

Abstract
Spontaneously arising (‘de novo’) mutations play an important role in medical genetics. For diseases with extensive locus heterogeneity – such as autism spectrum disorders (ASDs) – the signal from de novo mutations (DNMs) is distributed across many genes, making it difficult to distinguish disease-relevant mutations from background variation. We provide a statistical framework for the analysis of DNM excesses per gene and gene set by calibrating a model of de novo mutation. We applied this framework to DNMs collected from 1,078 ASD trios and – while affirming a significant role for loss-of-function (LoF) mutations – found no excess of de novo LoF mutations in cases with IQ above 100, suggesting that the role of DNMs in ASD may reside in fundamental neurodevelopmental processes. We also used our model to identify ~1,000 genes that are significantly lacking functional coding variation in non-ASD samples and are enriched for de novo LoF mutations identified in ASD cases.
Exome sequencing has allowed for the identification of de novo (newly arising) events and has already been effectively put to use in identifying causal variants in rare, Mendelian diseases. In the case of Kabuki syndrome, the observation of a de novo mutation (DNM) in MLL2 in 9 out of the10 patients strongly implicated the loss of MLL2 function as causal1. The conclusion that MLL2 is important in Kabuki syndrome etiology based on the de novo findings relies upon the unlikely accumulation of independent and infrequently occurring events in the vast majority of these unrelated cases. By contrast, DNMs play a smaller role in the pathogenesis of heritable complex traits, such as autism spectrum disorders (ASDs), and associated DNMs are spread across multiple genes. These differences in the etiologic architecture of complex traits make the task of identifying “causal” genes considerably more challenging. For example, recent exome sequencing studies demonstrated a significant excess of de novo loss-of-function (LoF) mutations in ASD cases, but lacked the ability to directly implicate more than a very few genes2–6.
The main complicating factor for interpreting the number of observed DNMs for a particular gene is the background rate of de novo mutation, which can vary greatly between genes. As more individuals are sequenced, multiple DNMs will inevitably be observed in the same gene by chance. However, if de novo mutation plays a role in a given disease, then we would expect to find that genes associated to disease should contain more DNMs than expected by chance.
Here, we develop a statistical model of de novo mutation in order to evaluate the findings from exome sequencing data. With this model, we establish a statistical framework to evaluate the rate of DNMs not only on a per-gene basis (in a frequentist manner analogous to common genome-wide association analysis), but also globally and by gene set. We further use this model to predict the expected amount of rare standing variation per gene and to detect those genes that are significantly and specifically deficient in functional variation – likely reflecting processes of selective constraint. Consequently, since selection has reduced standing functional variation in these genes, it is reasonable to hypothesize that mutations in these genes are more likely to be deleterious.
We used the mutational model along with our list of highly constrained genes to evaluate the relationship between de novo mutation and ASDs. Most of the families employed in these analyses were included in a set of previous studies of de novo mutation, which reported an overall excess of de novo LoF mutations in ASD cases, as well as multiple DNMs in specific genes2–5. We build on those studies to examine the aggregate rates of DNMs, the excess of multiply mutated genes, and the overlap of DNMs with gene sets, which highlights the complex relationship between intellectual functioning and the genetic architecture of ASD.
Results
Basis of the mutational model
Accurate estimation of the expected rate of de novo mutation in a gene requires a precise estimate of each gene’s mutability. While gene length is an obvious factor in a gene’s mutability, local sequence context is also a well-known source of mutation rate differences7. Accordingly, we extended a previous model of de novo mutation based on sequence context and developed gene-specific probabilities for different types of mutation: synonymous, missense, nonsense, essential splice site, and frameshift (Online Methods and Supplementary Fig. 1)3. All probabilities of mutation can be found in Supplementary Table 1. Underscoring the importance of the sequence context factors in the model, this genome-wide rate yields an expected mutation rate of 1.67×10−8 for the exome alone. Using counts of rare (minor allele frequency < 0.001) synonymous variants identified in the NHLBI’s Exome Sequencing Project (ESP), we found that our per-gene probabilities of mutation were significantly more correlated (r=0.940) with these counts than gene length alone (p < 10−16; Online Methods).
Having established accurate per-gene probabilities of mutation, we could then investigate the rates and distribution of DNMs found in sequencing studies. Specifically, we wished to systematically assess a) whether cases had genome-wide excesses of certain functional categories of de novo mutation; b) whether individual genes could be associated via de novo mutation with genome-wide statistical significance; c) whether specific sets of genes collectively showed significant enrichment of de novo mutations and d) whether there were genome-wide excesses of genes with multiple de novo mutations. Below we demonstrate the utility of the statistical framework to address all of these questions with respect to recently generated autism and intellectual disability family exome sequencing data.
Identifying genes under selective constraint
There has been a long standing interest in identifying genes in the human genome that are sensitive to mutational changes, as these genes would be the most likely to contribute to disease. Recent work made use of the ESP data to create a metric evaluating the proportion of common functional variation in each gene, thereby identifying genes that appeared to be intolerant of mutation8. Along these lines, we correlated our calculated per-gene probabilities of mutation with the observed counts of rare missense variants in the ESP data set. In contrast to the high consistency between predicted synonymous mutation rates and observed synonymous counts (expected if the category is under no specific selection), we observed a significant number of genes with severe deficit of missense variants compared to the expectation generated from predicted mutation rates. Such a deficit is consistent with strong evolutionary constraint: when damaging mutations arise, they are quickly removed from the population by purifying selection. To avoid erroneously identified constrained genes, we removed 134 genes with either significantly elevated or depressed synonymous and nonsynonymous rates (both p < 0.001; Online Methods).
Comparing both the synonymous and missense predictions of our model to the ESP data set, we identified a list of excessively constrained genes (p < 0.001) that represent roughly 5% of all genes (Supplementary Table 2). A high proportion of the most significantly constrained genes (missense constraint p < 1×10−6) had autosomal or X-linked dominant, largely sporadic, Mendelian disease entries listed in OMIM (n=27/86). By contrast, a set of genes for which the missense constraint was very close to expectation (n=111, −0.01 < Z < 0.01) had only two de novo or dominant disease inheritance entries in OMIM, which was significantly different from the highly constrained set (p < 10−8). For the 86 most highly constrained genes, no autosomal recessive Mendelian disorders have been documented. However, 11 of the 111 average constrained genes have been identified as causes of autosomal recessive Mendelian disorders (p < 0.003), underscoring the lack of strong constraint induced by recessive inheritance models.
Mutation rates for ASD and ID
We applied the model to two primary data sets: published results from ASD sequencing studies2–6 with a collection of additional unpublished ASD trios, and published results from patients with severe intellectual disability9,10. Table 1a shows the comparison between the predicted number of mutations per exome and the observed data from the 1,078 ASD cases as well as 343 sequenced unaffected siblings2–6. The model’s predictions match the observed data for the unaffected siblings well, but the cases show a significant excess of de novo LoF mutations consistent with the findings of the individual sequencing studies (p=2.05×10−7). Using our model to simulate null DNM sets, we found that there are significantly more genes with two or more de novo LoF mutations than would be expected by chance (p < 0.001, 6 observed when less than one was expected; Supplementary Table 3). Importantly, while we do not observe a global excess of de novo missense mutations, we do observe an excess of genes with two or more functional (LoF or missense) de novo mutations (observed 48 such genes when the average expected is 27; p < 0.001) and genes with two or more de novo missense mutations alone (observed 33 such genes when average expectation was 21, p=0.007 for missense, Table 1b). No such excess of genes containing multiple DNMs was seen in the unaffected siblings (Table 1b). Of note, our framework also supports the assessment of many other weightings and combinations of alleles – such as missense variants only (optimal for pure gain-of-function disease models), predicted damaging missense variants only, and exact probability estimates for specific combinations of LoF and missense variants - than those shown above.
Table 1.
Evaluation of the rates of de novo mutations in ASD cases and unaffected siblings. The observed and expected rate of mutations by type per exome for unaffected siblings2 and ASD cases, including some unpublished US and Finnish trios2–6 (a). (b) The number of genes with multiple de novo mutations in unaffected siblings and ASD cases across studies. The average number of expected genes with multiple de novo mutations was determined by simulation. LoF = Loss-of-function. DNMs = de novo mutations.
| a) Genome-wide excesses of mutational events | ||||
|---|---|---|---|---|
|
|
||||
| Unaffected Siblings | ||||
|
|
||||
| Mutation Type | Observed events per exome | Expected events per exome | p-value | |
|
|
||||
| Synonymous | 0.21 | 0.27 | 0.0218 | Two-tailed |
| Missense | 0.61 | 0.62 | 0.8189 | Two-tailed |
| Loss-of-Function | 0.09 | 0.09 | 0.4508 | One-tailed |
|
|
||||
| n = 343 families | ||||
|
|
||||
|---|---|---|---|---|
| ASD Cases | ||||
|
|
||||
| Mutation Type | Observed events per exome | Expected events per exome | p-value | |
|
|
||||
| Synonymous | 0.25 | 0.27 | 0.1065 | Two-tailed |
| Missense | 0.64 | 0.62 | 0.5721 | Two-tailed |
| Loss-of-Function | 0.13 | 0.09 | 2.05E-07 | One-tailed |
|
|
||||
| n = 1,078 families | ||||
| b) Genome-wide excesses of multiply hit genes | |||
|---|---|---|---|
|
|
|||
| Unaffected Siblings | |||
|
| |||
| Mutation Type | Observed genes with 2+ DNMs | Average expected genes with 2+ DNMs | p-value |
| Synonymous | 0 | 0.5 | 1.0 |
| Missense | 5 | 2.5 | 0.1049 |
| Loss-of-Function | 0 | 0.04 | 1.0 |
| LoF+missense | 6 | 3 | 0.0779 |
|
| |||
| n = 343 families | |||
|
|
|||
|---|---|---|---|
| ASD Cases | |||
|
| |||
| Mutation Type | Observed genes with 2+ DNMs | Average expected genes with 2+ DNMs | p-value |
| Synonymous | 4 | 3.8 | 0.5186 |
| Missense | 33 | 21.4 | 0.0070 |
| Loss-of-Function | 6 | 0.5 | < 0.001 |
| LoF+missense | 48 | 27.2 | < 0.001 |
|
| |||
| n = 1,078 families | |||
Table 2 lists some of the genes that have two or more LoF de novo mutations across the 1,078 ASD subjects. The results for all genes can be found in Supplementary Table 4. A conservative significance threshold of 1×10−6 was used, correcting for 18,271 genes and two tests. Considering this set of 1,078 trios as a single experiment, two genes – DYRK1A and SCN2A – exceeded this conservative genome-wide significance for more de novo LoF mutations than predicted. SCN2A also had significantly more functional de novo mutations than expected. CHD8, with three de novo LoF mutations and one missense, was very close to the significance threshold in these studies (p=1.76×10−6 for LoF; p=3.20×10−5 for functional). However, a recent targeted sequencing study found 7 additional CHD8 de novo LoF mutations in ASD cases11. This brought the total number of de novo LoF mutations in CHD8 to 10, which was highly significant (p=8.38×10−20 when accounting for the total number of trios – 2,750 – examined in the combination of the targeted and exome-wide study). These results offer the encouraging point that, as with GWAS, larger collaborative trio exome efforts will define unambiguous risk factors. It is important to note, however, that not all genes with a large number of de novo mutations in them had significant p-values. For example, TTN had four missense DNMs in ASD cases, but a p-value that is not even nominally significant due to the enormous size of the gene (p=0.18). Even having two de novo LoF mutations was on occasion not enough to provide compelling significance (POGZ, two frameshifts, p=8.93×10−5). In comparison, none of the genes found to contain multiple DNMs in the unaffected siblings crossed the significance threshold (Supplementary Table 5).
Table 2.
Individually significant genes identified from the analysis of de novo mutations in ASD cases. Genes with multiple loss-of-function (LoF) de novo mutations across 1,078 ASD cases. LoF mutations include nonsense, frameshift, and splice site-disrupting mutations. “# LoF Expected” refers to the expected number of de novo LoF mutations based on the probability of mutation for the gene as determined by our model. The genome-wide significance threshold is 1×10−6
| Gene | Mutations | # LoF Observed | # LoF Expected | p-value |
|---|---|---|---|---|
| DYRK1A | nonsense, splice, frameshift | 3 | 0.0072 | 6.15E-08 |
| SCN2A | nonsense, nonsense, frameshift | 3 | 0.0178 | 9.20E-07 |
| CHD8 | nonsense, splice, frameshift | 3 | 0.0221 | 1.76E-06 |
| KATNAL2 | splice, splice | 2 | 0.0049 | 1.19E-05 |
| POGZ | frameshift, frameshift | 2 | 0.0133 | 8.93E-05 |
| ARID1B | frameshift, frameshift | 2 | 0.0178 | 1.57E-04 |
These analyses were also applied to the results from the sequencing studies of moderate to severe (IQ < 60) intellectual disability9,10. Intellectual disability, like ASD, showed a significant excess of LoF DNMs (p=6.49×10−7; Table 3a). Even with a much smaller sample size (n=151), there were genes with significantly more LoF and functional DNMs than predicted by the model (Table 4). The intellectual disability data also have significantly more genes with multiple de novo missense, LoF, and functional mutations than predicted (p=0.009 for missense, p < 0.001 for LoF and functional).
Table 3.
Evaluation of the rates of de novo mutations in cases with intellectual disability. (a) The observed and expected rate of mutations by type per exome for cases of intellectual disability (ID)9,10. (b) The number of genes with multiple de novo mutations in intellectual disability cases across studies. The average number of expected genes with multiple de novo mutations was determined by simulation. LoF = Loss-of-function. DNMs = de novo mutations.
| a) Genome-wide excesses of mutational events | ||||
|---|---|---|---|---|
|
|
||||
| ID Cases | ||||
|
|
||||
| Mutation Type | Observed events per exome | Expected events per exome | p-value | |
|
|
||||
| Synonymous | 0.19 | 0.27 | 0.0267 | Two-tailed |
| Missense | 0.70 | 0.62 | 0.2380 | Two-tailed |
| Loss-of-Function | 0.24 | 0.09 | 6.49E-07 | One-tailed |
|
|
||||
| n = 151 families | ||||
| b) Genome-wide excesses of multiply hit genes | |||
|---|---|---|---|
|
|
|||
| ID Cases | |||
|
| |||
| Mutation Type | Observed genes with 2+ DNMs | Average expected genes with 2+ DNMs | p-value |
| Synonymous | 1 | 0.09 | 0.0879 |
| Missense | 3 | 0.5 | 0.0090 |
| LoF | 2 | 0.01 | < 0.001 |
| LoF+missense | 6 | 0.6 | < 0.001 |
|
| |||
| n = 151 families | |||
Table 4.
Individually significant genes identified from the analysis of de novo mutations from patients with intellectual disability. Genes with multiple functional de novo mutations across 151 cases of intellectual disability (ID)9,10. Loss-of-function (LoF) mutations include nonsense, frameshift, and splice site-disrupting mutations. The genome-wide significance threshold is 1×10−6. The number of mutations is either compared to the expected number for LoF only or for both LoF and missense, as indicated by the “# DNMs Expected” and “Test” columns.
| Gene | Mutations | #LoF | #Missense | # DNMs Expected | p-value | Test |
|---|---|---|---|---|---|---|
| SYNGAP1 | splice/frameshift/frameshift | 3 | 0 | 0.0017 | 8.15E-10 | LoF |
| SCN2A | missense/nonsense/frameshift/frameshift | 3 | 1 | 0.0025 | 2.56E-09 | LoF |
| SCN2A | missense/nonsense/frameshift/frameshift | 3 | 1 | 0.0187 | 5.01E-09 | LoF+mis |
| STXBP1 | missense/missense/splice | 1 | 2 | 0.0071 | 5.87E-08 | LoF+mis |
| TCF4 | missense/missense | 0 | 2 | 0.0069 | 2.39E-05 | LoF+mis |
| GRIN2A | missense/missense | 0 | 2 | 0.0162 | 1.34E-04 | LoF+mis |
| TRIO | missense/missense | 0 | 2 | 0.0333 | 5.60E-04 | LoF+mis |
In our ASD sample, we then investigated the rate of de novo events as a function of IQ; roughly 80% of this sample had an IQ assessment attempted. We found that the rate of de novo LoF mutation in ASD cases with a measured IQ above average was no different than expectation (IQ ≥ 100; n=229; 0.08 de novo LoF mutations per exome compared to expected 0.09, p=0.59). By contrast, the rate in the rest of the sample was substantially higher than expectation (n=572; rate of 0.17 de novo LoF mutations per exome, p=1.17×10−10). Furthermore, when directly compared (rather than to our expectation), these two groups were significantly different from each other, confirming a difference in genetic architecture among ASDs as a function of IQ (Supplementary Table 6, p < 0.001). These conclusions are unchanged in separate analyses of nonverbal and verbal IQ as well as full scale IQ (Supplementary Table 6).
Gene set enrichment
Given the significant global excess of de novo LoF mutations in ASD cases, we wanted to evaluate whether the set of genes harboring de novo LoF mutations had significant overlap with several sets of genes proposed as relevant to autism or describing biochemical pathways. We used the probabilities of mutation to determine the fraction of LoF mutations expected to fall into the given gene set. We then used the binomial distribution to evaluate the number of observed LoF mutations overlapping the set compared to the established expectation. When we applied this analysis to a set of 112 genes reported as disrupted in individuals with ASD or autistic features, we observed no enrichment of de novo LoF mutations (Fig. 1, “Betancur”)12. By contrast, we applied this analysis to a recent study of 842 genes found to interact with the Fragile X mental retardation protein (FMRP) in vivo and found a highly significant overlap (2.3-fold enrichment, p < 0.0001, Fig. 1)2,13. This enrichment with the targets of FMRP holds even when removing the DNMs identified in the Iossifov et al study that initially reported an enrichment of DNMs in ASD cases with FMRP-associated genes (2.5-fold enrichment, p < 0.0001)2.
Figure 1.
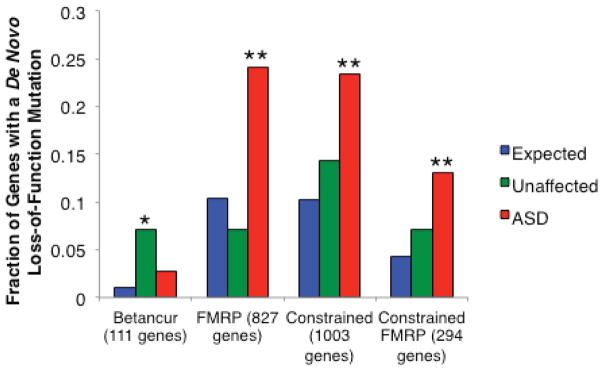
The expected and observed fraction of genes with a de novo loss-of-function mutation in ASD cases and unaffected controls for four gene sets of interest2–6,10,15. “Betancur” refers to a set of genes reported as disrupted in individuals with ASD or autistic features; of the 112 on the list, we could evaluate 11112. “FMRP” refers to the genes whose mRNAs are bound and regulated by the Fragile X Mental Retardation Protein (FMRP), as identified by Darnell and colleagues13. The “constrained” category is a set of 1,003 genes that we defined as significantly lacking rare missense variation, indicating intolerance to mutation. The targets of FMRP that are also considered constrained by our metric make up the “Constrained FMRP” category. * indicates p < 0.01; ** indicates p < 10−4.
We then evaluated the group of individuals from the ASD studies who had a de novo LoF event in one of the targets of FMRP. On average, these cases were enriched for having a measured IQ < 100 (Fisher’s exact p=4.01×10−4; Supplementary Table 7) as well as significantly reduced male:female ratio (p=0.02; Supplementary Table 8) as compared to the remaining sequenced cases (Supplementary Note). These individuals represent about 3% of the total sample, when at most a 1% overlap would be expected. The estimated odds ratio (OR) of de novo LoF events in the set of FMRP target genes was around 6, very similar to the OR estimated for large CNVs that disrupt multiple genes14. In addition, the OR for the published cases of moderate to severe intellectual disability noted above (IQ < 60; not ascertained for ASDs) having a de novo LoF event in the set of FMRP targets was roughly 10.
The same analysis was applied to the list of de novo LoF events from unaffected siblings of ASD cases and additional control individuals (n=647)2,4,5,15. There was a significant enrichment when evaluating the overlap with the set of autism related genes (p=0.0095, Fig. 1). However, no significance was observed for the overlap with the in vivo targets of FMRP. The list of de novo LoF mutations from the intellectual disability individuals, on the other hand, was significant for both sets (Supplementary Fig. 2). Even the de novo missense mutations found in the intellectual disability cases showed significant overlap with both sets under study (p=0.02 for autism-related genes, p < 0.0001 for the targets of FMRP, Supplementary Fig. 2).
Evaluating constrained genes
We further applied the enrichment analysis to our set of constrained genes and found that they contained more de novo LoF mutations than expected by chance (2.3-fold enrichment, p < 0.0001, Fig. 1). A greater fold enrichment was observed when focusing on the subset of constrained genes that were also identified in the FMRP study (3.0-fold enrichment, p < 0.0001, Fig. 1)13. We note that the FMRP targets have a significant overlap with the constrained set of genes (odds ratio = 1.29, p < 0.0001), which is consistent with the report that the targets of FMRP are under greater purifying selection than expected2. All enrichments were demonstrated to be independent of gene size (Supplementary Note).
The genes that contained a de novo missense or LoF mutation in the cases of intellectual disability also showed a significant enrichment for both the constrained gene set and the set of constrained targets of FMRP (p < 0.0001 for all lists). In comparison, no enrichment was found with either set and the list of genes that had a de novo LoF mutation in unaffected siblings and control individuals.
In addition to treating constraint as a dichotomous trait, we also evaluated the missense Z score for each of the genes with a de novo LoF mutation. We found that the distribution of missense Z scores for genes with a de novo LoF mutation in unaffected individuals was no different from the overall distribution of scores (Fig. 2; Wilcoxon p=0.8325). By contrast, both the genes with a de novo LoF mutation in ASD and intellectual disability cases had values significantly shifted towards high constraint (Wilcoxon p < 10−6 for both). Furthermore, we compared the distribution of Z scores between each of the three groups. Both the ASD and intellectual disability distributions were significantly different from the distribution of missense Z scores for unaffected individuals (p=0.0148 and 0.0012, respectively). The intellectual disability missense Z scores were also significantly higher than the ASD values (p=0.0319).
Figure 2.
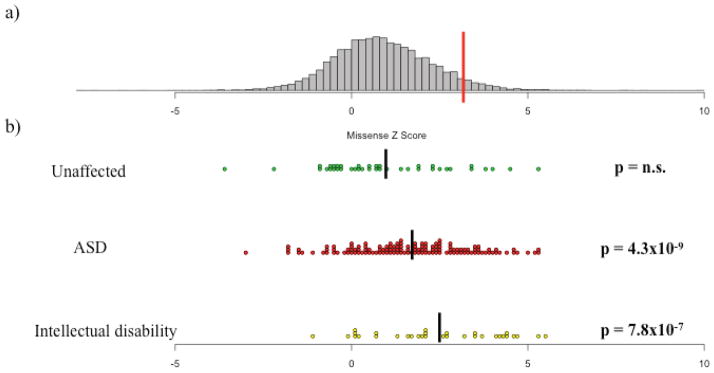
The distribution of missense Z scores and Z scores of de novo loss-of-function mutations identified in unaffected individuals, autism spectrum disorder (ASD) cases, and intellectual disability cases. (a) The distribution of missense Z scores. The red line indicates a Z score of 3.09, or the threshold for inclusion into the set of 1,003 constrained genes. (b) The missense Z scores for genes containing de novo LoF in unaffected individuals, ASD cases, and intellectual disability cases2–6,9,10,15. Black bars indicate the mean Z score of each group: 0.94, 1.68, and 2.46 for unaffected individuals, ASD cases, and intellectual disability cases, respectively. While the missense Z scores of the de novo LoF mutations found in unaffected siblings matched the overall distribution (Wilcoxon p=0.8325, n.s. = not significant), de novo LoF mutations found in both ASD and intellectual disability cases were significantly shifted towards more extreme constraint values (p < 10−6 for both). All p-values for deviation from the overall distribution are listed on the right side of the figure in bold. In addition, the distribution of missense Z scores between each of the three de novo lists were all individually significant at p < 0.05.
When evaluating the ASD cases split by IQ group, we found no enrichment of de novo LoF-containing genes with either constrained genes and targets of FMRP in the group with IQ ≥ 100 (p > 0.5 for both sets of genes) but very strong enrichment in the set with IQ < 100 (p < 0.0001 for both sets of genes). These results reinforce the variable contribution of de novo LoF mutations across subsets of ASD cases.
Comparison of constrained genes with existing methods
Identifying constrained genes by comparing observed nonsynonymous sites to expectation is conceptually similar to the traditional approach of detecting selective pressure by comparing observed nonsynonymous sites to observed synonymous sites (e.g. dN/dS) that has been used extensively. Our approach should in principle achieve greater statistical power to detect constrained genes; comparison of an observation to expectation is statistically more powerful than contrasting that observation with a generally smaller second observation – the number of observed synonymous variants. In order to investigate this claim, we identified genes that had significant evidence for selective constraint using the dN/dS metric (i.e. their ratio of synonymous and nonsynonymous sites deviated the genome-wide average at p < 0.001, Supplementary Note). There were only 377 of these genes, over half of which overlapped with the constrained gene list defined by our method (n=1003, overlap 237 genes). The genes identified as significantly constrained by only our metric – the top 10 of which include RYR2, MLL, MLL2, and SYNGAP1 – are still significantly enriched for known causes of autosomal and X-linked dominant forms of Mendelian disease (p=5 × 10−4). We therefore conclude that the model-based approach to identifying constrained genes adds substantial power to traditional approaches – the importance of this increased power to detect constraint in further articulated in the ASD and ID analyses below.
Several groups have previously published approaches, and specific gene sets from them, that are also aimed at identifying genes under excessive purifying selection or generally intolerant of functional mutation. Bustamante et al16 expanded on the McDonald-Kreitman framework17 contrasting fixed differences in the primate lineage to polymorphic differences in humans to identify a set of genes under weak negative selection, while more recently Petrovski et al8 utilized the excess of rare versus common missense variation within humans to flag genes intolerant of functional variation. We found a reasonable correlation between our metric of constraint and Petrovski’s Residual Variation Intolerance Score (RVIS; Supplementary Fig. 3)8. A comparison of these approaches as applied to prioritization of known haploinsufficient genes as well as the autism de novo LoF mutations described here are provided in the Supplementary Note, demonstrating the two human-only approaches (constraint and RVIS) performing better on these tasks of identifying severely impactful medical genetics lesions in modern humans (Supplementary Table 9). Intriguingly, both of these other approaches utilize independent information from each other and from our approach (which uses the absence of rare functional variation versus expectation within humans), raising the potential that composite scores employing all three sources of information pointing to which genes are most sensitive to heterozygous mutation could add further value.
Discussion
We have developed a framework for evaluating excesses of de novo mutations identified through exome sequencing. Even though this framework can be leveraged to evaluate excesses of mutations study-wide and in gene sets, the key focus is to evaluate the significance for individual genes. Given the small number of observed de novo events per gene, simple case-control comparisons cannot achieve any meaningful level of significance. For example, observing three de novo LoF mutations in a small gene in 1,000 case trios is perhaps quite compelling; however, a simple 3 to 0 comparison with 1,000 control trios yields no compelling statistical evidence (one-tailed p=0.125). Incidence of such extremely rare events, however, can be evaluated if the expected rate of such events is known. Sequencing large numbers of control trios to gather empirical rate estimates on a per-gene basis that are accurate is infeasible and inefficient. The calibrated model and statistical approach described here can achieve a close approximation of this ideal. Our method, therefore, offers the ability to evaluate the rate of rare variation in individual genes in situations where burden tests would fail.
Other groups have developed similar statistical frameworks11,18 – notably, the Epi4k consortium18 used the same base model we begin with3 to interpret event rates. Our model, however, has two primary strengths. First, our model of de novo mutation incorporates additional factors beyond sequence context that affect mutation rate. Both the depth of coverage – how many sequence reads were present on average – for each base and the regional divergence around the gene between humans and macaques independently and significantly improve the predictive value of our model (Supplementary Note). Second, given the high correlation between the number of rare synonymous variants in ESP and the probability of a synonymous mutation determined by our full model, we have a metric to evaluate the extent to which genes in the human genome show evidence of selective constraint. The list of 1,003 genes that we define as constrained contains an enrichment of genes known to cause severe human disease – an observation analogous to that recently made in using empirical comparison of common and rare rates of functional variation to evaluate intolerance8. In fact, site count deficits and site frequency shifts each contribute independent information to the definition of constraint and can in principle be combined in a composite test.
The results of our metric were compared to both the scores created by Petrovski and colleagues8 and loci identified as under negative selection by Bustamante et al 16. Overall, our metric and the residual variation intolerance scores defined by the Petrovski worked similarly well, reinforcing the benefits that could come from combining the two approaches. It is unsurprising that these methods outperform the evolutionary ones on the specific matter of genes intolerant to heterozygous mutation: longer term difference between polymorphism and fixed difference, more sensitive to weaker negative selection, require that mutations be tolerated well enough to become polymorphic in the first place whereas the absence of variation entirely will pick up the most strongly intolerant genes.
Ideally, we can conceptualize defining two metrics of genic constraint, one based on missense variants and the other based on LoF variants. With only 6,503 individuals in ESP, we are underpowered to determine significant deviations for most genes for the LoF variants. As sample size increases, our ability to calculate constraint improves. For example, if the sample size were to increase by an order of magnitude, we would be able to evaluate approximately 66% of genes using LoF variants. We therefore view the constrained gene list as a work in progress, to be updated when larger exome sequencing data sets become available.
Applying our statistical framework to de novo mutations from 1,078 ASD cases reveals that, while there is no global excess in de novo missense mutations, there are significantly more genes that contained multiple de novo missense mutations than expected. We also see significant overlap between the list of genes with a de novo LoF in ASD cases and the set of constrained genes that we defined. In addition, there is a significant overlap between the genes with a de novo LoF mutation and the targets of FMRP, as reported in Iossifov et al2. All of the significant signals in ASD – the global excess of de novo LoF mutations, the excess of genes with multiple functional de novo mutations, the overlap between the de novo LoF genes and both constrained genes and the targets of FMRP – are not found in the subset of ASD cases with IQ ≥ 100. The lack of signal in the IQ ≥ 100 indicates that genetic architecture among ASDs varies as a function of IQ. Overall, the probabilities of mutation defined by our full model and list of constrained genes can be used to critically evaluate the observed DNMs from sequencing studies and aid in the identification of variants and genes that play a significant role in disease.
Online Methods
De novo mutation information
Published de novo mutations were collected for both autism spectrum disorders (ASD)2–6 and severe intellectual disability9,10. Updated de novo calls were provided from two of the ASD studies3,5. Details about sample collection, sequencing, and variant processing can be found in the separate studies.
Additional sequencing
Exome sequencing of the additional families (n=129) was performed at the Broad Institute. Exons were captured using the Agilent 38 Mb SureSelect v2. After capture, another round of LM-PCR was performed to increase the quantity of DNA available for sequencing. All libraries were sequenced using an IlluminaHiSeq2000. Data were processed with Picard (http://picard.sourceforge.net/), which uses base quality-score recalibration and local realignment at known indels19 and BWA20 for mapping reads to hg19. SNPs were called using GATK for all trios jointly19,21. The variable sites that we have considered in analysis are restricted to those that pass GATK standard filters. From this set of variants, we identified putative de novo mutations and validate them as previously described3.
Mutational model
We wanted to create an accurate model of de novo mutation for each gene. In order to do so, we extended a previous sequence context-based model of de novo mutation to derive gene-specific probabilities of mutation for each of the following mutation types: synonymous, missense, nonsense, essential splice site, and frameshift3. In brief, the local sequence context was used to determine the probability of each base in the coding region mutating to each other possible base and then determine the coding impact of each possible mutation. These probabilities of mutation were summed across genes to create a per-gene probability of mutation for the aforementioned mutation types (see Supplementary Note for more details). Here, we applied the method to exons and immediately flanking essential splice sites, but note that the framework is applicable to non-genic sequences. While fitting the expected rates of mutation to observed data, we added a term for local primate divergence across 1 Mb (to capture additional unmeasured sources of regional mutational variability) and another for the average depth of sequence of each nucleotide (to capture inefficiency of variant discovery at lower sequencing depths); both terms significantly improved the fit of the model to observed data (details in Supplementary Note). We also investigated a regional replication timing term22, but found no evidence for it significantly improving the model (Supplementary Note).
To evaluate the predictive value of the model of de novo coding mutations, we extracted synonymous variants that were seen 10 times or fewer in the 6,503 individuals in the NHLBI’s Exome Sequencing Project (ESP) and compared the number of these rare variants in each gene to 1) the length of the gene and 2) the probability of a synonymous mutation for that gene determined by our model. While gene length alone showed a high correlation (0.880), our full model showed a significantly greater correlation (0.940, p < 10−16). Of note, the stochastic variability of counts from NHLBI ESP is such that if the model were perfect, the correlation to any instance of these data would be 0.975, indicating that little additional gene-to-gene variability remains to be explained. The relative rates of different types of coding mutations was quite similar to previous work based on primate substitutions23. With this calibrated model of relative mutability, we determined the absolute expected mutation rate per gene by applying a genome-wide mutation rate of 1.2×10−8 per base pair per generation (Supplementary Note)24,25.
Removing potential false positive constrained genes
In order to identify genes that appeared to be significantly constrained, we used our probabilities of mutation to predict the expected amount of synonymous and nonsynonymous variation in the NHLBI’s ESP data. Those genes that had the expected amount of synonymous variation, but were significantly (p < 0.001) deficient for missense variation were labeled as constrained. To ensure that genes were not nominated as being constrained erroneously, we excluded from all analyses 134 genes where the observed synonymous and nonsynonymous rates were both significantly elevated or significantly depressed (both p < 0.001). Upon inspection, this list contained a number of genes that contained an internal duplication (e.g. FLG), a nearby pseudogene (e.g. AHNAK2), and a number of cases where recent duplications and/or annotation errors have led to the same sequence being assigned to two genes (e.g. SLX1A and SLX1B). These are all scenarios where standard exome processing pipelines systematically undercall variation – reads are unmapped due to uncertainty of which gene to assign them to – or overcall false variants owing to read misplacement. This further suggests that a byproduct of this analysis framework is the identification of a residual set of challenging genes for current exome sequencing pipelines.
Supplementary Material
Acknowledgments
All data from published studies are available in the respective publications. All newly generated data and computational tools used in this paper will be available online as downloadable material. We have also constructed a website to query genes that provides information on constraint and the de novo mutations found in the specified gene across published studies of de novo mutation. We would like to thank E. Daly and M. Chess for their contributions to data analysis and the construction of the website, respectively. We acknowledge the following resources and families who contributed to them: the National Institute of Mental Health (NIMH) repository (U24MH068457); Autism Genetic Resource Exchange (AGRE) Consortium, a program of Autism Speaks (1U24MH081810 to Clara M. Lajonchere); The Autism Simplex Collection (TASC) (grant from Autism Speaks); Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (grant from the Simons Foundation); The Autism Consortium (grant from the Autism Consortium). This work was directly supported by NIH grants R01MH089208 (MJD), R01MH089025 (JDB), R01MH089004 (GDS), R01MH089175 (RAG), and R01MH089482 (JSS) and supported in part by NIH grants P50HD055751 (EHC), R01MH057881 (BD), and R01MH061009 (JSS). We acknowledge partial support from U54 HG003273 (RAG) and U54 HG003067 (E. Lander). We thank Thomas Lehner (NIMH), Adam Felsenfeld (NHGRI), and Patrick Bender (NIMH) for their support and contribution to the project. EB, JDB, BD, MJD, RAG, KR, AS, GDS, and JSS are lead investigators in the ARRA Autism Sequencing Collaboration (AASC). We would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls on the web: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Footnotes
URLs: Online Mendelian Inheritance in Man (http://omim.org/); Exome Variant Server (http://evs.gs.washington.edu/EVS/); site to query constraint information and de novo mutations from published studies (http://atgu.mgh.harvard.edu/webtools/gene-lookup/).
Author Contributions: Conceived of and designed the mutational model and constraint methods: K.E.S., B.M.N., M.J.D. Executed the analyses: K.E.S., E.B.R. Contributed to analysis concepts and methods: K.E.S., E.B.R., L.M.M., J.A.K., S.M., A.K., D.P.W., D.G.M., S.M.P., J.D.B., B.D., K.Ro. Contributed autism sequencing, evaluation, and manuscript comments: K.E.S., S.J.S., C.S., A.S., K.Re., S.G.B., M.d.P., A.P., E.B., J.B.P., E.H.C., R.A.G., G.D.S., J.S.S., B.D., K.Ro., B.M.N., M.J.D. Primary writing: K.E.S., E.B.R, B.M.N., M.J.D.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Ng SB, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature genetics. 2010;42:790–3. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iossifov I, et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–9. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonarakis SE. eLS. John Wiley & Sons, Ltd; 2006. CpG Dinucleotides and Human Disorders. [Google Scholar]
- 8.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Ligt J, et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. New England Journal of Medicine. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 10.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. The Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 11.O’Roak BJ, et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science. 2012 doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Betancur C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 13.Darnell JC, et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu B, et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nature genetics. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 17.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–4. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 18.Epi KC P. Epilepsy Phenome/Genome. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, et al. The Genome Analysis Toolkit: a Map Reduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koren A, et al. Differential Relationship of DNA Replication Timing to Different Forms of Human Mutation and Variation. The American Journal of Human Genetics. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kryukov GV, Pennacchio LA, Sunyaev SR. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. The American Journal of Human Genetics. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell CD, et al. Estimating the human mutation rate using autozygosity in a founder population. Nature genetics. 2012;44:1277–81. doi: 10.1038/ng.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nature genetics. 2011;43:712–4. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.