

Abstract
The ectodomain of anthrax toxin receptor 2 (ANTXR2) is composed of a von Willebrand factor A (VWA) domain that binds to anthrax toxin protective antigen (PA) and a newly defined immunoglobulin-like (Ig) domain, in which the disulfide bonds are required for PA pore formation and for the folding of ANTXR2. While the VWA domain has been well characterized, the structure and function of the whole ectodomain (VWA-Ig) are poorly defined, which is mainly due to the limited production of the soluble recombinant protein of the ectodomain. In the present study, the ANTXR2 ectodomain was fused to the C-terminus of bacterial Trigger Factor (TF), a chaperone that mediates the ribosome-associated, co-translational folding of newly synthesized polypeptides in E. coli. Under the control of a cold shock promoter, the fusion protein was overly expressed as a dominant soluble protein at a low temperature in the oxidative cytoplasm of Origami B cells, where formation of the disulfide bonds is favored. Through a series of chromatography, the ANTXR2 ectodomain was purified into homogeneity. The purified ectodomain is functional in binding to PA and mediating PA pore formation on the liposomal membranes, and the yield is applicable for future biochemical and structural characterization.
Keywords: anthrax toxin receptor 2, ectodomain, disulfide bonds, Trigger Factor
Introduction
A common way for pathogenic bacteria to overcome the host immune defense system is to deliver toxins into the cytoplasm of host cells and disrupt the key cellular metabolic pathways. Most of the intracellularly acting bacterial toxins enter into host cells through receptor-mediated endocytosis [1]. Study of the roles of receptors in toxin action is of interest in understanding bacterial pathogenesis and in developing novel therapeutics against infection.
Anthrax toxin, the major virulence factor of Bacillus anthracis, is a tripartite system, composed of two catalytic moieties, edema factor (EF) and lethal factor (LF), and a receptor-binding/pore-forming moiety, protective antigen (PA) [2]. PA (83 kDa) binds to cell-surface receptors and is cleaved by furin or a furin-like protease to generate an active, 63-kDa form (PA63) [3]. PA63 oligomerizes into a heptameric or octameric receptor-bound prepore [4,5], which contains high-affinity binding sites for EF and LF [6]. The toxin-receptor complexes are internalized by receptor-mediated endocytosis, and the prepore moiety undergoes an acidic pH-dependent conformational rearrangement within the endosome to form a cation-selective, transmembrane pore [7,8]. The PA pore mediates translocation of EF and LF across the endosomal membrane into the cytosol, where, EF, an 89-kDa calmodulin-dependent adenylate cyclase, elevates levels of cAMP [9], and LF, a 90-kDa zinc protease, inactivates mitogen-activated proteins kinase kinases [10].
Anthrax toxin receptors play an essential role in anthrax toxin action. They provide the toxin with a high-affinity anchor on the membranes and a path of entry into the cells. Two cellular receptors for PA have been identified: ANTXR1 (or TEM8) [11] and ANTXR2 (or CMG2) [12]. Recently, it has been shown that the lethality of anthrax toxin for mice is primarily mediated by ANTXR2 and that ANTXR1 plays only a minor role [13]. ANTXR2 is a type-I transmembrane protein and comprises an extracellular von Willebrand Factor A domain (VWA, 38-218), a stalk region (219-318), a single-pass transmembrane domain, and a cytoplasmic domain (Figure 1A). While the VWA domain has been well characterized as a high-affinity PA-binding domain [14-16], until recently has the stalk region been defined as an immunoglobulin-like (Ig) domain, in which the disulfide bonds are essential for anthrax toxin action [17] (Figure 1B).
Figure 1. Schematic of anthrax toxin receptor 2 (ANTXR2).

A. ANTXR2 is composed of a PA-binding VWA domain (residues 38-218), an Ig domain (residues 219-318), a single-pass transmembrane domain (TM, 319-343) and a cytoplasmic domain (Cyto, 344-489). For simplicity, here VWA domain is termed as R218, and the whole ectodomain, VWA-Ig, is termed as R318. B. The amino acid sequence of ANTXR2 ectodomain, R318. The residues in the Ig domain are bolded and the Cys residues are highlighted.
ANTXR2 is ubiquitously expressed in different types of tissues [12], however, its physiological roles have not been fully elucidated. It has been shown that ANTXR2 binds to extracellular matrix components and is associated with angiogenesis [18]. Mutations in the ANTXR2 gene are linked to two human autosomal recessive diseases, juvenile hyaline fibromatosis and infantile systemic hyalinosis [19-22]. Most recently, new missense mutations in the Ig domain of ANTXR2 (e.g. V310F and C315W) were identified in the patients with hyaline fibromatosis syndrome, where the mutations affected folding and disulfide bond formation of the receptor in the ER, thereby leading to ER retention [23].
While the Ig domain of ANTXR2 plays an important role in both anthrax toxin action and cellular physiological processes, the structure of the whole ectodomain (here termed R318) is still not available. Compared to the VWA domain (here termed R218) that is highly soluble when expressed in E. coli, R318 is much less soluble, which is presumably due to the low efficiency of forming correct disulfide bonds in the Ig domain. In this study, we have developed a novel protocol that significantly increased the solubility and hence the yield of functional R318 protein.
Materials and Methods
Genes, Plasmids and E. coli Strains
The gene encoding the residues 38-318 of ANTXR2 was amplified by PCR using the primers listed in Table I, and cloned into pGEX4T-1 (GE healthcare), pMAL-c2x (NEB), pET22b (Novagen), pCOLD-TF (Takara), respectively. The resulting constructs were named as pET22b-R318-His6, pGEX4T-R318, pMal-c2x-R318 (± His6), and pCOLD-TF-R318(± His6), respectively. All of the constructs were confirmed by DNA sequencing. The constructs were then transformed into Origami B (DE3) cells (Novagen) and the colonies were selected by 100 μg/ml of carbenicillin on LB agar plates.
Table I.
Primers used in this study
| Primers | Sequence (5′–3′) | Restriction Sites | |
|---|---|---|---|
| pET22b-R318-His6 | F | GGTGGTCATATGTCCTGCAGACGAGCCTTTG | Nde I/Xho I |
| R | GGTGGTCTCGAGCCCGTTAGAACATTCTGTGGC | ||
| pMal-c2x-R318-His6 | F | AGATCTGAATTCTCCTGCAGACGAGCCTTTG | EcoR I/Hind III |
| R | ATGCAAGCTTGTGTCAATGGTGATGGTGATG ATGCCCGTTAGAACA TTCTGTGGC |
||
| pMal-c2x-R318 | F | AGATCTGAATTCTCCTGCAGACGAGCCTTTG | EcoR I/Hind III |
| R | ATGCAAGCTTGTGTCACCCGTTAGAACATTCTGTGGC | ||
| pGEX4T-R318 | F | AGATCTGAATTCTCCTGCAGACGAGCCTTTG | EcoR I/Xho I |
| R | GGTGGTCTCGAGTCACCCGTTAGAACATTCTGTGGC | ||
| pCOLD-TF-R318-His6 | F | GGTGGTCATATGTCCTGCAGACGAGCCTTTG | Nde I/Xho I |
| R | GGTGGTCTCGAGTCAATGGTGATGGTGATGA TGCCCGTTAGAACATTCTGTGGC |
||
| pCOLD-TF-R318 | F | GGTGGTCATATGTCCTGCAGACGAGCCTTTG | Nde I/Xho I |
| R | GGTGGTCTCGAGTCACCCGTTAGAACATTCTGTGGC |
Analysis of Protein Expression and Solubility in Small-Scale Cultures
pET22b-R318-His6, pGEX4T-R318, pMal-c2x-R318, and pCOLD-TF-R318 were transformed into Origami B cells, respectively. The fresh colonies were cultured in 5 ml of LB medium at 30 ºC for overnight. Next morning, the starting cultures were diluted (1/50) into 30 ml of LB medium and continued to grow at 37 ºC till OD600 reached 0.8 –1.0. Then the cultures were grown at 16 ºC for 30 minutes and the protein expression were induced by 1 mM of IPTG for 16-24 hours. After induction, the cells from 1 ml of the culture were collected and lysed by B-Per bacterial protein extraction reagent (Pierce) following the manufacture’s protocol. The whole lysates were further fractionated into soluble supernatant and insoluble pellet by centrifugation at 16, 000 x g for 10 minutes. Equivalent amounts of whole cell lysate, supernatant and pellet were subjected to SDS-PAGE, followed by Coomassie blue staining. Expression of MBP-R318 was also confirmed by western blotting using an HRP-conjugated anti-MBP antibody (Pierce).
Preparative Expression and Purification of R318
The Origami B (DE3) cells harboring pCOLD-TF-R318 were cultured in a 200 ml of LB medium at 30 ºC for overnight. Next morning, the starting cultures were diluted (1/50) into 4-L of LB medium and continued to grow at 37 ºC till OD600 reached 0.8–1.0. Then the cultures were grown at 16 ºC and the protein expression was induced by 1 mM of IPTG for 16-24 hours. Bacterial cells were harvested by centrifugation and the cell paste was re-suspended in Buffer A (20 mM TrisHCl pH 7.3, 300 mM NaCl, 10mM imidazole) with 1mg/ml of lysozyme and EDTA-free protease inhibitor cocktail (1 tablet/100 ml) (Sigma). The cells were lysed in a Vibracell VCX 750 Sonifier (Sonics). The whole lysates were fractionated into the soluble supernatant and the insoluble pellet by centrifugation at 15, 000 rpm for 60 minutes. The soluble supernatant was passed through a Nickel-charged Sepharose column (50 ml, GE Healthcare), washed with 3 column volumes (CV) of Buffer A, and then eluted with a linear gradient of Buffer B (Buffer A + 500 mM imidazole) on an AKTA FPLC (GE Healthcare). The eluted fractions of TF-R318 proteins were pooled, concentrated and applied to a size exclusion chromatography using a Superdex 200 column, in which the monomeric TF-R318 was separated from the oligomeric (TF-R318)n. The monomeric TF-R318 was incubated with Factor Xa (1 μg Factor Xa for 50 μg of TF-R318) for 2 hours at room temperature, and then was passed through a 5 ml Nickel-charged Sepharose column (GE Healthcare) to remove the His-tagged TF and the uncut TF-R318. The free R318 was collected in flow through. Finally the free R318 was purified into homogeneity by a size exclusion chromatography using a Superdex 75 column. Purification of R318 with a C-terminal His-tag (R318-His6) was similar as purification of R318, except that after cleavage by Factor Xa, R318- His6 was separated from the His-tagged TF by an ion exchange chromatography using a mono Q column (GE healthcare). Briefly, the mono Q column was pre-equilibrated with Buffer A (20 mM TrisHCl, pH 8.5), and the cleavage reaction mixture (Factor Xa, R318-His6 and His6-TF) was loaded into the column and eluted with a gradient (0-60%) of Buffer B (20 mM TrisHCl, 1 M NaCl, pH 8.5).
Purification of PA83, R218 and R218-His6
PA83 and R218 were expressed and purified as previously described [4,14,24]. GST-R218-His6 was expressed as a soluble protein in the cytoplasm of BL21 (DE3) cells. The cells were grown at 37 ºC till OD600 reached 0.8 – 1.0, and then the cells were transferred to 30ºC for 30 minutes, which was followed by induction with 1 mM IPTG for 3-6 hours. The cells were harvested, lysed, and fractionated into soluble and insoluble factions. The GST-R218-His6 proteins in the soluble fraction were first purified by an affinity chromatography using the 50 ml Nickel-charged Sepharose column (GE Healthcare) as described in the section of “Preparative Expression and Purification of R318”. The GST tag was cleaved by thrombin (1 unit thrombin for 50 μg of GST-R218-His6), and the uncut GST-R218-His6, free GST and thrombin were removed by passing through a glutathione-Sepharose 4B column (GE Healthcare), followed by a benzamidine column (GE healthcare). R218-His6 was collected in flow through and further purified by a size exclusion chromatography using a Superdex 75 column (GE Healthcare).
Gel Shift Assay For PA-Receptor Binding
The PA-receptor binding was performed in the buffer 20 mM Tris-HCl (pH 8.5), 150 mM NaCl, 1mM MgCl2, at room temperature for 30 mins. The purified R218 and R318 were incubated with PA83 as the indicated combinations with molar ratio of receptor/PA at 3:1. The samples were then subjected to native gel electrophoresis, followed by Coomassie blue staining.
Liposome Preparation
The liposomes containing ANTS/DPX were prepared as previously described [24,25]. Briefly, 1, 2-dioleoyl-sn-glycero-3 phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-{[N(5-amino-1-carboxypentyl) iminodiacetic acid]succinyl} (Nickel salt) (DOGs-NTA-Ni) were mixed at molar ratio 100:8, and then the mixer was dried under N2 gas to form a lipid film, followed by vacuum overnight to remove residual solvent. The resulting dry lipid film was rehydrated with 5 mM HEPES (pH 7.3), 50 mM ANTS and 50 mM DPX, followed by six freeze-thaw cycles. The liposome/dye solution was extruded through a 200-nm pore size polycarbonate filter (Nucleo-pore) in a mini-extruder (Avanti Polar Lipids). Finally, the liposomes were buffer exchanged in a G-25 column into the buffer 5 mM HEPES, 150 mM NaCl (pH 7.3).
Time-Lapse Intensity Measurement of ANTS/DPX Dequenching
The ANTS fluorescence dequenching assay for PA pore formation was adopted from a former report [25]. Briefly, PA heptamer (PA63) was pre-incubated with R218-His6 and R318-His6 with a molar ratio of receptor/PA at 3:1 in 200 μl of 20 mM Tris-HCl (pH 8.5), 150 mM NaCl, 1mM MgCl2, at room temperature for 30 mins. Then 100 μl of the liposomes containing ANTS/DPX were incubated with the protein mixture for an additional 30 mins. The protein/liposome mixture was diluted into 1.3 ml of 20 mM TrisHCl (pH8.5), 150 mM NaCl, 1 mM MgCl2 with continuous stirring in an ISS-K2 multiphase frequency and modulation fluorometer with excitation at 380 nm and emission at 520 nm. After the base line was stabilized, 300 μl of 1M NaAc (pH 5.0) was injected into the cuvette, and the fluorescence signal was monitored in real-time. Crossed polarizers on excitation and emission beams, and a 435-nm long path filter were used to reduce the background scatter.
Results
TF-R318 under the control of a cold promoter was expressed as a soluble protein in the cytoplasm of Origami B cells
While other expression systems (e.g. yeasts, insect cells and mammalian cells) have been developed for expressing eukaryotic protein, E. coli is still the most recommended expression system to obtain large quantity of recombinant proteins with relatively low cost. In earlier studies, R218 has been expressed as a GST-tagged recombinant protein and purified in soluble form with high yield from E. coli, which has led to successful determination of the crystal structure of R218 [14]. Unlike R218, however, R318 was essential insoluble when it was expressed as a GST-fusion protein in the cytoplasm of E. coli, even at the temperature as low as 15 ºC (data not shown). We hypothesize that the disulfide bonds in the Ig domain of R318 play an important role in protein folding and solubility, while the reducing environment of the cytoplasm disfavors formation of the disulfide bonds. Therefore, in an earlier study we cloned R318 into the pMal-p2x vector (NEB) and expressed the MBP-R318 fusion protein in the periplasm of E. coli BL21 (DE3) cells at 16 ºC, where disulfide bond formation is favored [17]. As expected, we were able to purify soluble and functional R318, but the yield was limited (< 1mg/4-L culture) and was not applicable for further biochemical and structural studies that usually require large quantity of the recombinant proteins. The low yield of R318 protein could be attributed to one or more of the factors, such as low efficiency of protein transport from the cytosol to the periplasm, the small space of the periplasm relative to the cytoplasm, and the low efficient disulfide bond formation, etc. [26]. To increase the yield of R318, we have recently tested the expression of R318 in the Origami B strain of E. coli (Figure 2). Origami B strain carries the trxB/gor mutations that delete the activities of glutathione reductase and thioredoxin reductase, which greatly enhance disulfide bond formation in the cytoplasm [27,28]. Moreover, Origami B strain contains characteristics of lacZY deletion mutants of BL21 that enable adjustable levels of protein expression by titrating IPTG concentrations. Surprisingly, when induced with 0.1 mM IPTG at 15 ºC, R318 with either a His-tag (R318-His6) or a GST-tag (GST-R318) was still insoluble (Figure 2A and B). MBP-R318 was expressed in a low level, and the western blot showed that only 10% of MBP-R318 was in soluble faction (Figure 2C). Subsequently, we cloned the gene encoding R318 into the pCOLD-TF (trigger factor) vector to express a TF-R318 fusion protein under the control of the cspA cold promoter. After induction with 1 mM IPTG at 16 ºC, the fusion protein was overexpressed in Origami B cells and the majority of TF-R318 was in soluble fraction (Figure 2D).
Figure 2. TF-R318 under the control of a cold promoter is expressed as the most soluble protein in the cytoplasm of Origami B cells.

R318 with different fusion tags were expressed in Origami B cells as indicated. A. R318-His6; B. GST-R318; C. MBP-R318; D. TF-R318. The cells were harvested, lysed and fractionated by centrifugation. The whole cell lysates (W), soluble fraction (S) and insoluble pellet (P) were subjected to SDS-PAGE, followed by Coomassie blue staining. The fusion proteins were marked with arrows. Expression of MBP-R318 was further confirmed by western blot using an HRP-conjugated anti-MBP antibody (C).
R318 was purified into homogeneity through a series of chromatography
The large-scale expression of TF-318 was performed in 4-L of LB medium, where TF-R318 in Origami B cells was induced at OD600 0.6-1.0 with 1 mM IPTG at 16 ºC for 16-24 hours. At such a low temperature, expression of most endogenous proteins was inhibited, but expression of TF-R318 under the control of the cspA cold promoter was highly activated. At the time of harvest, TF-R318 was account for about ¼ of the total soluble proteins (Table II and Figure 3B). The soluble lysate was first applied to a Nickel-charged Sepharose column, and the His-tagged TF-R318 was purified by immobilized-metal affinity chromatography (IMAC) (Figure 3A). The eluted TF-R318 appeared to be a mixture of soluble oligomeric TF-R318, termed (TF-R318)n, and monomeric TF-R318, which were further separated by a size exclusion chromatography using a Superdex 200 column (Figure 3C). In SDS-PAGE without the reducing agent, the oligomeric (TF-R318)n was run as a smear with various oligomeric forms, while in the presence of the reducing agent, the majority of the oligomeric (TF-R318)n was reduced and run as a single monomeric band, suggesting that majority of the oligomeric (TF-R318)n was formed by cross-linking of the inter-molecular disulfide bonds (Inserted figures in Figure 3C). The monomeric TF-R318 was cleaved by Factor Xa and passed through a Nickel-charged Sepharose column, in which the His-tagged TF and the uncut TF-R318 were retained in the column and the free R318 was collected in flow through. Finally, the free R318 was purified into homogeneity by a size exclusion chromatography using a Superdex 75 column (Figure 3D).
Table II.
Summary of purification of TF R318/R318
| Chromatography | Total Protein (mg) | TF-R318/R318 (mg) | Percent Yield (%) |
|---|---|---|---|
| Lysate (soluble fraction) | 475 ± 25 | 100 ± 10 | 100 |
| IMAC (nickel affinity) | 60 ± 2 | 50.2 ± 3 | 54.2 |
| Sizing exclusion: Superdex 200 | 10.2 ± 1 | 10 ± 0.5 | 10 |
| Sizing exclusion: Superdex 75 | 5.1 ± 2 | 5 ± 2 | 5 |
The results of this table are estimated from several repeats of purifications. Quantification of the proteins was performed by either using BCA protein assay or densitometric scans of Coomassie-stained bands. In either case, known concentrations of BSA were used as standards.
Figure 3. R318 was purified into homogeneity through a series of chromatography.

A. The N-terminally His-tagged TF-R318 was first purified by an immobilized-metal affinity chromatography (IMAC) as described in Material and Methods. The majority of TF-R318 was eluted at 30-60% of Buffer B. B. After purification by IMAC, the samples of whole cell lysate (W), supernatant (S), flow through (FT), wash and elute, were collected and applied to SDS-PAGE to analyze the purification efficiency. As a control, the cell lysates (W and S) before IPTG induction were also analyzed to show the IPTG-inducible expression of TF-R318. C. The TF-R318 purified by IMAC was concentrated and further purified by a size exclusion chromatography in a Superdex 200 column, in which the soluble oligomeric (TF-R318)n and the monomeric TF-R318 were separated. Insert Figures: the oligomeric (TF-R318)n and monomeric TF-R318 were analyzed by SDS-PAGE with or without DTT, followed by Coomassie blue staining. D. The monomeric TF-R318 was incubated with Factor Xa, and then passed through a 5 ml Nickel-charged Sepharose column to remove the His-tagged TF. The free R318 was collected in flow through. R318 was further purified by a size exclusion chromatography using a Superdex 75 column. R318 was eluted as a ~ 30 kDa monomeric protein and was purified to nearly homogeneity as shown in SDS-PAGE.
The purified R318 was functional in binding to PA83 and mediating PA63 pore formation on the liposomal membranes
To test if the purified R318 is functional, we first used the gel shift assay to test the binding of R318 to PA83 (Figure 4). In a native gel electrophoresis, the binding of R218 to PA83 resulted in formation of R218-PA83 complex that “shifted PA83” to a new position in the gel. Similar to R218, incubation of R318 with PA83 resulted in a shift of PA83 to a new position, suggesting that R318 was bound to PA83 and formed a complex. Next, we tested if the purified R318 is functional in mediating heptameric PA63 pore formation on the liposomal membranes. Here, instead of using the previously established K+ release assay [17,24,29], we have adopted a convenient, sensitive fluorescence dequenching assay to measure PA pore formation on the liposomal membranes (Figure 5). ANTS/DPX, the anion/cation fluorophore/quencher pair, is widely used for membrane leakage [25,30,31]. ANTS fluorescence is quenched by DPX inside the liposomes, and it is dequenched upon release into the medium. As expected, addition of Triton X-100 caused rapid membrane lysis and induced a sharp increase of ANTS fluorescence, while the buffer without proteins did not cause any release. PA63 alone caused a median level of ANTS fluorescence dequenching, which was due to pore formation on the liposomal membranes by a portion of the acidification-converted PA63 pores through random encounters with the membranes. Compared to PA63 alone, the PA63 complexed with R218-His6 induced a significantly higher fluorescence. This result was consistent with the previous result observed in the K+ release assay showing that reconstitution of PA63-R218 complex to the liposomal membranes through interaction of a His-tag on R218 with the Nickel-chelating lipids on the liposomal membranes greatly enhanced PA63 pore formation [24]. Thus, the ANTX/DPX fluorescence-dequenching assay faithfully replicated the results obtained from the K+ release assay, suggesting that it can be used as a novel assay for PA63 pore formation. In our recent studies ([12,17] and unpublished data), either disruption or mis-formation of the disulfide bonds in the Ig domain resulted in a non-functional R318 that failed to mediate PA63 pore formation on the membranes. Here, the His-tagged R318 mediated PA63 pore formation in a similar level as R218, which suggests that the purified R318 is functional in mediating PA63 pore formation and the disulfide bonds in the Ig domain are correctly formed (Figure 5).
Figure 4. The purified R318 is functional in binding to PA83.
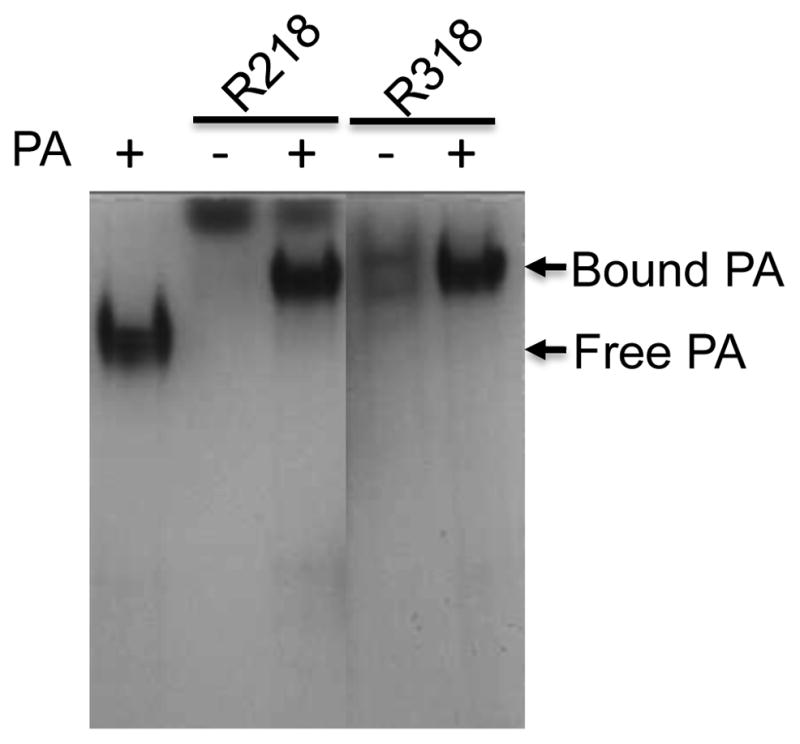
PA83 was incubated with R218 and R318 as indicated in the buffer 20 mM TrisHCl (pH 8.0), 150 mM NaCl, 1 mM MgCl2 for 1 hr. The samples were applied to native gel electrophoresis, followed by Coomassie blue staining.
Figure 5. The purified R318-His6 is functional in mediating PA63 pore formation at low pH.

The receptor-mediated PA63 pore formation was tested on the liposomal membranes using time-lapse intensity measurement of ANTS/DPX dequenching as described in Material and Methods. PA alone, buffer without proteins, and Triton X-100 (final concentration 0.06%) were used as negative or positive controls, respectively. The representative recordings of ANTS dequenching are shown in A. The fluorescence intensity at 60 seconds of post-acidification was quantified from two independent measurements and shown in B. Note: the fluorescence intensity induced by Triton X-100 was displayed in the y-axis on the right side of the graph.
Discussion
Our earlier study has shown that the disulfide bonds in the Ig domain of ANTXR2 are required for anthrax toxin pore formation and membrane translocation [17]. However, the mechanism how the disulfide bonds in the Ig domain regulate anthrax toxin action is still not clear. The structural and functional analysis of the Ig domain of ANTXR2 is significantly hindered due to the limited production of the recombinant ectodomain, R318. In the present study, we fused R318 to the bacterial chaperone Trigger Factor, and over-expressed the TF-R318 fusion protein at a low temperature under the control of a cold-shock promoter in the cytoplasm of Origami B cells, where formation of disulfide bonds is favored. This newly developed protocol has significantly increased the solubility of R318 and allowed us purify the functional R318 into homogeneity, which will facilitate biochemical and structural characterization of R318 in future.
When expressed as a GST-tagged protein in E. coli, R218 is soluble, but R318 is not soluble, suggesting that the Ig domain play a major role in the folding of the whole ectodomain. This is consistent with the recent study of the HFS patients that mutations in the Ig domain affect disulfide bond formation and folding, thereby leading to ER retention [22]. The insolubility of R318 in E. coli was apparently attributed to the reducing cytoplasm of E. coli, where disulfide bond formation is not favored. Considering the disulfide bonds as a major factor for protein folding, in the earlier study we co-expressed an MBP-R318 fusion protein with DsbC, a bacterial disulfide bond isomerase, in the periplasm of E. coli [17]. While we were able to purify soluble, functional R318 using this approach, the yield was too limited to be suitable for effective structural and functional analysis.
While the periplasm is regarded as an oxidative environment favoring disulfide bond formation, protein production in the periplasm is usually lower than that in the cytoplasm due to a number of reasons, especially when target protein has a poor solubility and requires expression at a low temperature with a low concentration of IPTG. Thus, Origami B strain that contains a modified oxidative cytoplasm becomes an attractive host for expressing target proteins that contain disulfide bonds. It was surprising that R318-His6, GST-R318 and MBP-R318 were still not soluble when expressed in Origami B cells, even with low IPTG induction and at a low temperature. This strongly suggests that in addition to the disulfide bonds, other factors also play an important role in the folding of R318 in E. coli. We have tried co-expression of R318 with the plasmid pG-KJE8 (Takara) that encodes a set of chaperones (dnaK-dnaJ-grpE-groES-groEL) in Origami B cells, but the solubility of R318 was not improved (data not shown). The observation that fusing R318 to the Trigger Factor significantly increased the solubility of R318 indicates that the TF-mediated co-translational folding [32] of the newly synthesized polypeptide of R318 is the rate-limiting step for producing correctly folded R318 in E. coli. It is worthy of mentioning that expression of a His-tagged R318 in the pCOLD vector (with the cspA cold shock promoter, but without TF fusion; Takara) did not produce soluble R318 (data not shown). This again strongly suggests that TF is the unique chaperone that is required for correct folding of R318 at the translational step. One can image that in the process of translation, TF mediates the folding of the backbone of the R318 polypeptide and brings the pairs of the Cys residues for disulfide bond formation in the Ig domain to close proximity, allowing formation of the disulfide bonds. And the formation of the disulfide bonds in turn stabilizes the folding of the protein backbones.
Expression of target protein at low temperature is a common approach to increase solubility of target protein in E. coli. However, there is a trade-off between solubility and productivity, which has to be carefully balanced. In this study, the trade-off was resolved by application of the cspA cold shock promoter that overly expressed TF-R318 at 16 ºC, a temperature where expression of the most endogenous proteins was repressed. Thus, TF-R318 was expressed as a dominant soluble protein in the cells, which greatly increased the productivity and facilitated purification.
While many techniques and host systems have been recently developed for expression of soluble target proteins, obtaining soluble, functional eukaryotic proteins containing multiple disulfide bonds in E. coli remains as a major challenge. This study provides an effective combination of expression vector and host strain, which may be applicable to expression and purification of other disulfide bond-containing proteins.
Highlights.
The ectodomain of human anthrax toxin receptor 2 (R318) was insoluble in E. coli.
R318 was cloned into a pCOLD-TF vector to produce a TF-R318 fusion protein.
TF-R318 was produced as a soluble protein in Origami B cells at 16 ºC.
R318 was purified into homogeneity through a series of chromatography.
Purified R318 was functional in binding and mediating anthrax toxin pore formation.
Acknowledgments
PJ, NL, DW, CX, JS conceived and performed the experiments. PJ and JS wrote the manuscript. This study is supported by the NIH grant SC1GM095475 (to J. Sun), UTEP new faculty startup fund (to J. Sun), the grants from the National Center for Research Resources (5G12RR008124), and the National Institute on Minority Health and Health Disparities (G12MD007592).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lin AEJ, Guttman JA. Hijacking the endocytic machinery by microbial pathogens. Protoplasma. 2010;244:75–90. doi: 10.1007/s00709-010-0164-2. [DOI] [PubMed] [Google Scholar]
- 2.Young JAT, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 3.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 4.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- 5.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, et al. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA. 2002;99:7045–7048. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacy DB, Collier RJ. Structure and function of anthrax toxin. Curr Top Microbiol Immunol. 2002;271:61–85. doi: 10.1007/978-3-662-05767-4_4. [DOI] [PubMed] [Google Scholar]
- 8.Collier RJ, Young JAT. Anthrax toxin. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 9.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 11.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 12.Scobie HM, Rainey GJA, Bradley KA, Young JAT. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu S, Crown D, Miller-Randolph S, Moayeri M, Wang H, Hu H, et al. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci USA. 2009;106:12424–12429. doi: 10.1073/pnas.0905409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacy DB, Wigelsworth DJ, Scobie HM, Young JAT, Collier RJ. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci USA. 2004;101:6367–6372. doi: 10.1073/pnas.0401506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 16.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Collier RJ. Disulfide bonds in the ectodomain of anthrax toxin receptor 2 are required for the receptor-bound protective-antigen pore to function. PLoS ONE. 2010;5:e10553. doi: 10.1371/journal.pone.0010553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, et al. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci. 2001;114:2755–2773. doi: 10.1242/jcs.114.15.2755. [DOI] [PubMed] [Google Scholar]
- 19.Dowling O, Difeo A, Ramirez MC, Tukel T, Narla G, Bonafe L, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73:957–966. doi: 10.1086/378781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73:791–800. doi: 10.1086/378418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JYY, Tsai YM, Chao SC, Tu YF. Capillary morphogenesis gene-2 mutation in infantile systemic hyalinosis: ultrastructural study and mutation analysis in a Taiwanese infant. Clinical and Experimental Dermatology. 2005;30:176–179. doi: 10.1111/j.1365-2230.2004.01698.x. [DOI] [PubMed] [Google Scholar]
- 22.Deuquet J, Lausch E, Superti-Furga A, van der Goot FG. The dark sides of capillary morphogenesis gene 2. Embo J. 2011;31:3–13. doi: 10.1038/emboj.2011.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deuquet J, Lausch E, Guex N, Abrami L, Salvi S, Lakkaraju A, et al. Hyaline fibromatosis syndrome inducing mutations in the ectodomain of anthrax toxin receptor 2 can be rescued by proteasome inhibitors. EMBO Mol Med. 2011;3:208–221. doi: 10.1002/emmm.201100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J, Vernier G, Wigelsworth DJ, Collier RJ. Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J Biol Chem. 2007;282:1059–1065. doi: 10.1074/jbc.M609869200. [DOI] [PubMed] [Google Scholar]
- 25.De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. Mycobacterium tuberculosis ESAT-6 Exhibits a Unique Membrane-interacting Activity That Is Not Found in Its Ortholog from Non-pathogenic Mycobacterium smegmatis. J Biol Chem. 2012;287:44184–44191. doi: 10.1074/jbc.M112.420869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgiou G, Segatori L. Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr Opin Biotechnol. 2005;16:538–545. doi: 10.1016/j.copbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Bessette PH, Aslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy R, Weiss R, Chen G, Iverson BL, Georgiou G. Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Protein Expr Purif. 2001;23:338–347. doi: 10.1006/prep.2001.1520. [DOI] [PubMed] [Google Scholar]
- 29.Sun J, Lang AE, Aktories K, Collier RJ. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci USA. 2008;105:4346–4351. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellens H, Bentz J, Szoka FC. pH-induced destabilization of phosphatidylethanolamine-containing liposomes: role of bilayer contact. Biochemistry. 1984;23:1532–1538. doi: 10.1021/bi00302a029. [DOI] [PubMed] [Google Scholar]
- 31.Ellens H, Bentz J, Szoka FC. H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry. 1985;24:3099–3106. doi: 10.1021/bi00334a005. [DOI] [PubMed] [Google Scholar]
- 32.Maier T, Ferbitz L, Deuerling E, Ban N. A cradle for new proteins: trigger factor at the ribosome. Curr Opin Struct Biol. 2005;15:204–212. doi: 10.1016/j.sbi.2005.03.005. [DOI] [PubMed] [Google Scholar]