All values are expressed as means ± SEM. Student's paired t test was used to compare the effects of a drug treatment on INMDA or IA. Between-group (e.g. MK801 vs. glycine) differences were compared using either unpaired t tests or analysis of variance repeated measures (ANOVA-RM). Where the F ratio was significant, post hoc comparisons were completed using the Bonferroni post hoc test or Dunnett's multiple comparison test when comparing values against a basal control. Pearson's correlation test was used to determine if correlations existed between two parameters. Differences were considered statistically significant at P < 0.05 and n refers to the number of cells. All statistical analyses were conducted using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Results
NMDAR activation inhibits the magnitude of IA
Whole-cell patch clamp recordings were obtained from 204 SON MNCs (Fig. 1A). We found that following bath-applied NMDA (30 μm, Fig. 1B), IA in MNCs was significantly inhibited (Fig. 1C and D, n = 12). Mean normalized I–V plots showed that the voltage-dependent activation properties of IA were not affected following NMDAR activation (Fig. 1E, half-activation potential (V½): −8.26 ± 1.97 mV and −10.94 ± 1.79 mV before and after NMDA, respectively; P = 0.2, paired t test). Moreover, once IA was evoked, a similar degree of NMDA-mediated inhibition was observed at all voltages measured (−40 mV to +20 mV, F = 0.588, P > 0.5, repeated measures ANOVA, Fig. 1F), indicating that the NMDA-mediated inhibition of IA was voltage independent. Importantly, we found that blockade of NMDARs per se (100 μm AP5) increased the basal magnitude of IA (control: 134.46 ± 22.34 pA pF–1, AP5: 176.90 ± 36.14 pA pF–1, P = 0.05 paired t test, n = 10).
To better study the time course of the NMDA effects, we focally and transiently puffed NMDA (20 μm, 1 p.s.i., 5 s duration, n = 11) directly onto the recorded neuron, while IA was repetitively evoked (single depolarizing pulse to 0 mV, 1 s interval) before, during and after NMDAR activation (Fig. 2A). IA was significantly inhibited 2 s following the initiation of the NMDA stimulus, returning to control levels at 12 s (Fig. 2B, F = 7.443, P < 0.0001, repeated measures ANOVA). The mean and individual IA density before and after NMDA application is shown in Fig. 2C. Finally, we found no correlation between the degree of IA inhibition and INMDA magnitude (Fig. 2D, Pearson's correlation r2: 0.0864 P > 0.05, Pearson's correlation test). The NMDA-mediated inhibition of IA persisted in a subset of recordings obtained with a normal Mg2+ ACSF (1 mm MgSO4) (%NMDA–IA peak inhibition = 3.7 ± 1.1, n = 5), although as expected (given that recordings were obtained at a holding potential of −70 mV), the effect was substantially smaller when compared to that observed in a low Mg2+ ACSF (∼20% inhibition, see Fig. 2B). Collectively, our results support the presence of a negative functional coupling between NMDARs and A-type channels in hypothalamic MNCs.
Figure 2. Transient NMDA receptor activation inhibits IA.
A, transient inward shift in Iholding following a focal brief application of NMDA (20 μm, 5 s) to an MNC. The lower traces show IA before (0 s), at the end (5 s) and after (15 s) NMDA application. IA was isolated electronically by digital subtraction of currents evoked from two separate protocols, in which preconditioning steps of either −80 mV and −10 mV were used, to fully evoke, and to completely inactivate IA, respectively. B, plot of the mean percentage NMDA-evoked inhibition of IA amplitude as a function of time (n = 11). C, mean (left) and before–after (right) plots of IA density before (open symbols) and 3.5 s after (filled symbols) initiation of transient NMDAR activation. D, plot of the percentage inhibition of IA as a function of INMDA, depicting a lack of correlation between the two parameters (r2 = 0.0864). *P < 0.05 vs. baseline IA (Dunnett's post hoc test); ***P < 0.0001 (paired t test).
Astrocytes influence the NMDAR–IA functional coupling via glutamate-mediated activation of extrasynaptic NMDARs
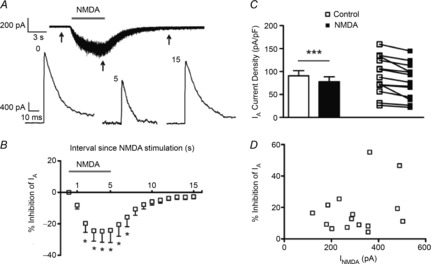
We recently showed that astrocytes regulate ambient glutamate levels and the degree of eNMDAR activation in MNCs via the activity of GLT1 glutamate transporters (Fleming et al. 2011; Potapenko et al. 2013). Thus, we explored here whether astrocytes indirectly affect IA magnitude via this mechanism. As previously reported (Fleming et al. 2011), GLT1 blockade (300 μm dihydrokainate, DHK) induced an inward shift in Iholding (Fig. 3A, DHK-mediated current density: 3.15 ± 0.94 pA pF–1), which we previously showed to be blocked by AP5 (Fleming et al. 2011). DHK resulted in a significant inhibition of IA (P < 0.05, n = 13, Fig. 3B), an effect that was voltage independent (Fig. 3C, F = 2.271, repeated measures ANOVA) and did not involve a change in the voltage-dependent activation properties of IA (Fig. 3D, V½ activation: −10.06 ± 2.18 mV and −9.69 ± 2.20 mV before and after DHK, respectively; P = 0.85, paired t test).
Figure 3. IA magnitude is inhibited following blockade of astrocyte GLT1 transporters.
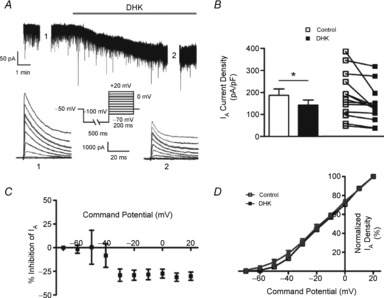
A, DHK-induced inward shift in Iholding in an MNC. Lower traces show IA before (1) and during (2) DHK application. B, mean (left) and before–after (right) plots of IA density (at 0 mV) before (open symbols) and after (filled symbols) DHK (300 μm) (n = 13). C, plot of mean percentage inhibition of IA in the presence of DHK vs. the command potential. D, mean plots of the normalized IA density vs. the command potential before (open squares) and during (filled squares) application of DHK. *P < 0.05 (paired t test).
To confirm the contribution of eNMDARs to the NMDA–IA coupling, experiments were repeated in conditions in which we selectively blocked either synaptic or extrasynaptic NMDARs. To selectively block sNMDARs, we used a well-characterized approach based on the combined use of the NMDA open channel blocker MK801 (20 μm) with 4-AP (2.5 mm) and bicuculline (20 μm), to chemically evoke synaptic release of glutamate (Hardingham et al. 2002; Jung et al. 2008). In a set of control experiments, we found that at this concentration, 4-AP evoked a 30.0 ± 5.6% inhibition of IA (n = 5, P < 0.01), which within 10 min after washout recovered almost completely (90.6 ± 5.6% of baseline magnitude, n = 5). We found that blockade of sNMDARs did not affect the DHK-induced shift in Iholding and DHK-induced IA inhibition (n = 9, P < 0.05, paired t test, Fig. 4A, C, D and E). Conversely, removal of glycine from the ACSF, a critical co-agonist of extrasynaptic (but not synaptic) NMDARs (Papouin et al. 2012) prevented the DHK-induced IA inhibition and largely reduced the DHK-evoked INMDA (n = 7, P = 0.89, paired t test, Fig. 4B, C, D and E). In fact, when results were plotted as percentage changes within each individual cell, a tendency for an enhancement of IA was observed, which was significantly different to the inhibition observed in cells treated with MK801 (Fig. 4D). The DHK-mediated inhibition of IA was almost completely blocked by the broad-spectrum glutamate receptor blocker kynurenic acid (138.68 ± 19.43 and 138.91 ± 23.37 pA pF–1 before and after DHK, respectively, P > 0.8, paired t test, n = 11). Taken together, these results indicate that the eNMDAR–IA functional coupling stands as a functionally relevant signalling mechanism by which astrocytes influence membrane excitability in MNC neurons.
Figure 4. Astrocyte GLT1 transporters influence IA via activation of extrasynaptic NMDA receptors.
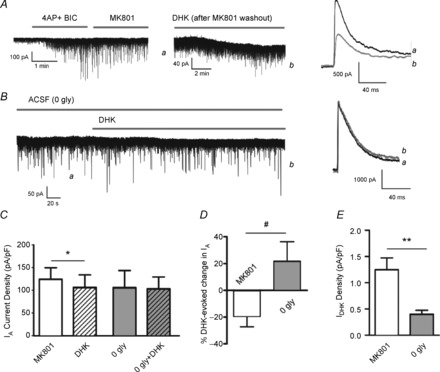
A, blockade of synaptic NMDA receptors (sNMDARs) with a combined application of 4-AP (2.5 mm) and bicuculline (BIC; 20 μm), followed by MK801 (20 μm) did not affect the DHK-evoked inhibition of IA (right panel). B, blockade of eNMDARs (0 gly ACSF) blunted the DHK-induced shift in Iholding (left panel) and the inhibition of IA (right panel). Mean IA density (C), mean percentage IA change (D), and mean shift in Iholding density (E) evoked by DHK are shown for 4-AP/BIC/MK801 (n = 9) and 0 glycine (n = 7) groups. *P < 0.05, paired t test; **P < 0.01, unpaired t test; #P < 0.05, unpaired t test.
Blunted evoked NMDA–IA coupling in dehydrated rats
We recently showed that during dehydration, a condition known to lead to astrocyte process retraction and diminished astroglial ensheathment of MNCs (Hatton et al. 1984; Tweedle & Hatton, 1984; Tasker et al. 2002), GLT1-mediated glutamate uptake was blunted, resulting in elevated ambient glutamate levels, enhanced eNMDAR activation and increased MNC firing activity (Fleming et al. 2011). Thus, to further assess the contribution of astrocytes to the regulation of the eNMDA–IA functional coupling, we repeated a set of experiments in MNCs from 48 h-dehydrated rats, in which we assessed changes in IA magnitude following transient activation of NMDARs. We found that while transient NMDAR activation still evoked a significant inhibition of IA, this effect was significantly blunted when compared to control rats subjected to the same experimental protocol (i.e. data points shown in Fig. 2B; 2-way ANOVA = experimental group × IA sweep; Fgroup: 5.6, P < 0.0001; 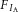 sweep: 21.2, P = 0.0001; interaction: F = 1.8, P = 0.03; Fig. 5). No differences in the magnitude of the evoked INMDA were observed between MNCs in control and dehydrated rats.
sweep: 21.2, P = 0.0001; interaction: F = 1.8, P = 0.03; Fig. 5). No differences in the magnitude of the evoked INMDA were observed between MNCs in control and dehydrated rats.
Figure 5. Blunted evoked NMDA–IA coupling in dehydrated rats.

Plot of the mean percentage NMDA-evoked inhibition of IA amplitude as a function of time obtained from MNCs (n = 11) recorded from dehydrated (48 h water-deprived) rats. Representative traces of IA recorded at the time points indicated are shown. *P < 0.05 vs. baseline IA (Dunnett's post hoc test).
The eNMDAR–IA functional coupling requires an increase in intracellular calcium and activation of PKC
We found that inhibition of IA, evoked either by bath-applied DHK or by a transient NMDA puff, was largely blunted in neurons dialysed with the fast Ca2+ chelator BAPTA (10 mm) (Fig. 6A, B, C and D). No differences in the magnitude of the evoked INMDA or IDHK were observed between MNCs dialysed with or without BAPTA (Fig. 6E). A similar block of the NMDA–IA functional coupling was found in MNCs dialysed with the slower Ca2+ chelator EGTA (5 mm) (97.32 ± 16.67 and 91.35 ± 14.23 pA pF–1, before and after NMDA, respectively, P > 0.41, paired t test, n = 10).
Figure 6. The eNMDAR-mediated inhibition of IA is Ca2+ dependent.
A, representative trace showing a DHK-induced inward shift in Iholding and lack of IA inhibition in an MNC dialysed with BAPTA (10 mm). B, a focal and brief application of NMDA (20 μm, 5 s) also failed to inhibit IA in an MNC dialysed with BAPTA. IA was isolated electronically by digital subtraction of currents evoked from two separate protocols, in which preconditioning steps of either −80 mV and −10 mV were used, to fully evoke, and to completely inactivate IA, respectively. C, mean (left) and before–after (right) plots of IA density (at 0 mV) before (open symbols) and after (filled symbols) DHK (300 μm) application in the presence of BAPTA in the patch pipette (n = 8). D, mean (left) and before–after (right) plots of IA density (at 0 mV) before (open symbols) and after (filled symbols) NMDA (20 μm) application in the presence of BAPTA in the patch pipette (n = 6). E, mean NMDA and DHK-evoked current density with and without BAPTA in the patch pipette.
To determine if the NMDAR–[Ca2+]i-mediated inhibition of IA involved a Ca2+-dependent, protein kinase C (PKC)-dependent pathway, we carried out a similar experiment in which NMDARs were transiently activated in MNCs dialysed with the PKC inhibitor chelerythrine chloride (10 mm). We found that while NMDAR activation still significantly inhibited IA magnitude (135.68 ± 25.49 and 125.74 ± 22.45 pA pF–1, before and after NMDA, respectively, n = 9, P = 0.02 paired t test), the degree of inhibition was significantly smaller than that observed in MNCs under control conditions (see Fig. 2C): control −24.7 ± 7.0%; PKC: −6.0 ± 1.6%, n = 11 and 9, respectively, P = 0.03 unpaired t test).
Activation of eNMDAR enhances the steady-state voltage-dependent inactivation of IA
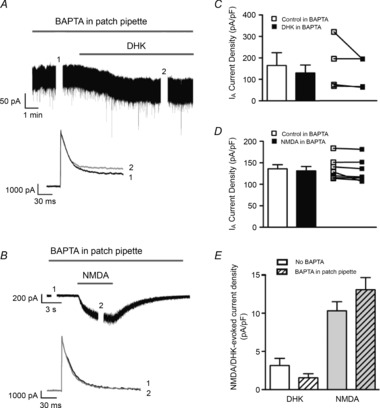
We previously showed that an increased voltage-dependent inactivation of IA was a main factor contributing to blunted IA magnitude in hypothalamic neurons during hypertension (Sonner et al. 2008, 2011), a pathological condition known to also involve an increased NMDAR activity within the hypothalamus (Li & Pan, 2007; Li et al. 2008). Thus, we explored whether the eNMDAR–IA functional coupling involved changes in IA inactivation properties. To evoke variable amounts of IA inactivation, neurons were depolarized using a command pulse to 10 mV, from a range of conditioning steps (−120 to −30 mV, 10 mV increments, 300 ms duration, Fig. 7A). Blockade of GLT1 transporters shifted the voltage dependence of inactivation of IA towards a more hyperpolarized membrane potential (leftward shift, Fig. 7B), reflected also as a significant hyperpolarization of the V½ inactivation potential (P < 0.01, paired t test, Fig. 7C). This is indicative of a higher degree of IA inactivation at any given Vm.
Figure 7. Blockade of astrocyte GLT1 transporters increases IA steady-state inactivation.
A, representative example of IA following voltage-dependent inactivation protocols (inset) before and during application of DHK in an MNC. B, plot of normalized IA amplitude vs. inactivating conditioning voltage steps from the same neuron shown in A, before and during DHK, showing a clear hyperpolarizing shift in the curve in DHK. C, mean (left) and before–after (right) plots of IA half-inactivation potential before and during DHK-mediated activation of NMDARs (n = 6). D, mean DHK-mediated change in V½ inactivation in the presence and absence of BAPTA in the patch pipette. ***P < 0.0001, unpaired t test; **P < 0.01, paired t test.
Finally, in MNCs dialysed with BAPTA, DHK failed to significantly shift the V½ inactivation (−39.87 ± 6.5 mV and −35.01 ± 4.6 mV before and after DHK, respectively, P > 0.5, unpaired t test, n = 9, see Fig. 7D).
The eNMDAR–IA coupling enhances repetitive firing discharge of MNCs
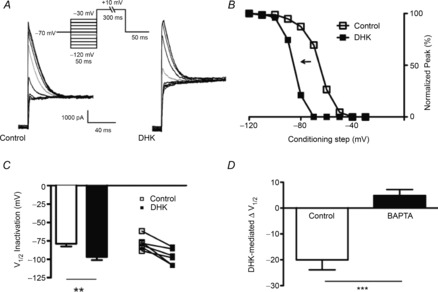
To determine the functional consequences of the GLT1–eNMDAR–IA signalling, we measured repetitive firing activity properties of MNCs before and after GLT1 blockade. The mean input resistance of neurons in this subset of experiments was 569.2 ± 33.6 MΩ. We found that the input–output function following bath-applied DHK was significantly strengthened, resulting in an overall increased number of evoked action potentials as a function of the injected current (2-way ANOVA = drug × current injection; Fdrug: 24.02, P < 0.0001; Fcurrent: 36.55, P < 0.0001; interaction: F = 0.95, P = 0.5, n = 9, Fig. 8A and C). In a different set of recordings, we found that DHK effects were prevented in the presence of the A-type K+ channel blocker 4-AP (2.5 mm) (2-way ANOVA = drug × current injection; Fdrug: 0.5, P = 0.5; Fcurrent: 88.2, P < 0.0001; interaction: F = 0.5, P = 0.8, n = 6, Fig. 8D). Similarly, depolarizing steps of increasing duration (175 pA, from 200 ms to 1800 ms, 200 ms increment) evoked more spikes in the presence of DHK (2-way ANOVA = drug × current injection; Fdrug: 24.0, P < 0.0001; Fduration: 36.6, P < 0.0001; interaction: F = 0.9, P = 0.5, n = 9, Fig. 8F and G), an effect prevented by 4-AP (2-way ANOVA = drug × current injection; Fdrug: 0.24, P = 0.6; Fduration: 11.3, P < 0.0001; interaction: F = 0.02, P = 1.0, n = 6, Fig. 8H).
Figure 8. The eNMDAR–IA functional coupling enhances MNC repetitive firing discharge.
A, sample of firing activity evoked in an MNC in control ACSF. B, sample traces from the same neuron (150 pA step) before and during application of DHK. Note the reduced delay to the first spike (duration between two arrows) and the increased number of evoked action potentials in DHK. C, plot of the mean number of evoked spike numbers vs. current step amplitude before and during DHK application (n = 9). D, similar plot to that in C but in the presence of 2.5 mm 4-AP (n = 6). E, bar graph summarizing the mean delay to the first evoked spike (150 pA step) in DHK in the presence and absence of 4-AP. F, sample of firing activity evoked by successively longer depolarizing pulses (200 ms increments) in an MNC in control ACSF. G, plot of the mean number of evoked spikes vs. pulse duration before and during DHK application (n = 9). H, similar plot to that in F but in the presence of 4-AP (n = 6). I, representative traces (175 pA, 400 ms step) before and during DHK application. J, bar graph summarizing the mean delay to the first evoked spike in DHK (200 ms step duration). *P < 0.05 vs. respective control (Bonferroni's post hoc test); #P < 0.05 vs. control (paired t test).
We found that the latency to the first evoked spike, a property in MNCs largely determined by the transient IA activation (Bourque, 1988) was significantly shortened during DHK (P < 0.05, paired t test, Fig. 8E and J), an effect prevented in the presence of 4-AP (Fig. 8E). Spike threshold did not change significantly in the presence of DHK (Δ −2.2 ± 14.9 mV).
The increased evoked firing activity following NMDAR activation was also observed following a transient, focal application of NMDA (20 μm, 5 s, n = 17, Fig. 9A–C), which occurred concomitantly with a diminished delay to the first evoked spike (Fig. 9B, before NMDA: 0.13 ± 0.01 s; after NMDA: 0.09 ± 0.01 s, P < 0.01, paired t test).
Figure 9. Transient activation of NMDARs potentiates subsequently evoked firing activity.
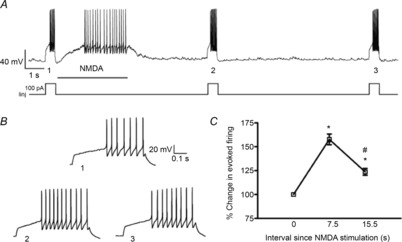
A, sample of repetitive firing activity evoked before and after transient activation of NMDA receptors (20 μm, 3.5 s) in an MNC. Iinj: current injected. B, the evoked firing for the periods 1 and 2 and 3 from panel A are shown at an expanded time scale. Note the shorter delay to the first evoked spike during the second depolarization (2) and the partial recovery during the third depolarization (3). C, plot of mean percentage changes in the evoked number of spikes following transient NMDAR activation (n = 17). *P < 0.001 vs. basal depolarization; #P < 0.01 vs. 7.5 s (Bonferroni's post hoc test).
Discussion
We report here a novel signalling mechanism that involves interactions among three major components influencing neuronal and network activity: astrocytes, intrinsic membrane properties and synaptic mechanisms. Specifically, our studies show that astrocytes, via the activity of GLT1 transporters, influence the degree of activation of a functional coupling between eNMDARs and the voltage-gated K+ current IA. Moreover, we show that this functional coupling enhances NMDAR efficacy, and increases hypothalamic neuronal activity in response to membrane depolarization.
Extrasynaptic NMDA receptor activation under astrocyte control inhibits the magnitude of IA
Our primary finding in this work is a significant inhibition of IA following NMDAR activation. Similar to other CNS neurons (Hardingham & Bading, 2010), we recently reported two molecularly and functionally distinct pools of NMDARs in MNCs: synaptic and extrasynaptic NMDARs (Fleming et al. 2011; Potapenko et al. 2013; Fig. 10). Synaptic NMDARs display a high affinity for glutamate, and rapidly desensitize following sustained activation. Conversely, eNMDARs display lower affinity, but do not desensitize in the presence of sustained glutamate levels. Our results support a major contribution of eNMDARs to the NMDAR–IA coupling. Even though focal and bath-applied NMDA accessed both types of NMDARs, the rapid inactivation of sNMDARs probably minimized their participation in this phenomenon. Moreover, we recently showed that eNMDARs in hypothalamic neurons, including MNCs, are tonically activated by extracellular glutamate, whose levels are tightly controlled by astrocyte GLT1 glutamate transporters. Thus, blockade of GLT1 function resulted in a build-up of extracellular glutamate and activation of eNMDARs, leading to a persistent tonic current (tonic INMDA) and a concomitant increase in neuronal firing discharge (Fleming et al. 2011). Here, we found that, similar to direct NMDA application, astrocyte GLT1 blockade lead to IA inhibition. The fact that this effect persisted when synaptic NMDARs were blocked (i.e. 4-AP/MK801 protocol) (Hardingham et al. 2002; Jung et al. 2008; Hardingham & Bading, 2010), but was blunted when eNMDARs were rendered inactive by removing the eNMDAR (but not sNMDAR) co-agonist glycine (Papouin et al. 2012), further supports a contribution of eNMDARs to the NMDA–IA functional coupling. This is in agreement with a recent study in cultured hippocampal neurons showing that activation of extrasynaptic (but not synaptic) NMDARs resulted in the downregulation of Kv4.2 channel subunits expression (Lei et al. 2010). However, whether activation of synaptic NMDARs during robust and synchronous synaptic activity can lead to IA inhibition was not explored in this study. The importance of this mechanism is further supported by the fact that blockade of NMDARs per se increased the basal magnitude of IA, suggesting that persistent activation of a small proportion of eNMDARs by ambient glutamate is sufficient to continuously suppress IA activity.
Figure 10. Schematic model showing the proposed functional coupling between eNMDARs and IA, via Ca2+-dependent activation of PKC.
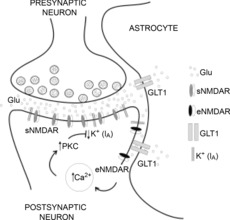
The model depicts the role of astrocyte GLT1 transporters in regulating the ability of ambient extracellular glutamate (Glu) to activate eNMDARs, which in a Ca2+- and PKC-dependent manner results in inhibition of IA.
It could be argued that the inhibition of IA following prolonged NMDAR activation was due to changes in overall membrane conductance during sustained NMDAR activation. Several pieces of evidences argue against this, however, including: (a) that transient NMDAR activation leads to a similar IA inhibition observed during sustained NMDAR activation; (b) the fact that the NMDA-mediated inhibition of IA was blocked by different treatments (i.e. BAPTA, EGTA, PKC inhibition) in which the relatively large change in basal NMDA-mediated change in conductance was still present; and (c) a lack of a significant correlation between the magnitude of the evoked INMDA and the associated percentage changes in IA. Taken together, these results show that activation of eNMDARs inhibits IA, and underscore the ability of astrocytes to influence the degree of activation and efficacy of the eNMDAR–IA coupling.
The eNMDAR–IA functional coupling is Ca2+- and PKC-dependent
We found that the eNMDAR–IA functional coupling was blocked when cells were dialysed with a slow (EGTA) or a fast-acting (BAPTA) Ca2+ chelator, supporting the contention that NMDAR- IA inhibition was dependent on a rise of intracellular Ca2+. The fact that similar results were obtained with both Ca2+ chelators suggests that the functional coupling did not involve a local Ca2+ microdomain. This argues against an intimate physical interaction between NMDARs and A-type channels, and suggests that brief and transient activation of sNMDARs during physiological levels of synaptic activity may not generate a sufficiently large rise in global Ca2+ to inhibit IA. However, alternative approaches such as co-immunoprecipitation are needed to more conclusively determine whether eNMDARs or sNMDARs and IA channel subunits are physically coupled in these neurons. We also found the NMDA–IA functional coupling to be blunted in MNCs dialysed with a PKC blocker. Given previous studies showing eNMDAR-mediated Ca2+-dependent activation of PKC (Sun & Liu, 2007), and that Ca2+-dependent PKC activity influenced IA properties, including inactivation (Ritter et al. 2012), it is then reasonable to speculate that the NMDAR–IA coupling in SON neurons involved a Ca2+-dependent activation of PKC. While NMDAR activation did not affect IA voltage-dependent activation properties, it increased IA voltage-dependent steady-state inactivation at membrane potentials (Vm) ≥−80 mV. Thus, as we recently reported in presympathetic PVN neurons of hypertensive rats (Sonner et al. 2008), the enhanced steady-state inactivation at these Vm values could account, at least in part, for the diminished IA availability and magnitude following NMDAR activation. It is important to note, however, that the NMDA–IA functional coupling was still observed when steady-state inactivation was completely removed (see Fig. 6A), suggesting that a factor other than inactivation (e.g. A-type channel internalization; Kim et al. 2007), could also contribute to the NMDAR-mediated IA inhibition.
Functional implications of the NMDAR–IA coupling
Besides influencing interspike intervals during repetitive firing, activation of IA in MNCs underlies a transient outward rectification that delays the onset of spiking upon membrane depolarization (Hu & Bourque, 1992; Luther & Tasker, 2000). Within this context, we found that astrocyte GLT1 blockade or transient pharmacological NMDAR activation both diminished the delay to spike onset, increasing in turn the number of evoked spikes. These effects were blocked by the A-type K+ channel blocker 4-AP. Pharmacological block of IA with 4-AP would be expected per se to diminish the delay to the first evoked spike (Fisher et al. 1998), and to increase firing activity. In our studies, however, we did not observe a significant difference in these parameters when comparing independent recordings obtained from different cells in the presence or absence of 4-AP. This was probably due to cell–cell variability in resting membrane potential and cell input resistance. This limitation in our experimental approach, however, did not preclude our ability to clearly demonstrate that 4-AP efficiently occluded DHK effects on firing activity. Similar results and conclusions to ours were obtained in comparable studies that evaluated the effects of other neurotransmitters/receptors, including activation of metabotropic glutamate receptors (Schrader & Tasker, 1997) and angiotensin II (Ma et al. 2006).
Taken together, our results indicate that in addition to its effect via direct membrane depolarization, the negative functional coupling to IA further contributes to NMDA-mediated increased membrane excitability and firing discharge in MNCs. Moreover, our studies show that the efficacy of this process is modulated by the local astrocyte microenvironment. In this sense, we recently showed that activity-dependent retraction of astrocyte processes during dehydration resulted in blunted GLT1 glutamate buffering and enhanced activation of eNMDARs (Fleming et al. 2011). Here, we show that this physiological challenge also led to an enhanced tonic activation of the NMDAR–IA signalling pathway in MNCs, resulting in (a) a basally diminished IA magnitude, and (b) an occluded response to an additional NMDAR activation. These results together support an increased tonic occupancy of eNMDARs and a concomitant inhibition of IA due to build-up of extracellular glutamate secondary to blunted GLT1 efficacy during dehydration (Fleming et al. 2011).
The NMDA–IA coupling may not only have functional consequences at the single cell level, but also at the entire population level. Similar to the hippocampus (Kim et al. 2005; Chen et al. 2006), A-type K+ channels in hypothalamic neurons modulate dendritic excitability, and we showed that their inhibition facilitates dendritic Ca2+ signalling and propagation (Sonner et al. 2011). A rise in dendritic Ca2+ in MNCs results in the local release of oxytocin and vasopressin (Ludwig & Leng, 2006), a phenomenon that can be locally initiated by NMDAR activation (de Kock et al. 2004; Son et al. 2013). Thus, by inhibiting IA and enhancing Ca2+ signalling, NMDAR activation would further potentiate dendritic release. This effect is expected to have important functional implications in hypothalamic information processing. Dendritic peptide release is not only involved in the autoregulation and optimization of MNC firing activity (Ludwig & Leng, 1997; Gouzenes et al. 1998), but we recently showed this to be a critical mechanism mediating neurosecretory-to-presympathetic interpopulation crosstalk during physiological challenges (Son et al. 2013). Thus, it will be important in future studies to determine the impact of the bidirectional NMDA–IA functional coupling in the generation of multimodal homeostatic hypothalamic responses.
In summary, our studies support a novel signalling mechanism involving a functional coupling between extrasynaptic NMDARs and transient A-type K+ channels, whose degree of activation is modulated by local astrocytes, and which plays an important role in the regulation of magnocellular neurosecretory neuronal excitability.
Glossary
- 4-AP
4-aminopyridine
- AP5
dl-2-amino-5-phosphonopentanoic acid
- DHK
dihydrokainic acid
- DIC
diffraction interference contrast
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- eNMDARs
extrasynaptic NMDARs
- IA
A-type potassium current
- Iholding
holding current
- IKDR
delayed-rectifier current
- INMDA
NMDA current
- MNC
magnocellular neurosecretory cell
- NMDAR
NMDA receptor
- PKC
protein kinase C
- sNMDAR
synaptic NMDAR
- SON
supraoptic nucleus
- SR
series resistance
- Vm
membrane potential
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Both authors contributed to the conception and design of the experiments, collection, analysis and interpretation of data, and drafting the article.
Funding
This work was supported by a National Heart, Lung, and Blood Institute Grant NIH HL112225 to J.E.S.
References
- Araque A, Navarrete M. Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci. 2010;365:2375–2381. doi: 10.1098/rstb.2009.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong WE, Smith BN. Tuberal supraoptic neurons–II. Electrotonic properties. Neuroscience. 1990;38:485–494. doi: 10.1016/0306-4522(90)90044-5. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Linn DM, Halmos KC, Tasker JG. Increased tonic activation of presynaptic metabotropic glutamate receptors in the rat supraoptic nucleus following chronic dehydration. J Physiol. 2003;551:815–823. doi: 10.1113/jphysiol.2003.042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW. Transient calcium-dependent potassium current in magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol. 1988;397:331–347. doi: 10.1113/jphysiol.1988.sp017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalis M, Dayanithi G, Nordmann JJ. The role of patterned burst and interburst interval on the excitation-coupling mechanism in the isolated rat neural lobe. J Physiol. 1985;369:45–60. doi: 10.1113/jphysiol.1985.sp015887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Burnashev N, Lodder JC, Mansvelder HD, Brussaard AB. NMDA receptors induce somatodendritic secretion in hypothalamic neurones of lactating female rats. J Physiol. 2004;561:53–64. doi: 10.1113/jphysiol.2004.069005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher TE, Voisin DL, Bourque CW. Density of transient K+ current influences excitability in acutely isolated vasopressin and oxytocin neurones of rat hypothalamus. J Physiol. 1998;511:423–432. doi: 10.1111/j.1469-7793.1998.423bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming TM, Scott V, Naskar K, Joe N, Brown CH, Stern JE. State-dependent changes in astrocyte regulation of extrasynaptic NMDA receptor signalling in neurosecretory neurons. J Physiol. 2011;589:3929–3941. doi: 10.1113/jphysiol.2011.207340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Hernandez J, Galarraga E, Pineda JC, Bargas J. Patterns of excitatory and inhibitory synaptic transmission in the rat neostriatum as revealed by 4-AP. J Neurophysiol. 1994;72:2246–2256. doi: 10.1152/jn.1994.72.5.2246. [DOI] [PubMed] [Google Scholar]
- Giaume C. Astroglial wiring is adding complexity to neuroglial networking. Front Neuroenergetics. 2010;2:129. doi: 10.3389/fnene.2010.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Mulligan SJ, MacVicar BA. Astrocyte control of the cerebrovasculature. Glia. 2007;55:1214–1221. doi: 10.1002/glia.20543. [DOI] [PubMed] [Google Scholar]
- Gouzenes L, Desarmenien MG, Hussy N, Richard P, Moos FC. Vasopressin regularizes the phasic firing pattern of rat hypothalamic magnocellular vasopressin neurons. J Neurosci. 1998;18:1879–1885. doi: 10.1523/JNEUROSCI.18-05-01879.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Perlmutter LS, Salm AK, Tweedle CD. Dynamic neuronal-glial interactions in hypothalamus and pituitary: implications for control of hormone synthesis and release. Peptides. 1984;5(Suppl. 1):121–138. doi: 10.1016/0196-9781(84)90271-7. [DOI] [PubMed] [Google Scholar]
- Hu B, Bourque CW. NMDA receptor-mediated rhythmic bursting activity in rat supraoptic nucleus neurones in vitro. J Physiol. 1992;458:667–687. doi: 10.1113/jphysiol.1992.sp019440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SC, Kim J, Hoffman DA. Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron. 2008;60:657–671. doi: 10.1016/j.neuron.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Deng P, Li Y, Xu ZC. Downregulation of Kv4.2 channels mediated by NR2B-containing NMDA receptors in cultured hippocampal neurons. Neuroscience. 2010;165:350–362. doi: 10.1016/j.neuroscience.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Pan HL. Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension. 2007;49:916–925. doi: 10.1161/01.HYP.0000259666.99449.74. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL. Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol. 2008;586:1637–1647. doi: 10.1113/jphysiol.2007.149732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Leng G. Autoinhibition of supraoptic nucleus vasopressin neurons in vivo: a combined retrodialysiselectrophysiological study in rats. Eur J Neurosci. 1997;9:2532–2540. doi: 10.1111/j.1460-9568.1997.tb01682.x. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. 2006;7:126–136. doi: 10.1038/nrn1845. [DOI] [PubMed] [Google Scholar]
- Luther JA, Tasker JG. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2000;523:193–209. doi: 10.1111/j.1469-7793.2000.t01-1-00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Bielefeldt K, Tan ZY, Whiteis CA, Snitsarev V, Abboud FM, Chapleau MW. Dual mechanisms of angiotensin-induced activation of mouse sympathetic neurons. J Physiol. 2006;573:45–63. doi: 10.1113/jphysiol.2006.106716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen R, Hu B, Renaud LP. Regulation of spontaneous phasic firing of rat supraoptic vasopressin neurones in vivo by glutamate receptors. J Physiol. 1995;484:415–424. doi: 10.1113/jphysiol.1995.sp020674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Tweedle CD, Hatton GI. Neuronalglial plasticity in the supraoptic dendritic zone: dendritic bundling and double synapse formation at parturition. Neuroscience. 1984;13:769–779. doi: 10.1016/0306-4522(84)90095-2. [DOI] [PubMed] [Google Scholar]
- Potapenko ES, Biancardi VC, Zhou Y, Stern JE. Astrocytes modulate a postsynaptic NMDA-GABAA-receptor crosstalk in hypothalamic neurosecretory neurons. J Neurosci. 2013;33:631–640. doi: 10.1523/JNEUROSCI.3936-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DM, Ho C, O'Leary ME, Covarrubias M. Modulation of Kv3.4 channel N-type inactivation by protein kinase C shapes the action potential in dorsal root ganglion neurons. J Physiol. 2012;590:145–161. doi: 10.1113/jphysiol.2011.218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Schrader LD, Tasker JG. Modulation of multiple potassium currents by metabotropic glutamate receptors in neurons of the hypothalamic supraoptic nucleus. J Neurophysiol. 1997;78:3428–3437. doi: 10.1152/jn.1997.78.6.3428. [DOI] [PubMed] [Google Scholar]
- Son SJ, Filosa JA, Potapenko ES, Biancardi VC, Zheng H, Patel KP, Tobin VA, Ludwig M, Stern JE. Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron. 2013;78:1036–1049. doi: 10.1016/j.neuron.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonner PM, Filosa JA, Stern JE. Diminished A-type potassium current and altered firing properties in presympathetic PVN neurones in renovascular hypertensive rats. J Physiol. 2008;586:1605–1622. doi: 10.1113/jphysiol.2007.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonner PM, Lee S, Ryu PD, Lee SY, Stern JE. Imbalanced K+ and Ca2+ subthreshold interactions contribute to increased hypothalamic presympathetic neuronal excitability in hypertensive rats. J Physiol. 2011;589:667–683. doi: 10.1113/jphysiol.2010.198556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE. Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2001;537:161–177. doi: 10.1111/j.1469-7793.2001.0161k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE, Armstrong WE. Reorganization of the dendritic trees of oxytocin and vasopressin neurons of the rat supraoptic nucleus during lactation. J Neurosci. 1998;18:841–853. doi: 10.1523/JNEUROSCI.18-03-00841.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu SJ. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Di S, Boudaba C. Functional synaptic plasticity in hypothalamic magnocellular neurons. Prog Brain Res. 2002;139:113–119. doi: 10.1016/s0079-6123(02)39011-3. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Oliet SH, Bains JS, Brown CH, Stern JE. Glial regulation of neuronal function: from synapse to systems physiology. J Neuroendocrinol. 2012;24:566–576. doi: 10.1111/j.1365-2826.2011.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA. Activity-dependent neuronal-glial and synaptic plasticity in the adult mammalian hypothalamus. Neuroscience. 1993;57:501–535. doi: 10.1016/0306-4522(93)90002-w. [DOI] [PubMed] [Google Scholar]
- Tweedle CD, Hatton GI. Synapse formation and disappearance in adult rat supraoptic nucleus during different hydration states. Brain Res. 1984;309:373–376. doi: 10.1016/0006-8993(84)90607-3. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]