

Abstract
Purpose.
Prostaglandin J2 (PGJ2) is neuroprotective in a murine model of nonarteritic anterior ischemic optic neuropathy (NAION). After assessing for potential toxicity, we evaluated the efficacy of a single intravitreal (IVT) injection of PGJ2 in a nonhuman primate model of NAION (pNAION).
Methods.
We assessed PGJ2 toxicity by administering it as a single high-dose intravenous (IV) injection, consecutive daily high-dose IV injections, or a single IVT injection in one eye of five adult rhesus monkeys. To assess efficacy, we induced pNAION in one eye of five adult male rhesus monkeys using a laser-activated rose bengal induction method. We then injected the eye with either PGJ2 or phosphate-buffered saline (PBS) intravitreally immediately or 5 hours post induction. We performed a clinical assessment, optical coherence tomography, electrophysiological testing, fundus photography, and fluorescein angiography in all animals prior to induction and at 1 day, 1 week, 2 weeks, and 4 weeks after induction. Following analysis of the first eye, we induced pNAION in the contralateral eye and then injected either PGJ2 or PBS. We euthanized all animals 5 weeks after final assessment of the fellow eye and performed both immunohistochemical and light and electron microscopic analyses of the retina and optic nerves.
Results.
Toxicity: PGJ2 caused no permanent systemic toxicity regardless of the amount injected or route of delivery, and there was no evidence of any ocular toxicity with the dose of PGJ2 used in efficacy studies. Transient reduction in the amplitudes of the visual evoked potentials and the N95 component of the pattern electroretinogram (PERG) occurred after both IV and IVT administration of high doses of PGJ2; however, the amplitudes returned to normal in all animals within 1 week. Efficacy: In all eyes, a single IVT dose of PGJ2 administered immediately or shortly after induction of pNAION resulted in a significant reduction of clinical, electrophysiological, and histological damage compared with vehicle-injected eyes (P = 0.03 for both VEP and PERG; P = 0.05 for axon counts).
Conclusions.
In nonhuman primates, PGJ2 administered either intravenously or intravitreally produces no permanent toxicity at even four times the dose given for neuroprotection. Additionally, a single IVT dose of PGJ2 is neuroprotective when administered up to 5 hours after induction of pNAION.
Keywords: anterior ischemic optic neuropathy, white matter, ischemia, 15d-prostaglandin J2, PGJ2, intravitreal injection, neuroprotection
The prostaglandin agonist PGJ2 is non-toxic and neuroprotective in a non-human primate model of nonarteritic anterior ischemic optic neuropathy.
Introduction
Nonarteritic anterior ischemic optic neuropathy (NAION) is caused by inadequate blood supply to the optic nerve head (ONH; i.e., the optic disc) and is the leading cause of sudden optic nerve-related vision loss in individuals over 50 years old.1 In the United States, it affects 2.3 to 10.2 per 100,000 people over 50 years of age,2,3 resulting in up to over 10,000 new cases per year. Very little is understood concerning the pathophysiology of the disease, and there currently is no medical or surgical treatment that has been proven to be consistently beneficial.4
Reproducible rodent and nonhuman primate models of NAION (rNAION, pNAION) have been developed by intravenous (IV) injection of rose bengal (RB) followed by intraocular laser photoactivation of the dye at the ONH that produces an optic neuropathy that, despite having a different etiology, clinically, electrophysiologically, and histopathologically resembles human NAION.5–8 These models provide an invaluable resource because they can be used to analyze critically the mechanisms resulting in postischemic ON damage and because they can be used as tools in the evaluation of potential neuroprotective and neuroreparative therapies for patients who experience NAION.
15-deoxy 12, 14 delta prostaglandin J2 (PGJ2) is a naturally occurring prostaglandin that has been found to be neuroprotective in a number of murine models of stroke, both in vitro and in vivo.9,10 PGJ2 influences at least two major pathways, both of which are key modulators of inflammation. First, PGJ2 is an intrinsic antagonist of the nuclear transcription factor nuclear factor kappa beta (NFκB),11 upregulation of which is the major central factor associated with both early cytokine-related and later cellular inflammation.12 Second, PGJ2 is the major ligand for activation of the nuclear factor peroxisomal proliferator activated receptor-gamma (PPARγ). In the brain, PPARγ expression occurs in microglia and astrocytes, two cell types that play an important role in inflammation, and systemic administration of 15d-PGJ2 results in a PPARγ-dependent decrease in neuronal apoptosis and necrosis in a murine model of brain stroke.10 Thus, it is not surprising that PPARγ agonists are associated with neuroprotection and reduced degenerative neuroinflammation.13,14
We previously reported that PGJ2, whether systemically administered or directly injected into the eyes of adult rats immediately following induction of our rNAION model, results in electrophysiological and histopathological evidence of preservation of optic nerve function as well as preservation of both retinal ganglion cells (RGCs) and RGC function, compared with control animals injected with phosphate-buffered saline (PBS) alone 30 days post injection.15 Visual evoked potentials (VEPs) obtained 7 days after a single intravitreal (IVT) injection of PGJ2 in rNAION-induced eyes had amplitudes similar to baseline measurements.15 Thirty days post induction, electron microscopic analysis of optic nerves from PGJ2-treated eyes demonstrated significant preservation of axons and minimal demyelination compared with eyes injected with PBS. RGC counts revealed significant RGC preservation in PGJ2-treated eyes compared with PBS-injected. Similar results also were reported after systemic injection of PGJ2.10
Despite the encouraging results described above in our rNAION model, a major problem with the assessment of potential treatments of ON ischemia as well as ischemia in other parts of the central nervous system (CNS) is that, to date, treatments successful in murine models have rarely been successful in human clinical trials. One reason is that rodent and primate physiologic responses can be considerably different.16 Thus, it is our opinion that the only way truly to determine if a drug is neuroprotective in humans is to test it in humans or, initially, in a species that responds in a similar manner. For this reason, we elected to test the efficacy of PGJ2 in our pNAION model.
Methods
Animals
All animal protocols were approved by the University of Maryland Institutional Animal Care and Utilization Committee (IACUC) and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. For induction of pNAION, male rhesus monkeys (Macaca mulatta, age 4–6 years, 6–14 kg) were anesthetized with a mixture of ketamine (10 mg/kg) and xylazine (2 mg/kg). For subsequent assessments, the animals were anesthetized initially with intramuscular (IM) ketamine, followed by an IM injection of atropine to reduce oral secretions. After initial anesthetization, the animals (n = 10) were intubated, and assessments performed while the animals were supported with a continuous infusion of either isoflurane (n = 2) or propofol (n = 8). Propofol was a far better agent than isoflurane for obtaining in vivo electrophysiological measures because isoflurane suppresses cortical electrical responsiveness.17 Intermittent IV or IM injections of ketamine were used throughout the assessment to reduce spontaneous eye movements.
PGJ2 Toxicology
Five animals (T1–T5) underwent toxicological studies of PGJ2 before efficacy experiments were begun. Prior to administration of PGJ2, both eyes of each animal were assessed clinically as well as electrophysiologically with VEPs, pattern electroretinograms (PERGs), ganzfeld ERGs, color photography, red-free photography, and fluorescein angiography (FA). Additionally, blood obtained by venipuncture was used to assess each animal's systemic biochemical (i.e., electrolyte), hepatic, and hematologic status. The dosages of PGJ2, routes and length of times of administration, and results are illustrated in Figure 1. Briefly, two animals (T1, T2) were injected with a single IV dose of PGJ2 (T1 = 40 μg/kg; T2 = 125 μg/kg). The animals then were assessed for changes in behavior and examined clinically, electrophysiologically, and with blood drawn for electrolytes, liver enzymes, and hematology at days 1 and 3 and then weekly for up to 4 weeks for potential PGJ2-induced toxicity. Two months after completion of these studies, one of these animals (T1) and a third animal (T3) were injected with five consecutive daily IV doses of PGJ2 (T1 = 40 μg/kg daily; T3 = 100 μg/kg daily) and assessed in the same manner.
Figure 1.
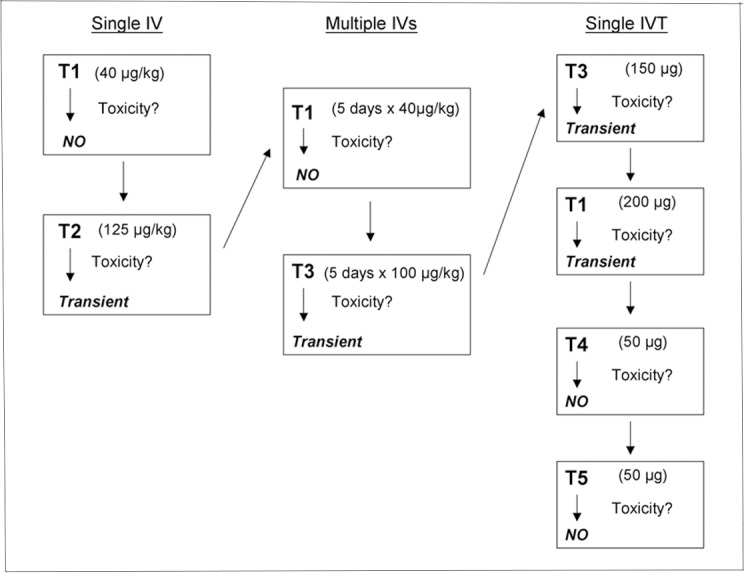
Schematic diagram of PGJ2 toxicity protocol showing doses, routes of administration, length of administration, and whether or not we noted any evidence of transient toxicity (no animal experienced permanent toxicity).
Several months after completion of the assessments of the animals given the five consecutive daily IV (systemic) injections, four animals (T1, T3, T4, and T5) were injected with a single IVT dose of PGJ2 (T1 = 200 μg; T3 = 150 μg; T4 = 50 μg; T5 = 50 μg). The volume injected was the same for all doses, and all injections were performed under aseptic conditions using a 31-gauge needle attached to an insulin syringe (BD-) and inserted through the pars plana. A trans-corneal paracentesis was performed within 1 minute after injection to reduce intraocular pressure, and antibiotic ointment was applied immediately after the injection. All four animals were again assessed clinically, electrophysiologically, and hematologically for up to 4 weeks following the IVT injection.
pNAION Induction
We induced pNAION in five animals (E1–E5) for our efficacy studies. Although we induced pNAION in both eyes of all five animals, these inductions were sequential, not simultaneous, with the second eye induced 5 weeks after the first eye. The procedure for induction of pNAION has been described previously.8 Briefly, after anesthetization with ketamine and xylazine, the pupil of one of the monkey's eyes was dilated with a combination of 1% tropicamide and 2.5% phenylephrine. The animal then was placed in front of a slit-lamp biomicroscope fitted with an ophthalmic neodymium-yttrium-aluminum-garnet (Nd-YAG) frequency-doubled diode laser (532 nm: Iridex, Mountain View, CA, USA). For pNAION induction, we replaced the 209-μm-diameter laser fiber optic cable with a 500-μm-diameter fiber optic cable that had SMA adapter ends. This alteration enabled us to use the standard 532-nm slit-lamp adapter to generate a 1.06-mm spot size using the 200-μm spot size setting. The optic disk in the eye to be lasered was visualized using a Glasser monkey contact lens (Ocular Instruments, Inc., Bellevue, WA, USA), and pNAION was induced by injecting RB intravenously in a dose of 2.5 mg/kg of lean body weight, followed 30 seconds later by dye activation at the optic disk using the 1.06-mm spot size, at 200 mW, for times ranging from 8 to 9 seconds. With equivalent induction times, laser power and dye administration, the generated lesion was consistent between eyes of a given animal (data not shown). Thus, we were able to generate submaximal lesions that enabled us to use one eye of each animal as a vehicle control, and the other for PGJ2 treatment. All animals had preserved visual function after each induction, as assessed by overall behavior, pupillary responses to light stimulation, and electrophysiologic assessment. Thus, despite being visually impaired from pNAION induction in both eyes, the animals were not blinded during the course of this study. This approach considerably reduced the total number of animals needed for the study and enabled us to compare directly responses between eyes of the same animal.
Neuroprotection Experiments, IVT Injection
Eyes were injected with PGJ2 or vehicle either 15 minutes (E1–E3) or 5 hours (E4, E5) after pNAION induction. Prior to injection, eyes were prepped three times with 5% povidone iodine and anesthetized with viscous ophthalmic tetracaine (Tetravisc). Topical ciprofloxacin drops were instilled, and the induced eye received an IVT injection consisting of either 50 μL of sterile filtered PBS 20% ethanol (vehicle) or 50 μg of sterile filtered PGJ2 dissolved in 50 μL of PBS 20% ethanol (PGJ2). The 50 μg dose of PGJ2 was chosen for our efficacy experiments based on the dosage that we previously used in our rNAION model15 and the difference in size between adult rat and adult rhesus monkey eyes. Five weeks following pNAION induction and injection of the first eye, pNAION was induced in the contralateral eye, and PGJ2 or vehicle administered, using the same technique. Neither the individual who performed the induction nor the individual who performed the IVT injections was masked as to which substance was being injected; however, the individual who performed the injections did not participate in the subsequent assessments of the animals. In addition, the individuals who performed the assessments could not be masked as to the substance injected because IVT PGJ2 causes slight clouding of the ocular media for several days following the injection.
Optical Coherence Tomography
Optical coherence tomography (OCT) was used to assess the thickness of the peripapillary retinal nerve fiber layer (PRNFL), volume of the peripapillary retina, and volume of the macula in four of the five animals (E2–E5): the OCT instrument was obtained after the first animal, E1, was treated). We used a Heidelberg Spectralis spectral-domain HRA + OCT (SD-OCT) instrument (Heidelberg Engineering, Heidelberg, Germany) with 5.4b-US software and equipped with an automated real time eye-tracking system (ART). Before imaging, each animal's pupils were dilated with topical 2.5% phenylephrine and 1.0% tropicamide. For assessment of the PRNFL, the circular scan mode was employed, which uses a circle measuring 3.5 mm in diameter. One hundred images were averaged. A minimum of three scans was obtained for each eye at the same location, manually segmented, and the average value determined. The posterior border of Bruch's membrane was used as the outer boundary for all retinal thickness measurements. For total peripapillary and retinal thickness, we obtained ON and macular volume scans with 30 images averaged per frame, and image spacing between scans of 120 μm. At least two sets of measurements were obtained at each site. The set with the best quality was used for manual segmentation. All manual segmentation was performed by the same investigator (NRM). After segmentation, the thickness of the PRNFL was determined by averaging the thicknesses of the six sectors (T, ST, SN, N, IN, and IT). The volumes of the peripapillary and macular retina were determined using the 3-mm Early Treatment Diabetic Retinopathy Study (ETDRS) circle.
Electrophysiology
Pattern VEPs and PERGs were performed prior to induction and at 1 day, 1, 2, and 4 weeks post induction of pNAION. Ganzfeld ERGs were performed at baseline, at 1 day, and at 4 weeks post induction. All recordings were performed with both of the animal's pupils dilated with topical 2.5% phenylephrine and 1.0% tropicamide.
Simultaneous PERG and VEP recordings were obtained with the monkey in the prone position and a Burian-Allen bipolar contact lens electrode placed on the eye. Potentials were recorded from each eye separately. To record simultaneous VEP and PERG waveforms, a Topcon fundus camera (TOPCON Corporation, Tokyo, Japan) was modified by inserting a 2-cm OLED screen from a head-mounted display into the split-viewer pathway.18 The input to the screen was an alternating (1.9 Hz) black-and-white checkerboard pattern having a luminance of 109 cd/m2 at a nominal contrast of 100%, generated by a LKC UTAS visual diagnostic system (LKC Technologies, Inc., Gaithersburg, MD, USA). The location of the screen was adjusted so that it was conjugate to the plane of the animal's pupil. In this way, when the monkey's retina was in focus, the image of the checkerboard was in focus on the monkey's retina. The macula was positioned using an infrared light source and an infrared-sensitive charge-coupled device camera to avoid bleaching visual pigment. The field size stimulated was 45°. A 32 × 32 matrix was used, producing a check size of 56′. At intervals during the testing, the position of the animal's eye was reassessed using the infrared camera, followed by a resting period of 5 to 7 minutes to reduce the effects of light exposure.
For VEP recordings, the active electrode (Oz) was placed above the inion in the midline over the shaved skull, the reference electrode was placed in the midline frontal position (Fz), and the ground electrode was placed on an arm using Grass gold surface electrodes and EC2 electrode paste (Grass Instruments, Warwick, RI, USA). Recordings were repeated at least 10 times per eye; 8 to 10 consistent recordings were averaged offline to produce a waveform, the parameters of which were used in comparative analyses. For analysis of PERGs, the amplitudes of P50 (N35 trough to P50 peak) and N95 (P50 peak to N95 trough) waves were measured.19
Ganzfeld ERGs were performed following 30 minutes of dark adaptation. A Burian-Allen bipolar electrode was placed in each eye and a ground electrode placed on an arm. Responses were elicited following the International Society for Clinical Electrophysiology of Vision (ISCEV) protocol.20 High and low band-pass filters were set at 0.3 Hz and 500 Hz, respectively. Oscillatory potentials were extracted using software developed by Severns et al.21 available in existing LKC software.
Optic Nerve Vascular Imaging
Clinical optic nerve vascular imaging was performed using FA. Fluorescein angiography was performed at baseline, 1 day, and at 1, 2, and 4 weeks post pNAION induction by intravenously injecting 0.30 mL of 25% fluorescein dye (AK-Fluor; AMP; Akorn, Decatur, IL, USA). Retinal and ON photographs were obtained using a Topcon fundus camera with a standard excitation filter transmitting blue-green light at 465 to 490 nm, the peak excitation range of fluorescein, and a barrier filter transmitting a narrow band of yellow at fluorescein's peak emission range of 520 to 530 nm.
Tissue Collection and Preparation
Animals were euthanized approximately 70 days following induction and injection of the second eye under deep surgical plane anesthesia by intracardiac saline perfusion followed subsequently with 4% paraformaldehyde in PBS (PF-PBS). Following dissection and postfixation in PF-PBS, ON tissues were prepared for standard histology either by paraffin embedding (7-μm-thick sections) or by cryoprotection in 30% sucrose and frozen section embedding (10-μm-thick sections) in optical cutting temperature compound. All nerves were embedded on end. After removal of the anterior segment, globes were incised to form a Maltese cross pattern, with the macula in the center of one of the arms. Each arm was bisected longitudinally, with half the arm saved for paraffin embedding and the other half for frozen section. Paraffin-embedded retinal sections (7-μm thick) were dewaxed and evaluated by staining with hematoxylin and eosin (H&E).
For transmission electron microscopy (TEM), ON tissue was postfixed in glutaraldehyde-paraformaldehyde buffer. All ONs were placed on end and divided into six pie-shaped sections, impregnated with uranyl acetate, and shadowed with osmium. Each section was then embedded on end in Araldite-Epon.
Immunohistochemistry
Seven-micrometer-thick paraffin sections of the ON were dewaxed and rehydrated. Following boiling citrate buffer (pH 6.0)-antigen retrieval, the sections were blocked with 2% donkey serum and incubated with mouse monoclonal antibody to neurofilaments (SMI312: Sternberger Monoclonals, Lutherville, MD, USA) for intact axons or rabbit anti-human glial fibrillary acidic protein (GFAP; ab7262: Abcam, Cambridge, MA, USA) for glial scarring at 1:1000 dilution overnight at 4°C. Slides then were washed and incubated with the appropriate fluorescent-labelled secondary donkey antibody (Jackson Immunoresearch, West Point, PA, USA) for 4 hours at room temperature, followed by extensive washing. Slides were mounted with aqueous mounting medium and examined at the appropriate wavelengths using an Olympus four-channel confocal microscope.
Electron Microscopic Analysis and Axon Quantification
Two hundred nanometer-thick cross-sections were prepared from each embedded ON section, floated onto copper mesh grids, and examined by TEM using a Tecnai FEI T12 electron microscope. One individual (AB) blinded to the substance injected in the eye from which the ON was obtained generated all axon counts. Counts were performed stereologically using 60 randomly selected axon fields after dissection of each monkey ON into six equivalent regions. Ten random sites from each region were imaged at 2100× to yield 60 131-μm2 fields from each ON (total 7825 μm2). A minimum of 600 axons was counted in each nerve. We calculated the coefficient of error (COE) for each nerve. The COE provides information on the accuracy of the population count used to supply stereologically obtained estimates and is a standard statistical value that is used extensively in stereological studies (www.stereology.info/coefficient-of-error/; provided in the public domain by MBF Bioscience, Williston, VT, USA). COEs were in the best-practice boundary range (0.05–0.18).22,23 This was particularly crucial given that, in normal adult macaques, the age-independent mean of ON axons is approximately 1,100,000, with a range of 785,532 to 1,280,474 and a mean difference of 0.6% (SD = 1.7%) between the two eyes.24
Results
PGJ2 Toxicity
PGJ2, whether injected intravenously as a single dose, intravenously in five daily doses, or intravitreally as a single dose was not associated with systemic toxicity as assessed by changes in daily animal behavior and serologic parameters in any of the five animals assessed. Specifically, there were no changes in animal behavior, liver enzymes showed no consistent or significant alterations after treatment, and there were no significant alterations in serum electrolytes or complete blood count and differential. No changes in the appearance of the ONs or retinas were detected in the eyes of any of the animals. In addition, no significant changes were detected in the ganzfeld ERG in any of the five animals after either IV or IVT administration of PGJ2; however, both VEP and PERG amplitude reductions were detected at 1 day in animal T2 after a single IV injection of 125 μg/kg of PGJ2, in animal T3 after five consecutive daily IV injections of 100 μg/kg of PGJ2, in animal T1 after a single IVT injection of 200 μg of PGJ2, and in animal T3 after a single IVT injection of 150 μg of PGJ2, suggesting that at these doses, both intravenously and intravitreally administered PGJ2 had a toxic effect on ON function. Nevertheless, all amplitudes recovered to baseline values within 7 days of the last injection, and neither of the animals that received an IVT injection of 50 μg of PGJ2, the dose ultimately chosen for efficacy studies, showed any clinical or electrophysiological evidence of ocular toxicity.
PGJ2 Efficacy
The Table describes the conditions and testing on the five animals used for efficacy experiments.
Table.
Animals Used in the Efficacy Studies and the Clinical Testing Performed on Them
|
Animal |
Injection Time Post pNAION for Both PGJ2 and Vehicle |
Eye Injected First/ Substance Injected |
OCT |
VEP/PERG |
Ganzfeld ERG |
Color Photos and FA |
H & I |
| E1 | <15 min | OD/PGJ2 | * | X | X | X | X |
| E2 | <15 min | OD/PGJ2 | † | † | X | † | X |
| E3 | <15 min | OD/PGJ2 | X | X | X | X | X |
| E4 | 5 h | OD/Vehicle | X | X | X | X | X |
| E5 | 5 h | OD/Vehicle | X | X | X | X | X |
H & I, histopathological and immunohistochemical assessment; OD, right eye; VEP/PERG, visual evoked potentials/pattern-evoked electroretinography.
Experiments carried out prior to acquisition of OCT machine.
Animal developed corneal opacity in one eye following IVT injection, preventing testing in that eye until 4 weeks post induction.
Clinical examination revealed that in all five animals assessed for efficacy of PGJ2 versus vehicle (E1–E5), optic disc and peripapillary swelling were substantially reduced in the PGJ2-treated eye, compared with the vehicle-injected eye (Fig. 2, Supplementary Figs. S1–S3).
Figure 2.

Representative color photographs of the optic discs of the vehicle-injected and PGJ2-treated eyes after pNAION (E4). In this animal, induction of pNAION was followed 5 hours later by a single IVT injection of either 50 μL of vehicle or 50 μL of PGJ2 (total dose = 50 μg). Note that there is much less swelling of the left optic disc than the right optic disc at both 1 day and 1 week post injection and that at 4 weeks post injection, there is more optic disc and PRNFL atrophy in the vehicle-injected eye than in the PGJ2-treated eye. Note also the peripapillary retinal hemorrhages present in the vehicle-injected eye but not in the PGJ2-treated eye. Optic disc photographs of three of the other animals are available as Supplementary Figures S1 through S3.
Fluorescein angiography revealed a variable amount of leakage of dye from the optic disc in all pNAION eyes. The leakage was maximal at day 1 and decreased over time such that within 4 weeks, there was variable late staining of all optic discs but no leakage (Fig. 3, Supplementary Figs. S4–S6).
Figure 3.
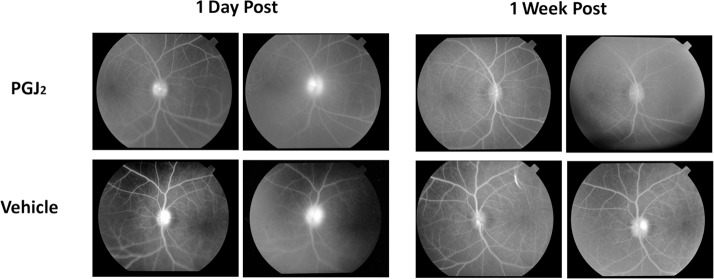
Fluorescein angiograms of the optic discs of the PGJ2-treated and vehicle-injected eyes in monkey E1 1 day and 1 week after induction of pNAION followed within 15 minutes by a single IVT injection of either 50 μL of PGJ2 (total dose = 50 μg) or 50 μL of vehicle. Note that there is less fluorescein leakage in the PGJ2-treated eye than in the vehicle-injected eye at both time periods.
All four animals assessed by SD-OCT (E2–E5) showed a variable amount of both peripapillary intraretinal edema and peripapillary subretinal fluid that gradually resolved over approximately 2 weeks (Fig. 4).
Figure 4.
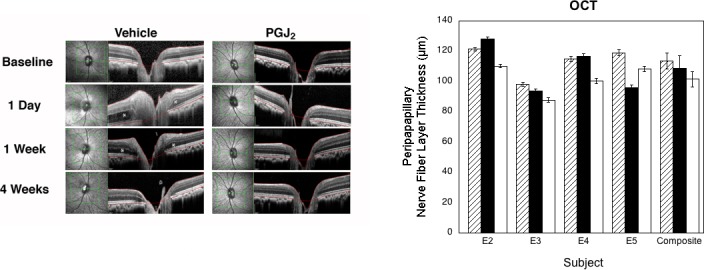
Results of OCT. Left, OCT of the vehicle-injected and PGJ2-treated eyes of animal E4 at baseline and at 1 day, 1 week, and 4 weeks post pNAION induction. Note that there is considerably more intraretinal and subretinal swelling at both 1 day and 1 week post pNAION in the vehicle-injected eye than in the PGJ2-treated eye (asterisks) and that there is better preservation of the retinal architecture in the PGJ2-treated eye than in the vehicle-injected eye at 4 weeks post pNAION induction. Right, PRNFL thickness for each animal (n = 4) and for the group as a whole at baseline (hatched bars) and 1 month post pNAION induction followed by IVT injection of either PGJ2 (black bars) or vehicle (white bars). (Vehicle versus baseline: P = 0.06; PGJ2 versus baseline: P = 0.63; PGJ2 versus vehicle: P = 0.03, Wilcoxon Rank Sum Test.) Error bars are ±1 SD except for composite, which is ±1 SE.
In all four of the animals assessed by SD-OCT two of which (E2, E3) had been injected immediately after induction and two of which (E4, E5) had been injected 5 hours after induction, the thickness of the PRNFL was markedly increased at postinduction day 1, was further increased at 1 week post induction, and then gradually thinned thereafter. In three of the four animals (E2–E4), the 4-week postinduction assessment revealed that the PRNFL was thicker in eyes that had been injected with PGJ2 than in eyes injected with vehicle. In the fourth animal (E5), the PRNFL at final analysis was thicker in the vehicle-injected eye than in the PGJ2-treated eye even though both electrophysiologic data and axon counts indicated more damage in the vehicle-injected eye than in the PGJ2-treated eye (see below). We have no explanation for this discrepancy; nevertheless, for the group as a whole, the difference in PRNFL thickness between the vehicle-injected and PG2-treated eyes was significant (vehicle versus baseline: P = 0.06; PGJ2 versus baseline: P = 0.63; PGJ2 versus vehicle: P = 0.03, Wilcoxon Rank Sum Test).
With respect to electrophysiological testing, three of the five animals (E1, E4, and E5) showed significantly less reduction in both the VEP amplitude and the amplitude of the N95 component of the PERG in the PGJ2-treated eye than in the vehicle-injected fellow eye at 4 weeks post induction (Fig. 5). None of the animals showed significant differences in the VEP and N95 amplitudes when baseline and PGJ2-treated eye measurements at 4 weeks were compared, nor were there any significant differences when the group was compared as a whole (For both VEP and PERG, vehicle versus baseline: P = 0.03; PGJ2 versus baseline: P = 0.44; PGJ2 versus vehicle, P = 0.03, Wilcoxon Rank Sum Test).
Figure 5.
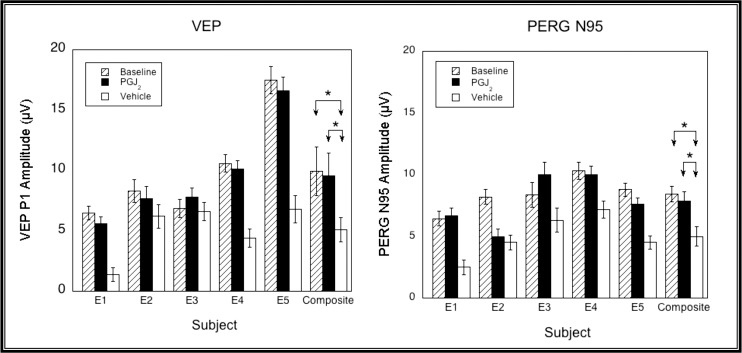
VEP (left) and PERG N95 (right) amplitudes measured at baseline (hatched bars), and 1 month post pNAION and injection with either PGJ2 (black bars) or vehicle (white bars). Error bars are ±1.96 SE. VEP amplitudes were significantly higher in three of five eyes (E1, E4, and E5) injected with PGJ2 compared with vehicle. PERG N95 amplitudes were significantly higher in three of five eyes (E1, E3, and E5) injected with PGJ2 compared with vehicle. Neither VEP nor PERG N95 amplitudes were significantly different from baseline in the PGJ2-treated eye of any animal or for the group as a whole (for both VEP and PERG, vehicle versus baseline: P = 0.03; PGJ2 versus baseline: P = 0.44; PGJ2 versus vehicle, P = 0.03, Wilcoxon Rank Sum Test).
In addition, there were strong linear correlations between the amplitudes of the VEP and N95 component of the PERG in each of these animals (E1, E2, E3, and E5) and in the aggregate (P < 0.01), which would be expected if axon loss resulted in eventual RGC loss (Fig. 6).
Figure 6.

PERG N95 and VEP amplitudes were significantly correlated in each animal (E1 [P < 0.02], E2, E3, E4, and E5 [P < 0.01, two-tailed test] and in the aggregate [P < 0.01, two-tailed test]). Note that the significance of the E4 correlation is based largely on the position of 1 data point. These data are consistent with RGC loss resulting from axon damage. Data from one eye (the eye with the greatest range of values) of each animal are plotted as are composite points from all five animals (lower right graph).
There were no abnormalities in the ganzfeld ERG in any of the eyes injected with vehicle or PGJ2 (Fig. 7).
Figure 7.

Ganzfeld electroretinogram in animal E4 1 month after induction of bilateral pNAION (inductions performed sequentially, 4 weeks apart) followed 5 hours later by either a single IVT injection of 50 μL of vehicle or 50 μL of PGJ2 (total dose = 50 μg). Note that there are no abnormalities of the amplitudes of either the a-wave or the b-wave in either the vehicle-injected or the PGJ2-treated eye. Similar results were seen in the other four animals used for assessment of efficacy (E1, E2, E3, and E5), whether the injections were performed immediately or 5 hours after induction of pNAION.
Histopathological Findings
All five animals assessed for efficacy of PGJ2 were euthanized at least 30 days after pNAION was induced in the second eye. Significant histological differences between all vehicle-injected and PGJ2-treated eyes were present in all animals (Fig. 8). H&E staining revealed increased regional cellular infiltration in the ON cross-sections of vehicle-treated eyes compared with PGJ2-treated eyes (Figs. 8A, 8D: compare 8A [vehicle-injected] with 8D [PGJ2-treated]). Gross structural differences in the individual septated regions also were noted, with increased cellularity present within individual compartments (compare 8A [PGJ2-treated] with 8D [vehicle-injected]). This suggested that PGJ2 treatment after pNAION-associated ischemia not only reduced overall damage but also altered the type of damage associated with the lesion, with increased focal damage in the ONs of vehicle-injected eyes, compared with the ONs of PGJ2-treated eyes.
Figure 8.

Comparison of ON immunostaining results following post pNAION vehicle versus PGJ2 injection in animal E1. (A–C) ON cross sections from vehicle-injected eye. (D–F) ON cross-sections from PGJ2-injected eye. (A, D) H&E staining (4× composites). (B, E) Intact axon immunostaining using SMI312 antibody. The ON from the vehicle-injected eye has axon loss in a dense regional pattern (B), whereas the ON from the PGJ2-treated eye has much less overall axon loss (E). (C, F) GFAP immunostaining. White lines in C and E encircle areas of greatest GFAP immunostaining, consistent with glial scarring. Glial scarring is considerably reduced in the ON from the PGJ2-treated eye, compared with the ON of the vehicle-injected eye (compare [C], vehicle-injected, with [F], PGJ2-treated) from the same animal. Similar findings were present in the eyes of the other four animals used for efficacy studies. Scale bar: 1 mm.
We confirmed the impressions made from observing the H&E ON staining patterns with analysis of ON-axon loss and scarring using immunohistochemical staining for intact axons (SMI312; Figs. 8B, 8E) and glial scarring (GFAP; Figs. 8C, 8F). In all five animals, ONs from vehicle-injected eyes had severe axon loss with dense regions of dropout (Fig. 8B) similar to the severity and pattern of loss identified in the only histologically assessed clinical case of NAION.25,26 In contrast, PGJ2-treated eyes had ONs with reduced axon loss, and the axon loss that was present was more diffuse (Fig. 8E). The difference in severity and pattern of axon loss between ONs from PGJ2-treated and vehicle-injected eyes was mirrored by the comparative differences in the glial scarring patterns seen between ONs from vehicle-injected and PGJ2-treated eyes (Figs. 8C, 8F). ONs from vehicle-injected eyes had larger areas of glial scarring and stronger GFAP signal (Fig. 8C) than did ONs from PGJ2-treated eyes (Fig. 8F).
Electron microscopic examination and quantitative analysis (Fig. 9) revealed that, regardless if eyes were injected immediately or 5 hours after induction of pNAION, the ONs of four of the five eyes injected with vehicle demonstrated increased axon loss and interaxonal debris, with reduced myelin sheath thickness, whereas the ONs of PGJ2-treated eyes showed significant preservation of axons compared with vehicle-injected eyes. Damaged/demyelinated (arrowheads) and dead axons (arrows) were most easily detectable by TEM. There was also increased myelin sheath thickness (compare small-caliber axons in Figs. 9A, 9B). Relative differences in axon preservation between eyes of each animal were evaluated using axon counts (results shown in Table in Fig. 9C). In every animal, ONs from PGJ2-treated eyes revealed increased axon preservation, compared with ONs of vehicle-injected eyes. This difference was statistically significant in four of the five animals (Fig. 9C: Table) and despite the significant variability in ON axons in normal animals, significant when the vehicle-injected and PGJ2-treated eyes were considered as a group (P = 0.05). Both the individual results and group results are summarized as a bar graph in Figure 9D. There was a 24.6% increase in relative axon preservation in the ONs of PGJ2-treated eyes compared with ONs of vehicle-injected eyes.
Figure 9.

Electron microscopic analysis of ONs of post pNAION vehicle-injected and PGJ2-treated eyes (n = 5). (A, B) Representative TEMs of animal E5. (A) Vehicle-injected ON. (B) PGJ2-treated ON. There is relative reduced axon packing density in the ON of the vehicle-injected eye compared with the PGJ2-treated eye. Although degenerating axons are seen in both ONs (arrowheads), there is increased interaxonal debris in the vehicle-injected nerve (compare [A] and [B]), and the myelin sheaths are thinner in the nerve from the vehicle-injected eye than in the ON from the PGJ2-treated eye. Demyelinated axons are seen in both nerves (arrowheads). (C) ON axon quantification in vehicle-injected and PGJ2-treated eyes. Sixty axon fields were randomly selected in each ON, and axons counted in each 135.5-um2 field, using a stereological frame (see Methods). Total counted axons are indicated in the first (vehicle-injected) and third (PGJ2-treated) columns. Mean axons/field are indicated in the second (vehicle-injected) and fourth (PGJ2-treated) columns. Significance between axon counts was determined using a two-tailed t-test for individual animals and Wilcoxon Rank Sum test for the group as a whole. PGJ2-treated eyes had more preserved axons than vehicle-injected eyes in all animals. The difference was significant in four animals (E1: P < 0.001; E2: P = 0.01; E4: P = 0.02; E5: P = 0.02) as well as for the group as a whole (P = 0.05). (D) Bar graph showing relative axon preservation in the five individual test animals as well as for the group as a whole (for vehicle-injected eyes, SD = 2.04 and SE = 1.01; for PGJ2-treated eyes, SD = 3.84 and SE = 1.34). Scale bar: 500 nm. Error bars are ±2 SE.
Discussion
In this study, we have correlated the full range of clinically available imaging and electrophysiological analyses with axon stereology in a nonhuman primate model of NAION. The close agreement among the different tests used to evaluate structure and function validated the use of any one of them to evaluate damage from NAION. We found that for most animals, PGJ2 treatment, whether given immediately post pNAION or 5 hours after, substantially reduced the amount of pNAION-associated damage compared with contralateral vehicle-injected control eyes. The majority of PGJ2-treated eyes showed reduced ON and PRNFL edema by fundus photography and OCT imaging, and reduced ON capillary leakage at 1 day and 7 days post NAION induction. None of the pattern VEP and PERG N95 amplitudes in PGJ2-treated eyes was statistically different than baseline values, suggesting that PGJ2 is strongly neuroprotective in eyes with pNAION compared with vehicle-injected controls. The relative functional preservation in PGJ2-treated ONs documented by electrophysiological findings was in close agreement with axon stereology. Thus, by all measures, a single IVT injection of PGJ2 was neuroprotective following sudden anterior ON ischemia when given either immediately after or within 5 hours post induction. PGJ2 is therefore the first candidate drug confirmed to be effective in the NAION models of both rodents and nonhuman primates, supporting its potential use in humans.
OCT studies have revealed that in many cases, human NAION is characterized not only by optic disc edema but also by the development of peripapillary retinal edema associated with intraretinal and subretinal fluid.27,28 We observed similar phenomena in our pNAION model, with PGJ2-treated eyes showing both less intraretinal edema and subretinal fluid and more rapid resolution of the edema and fluid than seen in vehicle-injected eyes.
PGJ2's neuroprotective effect is apparent when the IVT injection is given either immediately or even several hours following induction of pNAION. This suggests that a single injection of PGJ2 may be able to reduce significant visual loss in humans with NAION. Nevertheless, more work needs to be done to determine PGJ2's treatment time window before there is irreversible damage.
The mechanism by which PGJ2 limits ON damage and improves ON function is not entirely clear, but given our previous findings of significant inflammation in the area of ischemia in both our murine29 and our nonhuman primate26 models of NAION as well as in the only human case of NAION that has been evaluated,26 we believe that PGJ2 reduces early inflammation and, thus, limits the postischemia compartment syndrome previously documented in human NAION. This conclusion is suggested by differences in the severity and pattern of axon loss seen in the ONs of vehicle-injected versus PGJ2-treated eyes. PGJ2-treated eyes showed improved density of surviving axons, as well as a reduced level of glial scarring detectable by GFAP staining. PGJ2 also is released by pericytes under conditions of ischemia,30,31 and this autocrine activity may reduce the initial ON edema and its resultant ON compression.
If, in fact, PGJ2's neuroprotective effect results from its effects on postischemic inflammation, the mechanism is likely related to two specific actions. PGJ2 directly antagonizes NFκB activity and also acts as a PPARγ agonist in the CNS vasculature and microglia.11,13 Both mechanisms are involved in the regulation of early inflammatory response. Inflammation plays a major role in tissue plasticity and regeneration after ischemia.32 The inflammatory response can be a key variable in lesion severity33 and also may limit regenerative potential after ischemic damage.34 Our current results suggest that selective postischemic inflammatory control may be a promising target for neuroprotection.
Our toxicity studies were designed to evaluate PGJ2's gross systemic toxicity, by scheduled dose escalation in five animals. No changes in systemic biomarkers were detected by this route. No electrophysiologic changes were detectable when PGJ2 was systemically administered at 40 μg/kg; however, there was transient amplitude loss of the VEP and PERG N95 components that completely resolved within 1 week when an IV dose of 125 μg/kg was used. PGJ2 previously was reported to be neurotoxic when administered chronically in vivo35–37 and to oligodendrocyte precursors in vitro.38 However, neuronal toxicity in these studies required both high concentrations and multiple doses of PGJ235 or persistent exposure in tissue culture.38 Nevertheless, extended PGJ2 toxicity studies need to be performed prior to any human clinical trials.
Intravitreal administration of PGJ2 enables delivery of high local concentrations of the drug with reduced risk of systemic side effects.39–41 The 50-μg intraocular dose used in our efficacy studies was derived from the dose used in our previous study of IVT PGJ2 in rat eyes with rNAION.15 Specifically, the volume of the adult rat vitreous cavity is 60 μL, whereas the volume of the adult rhesus monkey vitreous cavity is 3500 μL (i.e., ~ 50× that of the rat). We had injected 1 μg IVT in our rats. Accordingly, we injected 50 μg in our primates. There were no changes in the VEP, PERG, or ganzfeld ERG after a single administration of this dose. There were transient reductions in the amplitudes of both the VEP and the N95 component of the PERG after single IVT injections of 150 μg and 200 μg were given in two animals, respectively. In both cases, the amplitudes returned to baseline values within 1 week post injection. These doses are 3 and 4 times higher, respectively, than the dose (50 μg) we used in our efficacy studies. Although these results do not totally eliminate the possibilities of long-term systemic or ocular toxicity from IVT PGJ2, they do suggest that PGJ2's therapeutic index is likely to be sufficient for early treatment of NAION, particularly given that PGJ2 would be used as a single dose for a serious, vision-disabling disease, rather than as a chronic treatment. More work is needed to elucidate further any potential deleterious effects, whether systemic or local, of intravitreally administered PGJ2.
This is the first study to use an experimental primate model of NAION to evaluate the potential therapeutic efficacy of a new agent for the treatment of human NAION. Because of the precious nature of primate resources, we wanted to maximize the information obtained from each individual animal and minimize the number of primates used for both the toxicity and the efficacy arms of the study. We therefore assessed several different doses of PGJ2 as well as different routes of administration of drug in five animals, allowing sufficient time between studies for the animals to recover from administration. The use of multiple functional tests (assessment of pupillary responses to light, VEP, PERG, ganzfeld ERG), in vivo imaging (IVFA, SD-OCT, fundus photos) and, ultimately, histological and immunohistochemical analyses of the eyes of all animals, compared with initial baseline analyses, further enabled us to identify and compare ON ischemia-induced changes in retinal and ON structure and function in each eye with a high degree of sensitivity. Our ability to predictably and reproducibly generate a NAION-type defect is one of the great strengths of the current pNAION model. It should be emphasized again that although we induced visual impairment in all pNAION-induced eyes, none of the animals was blinded; all continued to see and respond appropriately. Judicious use of the pNAION model for analysis of potential clinical NAION treatments enabled us to use each animal as its own control. This approach can minimize variability and reduce the number of animals needed to analyze treatment efficacy, for future preclinical analyses.
Acknowledgments
Supported by NEI Grant R01 EY019529 and a grant from the Hirschhorn Foundation.
Disclosure: N.R. Miller, None; M.A. Johnson, None; T. Nolan, None; Y. Guo, None; A.M. Bernstein, None; S.L. Bernstein, P
References
- 1. Arnold AC. Ischemic optic neuropathy. In: Miller NR, Newman NJ, Biousse V, Kerrison JB. eds Walsh and Hoyt's Clinical Neuro-Ophthalmology. 6th ed, Vol 1 Baltimore, MD: Lippincott-Williams & Wilkins; 2005; 349–384. [Google Scholar]
- 2. Johnson LN, Arnold AC. Incidence of nonarteritic and arteritic anterior ischemic optic neuropathy: population-based study in the State of Missouri and Los Angeles County, California. J Neuroophthalmol. 1994; 14: 38–44. [PubMed] [Google Scholar]
- 3. Hattenhauer MG, Leavitt JA, Hodge DO, Grill R, Gray DT. Incidence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1997; 123: 103–107. [DOI] [PubMed] [Google Scholar]
- 4. Atkins EJ, Bruce BB, Newman NJ, Biousse V. Treatment of nonarteritic anterior ischemic optic neuropathy. Surv Ophthalmol. 2010; 55: 47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernstein SL, Guo Y, Kelman SE, Flower RW, Johnson MA. Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2003; 44: 4153–4162. [DOI] [PubMed] [Google Scholar]
- 6. Goldenberg-Cohen N, Guo Y, Margolis F, Cohen Y, Miller NR, Bernstein SL. Oligodendrocyte dysfunction after induction of experimental anterior optic nerve ischemia. Invest Ophthalmol Vis Sci. 2005; 46: 2716–2725. [DOI] [PubMed] [Google Scholar]
- 7. Bernstein SL, Guo Y, Slater BJ, Puche A, Kelman SE. Neuron stress and loss following rodent anterior ischemic optic neuropathy in double-reporter transgenic mice. Invest Ophthalmol Vis Sci. 2007; 48: 2304–2310. [DOI] [PubMed] [Google Scholar]
- 8. Chen CS, Johnson MA, Flower RA, Slater BJ, Miller NR, Bernstein SL. A primate model of nonarteritic anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2008; 49: 2985–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin TN, Cheung WM, Wu JS, et al. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006; 26: 481–487. [DOI] [PubMed] [Google Scholar]
- 10. Nicholson JD, Puche AC, Guo Y, Weinreich D, Slater BJ, Bernstein SL. PGJ2 provides prolonged CNS stroke protection by reducing white matter edema. PLoS One. 2012; 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Straus DS, Pascual G, Li M, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000; 97: 4844–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ridder DA, Schwaninger M. NF-kappaB signalling in cerebral ischemia. Neuroscience. 2009; 158: 995–1006. [DOI] [PubMed] [Google Scholar]
- 13. Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006; 26: 811–820. [DOI] [PubMed] [Google Scholar]
- 14. Fong WH, Tsai HD, Chen YC, Wu JS, Lin TS. Anti-apoptotic actions of PPAR-gamma against ischemic stroke. Mol Neurobiol. 2010; 41: 180–186. [DOI] [PubMed] [Google Scholar]
- 15. Touitou V, Johnson MA, Guo Y, Miller NR, Bernstein SL. Sustained neuroprotection from a single intravitreal injection of PGJ2 in a rodent model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2013; 54: 7402–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernstein SL, Guo Y, Peterson K, Wistow G. Expressed sequence tag analysis of adult human optic nerve for NEIBank: identification of cell type and tissue markers. BMC Neurosci. 2009; 10: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mäkelä K, Hartikainen K, Rorarius M, Jäntti V. Suppression of F-VEP during isoflurane-induced EEG suppression. Electroencephalogr Clin Neurophysiol. 1996; 100: 269–272. [DOI] [PubMed] [Google Scholar]
- 18. Johnson MA, Slater BJ, Miller NR, Bernstein SL, Flower RW. A simple integrated system for electrophysiologic recording in animals. Doc Ophthalmol. 2009; 119: 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bach M, Halina M, Holder GE. Standard for pattern electroretinography: International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol. 2000; 101: 11–18. [DOI] [PubMed] [Google Scholar]
- 20. Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography. Doc Ophthalmol. 2004; 108: 107–114. [DOI] [PubMed] [Google Scholar]
- 21. Severns ML, Johnson MA, Bresnick GH. Methodological dependence of electroretinogram oscillatory potential amplitudes. Doc Ophthalmol. 1994; 86: 23–31. [DOI] [PubMed] [Google Scholar]
- 22. Li C, Yang S, Chen L, et al. Stereological methods for estimating the myelin sheaths of the myelinated fibers in white matter. Anat Rec. 2009; 292: 1648–1655. [DOI] [PubMed] [Google Scholar]
- 23. Kaplan S, Geuna S, Ronchi G, Ulkay MB, von Bartheld CS. Calibration of the stereological estimation of the number of myelinated axons in the rat scientific nerve: a multicenter study. J Neurosci Methods. 2010; 187: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cull GA, Reynaud J, Wang L, Cioffi GA, Burgoyne CF, Fortune B. Relationship between orbital optic nerve axons counts and retinal nerve fiber layer thickness measured by spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012; 53: 7766–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tesser RA, Niendorf ER, Levin LA. The morphology of an infarct in nonarteritic anterior ischemic optic neuropathy. Ophthalmology. 2003; 110: 2013–2035. [DOI] [PubMed] [Google Scholar]
- 26. Salgado C, Vilson F, Miller NR, Bernstein SL. Cellular inflammation in nonarteritic anterior ischemic optic neuropathy and its primate model. Arch Ophthalmol. 2011; 129: 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bellusci C, Savini G, Caronelli M, Carelli V, Sadun AA, Barboni P. Retinal nerve fiber layer thickness in nonarteritic anterior ischemic optic neuropathy: OCT characterization of the acute and resolving phases. Graefes Arch Clin Exp Ophthalmol. 2008; 246: 641–647. [DOI] [PubMed] [Google Scholar]
- 28. Hedges TR III, Vuong LN, Gonzalez-Garcia AO, Mendoza-Santiesteban CE, Amaro-Quierza ML. Subretinal fluid from anterior ischemic optic neuropathy demonstrated by optical coherence tomography. Arch Ophthalmol. 2008; 126: 812–815. [DOI] [PubMed] [Google Scholar]
- 29. Zhang C, Guo Y, Miller NR, Bernstein SL. Optic nerve infarction and post-ischemic inflammation in the rodent model of anterior ischemic optic neuropathy (rNAION). Brain Res. 2009; 126467–126475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005; 114: 100–109. [DOI] [PubMed] [Google Scholar]
- 31. Dore-Duffy P, Balabanov R, Beaumont T, Katar M. The CNS pericyte response to low oxygen: early synthesis of cyclopentenone prostaglandins of the J-series. Microvasc Res. 2005; 69: 79–88. [DOI] [PubMed] [Google Scholar]
- 32. Tuttolomondo A, Di Sciacca R, Di Raimondo D, Renda C, Pinto A, Licata G. Inflammation as a therapeutic target in acute ischemic stroke treatment. Curr Top Med Chem. 2009; 9: 1240–1260. [DOI] [PubMed] [Google Scholar]
- 33. Kucharova K, Chang Y, Boor A, Yong VW, Stallcup WB. Reduced inflammation accompanies diminished myelin damage and repair in the NG2 null mouse spinal cord. J Neuroinflammation. 2011; 8: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory moleculars. Exp Neurol. 1997; 148: 587–603. [DOI] [PubMed] [Google Scholar]
- 35. Ogburn KD, Bottiglieri T, Wang Z, Figueiredo-Pereira ME. Prostaglandin J2 reduces catechol-O-methyltransferase activity and enhances dopamine toxicity in neuronal cells. Neurobiol Dis. 2006; 22: 294–301. [DOI] [PubMed] [Google Scholar]
- 36. Arnaud LT, Myeku N, Figueiredo-Pereira ME. Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem. 2009; 110: 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pierre SR, Lemmens MA, Figueiredo-Pereira ME. Subchronic infusion of the product of inflammation prostaglandin J2 models sporadic Parkinson's disease in mice. J Neuroinflammation. 2009; 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiang Z, Lin T, Reeves SA. 15-d PGJ2 induces apoptosis of mouse oligodendrocyte precursor cells. J Neuroinflammation. 2007; 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Do DV, Nguyen QD, Shah SM, et al. An exploratory study of the safety, tolerability and bioactivity of a single intravitreal injection of vascular endothelial growth factor Trap-Eye in patients with diabetic macular oedema. Br J Ophthalmol. 2009; 93: 144–149. [DOI] [PubMed] [Google Scholar]
- 40. Lopez-Galvez MI, Pastor-Jimeno JC. [Efficacy and safety of intravitreal injection of triamcinolone acetonide as treatment for diffuse diabetic macular edema]. Arch Soc Esp Oftalmol. 2009; 84: 547–548. [DOI] [PubMed] [Google Scholar]
- 41. Kumar V, Ghosh B, Raina UK, Goel N. Efficacy and safety of one intravitreal injection of bevacizumab in diabetic macular oedema. Acta Ophthalmol. 2010; 88: e3 author reply e4. [DOI] [PubMed] [Google Scholar]