

ABSTRACT
Mycobacterial evolution involves various processes, such as genome reduction, gene cooption, and critical gene acquisition. Our comparative genome size analysis of 44 mycobacterial genomes revealed that the nonpathogenic (NP) genomes were bigger than those of opportunistic (OP) or totally pathogenic (TP) mycobacteria, with the TP genomes being smaller yet variable in size—their genomic plasticity reflected their ability to evolve and survive under various environmental conditions. From the 44 mycobacterial species, 13 species, representing TP, OP, and NP, were selected for genomic-relatedness analyses. Analysis of homologous protein-coding genes shared between Mycobacterium indicus pranii (NP), Mycobacterium intracellulare ATCC 13950 (OP), and Mycobacterium tuberculosis H37Rv (TP) revealed that 4,995 (i.e., ~95%) M. indicaus pranii proteins have homology with M. intracellulare, whereas the homologies among M. indicus pranii, M. intracellulare ATCC 13950, and M. tuberculosis H37Rv were significantly lower. A total of 4,153 (~79%) M. indicus pranii proteins and 4,093 (~79%) M. intracellulare ATCC 13950 proteins exhibited homology with the M. tuberculosis H37Rv proteome, while 3,301 (~82%) and 3,295 (~82%) M. tuberculosis H37Rv proteins showed homology with M. indicus pranii and M. intracellulare ATCC 13950 proteomes, respectively. Comparative metabolic pathway analyses of TP/OP/NP mycobacteria showed enzymatic plasticity between M. indicus pranii (NP) and M. intracellulare ATCC 13950 (OP), Mycobacterium avium 104 (OP), and M. tuberculosis H37Rv (TP). Mycobacterium tuberculosis seems to have acquired novel alternate pathways with possible roles in metabolism, host-pathogen interactions, virulence, and intracellular survival, and by implication some of these could be potential drug targets.
IMPORTANCE
The complete sequence analysis of Mycobacterium indicus pranii, a novel species of Mycobacterium shown earlier to have strong immunomodulatory properties and currently in use for the treatment of leprosy, places it evolutionarily at the point of transition to pathogenicity. With the purpose of establishing the importance of M. indicus pranii in providing insight into the virulence mechanism of tuberculous and nontuberculous mycobacteria, we carried out comparative genomic and proteomic analyses of 44 mycobacterial species representing nonpathogenic (NP), opportunistic (OP), and totally pathogenic (TP) mycobacteria. Our results clearly placed M. indicus pranii as an ancestor of the M. avium complex. Analyses of comparative metabolic pathways between M. indicus pranii (NP), M. tuberculosis (TP), and M. intracellulare (OP) pointed to the presence of novel alternative pathways in M. tuberculosis with implications for pathogenesis and survival in the human host and identification of new drug targets.
INTRODUCTION
The evolution of Mycobacterium species is usually driven by processes, including deletion (nonfunctional genes are deleted/inactivated and subsequently eroded), insertion (horizontal transfer and gene duplication), or a combination of these events, which aid in survival under different environmental conditions or geographic niches (1–8). In nature, the free-living species require larger genomes than parasitic species (9, 10). This trend is also clearly evident from analyses of mycobacterial genomes where a distinct pattern of decreasing genomic content is seen as one moves from nonpathogenic pathogens (NP) to opportunistic pathogens (OP) to true pathogens (TP). We therefore performed genome size analysis with 44 Mycobacterium strains (Table 1) that represented NP, OP, and TP, and our analysis revealed that NP strains on average are bigger than those of OP and TP strains. One of the largest genomes in the Mycobacterium genus is that of Mycobacterium smegmatis, a nonpathogenic mycobacterium with approximately 6,717 protein-coding genes (genome size, 6.9 Mb). On the other extreme is a true pathogenic mycobacterium, Mycobacterium leprae (the leprosy bacterium), with the smallest genome, of approximately 2,770 protein-coding genes (genome size, 3.3 Mb) and approximately 1,600 functional and 1,100 nonfunctional/inactive genes (11).
TABLE 1 .
Mycobacterial genomes selected for analysis
| Organism | KEGG name | Yr of sequencing | No. ofgenes | Genomesize (bp) | Pathogenicity |
|---|---|---|---|---|---|
| Mycobacterium smegmatis MC2 155 uid57701a | msm | 2006 | 6,938 | 6,988,209 | NP |
| Mycobacterium smegmatis MC2 155 uid171958 | msg | 2012 | 6,742 | 6,988,208 | NP |
| Mycobacterium vanbaalenii PYR 1a | mva | 2006 | 6,136 | 6,491,865 | NP |
| Mycobacterium sp. KMS | mkm | 2006 | 6,079 | 6,256,079 | NP |
| Mycobacterium sp. JLS | mjl | 2007 | 5,842 | 6,048,425 | NP |
| Mycobacterium gilvum PYR-GCKa | mgi | 2007 | 5,669 | 5,982,829 | NP |
| Mycobacterium sp. MCS | mmc | 2006 | 5,698 | 5,920,523 | NP |
| Mycobacterium gilvum Spyr1 | msp | 2010 | 5,552 | 5,783,292 | NP |
| Mycobacterium indicus prania | mid | 2012 | 5,318 | 5,589,007 | NP |
| Mycobacterium sp. JDM601 | mjd | 2011 | 4,398 | 4,643,668 | NP |
| Mycobacterium tuberculosis H37Ra | mra | 2007 | 4,084 | 4,419,977 | NP |
| Mycobacterium bovis BCG Pasteur 1173P2 | mbb | 2007 | 4,033 | 4,374,522 | NP |
| Mycobacterium bovis BCG Tokyo 172 | mbt | 2009 | 4,027 | 4,371,711 | NP |
| Mycobacterium bovis BCG Mexico | mbm | 2012 | 4,031 | 4,350,386 | NP |
| Mycobacterium rhodesiae | mrh | 2012 | 6,336 | 6,415,739 | OP |
| Mycobacterium chubuense | mcb | 2012 | 6,068 | 6,342,624 | OP |
| Mycobacterium sp. MOTT36Y | mmm | 2012 | 5,177 | 5,613,626 | OP |
| Mycobacterium intracellulare MOTT-64 | mir | 2012 | 5,297 | 5,501,090 | OP |
| Mycobacterium avium 104a | mav | 2006 | 5,313 | 5,475,491 | OP |
| Mycobacterium intracellulare MOTT-02 | mit | 2012 | 5,198 | 5,409,696 | OP |
| Mycobacterium intracellulare ATCC 13950a | mia | 2012 | 5,193 | 5,402,402 | OP |
| Mycobacterium abscessus ATCC 19977a | MAb | 2008 | 4,991 | 5,090,491 | OP |
| Mycobacterium massiliense | mmv | 2012 | 2,680 | 5,068,807 | OP |
| Mycobacterium avium paratuberculosis K-10a | mpa | 2004 | 4,399 | 4,829,781 | OP |
| Mycobacterium marinum Ma | mmi | 2008 | 5,570 | 6,660,144 | TP |
| Mycobacterium ulceransa | mul | 2006 | 5,062 | 5,805,761 | TP |
| Mycobacterium canettii | mce | 2011 | 3,982 | 4,482,059 | TP |
| Mycobacterium tuberculosis F11 | mtf | 2007 | 3,998 | 4,424,435 | TP |
| Mycobacterium tuberculosis UT205 | mtd | 2012 | 3,859 | 4,418,088 | TP |
| Mycobacterium tuberculosis H37Rv uid170532 | mtv | 2012 | 4,170 | 4,411,708 | TP |
| Mycobacterium tuberculosis H37Rv uid57777a | mtu | 1998 | 4,062 | 4,411,532 | TP |
| Mycobacterium tuberculosis RGTB423 | mti | 2012 | 3,670 | 4,406,587 | TP |
| Mycobacterium tuberculosis CCDC5180 | mtl | 2012 | 3,638 | 4,405,981 | TP |
| Mycobacterium tuberculosis CDC1551 | mtc | 2001 | 4,293 | 4,403,837 | TP |
| Mycobacterium tuberculosis KZN 605 | mtz | 2012 | 4,071 | 4,399,120 | TP |
| Mycobacterium tuberculosis CCDC5079 | mte | 2012 | 3,695 | 4,398,812 | TP |
| Mycobacterium tuberculosis CTRI-2 | mto | 2012 | 4,001 | 4,398,525 | TP |
| Mycobacterium tuberculosis KZN 1435 | mtb | 2009 | 4,107 | 4,398,250 | TP |
| Mycobacterium tuberculosis KZN 4207 | mtk | 2012 | 4,044 | 4,394,985 | TP |
| Mycobacterium africanum | maf | 2011 | 3,983 | 4,389,314 | TP |
| Mycobacterium tuberculosis RGTB327 | mtg | 2012 | 3,739 | 4,380,119 | TP |
| Mycobacterium bovis AF2122/97 a | mbo | 2003 | 4,001 | 4,345,492 | TP |
| Mycobacterium leprae TNa | mle | 2001 | 2,770 | 3,268,203 | TP |
| Mycobacterium leprae Br4923 | mlb | 2009 | 2,770 | 3,268,071 | TP |
Strain used for further analyses.
Mycobacterium indicus pranii (12) has been shown to have novel immunomodulatory properties (13–17) and proven therapeutic value in the treatment of leprosy (13, 14). The evolution of this clinically “benevolent” bacterium has been suggested to be at the point of transition to pathogenicity (12), despite earlier data from DNA sequence analysis of select genes, hsp70 (EU688981), gyrA (EU688980), and dnaJ (EU688982) of M. indicus pranii, which showed identity (99%) with corresponding genes of M. intracellulare. Comparative proteomic analyses of virulence factors of M. tuberculosis and their homologs in 12 different mycobacterial species, including M. indicus pranii, point toward gene cooption as an important mechanism in the evolution of mycobacteria (18). We now describe comparative proteomic analyses of 13 species of Mycobacterium, including M. indicus pranii (Table 2). The 13 Mycobacterium species were selected because they represented TP, OP, and NP. True pathogens, the most virulent mycobacteria, include Mycobacterium tuberculosis, the causative agent of human tuberculosis; Mycobacterium bovis, the causative agent of bovine tuberculosis; Mycobacterium leprae, the causative agent of leprosy, and a virulent nontuberculous mycobacterium (NTM), Mycobacterium ulcerans, which causes Buruli ulcers, which are the third most common mycobacterial disease in humans (19). Mycobacterium marinum, the causative agent of fish tank granuloma in humans and granulomatous lesions similar to those of M. tuberculosis in zebrafish, was also included in the true pathogen group for our analyses (20, 21). Opportunistic pathogens belong to the NTM group and cause pulmonary and other disseminated infections in immunocompromised individuals (22). Members of the Mycobacterium avium complex (MAC), Mycobacterium avium and Mycobacterium avium-M. intracellulare, cause opportunistic pulmonary infections in humans, whereas Mycobacterium avium subsp. paratuberculosis, the third member of the MAC group, is the suspected causative agent of Crohn’s disease in humans (22, 23). Mycobacterium abscessus is a rapid-growing mycobacterium which causes pulmonary and cutaneous infections in immunocompromised hosts (24). The nonpathogenic group includes Mycobacterium gilvum, Mycobacterium vanbaalenii, and Mycobacterium smegmatis, which rarely cause disseminated infections, even in immunocompromised individuals (25–27). Our results convincingly establish the very upstream evolutionary position of M. indicus pranii and also highlight some important differences in the metabolic pathway of M. tuberculosis H37Rv which are of possible significance in virulence and pathogenesis.
TABLE 2 .
The 13 Mycobacterium species included in the analyses
| Mycobacterium species | KEGG alias | Categorization based on virulence | NCBI RefSeq accession no. | No. of proteins |
|---|---|---|---|---|
| Mycobacterium tuberculosis H37Rv | MYCTU | True pathogen | NC_000962 | 4,003 |
| Mycobacterium bovis subsp. bovis AF2122/97 | MYCBO | True pathogen | NC_002945 | 3,918 |
| Mycobacterium leprae TN | MYCLE | True pathogen | NC_002677 | 1,605 |
| Mycobacterium ulcerans Agy99 | MYCUA | True pathogen | NC_005916, NC_008611 | 4,241 |
| Mycobacterium marinum M | MYCMM | True pathogen | NC_010604, NC_010612 | 5,452 |
| Mycobacterium avium 104 | MYCA1 | Opportunistic pathogen | NC_008595 | 5,120 |
| Mycobacterium intracellulare ATCC 13950 | MIA | Opportunistic pathogen | NC_016946 | 5,144 |
| Mycobacterium avium subsp. paratuberculosis K-10 | MYCPA | Opportunistic pathogen | NC_002944 | 4,350 |
| Mycobacterium abscessus ATCC 19977 | MYCAB | Opportunistic pathogen | NC_010394, NC_010397 | 4,941 |
| Mycobacterium indicus pranii MTCC 9506 | MIP | Nonpathogen | NC_018612 | 5,254 |
| Mycobacterium smegmatis MC2 155 | MYCS2 | Nonpathogen | NC_008596 | 6,717 |
| Mycobacterium gilvum PYR-GCK | MYCGI | Nonpathogen | NC_009338, NC_009339, NC_009340, NC_009341 | 5,579 |
| Mycobacterium vanbaalenii PYR-1 | MYCVP | Nonpathogen | NC_008726 | 5,979 |
RESULTS AND DISCUSSION
Reannotation of the M. indicus pranii proteome.
InterPro/Pfam domain knowledge for M. indicus pranii proteins was used to assign potential functions to 4,363 M. indicus pranii open reading frames (ORFs; ~83% of the M. indicus pranii proteome) (Fig. 1). Of the remaining 891 proteins, 164 were annotated using the phylogenetic classification of proteins encoded in complete genomes known as COG (Cluster of Orthologous Groups classification), but they failed to match with any domain in Pfam or InterPro. Previously, 3,870 (~70%) of M. indicus pranii ORFs were assigned a putative function on the basis of COG classification (Fig. 1). Out of 1,554 hypothetical proteins in M. indicus pranii based on the COG annotation, 656 have been assigned a putative function based on functional domain knowledge from the InterPro/Pfam database.
FIG 1 .

Comparative plot for annotation of M. indicus pranii (MIP) based on annotations in COG and InterPro/Pfam.
Interestingly, 60 proteins were found to have conflicting COG- and InterPro/Pfam-based annotations. In such ambiguous cases, the protein sequences were further submitted to analysis using GENE3D to further confirm the annotation. GENE3D upheld the Pfam/InterPro annotation for all except two cases (MIP_02898 and MIP_06278), for which no hit was found in GENE3D. COG and Pfam/InterPro annotations of these 2 proteins have no link in the existing literature or protein family knowledge (see Table S1 in the supplemental material). Thus, annotating a new proteome using Interpro not only provides better annotation coverage but also increases the confidence of annotation by providing in-depth knowledge regarding domains, motifs, and a structural annotation of the given protein sequence.
Comparative genome size analysis.
The complete genome sequences of the 44 mycobacterial species used in our analyses were available in the public domain. The Mycobacterium sp. MOTT36Y (5,613,626 bp) represents the OP group of mycobacteria closest to M. indicus pranii (5,589,007 bp) in terms of genome size. Among the OP group of mycobacteria, those closest to Mycobacterium intracellulare (28) (5,402,402 to 5,501,090 bp) are Mycobacterium sp. MOTT36Y (5,613,626 bp), Mycobacterium avium 104 (5,475,491 bp), and Mycobacterium abscessus ATCC 19977 (5,090,491 bp). It is interesting that based on the genome size, the genome of M. avium 104, an OP, fits between M. intracellulare MOTT-64 (5,501,090 bp) and M. intracellulare MOTT-02 (5,409,696 bp) (Fig. 2).
FIG 2 .
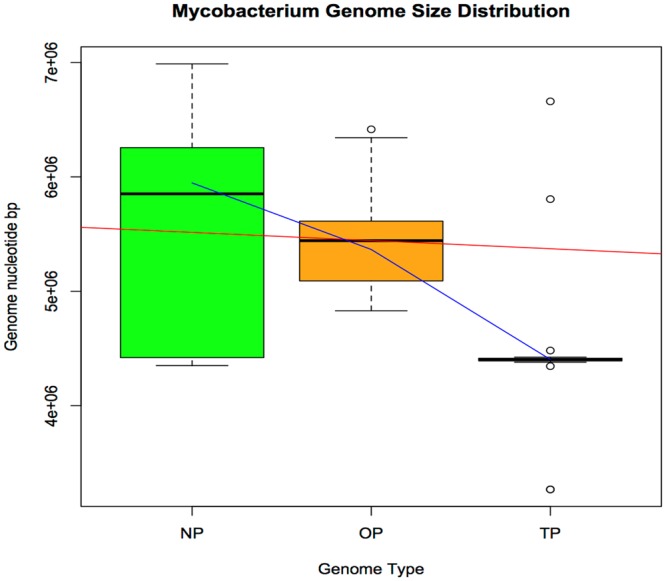
The genome size of NP mycobacteria is larger than that of OP and TP mycobacteria. The green box plot represents the NP genome sizes, the orange box represents OP genomes, and TP genomes are represented by the pink box, which is very tight. The red line is the regression line, and the blue line is the lowess line. The small circles denote outliers. The TP genomes were on average smaller yet variable in size. The genome plasticity for TP mycobacteria possibly highlights their abilities to evolve and survive under various environmental conditions.
Sequence-based functional analysis.
Homologs obtained by BLASTp analysis were assigned functional relationships by comparing their Interpro/Pfam functional domains. About 90% functional similarity between proteins can be observed if their sequences are at least 60% identical (29); neither the percentage of sequence identity nor expectation value can give complete insight into the relationship between two proteins (30). Taking these reports into account, we performed an analysis to establish functional assignments based on InterPro/Pfam domain hits to the sequence identity data of BLASTp between proteins of M. indicus pranii and 12 other Mycobacterium species (Table 2; Fig. 3A). Our analyses revealed that Interpro/Pfam hits indicated M. indicus pranii to be most closely related to members of the Mycobacterium avium complex, with M. intracellulare and M. avium 104 having 77.9% and 74.9% of proteins functionally similar to those in the M. indicus pranii proteome, respectively. The functional relatedness of homologs fits well into the upper left corner of the receiver operating characteristics (ROC) space, indicating high sensitivity and specificity, which qualifies the functional relatedness analyses as an optimal model (Fig. 3B).
FIG 3 .

(A) Homologs of M. indicus pranii proteins in 12 other Mycobacterium species were determined to be functionally related using InterPro/Pfam domain knowledge. (B) The ROC points are in the upper left corner, suggesting the findings are statistically significant. MIP, M. indicus pranii.
This analysis was further used to find a BLASTp sequence identity cutoff score below which no homologs shared a functional domain. In order to correlate the BLASTp sequence identities of homologs with their functional relatedness, numbers of true positives (tp), false positives (fp), true negatives (tn), and false negatives (fn) were distributed as per BLASTp sequence identity, ranging from 0 to 100% and using 10% as the bin size. When we plotted the true-positive rate (TPR) versus a range of lower sequence identity cutoffs (0 to 100%), it was observed that below a 20% sequence identity no homologs shared functional domains (see Fig. S1 in the supplemental material). This cutoff was further confirmed when we plotted the Matthews correlation coefficient (MCC) for a range of sequence identity cutoffs (0 to 100%). MCC values were zero, as calculated for the sequence identity cutoff of 20%, and exhibited a sharp rise at 30% sequence identity (see Fig. S2 in the supplemental material).
For further comparative analyses, a 20% sequence identity cutoff with an e value of <1e−4 was used to analyze BLASTp results. This cutoff was used to determine the number of homologous protein-coding genes shared between M. indicus pranii, M. intracellulare ATCC 13950, and M. tuberculosis H37Rv. Our analysis of homologous protein-coding genes shared between M. indicus pranii (NP), M. intracellulare ATCC 13950 (OP), and M. tuberculosis H37Rv (TP) revealed 4,995 (~95%) M. indicus pranii proteins had homology with M. intracellulare ATCC 13950, whereas homologies between M. indicus pranii, M. intracellulare ATCC 13950, and M. tuberculosis H37Rv were significantly lower. A total of 4,153 (~79%) M. indicus pranii proteins and 4,093 (~79%) M. intracellulare ATCC 13950 proteins exhibited homology with the M. tuberculosis H37Rv proteome, while 3,301 (~82%) and 3,295 (~82%) M. tuberculosis H37Rv proteins showed homology with M. indicus pranii and M. intracellulare ATCC 13950 proteomes, respectively (Fig. 4; see also Table S2 in the supplemental material).
FIG 4 .

Comparative genomics of selected mycobacterial genomes. The genomes of Mycobacterium indicus pranii (shown in green; an NP), Mycobacterium intracellulare ATCC 13950 (orange; an OP), and Mycobacterium tuberculosis H37Rv (pink; a TP) were selected for comparative genomic analyses. We used BLASTp, with a cutoff of 20% identity and e value of 10e–4, to determine the number of homologous protein-coding genes common between them (shown as edge labels between the nodes). The arrowhead represents the query genome, whereas the arrow tail represents the subject genome.
Comparative metabolic pathway analyses.
There were 387 enzymes (EC numbers) common between M. intracellulare ATCC 13950 and M. indicus pranii (part of the MAC complex). When these two genomes were compared to M. tuberculosis (part of the MTB complex), only 17 enzymes remained uniquely shared between the M. intracellulare ATCC 13950 and M. indicus pranii genomes (Fig. 5a). Compared to M. avium 104, only 12 enzymes remained uniquely shared between M. intracellulare ATCC 13950 and M. indicus pranii (Fig. 5b). Three enzymes, EC 1.8.7.1 (sulfite reductase [ferredoxin]) (31), EC 2.7.1.6 (galactokinase [phosphorylating]), and EC 5.4.2.8 (phospho mannose mutase) (32), were shared both between M. intracellulare ATCC 13950 and M. tuberculosis H37Rv and between M. intracellulare ATCC 13950 and M. avium 104. As these enzymes were absent from the M. indicus pranii genome and were shared between OP and TP, they may be linked to the pathogenesis of Mycobacterium tuberculosis.
FIG 5 .

The enzymatic similarities between M. indicus pranii (MIP), M. intracellulare ATCC 13950 (MIA), M. avium 104 (MYCA1), and M. tuberculosis H37Rv (MYCTU) highlight interesting enzymatic plasticity properties. M. intracellulare ATCC 13950 (OP; orange) shares three enzymes (EC 1.8.7.1, sulfite reductase [ferredoxin]; EC 2.7.1.6, galactokinase [phosphorylating]; EC 5.4.2.8, phosphor mannose mutase) with M. avium 104 (OP; blue) and M. tuberculosis H37Rv (TP; red), which are absent in M. indicus pranii (NP; green). M. intracellulare ATCC 13950 and M. indicus pranii share 17 enzymes between them that are absent in M. tuberculosis H37Rv (a), while they share 12 enzymes between them that are absent in M. avium 104 (b).
Although the genome sizes of OPs (M. intracellulare ATCC 13950 and M. avium 104) and TP (M. tuberculosis) are reduced compared to the genome size of M. indicus pranii (an NP), our analysis indicated that the OP and TP genomes have acquired few enzyme-coding genes. It is tempting to suggest a likely association between these acquired enzymes and the virulence of these OPs and TPs. One of the three shared enzymes, EC 1.8.7.1, which encodes a ferredoxin-dependent sulfite reductase (encoded by the nirA gene), is active during the dormant phase and has been reported to be a potential drug target for Mycobacterium tuberculosis (33).
Comparative metabolic pathway analysis (Fig. 6) between M. tuberculosis, M. intracellulare ATCC 13950, and M. indicus pranii showed the presence of alternate pathways, such as those in the fatty acid elongation pathway (fabH and fabK) and lipid biosynthesis. M. tuberculosis has acquired some novel pathways which involve 23 enzymes that are not present in M. indicus pranii or M. intracellulare ATCC 13950, such as those for butanoate metabolism, amino acid biosynthesis pathways, etc. (Fig. 6, shown in red). Alternate metabolic pathways must have evolved during mycobacterial evolution. M. tuberculosis H37Rv has few unique enzymes, which might be part of its evolutionary adaptation, and they thereby present potential drug targets. For example, gene Rv1771 (EC 1.1.3.8) is found in the ascorbate and aldarate metabolism pathways. Gene Rv3097c (EC 3.1.1.3) is an important precursor enzyme in the fatty acid pathway (Fig. 6b and c, highlighted by the cyan circle), which is absent in M. indicus pranii and M. intracellulare ATCC 13950. A few other examples include the genes Rv0069c (EC 4.3.1.17), Rv1905c (EC 1.4.3.3), Rv2192c (EC 2.4.2.18), Rv2006 (EC 3.2.1.28), Rv3393 (EC 3.2.2.1), and Rv0091 (EC 3.2.2.9), which are present in M. tuberculosis but absent in M. indicus pranii and M. intracellulare ATCC 13950. These genes might play an important role in the metabolism of M. tuberculosis as well as in the host-pathogen interaction and as a virulence factor.
FIG 6 .

A comparative metabolic pathway analysis between M. tuberculosis H37Rv, M. intracellulare ATCC 13950, and M. indicus pranii reveals the presence of novel pathways in Mycobacterium tuberculosis that are not present in M. indicus pranii or M. intracellulare ATCC 13950. The comparative analysis of metabolic enzymes present in M. tuberculosis H37Rv, M. intracellulare ATCC 13950, and M. indicus pranii based on KEGG pathways are shown. (a) The unique pathways in M. tuberculosis H37Rv are shown in red. The common pathways between M. intracellulare ATCC 13950 and M. indicus pranii are shown in pink. The common pathways present in these three organisms are shown in black. Note that M. tuberculosis H37Rv has acquired alternate pathways (red) for its survival. There are few common pathways between M. intracellulare ATCC 13950 and M. indicus pranii (pink) that are absent in M. tuberculosis H37Rv. (b) A section (circled) of the lipid biosynthesis subpathway (glycerolipid metabolism) highlights the presence of an alternate enzyme (EC 3.1.1.3, Rv3097c) that performs molecular transformations in M. tuberculosis H37Rv. (c) A section (circled) of the lipid biosynthesis subpathway (fatty acid metabolism) highlights an alternate enzyme (FabH gene) present in M. intracellulare ATCC 13950 and M. indicus pranii but absent in M. tuberculosis H37Rv.
Conclusions.
The COG method of proteome annotation is based on assignment of a sequence-based orthology, whereas function prediction tools like InterPro add to in-depth annotation of a gene by utilizing the domain and signature knowledge. We found that COG-based annotation of the M. indicus pranii proteome consisted of some ambiguous cases compared with other protein domain databases that are used for annotation. The combination of homology-based COG and a functional domain database like InterPro/Pfam provided the maximum coverage for annotating a proteome. The protein domain knowledge available using InterPro/Pfam and the Conserved Domains Database (CDD) can help associate sequence-based homologs with the functional orthologs. From the above approach, we found that among mycobacterial species, for a protein to be a homolog, its sequence identity should be above 20%. We have also highlighted here the importance of comparative genomics and protein domains by curating 60 misannotated M. indicus pranii genes in the public database (see Table S1 in the supplemental material).
Our comparative genomic and proteomic analyses of pathogenic and nonpathogenic mycobacterial species provided strong evidence suggesting that despite having identical rRNA genes (except for notable differences in the 23S rRNA gene) with M. intracellulare, M. indicus pranii (an NP with strong immunomodulatory properties) is a predecessor of the M. avium complex (12) and is at an evolutionarily transitory position with respect to a fast versus slow grower and as a saprophyte versus a seasoned pathogen (6, 12). During the process of evolution, M. indicus pranii evolved into M. intracellulare ATCC 13950 (an OP) when a few genes were deleted and a few enzyme-encoding genes were acquired (which may provide a common/evolutionary link between M. avium 104 [OP] and M. intracellulare ATCC 13950 [OP]). A similar pattern as with M. intracellulare ATCC 13950 is exhibited by Mycobacterium tuberculosis, where a large portion of genes with a conserved proline-glutamate (PE) motif or proline-proline-glutamate motif (PPE) family have been acquired (5), although most of them are still hypothetical proteins. Although we know that members of the PE/PPE gene family code for virulence factors (4, 34–36), it will be interesting if some of these hypothetical proteins have any enzymatic function, as until now only one PE protein, PE30 (Rv3097c), has been reported to exhibit enzymatic activity (37). Such proteins can be exploited for antituberculosis drug therapy. The host-pathogen interaction network between M. tuberculosis and humans (38) might provide some insight into the evolutionary pressure under which M. tuberculosis obtained a new set of pathways for its survival, which can be exploited again for antituberculosis interventions. Furthermore, our findings on the presence of alternative metabolic pathways in Mycobacterium tuberculosis pose important questions about their role in virulence and the consequent implications for designing new interventions against tuberculosis.
MATERIALS AND METHODS
Reannotation of the M. indicus pranii proteome.
The prediction of protein function domains for the ORFs of M. indicus pranii was carried out using InterPro (39, 40) and Pfam (41). The domain hits of individual proteins were compared to annotations of the COG database (42). As InterProscan uses CATH GENE3D version 3.3.0 (43), in cases of ambiguity the protein sequences were submitted to GENE3D v11.0 and the Domain Enhanced Lookup Time Accelerated BLAST (DELTA-BLAST) system (44) to confirm the annotation.
Functional relatedness of homologs.
A sequence identity above 60% between two proteins is required to have 90% functional similarity (29); however, neither the sequence identity nor the expectation value can give complete insight into the relationship between two proteins (30). We therefore tried to relate the functional similarity based on Interpro/Pfam domain hits to the coverage and sequence identity of BLASTp results between M. indicus pranii and other Mycobacterium species. To show the functional relationships between homologs, we assigned a value of 1 for a positive functional relationship if the homologs shared at least one InterPro/Pfam ID and a value of 0 if they shared none, indicating no functional relationship.
The statistical significance of our approach was determined using data from DELTA-BLAST, which returns the domain hits of a protein from the CDD. For each homolog pair, we assigned a value of 1 if they shared at least one conserved domain and a value of 0 if they did not share any conserved domain listed in the CDD. The findings from the above two lists (InterPro/Pfam and CDD) were compared to calculate the number of tp, tn, fp, and fn (see Chart S1 in the supplemental material), which were used to plot ROC curves.
In order to obtain a sequence identity cutoff below which no functional similarity or homologs were observed in the Mycobacterium genus (13 species included in the analyses), we plotted the number of tp against sequence identity (varying from 0% to 100%) for M. indicus pranii versus all 12 Mycobacterium other species (see Fig. S1 in the supplemental material). Furthermore, MCC values were calculated for sequence identity cutoffs ranging from 0 to 100% (see Fig. S2 in the supplemental material). We also performed all-against-all BLASTp-based homology searches for 13 Mycobacterium species, using a sequence identity cutoff of 20% and an e value of <1e−04 (45).
Comparative metabolic pathway analyses.
Analysis of metabolic enzymes was carried out based on the IUBMB EC numbers (46) in the KEGG database (47) (accessed in December 2012) for M. indicus pranii (387 EC enzymes), M. intracellulare ATCC 13950 (394 EC), M. avium 104 (413 EC), and M. tuberculosis (396 EC) genomes (48). Comparative metabolic pathway analysis between M. tuberculosis, M. intracellulare ATCC 13950, and M. indicus pranii was performed using iPath2.0 (49).
SUPPLEMENTAL MATERIAL
Chart summarizing the bases for statistical analyses. Download
Correlation of BLASTp sequence identities between homologs of M. indicus pranii proteins in 12 other Mycobacterium species with their functional relatedness (Interpro/Pfam). Plots of the number of true positives among the total number of homologs versus various sequence identities showed that below a 20% sequence identity cutoff, no functional similarity between homologs was observed for Mycobacterium species. Download
Correlation for BLASTp sequence identities between homologs of M. indicus pranii proteins in 12 other Mycobacterium species, with their functional relatedness (Interpro/Pfam). Mathews correlation coefficient values were plotted for various sequence identity cutoffs. MCC values are 0 for sequence identity cutoffs of 0 to 20% and show a steep rise at 30% sequence identity, suggesting that below the 20% sequence identity cutoff no functional similarity between homologs can be observed for Mycobacterium species. Download
List of ambiguous cases found while reannotating the M. indicus pranii proteome based on InterPro domains
Mycobacterial genomes selected for comparative genomics analysis
ACKNOWLEDGMENTS
We thank Dame J. M. Thornton, Director, EMBL-EBI, for her support. S.A.R. is a Research Scientist and Senior Research Fellow at EMBL-EBI, Wellcome Trust Genome Campus, Hinxton, United Kingdom, and thanks EMBL for funding. N.Z.E. and S.E.H. thank the Department of Biotechnology, Ministry of Science and Technology, Government of India (MoS&T, GoI) for a Center of Excellence grant. S.E.H. and A.K.T. are JC Bose National Fellows of the Department of Science and Technology (MoS&T, GoI).
S.A.R., A.K.T., and S.E.H. designed the study. S.A.R., Y.S., S.K., and J.A. analyzed and interpreted the data. S.A.R., Y.S., S.K., N.Z.E., A.K.T., and S.E.H. drafted the manuscript. All authors read and approved the final manuscript.
We declare we have no competing interests.
Footnotes
Citation Rahman SA, Singh Y, Kohli S, Ahmad J, Ehtesham NZ, Tyagi AK, Hasnain SE. 2014. Comparative analyses of nonpathogenic, opportunistic, and totally pathogenic mycobacteria reveal genomic and biochemical variabilities and highlight the survival attributes of Mycobacterium tuberculosis. mBio 5(6):e02020-14. doi:10.1128/mBio.02020-14.
REFERENCES
- 1. Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, Parsons LM, Pym AS, Samper S, van Soolingen D, Cole ST. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. U. S. A. 99:3684–3689. 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wirth T, Hildebrand F, Allix-Béguec C, Wölbeling F, Kubica T, Kremer K, van Soolingen D, Rüsch-Gerdes S, Locht C, Brisse S, Meyer A, Supply P, Niemann S. 2008. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4:e1000160. 10.1371/journal.ppat.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arnold C. 2007. Molecular evolution of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 13:120–128. 10.1111/j.1469-0691.2006.01637.x. [DOI] [PubMed] [Google Scholar]
- 4. Ahmed N, Dobrindt U, Hacker J, Hasnain SE. 2008. Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention. Nat. Rev. Microbiol. 6:387–394. 10.1038/nrmicro1889. [DOI] [PubMed] [Google Scholar]
- 5. Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 6:95. 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmed N, Saini V, Raghuvanshi S, Khurana JP, Tyagi AK, Tyagi AK, Hasnain SE. 2007. Molecular analysis of a leprosy immunotherapeutic bacillus provides insights into Mycobacterium evolution. PLoS One 2:e968. 10.1371/journal.pone.0000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rao KR, Kauser F, Srinivas S, Zanetti S, Sechi LA, Ahmed N, Hasnain SE. 2005. Analysis of genomic downsizing on the basis of region-of-difference polymorphism profiling of Mycobacterium tuberculosis patient isolates reveals geographic partitioning. J. Clin. Microbiol. 43:5978–5982. 10.1128/JCM.43.12.5978-5982.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmed N, Alam M, Rao KR, Kauser F, Kumar NA, Qazi NN, Sangal V, Sharma VD, Das R, Katoch VM, Murthy KJ, Suneetha S, Sharma SK, Sechi LA, Gilman RH, Hasnain SE. 2004. Molecular genotyping of a large, multicentric collection of tubercle bacilli indicates geographical partitioning of strain variation and has implications for global epidemiology of Mycobacterium tuberculosis. J. Clin. Microbiol. 42:3240–3247. 10.1128/JCM.42.7.3240-3247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17:589–596. 10.1016/S0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- 10. Ochman H, Davalos LM. 2006. The nature and dynamics of bacterial genomes. Science 311:1730–1733. 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- 11. Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honoré N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011. 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 12. Saini V, Raghuvanshi S, Khurana JP, Ahmed N, Hasnain SE, Tyagi AK, Tyagi AK. 2012. Massive gene acquisitions in Mycobacterium indicus pranii provide a perspective on mycobacterial evolution. Nucleic Acids Res. 40:10832–10850. 10.1093/nar/gks793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma P, Mukherjee R, Talwar GP, Sarathchandra KG, Walia R, Parida SK, Pandey RM, Rani R, Kar H, Mukherjee A, Katoch K, Benara SK, Singh T, Singh P. 2005. Immunoprophylactic effects of the anti-leprosy Mw vaccine in household contacts of leprosy patients: clinical field trials with a follow up of 8-10 years. Lepr. Rev. 76:127–143. [PubMed] [Google Scholar]
- 14. Talwar GP, Zaheer SA, Mukherjee R, Walia R, Misra RS, Sharma AK, Kar HK, Mukherjee A, Parida SK, Suresh NR, et al. etAl. 1990. Immunotherapeutic effects of a vaccine based on a saprophytic cultivable mycobacterium, Mycobacterium w in multibacillary leprosy patients. Vaccine 8:121–129. 10.1016/0264-410X(90)90134-8. [DOI] [PubMed] [Google Scholar]
- 15. Nyasulu PS. 2010. The role of adjunctive Mycobacterium w immunotherapy for tuberculosis. J. Exp. Clin. Med. 2:124–129. 10.1016/S1878-3317(10)60020-4. [DOI] [Google Scholar]
- 16. Gupta A, Geetha N, Mani J, Upadhyay P, Katoch VM, Natrajan M, Gupta UD, Bhaskar S. 2009. Immunogenicity and protective efficacy of “Mycobacterium w” against Mycobacterium tuberculosis in mice immunized with live versus heat-killed Mycobacterium w by the aerosol or parenteral route. Infect. Immun. 77:223–231. 10.1128/IAI.00526-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel N, Deshpande MM, Shah M. 2002. Effect of an immunomodulator containing Mycobacterium w on sputum conversion in pulmonary tuberculosis. J. Indian Med. Assoc. 100:191–193. [PubMed] [Google Scholar]
- 18. Singh Y, Kohli S, Sowpati DT, Rahman SA, Tyagi AK, Hasnain SE. 2014. Gene cooption in Mycobacteria and search for virulence attributes: comparative proteomic analyses of Mycobacterium tuberculosis, Mycobacterium indicus pranii and other mycobacteria. Int. J. Med. Microbiol. 304:742–748. 10.1016/j.ijmm.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 19. Demangel C, Stinear TP, Cole ST. 2009. Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 7:50–60. 10.1038/nrmicro2077. [DOI] [PubMed] [Google Scholar]
- 20. Berg RD, Ramakrishnan L. 2012. Insights into tuberculosis from the zebrafish model. Trends Mol. Med. 18:689–690. 10.1016/j.molmed.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 21. Wu TS, Chiu CH, Yang CH, Leu HS, Huang CT, Chen YC, Wu TL, Chang PY, Su LH, Kuo AJ, Chia JH, Lu CC, Lai HC. 2012. Fish tank granuloma caused by Mycobacterium marinum. PLoS One 7:e41296. 10.1371/journal.pone.0041296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cook JL. 2010. Nontuberculous mycobacteria: opportunistic environmental pathogens for predisposed hosts. Br. Med. Bull. 96:45–59. 10.1093/bmb/ldq035. [DOI] [PubMed] [Google Scholar]
- 23. Chiodini RJ, Chamberlin WM, Sarosiek J, McCallum RW. 2012. Crohn’s disease and the mycobacterioses: a quarter century later: causation or simple association? Crit. Rev. Microbiol. 38:52–93. 10.3109/1040841X.2011.638273. [DOI] [PubMed] [Google Scholar]
- 24. Mueller PS, Edson RS. 2001. Disseminated Mycobacterium abscessus infection manifesting as fever of unknown origin and intra-abdominal lymphadenitis: case report and literature review. Diagn. Microbiol. Infect. Dis. 39:33–37. 10.1016/S0732-8893(00)00211-X. [DOI] [PubMed] [Google Scholar]
- 25. Bohsali A, Abdalla H, Velmurugan K, Briken V. 2010. The non-pathogenic mycobacteria M. smegmatis and M. fortuitum induce rapid host cell apoptosis via a caspase-3 and TNF dependent pathway. BMC Microbiol. 10:237. 10.1186/1471-2180-10-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gupta AK, Katoch VM, Chauhan DS, Lavania M. 2012. Potential of Mycobacterium vanbaalenii as a model organism to study drug transporters of Mycobacterium tuberculosis, Mycobacterium marinum and Mycobacterium ulcerans: homology analysis of M. tuberculosis drug transporters among mycobacterial species. Infect. Genet. Evol. 12:853–856. 10.1016/j.meegid.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 27. Stanford JL, Gunthorpe WJ. 1971. A study of some fast-growing scotochromogenic mycobacteria including species descriptions of Mycobacterium gilvum (new species) and Mycobacterium duv alii (new species). Br. J. Exp. Pathol. 52:627–637. [PMC free article] [PubMed] [Google Scholar]
- 28. McDowall J, Hunter S. 2011. Interpro protein classification. Methods Mol. Biol. 694:37–47. 10.1007/978-1-60761-977-2_3. [DOI] [PubMed] [Google Scholar]
- 29. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. Interpro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40:D306–D312. 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301. 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22–28. 10.1093/nar/29.4.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lees J, Yeats C, Perkins J, Sillitoe I, Rentzsch R, Dessailly BH, Orengo C. 2012. Gene3D: a domain-based resource for comparative genomics, functional annotation and protein network analysis. Nucleic Acids Res. 40:D465–D471. 10.1093/nar/gkr1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. 2012. Domain enhanced lookup time accelerated BLAST. Biol. Direct 7:12. 10.1186/1745-6150-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian W, Skolnick J. 2003. How well is enzyme function conserved as a function of pairwise sequence identity? J. Mol. Biol. 333:863–882. 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 35. Joshi T, Xu D. 2007. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genomics 8:222. 10.1186/1471-2164-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12:85–94. 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 37. Thompson RH. 1962. Classification and nomenclature of enzymes. Science 137:405–408. 10.1126/science.137.3528.405. [DOI] [PubMed] [Google Scholar]
- 38. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114. 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou T. 2013. Computational reconstruction of metabolic networks from KEGG. Methods Mol. Biol. 930:235–249. 10.1007/978-1-62703-059-5_10. [DOI] [PubMed] [Google Scholar]
- 40. Yamada T, Letunic I, Okuda S, Kanehisa M, Bork P. 2011. iPath2.0: interactive pathway explorer. Nucleic Acids Res. 39:W412–W415. 10.1093/nar/gkr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim BJ, Choi BS, Choi IY, Lee JH, Chun J, Hong SH, Kook YH, Kim BJ. 2012. Complete genome sequence of Mycobacterium intracellulare clinical strain MOTT-36Y, belonging to the INT5 genotype. J. Bacteriol. 194:4141–4142. 10.1128/JB.00752-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raman K, Yeturu K, Chandra N. 2008. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst. Biol. 2:109. 10.1186/1752-0509-2-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarthy TR, Torrelles JB, MacFarlane AS, Katawczik M, Kutzbach B, Desjardin LE, Clegg S, Goldberg JB, Schlesinger LS. 2005. Overexpression of Mycobacterium tuberculosis manB, a phosphomannomutase that increases phosphatidylinositol mannoside biosynthesis in Mycobacterium smegmatis and mycobacterial association with human macrophages. Mol. Microbiol. 58:774–790. 10.1111/j.1365-2958.2005.04862.x. [DOI] [PubMed] [Google Scholar]
- 44. Schnell R, Sandalova T, Hellman U, Lindqvist Y, Schneider G. 2005. Siroheme- and [Fe4-S4]-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. J. Biol. Chem. 280:27319–27328. 10.1074/jbc.M502560200. [DOI] [PubMed] [Google Scholar]
- 45. Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. 2012. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94:110–116. 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 46. Kohli S, Singh Y, Sharma K, Mittal A, Ehtesham NZ, Hasnain SE. 2012. Comparative genomic and proteomic analyses of PE/PPE multigene family of Mycobacterium tuberculosis H37Rv and H37Ra reveal novel and interesting differences with implications in virulence. Nucleic Acids Res. 40:7113–7122. 10.1093/nar/gks465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tundup S, Mohareer K, Hasnain SE. 2014. Mycobacterium tuberculosis PE25/PPE41 protein complex induces necrosis in macrophages: role in virulence and disease reactivation? FEBS Open Bio 4:822–828. 10.1016/j.fob.2014/09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mishra KC, de Chastellier C, Narayana Y, Bifani P, Brown AK, Besra GS, Katoch VM, Joshi B, Balaji KN, Kremer L. 2008. Functional role of the PE domain and immunogenicity of the Mycobacterium tuberculosis triacylglycerol hydrolase LipY. Infect. Immun. 76:127–140. 10.1128/IAI.00410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumar D, Nath L, Kamal MA, Varshney A, Jain A, Singh S, Rao KV. 2010. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140:731–743. 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chart summarizing the bases for statistical analyses. Download
Correlation of BLASTp sequence identities between homologs of M. indicus pranii proteins in 12 other Mycobacterium species with their functional relatedness (Interpro/Pfam). Plots of the number of true positives among the total number of homologs versus various sequence identities showed that below a 20% sequence identity cutoff, no functional similarity between homologs was observed for Mycobacterium species. Download
Correlation for BLASTp sequence identities between homologs of M. indicus pranii proteins in 12 other Mycobacterium species, with their functional relatedness (Interpro/Pfam). Mathews correlation coefficient values were plotted for various sequence identity cutoffs. MCC values are 0 for sequence identity cutoffs of 0 to 20% and show a steep rise at 30% sequence identity, suggesting that below the 20% sequence identity cutoff no functional similarity between homologs can be observed for Mycobacterium species. Download
List of ambiguous cases found while reannotating the M. indicus pranii proteome based on InterPro domains
Mycobacterial genomes selected for comparative genomics analysis