

Abstract
Dopamine beta-hydroxylase (DBH) is the biosynthetic enzyme catalyzing formation of norepinephrine. Changes in DBH expression or activity have been implicated in the pathogenesis of cardiovascular and neuropsychiatric disorders. Genetic determination of DBH enzymatic activity and its secretion are only incompletely understood. We began with a genome-wide association search for loci contributing to DBH activity in human plasma. Initially, in a population sample of European ancestry, we identified the proximal DBH promoter as a region harboring three common trait-determining variants (top hit rs1611115, P = 7.2 × 10−51). We confirmed their effects on transcription and showed that the three variants each acted additively on gene expression. Results were replicated in a population sample of Native American descent (top hit rs1611115, P = 4.1 × 10−15). Jointly, DBH variants accounted for 57% of DBH trait variation. We further identified a genome-wide significant SNP at the LOC338797 locus on chromosome 12 as trans-quantitative trait locus (QTL) (rs4255618, P = 4.62 × 10−8). Conditional analyses on DBH identified a third genomic region contributing to DBH variation: a likely cis-QTL adjacent to DBH in SARDH (rs7040170, P = 1.31 × 10−14) on chromosome 9q. We conclude that three common SNPs in the DBH promoter act additively to control phenotypic variation in DBH levels, and that two additional novel loci (SARDH and LOC338797) may also contribute to the expression of this catecholamine biosynthetic trait. Identification of DBH variants with strong effects makes it possible to take advantage of Mendelian randomization approaches to test causal effects of this intermediate trait on disease.
INTRODUCTION
Dopamine β-hydroxylase (DBH) is the final enzyme in norepinephrine biosynthesis, catalyzing the oxidative hydroxylation of dopamine to norepinephrine in the noradrenergic nerve endings of the central and peripheral nervous systems (1). In the bloodstream, DBH enzymatic activity is abundant, emerging from both the sympathetic terminals and the adrenal medullary chromaffin cells (1). As a result of exocytosis, DBH is co-released with norepinephrine from synaptic vesicles into extracellular space and thus can be found in plasma and cerebrospinal fluid (CSF) (2,3). The enzymatic activity of plasma or CSF DBH corresponds to the level of DBH protein, with plasma and CSF DBH correlating highly in humans (4,5). As such, DBH is of high interest to both the neuropsychiatric and cardiovascular field. Changes in DBH activity and/or genetic variants in the DBH gene have been implicated in the pathophysiology of major depression (6), ADHD (7,8), Parkinson (9) and Alzheimer's disease (10,11) and PTSD (12,13), potentially through changes in central catecholamine levels, whereas altered sympathoadrenal activity is thought to be implicated in the pathogenesis of hypertension and cardiovascular disease (14,15).
In family and twin studies plasma DBH (pDBH) activity is highly heritable, relatively stable over time in the same person, and only minimally susceptible to environmental factors such as physical stress or drugs (16). Furthermore DBH activity shows highly variable inter-individual differences which are likely the result of genetic factors (5,17), with heritability estimates accounting for ∼80–90% of the variation.
Linkage analysis with non-DNA markers has identified a single quantitative trait locus (QTL) for DBH activity in a region on chromosome 9 (9q34) (18,19) and the DBH gene was later mapped to that region (20,21). Sequencing analyses by Zabetian et al. (22,23) further characterized the molecular structure of DBH and identified a SNP in the promoter region (rs1611115/C-970T/formerly C-1021T), which explained a large ∼35–52% inter-individual variation in pDBH activity, while functional polymorphisms (A197T in exon 3, A304S in exon 5 and R535C in exon 11) in the gene did only show a modest putative effect for R535C in these studies (see review in 16). Extended sequencing in the promoter region identified six common SNPs in the proximal promoter and showed functional properties in in vitro and in vivo experiments for rs1611115 and rs1989787 (C-2073T). A newer linkage study in families confirmed DBH as a major contributor of pDBH activity, but also suggested two additional loci, one in close proximity to DBH and the second on chromosome 20p12 (24).
Analysis of DBH levels in clinical populations reported racial differences in pDBH activities, with Blacks having lower levels than Whites (25). Genetic studies on the DBH locus, initially performed in populations of European ancestry, have then been extended to include subjects of African and Asian descent and confirmed rs1611115 as the polymorphism with the strongest effect (22,26).
Here, we performed the first genome-wide association study (GWAS), with goals to: (1) replicate and extend previous findings on DBH locus variation and its effect on pDBH activity, (2) extend the search to identify additional, trans-QTLs for pDBH activity levels and (3) expand ancestry studies to include subjects of Native American descent and Hispanic ethnicity. In addition, we further examined functional properties of genetic markers in the DBH promoter region displaying peak-association with plasma DBH activity, in transfected chromaffin cells as well as in vivo. We show that DBH variants with strong effects may be used in a Mendelian randomization (MR) approach to test causal effects of this intermediate trait on disease, such as cardiovascular and neuropsychiatric symptoms and disorders.
RESULTS
Genome-wide association study in subjects of European ancestry
An initial GWAS for plasma DBH activity was performed with genotypes of 341 subjects of European ancestry (European Americans, EAs). The mean pDBH level in the 341 EAs was 11.44 IU/L [standard deviation (SD) = 6.95] (Supplementary Material, Fig. S1). Genotypes underwent rigorous quality control and included a final set of 7 871 575 markers obtained by genotyping and imputation. Linear regression under an additive genetic model, incorporating appropriate covariates, resulted in a low genomic control inflation factor of λGC = 1.002. A quantile–quantile (QQ) plot is shown in Supplementary Material, Figure S2A. A table with all GWAS results is available in the Supplementary Material, Table S1.
Our analyses identified the DBH locus as genome-wide significant with the top hit for a directly genotyped SNP rs1611115 at P = 7.2 × 10−51 (Fig. 1A and Table 1). A regional association plot of the DBH locus showed 34 genome-wide significant DBH SNPs within the same linkage disequilibrium (LD) block (Fig. 1B). Of these, one SNP was found in an exon (synonymous SNP exm793933, P = 1.023 × 10−27), 22 were intronic and 11 were located upstream of DBH, including 3 common SNPs within a 3 kb region of the promoter (rs1076150, rs1989787 and rs1611115, shown in detail in Table 1, top part). Two of these promoter SNPs (rs1989787 and rs1611115) were known to be functional (see 14 and 15) and the functionality of rs1076150 was investigated below. The proportion of variability explained (R2) by the DBH gene, based on five highly significant DBH SNPs in low LD with each other plus the three (putative) functional promoter SNPs, was 0.569.
Figure 1.
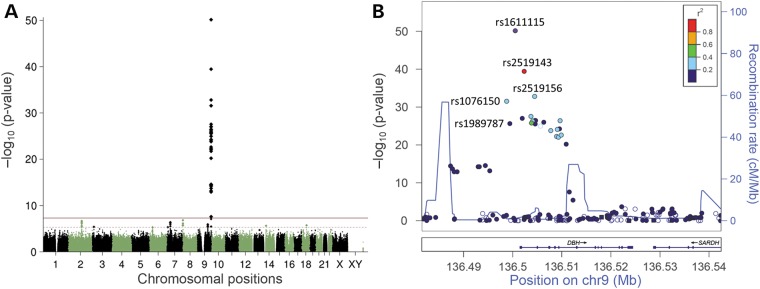
Results of the GWAS of plasma DBH activity in 341 subjects of European origin. (A) Manhattan plot showing the −log10 (P-values) for SNP associations with plasma DBH activity across the genome. The red horizontal line represents the genome-wide significance threshold at P < 5 × 10−8 and the dashed line represents suggestive evidence for association at P < 5 × 10−6. (B) Regional association plot, showing significant regions in DBH on chromosome 9. Directly genotyped SNPs are indicated by an asterisk (*). The SNPs are color coded based on the linkage disequilibrium with the most significant SNP rs1611115.
Table 1.
Most significant hits in the genome-wide association study
| Allele |
EA GWAS |
NA descent GWAS |
Meta-analysisa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | CHR | BP | Gene | Location | 1/2b | Allele 1 freq. | Effect size | SE | P | Allele 1 freq. | Effect size | SE | P | Q | Effect size | P |
| rs1076150c | 9 | 136498761 | DBH | Upstream | T/C | 0.512 | −0.947 | 0.072 | 2.74E−32 | 0.710 | −0.779 | 0.163 | 7.67E−06 | 0.35 | −0.920 | 1.38E−44 |
| rs1989787 | 9 | 136499412 | DBH | Upstream | T/C | 0.312 | 0.924 | 0.079 | 2.13E−26 | 0.196 | 0.747 | 0.191 | 1.92E−04 | 0.39 | 0.898 | 1.50E−34 |
| rs1611115c | 9 | 136500515 | DBH | Upstream | T/C | 0.248 | −1.265 | 0.070 | 7.20E−51 | 0.317 | −1.195 | 0.125 | 4.10E−15 | 0.63 | −1.248 | 4.60E−92 |
| rs7540659 | 1 | 100196119 | FRRS1 | Intron | T/A | 0.341 | −0.024 | 0.095 | 0.801 | 0.462 | −0.793 | 0.160 | 3.64E−06 | 0.00 | −0.398 | 0.301 |
| rs60674788 | 2 | 35027196 | CR617033 | Downstream | C/G | 0.257 | −0.403 | 0.101 | 8.11E−05 | 0.354 | −0.536 | 0.179 | 0.004 | 0.52 | −0.435 | 7.64E−07 |
| rs4459781 | 2 | 134204665 | NCKAP5 | Intron | C/T | 0.281 | −0.499 | 0.094 | 2.15E−07 | 0.215 | −0.098 | 0.209 | 0.641 | 0.08 | −0.431 | 5.12E−07 |
| rs77518496 | 2 | 143629286 | KYNU | Upstream | G/A | 0.032 | −0.012 | 0.232 | 0.959 | 0.115 | 1.252 | 0.253 | 3.88E−06 | 0.00 | 0.616 | 0.330 |
| rs2351772 | 2 | 204079313 | NBEAL1 | Intron | C/T | 0.418 | 0.346 | 0.091 | 1.71E−04 | 0.527 | 0.441 | 0.149 | 0.004 | 0.58 | 0.372 | 1.61E−06 |
| rs112239800 | 2 | 232517876 | BC069004 | Downstream | G/A | 0.102 | 0.509 | 0.149 | 7.38E−04 | 0.115 | 1.079 | 0.268 | 1.33E−04 | 0.06 | 0.645 | 7.67E−07 |
| rs13095328 | 3 | 15226050 | DIVA | Intron | C/T | 0.100 | −0.669 | 0.143 | 3.97E−06 | 0.059 | 0.634 | 0.354 | 0.077 | 0.00 | −0.058 | 0.929 |
| rs3774729c | 3 | 63982082 | ATXN7 | Exon | A/G | 0.323 | 0.000 | 0.086 | 0.996 | 0.290 | 0.869 | 0.164 | 9.60E−07 | 0.00 | 0.424 | 0.329 |
| rs56030924 | 3 | 63995563 | AK023371 | Intron | A/G | 0.286 | 0.017 | 0.090 | 0.848 | 0.269 | 0.885 | 0.164 | 6.39E−07 | 0.00 | 0.440 | 0.310 |
| rs831692 | 3 | 64003983 | PSMD6 | Intron | A/G | 0.310 | 0.031 | 0.089 | 0.731 | 0.288 | 0.893 | 0.166 | 6.65E−07 | 0.00 | 0.451 | 0.296 |
| rs56237630 | 3 | 64049375 | PRICKLE2 | Downstream | A/C | 0.145 | −0.002 | 0.119 | 0.988 | 0.214 | 0.941 | 0.182 | 1.52E−06 | 0.00 | 0.460 | 0.329 |
| rs12639432c | 3 | 134770520 | EPHB1 | Intron | T/C | 0.302 | 0.339 | 0.091 | 2.44E−04 | 0.462 | 0.546 | 0.150 | 4.71E−04 | 0.24 | 0.395 | 4.16E−07 |
| rs7779937 | 7 | 10971712 | NDUFA4 | Downstream | A/G | 0.048 | −0.922 | 0.198 | 4.74E−06 | 0.016 | 0.643 | 0.660 | 0.333 | 0.02 | −0.266 | 0.731 |
| rs13242648 | 7 | 35777951 | CR595224 | Downstream | T/A | 0.196 | 0.554 | 0.107 | 3.92E−07 | 0.136 | −0.456 | 0.231 | 0.051 | 0.00 | 0.070 | 0.890 |
| rs12701456 | 7 | 35827802 | SEPT7 | Upstream | C/T | 0.196 | 0.547 | 0.107 | 5.03E−07 | 0.132 | −0.389 | 0.231 | 0.096 | 0.00 | 0.101 | 0.828 |
| rs13255006 | 8 | 1989315 | MYOM2 | Upstream | C/G | 0.319 | 0.500 | 0.093 | 1.52E−07 | 0.172 | −0.034 | 0.216 | 0.876 | 0.02 | 0.268 | 0.310 |
| rs1338730 | 9 | 103520981 | MURC | Downstream | C/T | 0.402 | −0.419 | 0.085 | 1.15E−06 | 0.253 | −0.404 | 0.193 | 0.039 | 0.94 | −0.417 | 7.40E−08 |
| rs823919 | 9 | 104662606 | GRIN3A | Upstream | A/G | 0.124 | 0.582 | 0.127 | 6.56E−06 | 0.172 | 0.283 | 0.215 | 0.191 | 0.23 | 0.505 | 3.98E−06 |
| rs7857468 | 9 | 136585380 | SARDH | Intron | A/C | 0.195 | 0.544 | 0.099 | 8.09E−08 | 0.170 | 0.142 | 0.236 | 0.549 | 0.12 | 0.484 | 1.19E−07 |
| rs10795764 | 10 | 10238394 | BC032914 | Downstream | C/T | 0.434 | 0.093 | 0.083 | 0.265 | 0.559 | 0.750 | 0.133 | 2.33E−07 | 0.00 | 0.413 | 0.208 |
| rs870553 | 10 | 133970542 | JAKMIP3 | Intron | G/A | 0.010 | −1.210 | 0.417 | 0.004 | 0.059 | −1.262 | 0.332 | 2.75E−04 | 0.92 | −1.242 | 1.77E−06 |
| rs112825992 | 10 | 134008571 | DPYSL4 | Intron | T/C | 0.009 | −1.252 | 0.450 | 0.006 | 0.055 | −1.209 | 0.320 | 2.92E−04 | 0.94 | −1.223 | 2.65E−06 |
| rs4255618 | 12 | 131837477 | LOC338797 | Intron | C/A | 0.353 | 0.388 | 0.088 | 1.26E−05 | 0.322 | 0.502 | 0.154 | 0.002 | 0.52 | 0.416 | 4.62E−08 |
| rs8013529c | 14 | 23649792 | SLC7A8 | Intron | G/A | 0.139 | −0.566 | 0.118 | 2.37E−06 | 0.059 | 0.320 | 0.328 | 0.332 | 0.01 | −0.176 | 0.689 |
| rs12595689 | 15 | 86009293 | AKAP13 | Intron | C/G | 0.085 | 0.010 | 0.158 | 0.952 | 0.102 | −1.198 | 0.222 | 5.88E−07 | 0.00 | −0.584 | 0.333 |
| rs117711052 | 17 | 74305308 | QRICH2 | Upstream | C/G | 0.024 | 1.155 | 0.278 | 4.12E−05 | 0.016 | 1.567 | 0.627 | 0.014 | 0.55 | 1.223 | 1.48E−06 |
| rs115172145 | 17 | 74310984 | PRPSAP1 | Intron | C/T | 0.024 | 1.144 | 0.278 | 4.77E−05 | 0.016 | 1.567 | 0.627 | 0.014 | 0.54 | 1.213 | 1.74E−06 |
| rs7228140 | 18 | 45907244 | ZBTB7C | Intron | C/T | 0.046 | −0.941 | 0.194 | 1.88E−06 | 0.071 | 0.439 | 0.313 | 0.165 | 0.00 | −0.273 | 0.692 |
| Conditional analysisd | ||||||||||||||||
| rs7857468 | 9 | 136585380 | SARDH | Intron | A/C | 0.195 | 0.500 | 0.065 | 2.38E−13 | 0.170 | 0.489 | 0.155 | 0.002 | 0.946 | 0.498 | 1.15E−16 |
| rs7040170c | 9 | 136586367 | SARDH | Intron | G/A | 0.221 | 0.439 | 0.062 | 7.82E−12 | 0.177 | 0.456 | 0.153 | 0.004 | 0.918 | 0.442 | 1.31E−14 |
aRandom-effects models were used for SNPs with significant heterogeneity Q values (bold), otherwise fixed-effects models.
bAllele 1 is the coding allele.
cDirectly genotyped SNP.
dRegression analyses including DBH SNPs rs1076150, rs1989787 and rs1611115 as covariates.
P-values in bold meet suggestive (P < 5.0E−06) or genome-wide significance (P < 5.0E−08).
No other chromosomal region reached genome-wide significance. However, there were 10 regions which showed suggestive evidence (P < 5 × 10−6) in EAs. For each of these, the SNP with the lowest P-value is presented in Table 1 (middle part) and Supplementary Material, Figure S3A.
Replication of the GWAS in subjects of Native American ancestry
To replicate our findings we performed a second GWAS on subjects of Native American descent (NAs), including subjects with varying degrees of NA admixtures as typically seen in Hispanic subjects (n = 91). The mean pDBH level in 93 NAs was 10.2 IU/l (SD = 6.94) and was not significantly different from pDBH levels in EAs (P > 0.29). The genomic control inflation λGC was 1.009 (a QQ-plot is shown in Supplementary Material, Fig. S2B). A table with all GWAS results is available in the Supplementary Material, Table S2. Replicating our results in EAs, we confirmed the DBH locus to be highly significant, with the same top hit rs1611115 at P = 4.10 × 10−15 (Table 1 and Supplementary Material, Fig. S4A). A regional association plot of the DBH locus showed an additional five intronic genome-wide significant SNPs within the same LD-block (Supplementary Material, Fig. S4B). The proportion of variability explained (R2) by the DBH locus, based on four independent (LD < 0.5), highly significant DBH SNPs (including the three promoter SNPs), was 0.57.
We did not identify other genome-wide significant regions in this small NA population. Eight other loci showed suggestive evidence for association with pDBH activity (P < 5 × 10−6). For each of these regions the SNP with the lowest P-value is presented in Table 1 (middle part) and Supplementary Material, Figure S3B.
Meta-analysis of EA and NA GWAS
An inverse variance weighted meta-analysis of the EA and NA GWAS results indicated no significant heterogeneity (Q) at the DBH locus and resulted in highly significant associations for the promoter region of this locus with the top hit rs1611115 at P = 4.60 × 10−92, as well as rs1076150 (T-2734C) and rs1989787, at P = 1.38 × 10−44 and P = 1.50 × 10−34, respectively (Table 1, right side and Supplementary Material, Fig. S5A). A complementary pooled analysis (mega-analysis) of the EA and NA subjects for the three promoter SNPs showed comparable results (Supplementary Material, Fig. S5B). A C to T transition progressively diminished pDBH activity for rs1076150 and rs1611115, while increasing pDBH activity for rs1989787. In each case, SNP allele effects on trait seemed to be additive, with intermediate effects for SNP heterozygotes, confirmed by the fact that recessive and dominant genetic models were less significant than the additive model for these three SNPs (data not shown).
In addition to the DBH locus, the meta-analysis showed a genome-wide significant association for intronic SNP rs4255618 in LOC338797 on chromosome 12 (P = 4.62 × 10−8). A BLAST search (on BLASTN_2.2.28+ at NCBI) of the RNA-coding region (∼20 kb) of this uncharacterized locus showed no homology to DBH. In addition, seven new loci reached suggestive evidence for association in the meta-analysis (top hits for these loci are shown in Table 1). The proportion of variability explained (R2) by the DBH locus, based on seven highly significant DBH SNPs in low LD with each other (including the three promoter SNPs), was 0.57. Adding rs4255618 in LOC338797 to the DBH model significantly increased R2 to 0.59 (LR test P = 6.09 × 10−5) in a joint analysis of EA and NA subjects.
Conditional analysis on the DBH locus
Because of the strong effect of the DBH locus on pDBH activity, we repeated the GWAS conditioned on the three DBH peak functional promoter SNPs rs1076150, rs1989787, and rs1611115 in EAs, NAs and the meta-analysis to test for additional, DBH-independent loci (Supplementary Material, Fig. S6). The SARDH locus, adjacent to DBH and previously showing suggestive evidence for association, became genome-wide significant in EAs with an imputed top hit for rs7857468 (P = 2.38 × 10−13). Rs7857468 replicated in NAs with a nominally significant P = 0.002, resulting in a meta-analysis P-value = 1.15 × 10−16 (Table 1, bottom part). Results for the most significant directly genotyped SNP in SARDH (rs7040170, P = 1.31 × 10−14) are also shown. Regional association plots of the conditioned GWAS results in EAs and NAs for the DBH and neighboring SARDH loci are shown in Supplementary Material, Figure S6B and D. Adding the SARDH SNP to the LOC338797 and DBH model significantly increased R2 to 0.648 (LR test P = 8.13 × 10−16) in a joint analysis of EA and NA subjects. The conditional analysis did not result in stronger results for the loci showing suggestive evidence in the primary analyses (Supplementary Material, Fig. S6A and C).
Functional analysis of variant C-2734T and four naturally occurring haplotypes in the DBH promoter
Functional analyses of the promoter variants rs1611115 and rs1989787 have previously been published by our group (14,15). Here we extend these analyses to the third promoter variant rs1076150, identified in the GWAS with a highly significant effect. Using the same six common promoter SNPs (minor allele frequency MAF > 0.05) as in previous work, we constructed luciferase promoter plasmids for four common, naturally occurring six-SNP haplotypes from the BAC promoter insert. The promoter activity of these four natural haplotypes (HAPs 1–4), measured as a function of luciferase expression in chromaffin cells is shown in Supplementary Material, Figure S7. We found that genotypic variations showed a significant overall effect (F = 33.8, P < 0.001), with haplotypes showing different DBH promoter/luciferase reporter activities (expressed as Firefly/Renilla ratio). To evaluate the individual effect of the rs1076150 SNP we constructed mutant variants on balanced backgrounds for two of the four haplotypes (HAP2 and HAP4), differing only at the desired −2734 position. When compared with the T allele, the C allele displayed higher expression on two different backgrounds (HAP2: P = 0.0047 and HAP4: P = 0.0098) (Fig. 2).
Figure 2.

In vitro effects of human DBH promoter variant C-2734T (rs1076150): Balanced mutants on two haplotype backgrounds (HAP2, HAP4) yield consistent (C > T) effects on transcription in chromaffin cells. Strength of the promoter variants is shown as luciferase activity in PC12 cell type (mean ± SEM). P-values are result of C versus T variant comparison for each haplotype background by ANOVA.
Bioinformatics of variant promoter motifs
In order to further investigate the functional properties of the DBH promoter variant rs1076150, we used bioinformatics tools (CONSITE and MotifLab) for the analysis of regulatory sequences. Both tools predicted that at position −2734 (upstream from the translation start site), SNP rs1076150 disrupted a binding motif for the transcription factor Snai1. As indicated in Supplementary Material, Figure S8, the match and binding score for the C-allele were predicted to be higher than for the T allele, possibly resulting in different expression levels of the DBH protein. For a complete characterization of the DBH promoter region, the computational molecular predictions and proposed mechanistic consequences of disrupted transcription factor binding motifs for the other two functional promoter variants rs1611115 and rs1989787 were added in Supplementary Material, Figure S8.
In vivo effects of functional DBH promoter haplotypes on human pDBH activity
We further evaluated the directional effects of the three functional SNPs (rs1076150 → rs1989787 → rs1611115) in the DBH promoter region (which showed the highest associations with pDBH activity in the GWAS) in a haplotype analysis in the combined 434 EA and NA subjects. First, we considered haplotype homozygotes for the four naturally occurring diploid haplotypes (Fig. 3A), and noted significant differences in pDBH activity with a plasma activity rank order of: CTC>CCC>TCC>TCT (P = 1.84 × 10−29). Finally we analyzed the effects of haplotype copy number on pDBH activity for the four haplotypes (Fig. 3B). The results were internally consistent with those for haplotype homozygotes, showing that increasing CTC copy number progressively elevated pDBH activity (P = 7.49 × 10−32), with reciprocal effects for haplotype TCT copy number (P = 2.96 × 10−66). Corresponding individual SNP effects are also shown in Supplementary Material, Figure S5A.
Figure 3.

In vivo effects of DBH promoter functional variants T-2734C (rs1076150), C-2073T (rs1989787) and C-970T (rs1611115) on plasma DBH activity (IU/l). (A) DBH promoter diploid haplotype (rs1076150 → rs1989787 → rs1611115) effect on pDBH activity (IU/l). Only subjects homozygous for a given haplotype (rs1076150 → rs1989787 → rs1611115) are shown. (B) Effect of DBH promoter haplotype (rs1076150 → rs1989787 → rs1611115) copy number (0, 1, or 2 copies per genome) on pDBH activity (IU/l, adjusted mean ± SEM).
Application of the MR test using genetic variants in DBH
PTSD re-experiencing symptoms were assessed post-deployment in 402 subjects with available pDBH levels and ranged from 0 to 29 (mean = 5.87). Re-experiencing symptoms were significantly associated with pDBH (beta = 0.13, P = 0.012), making a MR analysis applicable. The MR estimate of the association of pDBH and re-experiencing symptoms was significant (beta = 0.21, P = 0.002), indicating that pDBH is a causal component in the development of re-experiencing symptoms.
DISCUSSION
Dopamine β-hydroxylase as an essential part of the catecholamine biosynthetic pathway, converts dopamine to norepinephrine. DBH is encoded by a single gene located on chromosome 9q34 and its enzymatic activity is expressed both in plasma and CSF. The effects of this cis-QTL on plasma, serum and/or CSF DBH activity have been previously investigated in isolation (14,22,24), but to date no genome-wide association studies have been reported on DBH activity. Here, we present the first GWAS of plasma DBH levels and further characterize transcriptional control of the DBH gene.
Our GWAS was first performed in subjects of EA ancestry. We replicated the DBH locus as major contributor to pDBH activity, explaining ∼57% of the variability in EAs. As found by others, rs1611115 was the most significant polymorphism in this gene (22), with a P < 7.2 × 10−51, by far exceeding the genome-wide significance threshold of P < 5 × 10−8, and another 33 SNPs (some of them with independent effects) at this locus met genome-wide significance. No other loci were found to be genome-wide significant in this relatively small sample of 341 EAs, but 10 loci reached suggestive evidence of association with pDBH at P < 5 × 10−6 and await further replication in larger datasets. However, none of these loci were located on 20p12, a trans-QTL suggested in a linkage study by (24). The often poor correspondence between the susceptibility loci identified in genetic linkage and genome-wide association studies may be due in part to allelic heterogeneity, which reduces power in GWAS compared to linkage analyses (27).
Genetic association studies on the DBH locus have compared the three main ancestry groups from Europe, Africa and Asia. EAs were reported to have higher mean pDBH levels as compared to Japanese (22) and Africans from Nigeria (14,25). The promoter SNP rs1611115 was consistently reported as the most significant candidate SNP in DBH across studies and ancestral groups (14,22,23,28). Here, we extend this work to include subjects of genetically determined Native American descent, typically self-identifying as either Native American or Hispanic in our study. We found no difference in pDBH activity levels between our EA and NA subjects. The GWAS replicated the DBH locus with the same top hit (rs1611115 at P = 4.1 × 10−15) and consistent effect size estimates (R2 = 0.59 and 0.57 in EAs, respectively) in this even smaller sample of 93 subjects.
Increasing our power to detect additional loci by combining the relatively small number of EA and NA subjects in a meta-analysis, we identified LOC338797 (rs4255618) on chromosome 12q at P = 4.62 × 10−8, meeting the traditional genome-wide significance threshold of 5 × 10−8. However, genotype imputations based on 1000 Genomes Project reference data are increasing the effective number of independent tests and more stringent thresholds have recently been suggested (e.g. 1 × 10−8 for all common SNPs) (29). Irrespective of the specific threshold selected, the relevance of LOC338797 and all findings showing suggestive evidence of association have to be confirmed through independent replication of these results. LOC338797 seems to encode a 4-exon, previously uncharacterized 1794-base lncRNA, but the RNA-coding region bears no homology to DBH itself, and its role in DBH remains to be determined. However, adding LOC338797 to our genetic model of DBH only marginally increased the percent trait variability explained (from 57 to 59% in the combined analysis).
An additional analysis conditioned on the DBH locus promoter SNPs, to mask its strong effect on trait, identified sarcosine dehydrogenase SARDH, a gene adjacent to DBH, as an apparently independent, genome-wide significant hit in EAs. Its top hit rs7857468 was nominally replicated in NAs, leading to an overall P-value of 1.15 × 10−16, and further improving our model to explain 65% of overall variability in pDBH activity. SARDH encodes an enzyme localized to the mitochondrial matrix that catalyzes the oxidative demethylation of sarcosine. Even though adjacent to (and within 86.6 kb of) DBH, the conditional peak SARDH markers displayed little LD with the DBH promoter, as judged by marker-on-marker LD (R2 < 0.2) as well as a cM/Mb recombination boundary peak (Supplementary Material, Fig. S6B and D). However, analysis of the local chromosomal region by Chromatin conformation capture (or Hi-C, (30)) in human ES cells as well IMR-90 fibroblasts revealed that both DBH and SARDH inhabit the same topological domain, bounded by insulator/barrier (CTCF motif) elements. Thus, it is conceivable that the SARDH region harbors a 3′ transcriptional enhancer for DBH expression.
Mechanisms underlying DBH expression and secretion into plasma and CSF have invoked continuing interest among a broad range of investigators. One genetic variant in particular (rs1611115) has been widely investigated and ultimately documented (14) as a functional variant in the DBH promoter (14,22). We previously conducted systematic polymorphism discovery across the human DBH locus, and probed the functional consequences of two promoter variants (rs1989787 and rs1611115). We showed that rs1611115 disrupted consensus transcriptional motifs for n-MYC and MEF-2 (14) and rs1989787 for c-FOS (15), and that trans-activation of these variants by the corresponding transcription factors resulted in changes in DBH expression. The effects of variant rs1076150 on transcription reported here are novel, and allowed us to evaluate the effects upon gene expression of all three functional variants simultaneously. Here, we present an overview of properties of all three major functional variants in the proximal DBH promoter (Supplementary Material, Fig. S8). We found additive effects of each functional SNP upon DBH secretion into plasma (Supplementary Material, Fig. S5), and noted that the activity of contributory SNP alleles summated to give rise to a spectrum of promoter haplotype activities (Fig. 3A and B).
Genetic variants in DBH and/or pDBH activity have been directly implicated in mechanisms leading to increased susceptibility to disease. As the final enzyme in norepinephrine biosynthesis, DBH plays a role in differential availability of dopamine and norepinephrine. Consequently, DBH is involved in mechanisms underlying disorders associated with changes in the noradrenergic system (31–35). For example, our most significant DBH variant (rs1611115) is influencing heritable ‘intermediate phenotypes’ (e.g. autonomic and renal traits) as physiological risk traits in later development of hypertension (e.g. the T allele was found to decrease urine epinephrine excretion and basal blood pressure) (14,15) and progressive renal disease (36). In addition, biological and genetic studies suggest associations of low DBH levels with psychotic symptoms, and with mental disorders such as schizophrenia, depression, attention deficit hyperactivity disorder and alcoholism (see review 16). However, large GWAS on cardiovascular and psychiatric disorders (e.g. as reported by Ricopili) did not replicate strong effects for genetic variants in DBH.
The large proportion of DBH heritability that can be explained by a small number of genetic markers, in combination with the potentially important role of this intermediate phenotype for both psychiatric and cardiovascular disorders is unique and may represent a useful methodological tool to develop and test genetic epidemiological methods (37,38). To this end, we have applied genetic markers in DBH to the MR approach to investigate a potential causal effect of the pDBH and PTSD association previously reported (12,13). Our preliminary results on the effect of pDBH on PTSD re-experiencing symptoms indeed support this causal relation, but these findings will need to be confirmed in larger studies.
In conclusion, a first GWAS on pDBH activity identified the DBH gene as the principal locus determining pDBH levels in both EA and NA populations, explaining 57% of the variability. Two additional novel loci, SARDH and LOC338797, explaining combined an additional 8% of overall variability, were identified here and will have to be replicated in independent studies. Compared with other GWAS studies, the effects reported here were detected in relatively small datasets. Future studies on larger datasets may discover additional loci of smaller effects. Further, we demonstrated the potential application of strong genetic predictors of intermediate phenotypes such as DBH to the investigation of the disease etiology in the context of PTSD.
In perspective, the characterization of DBH activity and its underlying genetic regulation has positioned us uniquely for future studies of ‘intermediate phenotypes’, potentially leading to discovery of causal variants in complex genetic traits and disorders such as found in the psychiatric and cardiovascular fields.
MATERIALS AND METHODS
Subjects and biological sample collection
Participants were recruited from the Marine Resiliency Study (MRS), a large, prospective study of post-traumatic stress disorder (PTSD) involving active-duty United States Marines bound for deployment to Iraq or Afghanistan (39). The protocols for these studies were approved by the University of California-San Diego Institutional Review Board (IRB Protocols #070533, #110770X), and all subjects provided written informed consent to participate. Here we evaluated a subgroup of the MRS with available genotype and pDBH activity phenotype data, including 532 healthy, unrelated males from four different battalions (cohorts) assessed at pre-deployment. Following a 7-month deployment to a combat zone, post-traumatic stress symptoms were evaluated using a structured diagnostic interview, the Clinician Administered PTSD Scale (CAPS; (40–43)). Inter-rater reliability in MRS for the CAPS total score was high (Intraclass correlation coefficient = 0.99). Re-experiencing symptoms (CAPS-B symptom cluster) were used here. Initially, ethnicity and race were established by self-report, including information on geographic origin of both parents. The cohort studied here included 86% Caucasian and 22% Hispanic subjects, with a mean (±SD) age of 22.41 ± 3.23 years (range 18–41), typical for the overall MRS participants.
Blood was sampled from an antecubital vein for preparation of heparinized plasma (for assay of pDBH activity) and EDTA-anticoagulated blood (for preparation of genomic DNA). Heparinized blood from lithium heparin tubes was kept on ice prior to centrifugation and plasma was stored at −70°C prior to thawing for assays in batch. Genomic DNA was prepared from 1–2 ml blood leukocytes and diluted to a standard concentration of 50 ng/µl for genotyping.
Genotyping, quality control procedures and genotype imputations
Genotyping of 2585 DNA samples (532 with pDBH activity measures) was carried out by Illumina (http://www.illumina.com/) using the HumanOmniExpressExome array (HOEE 8v1_A) with 951 117 loci. Initial allele calling was performed by Illumina in GenomeStudio (V2011.1) and resulted in a sample success rate of 99.65%, a locus success rate of 99.86%, a genotype call rate of 99.88%, with reproducibility including 28 replicate DNA sample pairs of >99.99%. Additional data cleaning was performed in PLINK v1.07 (44) using standard procedures. SNPs were excluded if the call rate was <95%, if they violated Hardy–Weinberg Equilibrium (P < 1 × 10−6), or if they showed plate effects (P-value < 1 × 10−8 for any one plate or < 1 × 10−4 for two or more plates). Sample ID was confirmed by evaluating concordance between 31 overlapping genotypes from the HOEE array and those from an initial ‘fingerprinting’ panel including 41 ancestry-informative markers (AIMs) (45), resulting in the exclusion of one sample (overall concordance rate >0.99). Unexpected familial relationships were identified using pairwise identical-by-descent estimation and two subjects from sib-pairs were removed. Sample heterozygosity was between 0.211 and 0.302 and no excessive high or low samples were identified. The final dataset included 851 541 markers genotyped in 2548 individuals with a genotyping rate of >0.998.
Imputations were performed with standard protocols using the default parameters in IMPUTE2 v2.2.2, using 1000 Genomes Phase 1 integrated variant set haplotypes for the autosomes and the interim set for the X chromosome. Prior to imputation, genetic markers that had exceedingly rare alternative alleles (minor allele frequency MAF < 0.0002) were excluded. Next, genomes were divided into ∼5 Mb segments, and phasing and imputed genotypes were calculated for each. Imputed markers with low imputation quality values (Info value ≤0.5) were excluded. GTOOL v0.7.0 was used to convert genotype probabilities into calls for markers with probabilities >90% (genotypes were called missing if the posterior probability of any genotype was ≤90%), resulting in a total of 24 068 319 successfully imputed polymorphic markers, and a total of 24 919 860 genotyped and imputed markers for association analyses.
Ancestry assessment and control for genetic background heterogeneity
Ancestry was determined using genetic information as described in (45). In brief, genotypes of 1783 AIMs were used to determine a subject's ancestry at the continental level for the seven geographic regions Africa, Middle East, Europe, Central/South Asia, East Asia, Americas and Oceania. Ancestry estimates were determined using STRUCTURE v2.3.2.1. (46) at K = 7, including prior population information of the HGDP reference set (47). Based on these ancestry estimates, MRS subjects included here were placed into two main ancestral groups: subjects with >95% European ancestry were grouped with EAs (N = 341); and subjects with >5% Native American ancestry (and <10% African, and <5% each Central Asian, East Asian and Oceanic ancestry) as Native American descendants (NAs) (N = 93). A very wide range of Native American ancestry proportions is typical for subjects of self-reported Hispanic and Native American ethnicity/race (e.g. (48,49). Subjects with other ancestral backgrounds were not analyzed here (N = 98).
GWAS was performed separately in 341 EAs and 93 NAs. To control for additional genetic background heterogeneity within the two ancestral groups, and varying degrees of EA admixture within the NAs, principal component analyses (PCA) implemented in the EIGENSTRAT software (50) based on 10 000 random, autosomal SNPs were performed. The first 3 Eigenstrat-derived PCAs were included each as covariates in the association analyses.
Functional effects of trait-associated DBH promoter variants (rs1076150, rs1989787, rs1611115): promoter/luciferase reporter activity assays
Human DBH promoter/reporter plasmids were constructed from BAC genomic clone (RP11-317B10) obtained from CHORI (http://bacpac.chori.org) as described before. The DBH promoter region (extending distally from −3000 to +51 bp) containing six common polymorphic sites was excised from the BAC clone and inserted into the upstream/polylinker region of firefly luciferase reporter plasmid pGL3-Basic (Promega; Madison, WI, USA). Common naturally occurring haplotypes and additional variants were made by site-directed mutagenesis (QuikChange, Stratagene (Agilent), Santa Clara, CA, USA), verified by dideoxy sequencing, and co-transfected with Renilla luciferase expression plasmid pRL-TK (Herpes simplex virus thymidine kinase promoter driving Renilla luciferase, Promega) as a transfection efficiency control, into PC12 pheochromocytoma cells (at ∼50–60% confluence, 1 day after 1:4 splitting) as previously described (14). Firefly and Renilla luciferase activities in cell lysates were measured 16 h post-transfection, and results were presented as Firefly/Renilla luciferase activity ratio (‘Stop & Glo’; Promega, Madison, WI, USA).
Biochemical properties of plasma DBH
Plasma DBH activity was measured in 25 µl of heparinized plasma by a modified Nagatsu/Udenfriend spectrophotometric method (51), and reported as IU/l (IU/l=μmol/min/l plasma at 37°C, protocol available online at http://hypertension.ucsd.edu/). This method is based on a conversion of the synthetic DBH substrate tyramine by DBH (in the presence of Cu2+, N-ethylmaleimide and fumarate) to octopamine, which is then oxidized to parahydroxybenzaldehyde by sodium periodate. The oxidation is terminated by sodium metabisulfite, and the end product parahydroxybenzaldehyde is quantified by its absorbance at 330 nm in the ultraviolet spectrum. The mean plasma DBH activity inter-assay coefficient of variation was 12.8%. The mean plasma DBH level in 532 subjects was 10.86 IU/l (SD = 6.77) and ranged from 0.01 to 37.41 IU/l (Supplementary Material, Fig. S1).
Bioinformatic analyses
Computational prediction and motif discovery for transcription factors in the promoter region of DBH where candidate SNPs were positioned was made using web interface tools CONSITE (52) and graphical interface MotifLab (53), available at (http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite/) and (http://tare.medisin.ntnu.no/motiflab/), respectively. For both tools, predictions were based on position weight matrices for binding sites annotated in JASPAR and TRANSFAC databases. Motifs from consensus sequences, whose score was higher than 80% for binding to a motif containing a target SNP, were considered candidates.
Statistical analyses
Plasma DBH levels were square-root transformed to conform to normality (P> 0.74, Kolmogorov–Smirnov test). GWAS of transformed plasma DBH levels was performed in EA (N = 341) and NAs (N = 93) separately using linear regression under an additive genetic model with covariates age, cohort (three dummy coded variables), and three PCAs as implemented in PLINK. SNPs were pruned to a minor allele frequency (MAF) ≥0.01 in the combined dataset, which resulted in the inclusion of 7 871 575 SNPs. Genome-wide significance was set to P < 5 × 10−8 and suggestive evidence for association was considered at P < 5 × 10−6. Meta-analyses on the EA and NA results were performed in PLINK, using a fixed-effects model for SNPs with no significant heterogeneity (I) and a random-effects model when heterogeneity was significant (Cochrane's Q statistic). Conditional analyses on the DBH locus were performed to identify additional genetic associations by including the three DBH peak promoter SNPs rs1076150, rs1989787 and rs1611115 as additive covariates. Percent variability explained (R2) by a SNP or multiple SNPs in a gene were calculated using a linear regression in R 3.0.0, using the—clump function in PLINK to generate a list of highly significant SNPs in low LD for each gene with genome-wide significant SNPs. QQ plots and Manhattan plots were made using R 3.0.0. LocusZoom 1.2 (54) was used to construct regional association plots, including recombination information from HapMap phase II CEU. SG-ADVISER (http://genomics.scripps.edu/ADVISER/) was used for SNP annotations.
Analysis of variance (ANOVA) was used to compare luciferase reporter activity between different DBH haplotypes in vitro, and linear regression models and ANOVA based on an additive genetic model, with age, cohort and three PCAs as covariates were used for in vivo experiments to test for associations of haplotypes with DBH enzymatic activity in plasma using IBM SPSS Statistics, v.20.
Associations between pDBH levels, CAPS total score and symptom cluster B were tested in the combined EA (N = 341) and NA (N = 93) sample. To account for the non-normal distribution of CAPS scores, a zero-inflated negative binomial distribution (ZINB) regression was used (55), with additional covariates age, cohort (three dummy coded variables), and three PCAs based on continental ancestry. Associations between DBH SNPs and CAPS scores were tested under an additive genetic model.
Instrumental variable analysis. To demonstrate the utility of strong genetic effects on intermediate phenotypes for application to a MR approach, an association of pDBH with post-deployment PTSD re-experiencing symptoms was tested, using a ZINB regression (55), with additional covariates age, cohort and PCs. Following the determination of a significant association, the DBH SNP with the strongest effect (rs1611115) on pDBH was used as an instrument to test if pDBH is in the causal pathway to disease development (i.e. PTSD). MR estimates for the effect of pDBH on CAPS were then derived using a control function approach (56) an ordinary least squares regression of pDBH levels on rs1611115 was performed, including covariates age, cohort and PCs, followed by a ZINB regression of the CAPS score on pDBH, including the residuals from the first regression and age, cohort and PCs as covariates.
SUPPLEMENTARY MATERIAL
FUNDING
C.M.N. is supported by National Institutes of Health grants R01MH093500, U01 MH092758, by the Marine Corps and Navy Bureau of Medicine and Surgery (BUMED). D.T.O’.C. is supported by National Institutes of Health grant DK094894. M.M. is supported in part by the Croatian Science Foundation and R01MH093500. D.G.B. is supported to this in part by the Marine Corps and Navy Bureau of Medicine and Surgery (BUMED), VA Health Services Research and Development (VAHSR&D), VA Clinical Research and Development (VA CSR&D), NIMH R01MH093500 and the VA Center of Excellence for Stress and Mental Health (CESAMH).
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge special assistance from members of VA Center of Excellence for Stress and Mental Health, VA San Diego Research and Fiscal Services and the 1st Marine Division and Navy Medicine at 29 Palms and at Camp Pendleton.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Kim C.H., Zabetian C.P., Cubells J.F., Cho S., Biaggioni I., Cohen B.M., Robertson D., Kim K.S. Mutations in the dopamine beta-hydroxylase gene are associated with human norepinephrine deficiency. Am. J. Hum. Genet. 2002;108:140–147. [PubMed] [Google Scholar]
- 2.De Potter W.P., De Schaepdryver A.F., Smith A.D. Release of chromogranin A and dopamine-beta-hydroxylase from adrenergic nerves during nerve stimulation. Acta. Physiol Scand. Suppl. 1970;357:8. [PubMed] [Google Scholar]
- 3.Weinshilboum R.M., Thoa N.B., Johnson D.G., Kopin I.J., Axelrod J. Proportional release of norepinephrine and dopamine hydroxylase from sympathetic nerves. Science. 1971;174:1349–1351. doi: 10.1126/science.174.4016.1349. [DOI] [PubMed] [Google Scholar]
- 4.O'Connor D.T., Cervenka J.H., Stone R.A., Levine G.L., Parmer R.J., Franco-Bourland R.E., Madrazo I., Langlais P.J., Robertson D., Biaggioni I. Dopamine beta-hydroxylase immunoreactivity in human cerebrospinal fluid: properties, relationship to central noradrenergic neuronal activity and variation in Parkinson’s disease and congenital dopamine beta-hydroxylase deficiency. Clin. Sci. (Lond.) 1994;86:149–158. doi: 10.1042/cs0860149. [DOI] [PubMed] [Google Scholar]
- 5.Weinshilboum R.M., Raymond F.A., Elveback L.R., Weidman W.H. Serum dopamine-beta-hydroxylase activity: sibling–sibling correlation. Science. 1973;181:943–945. doi: 10.1126/science.181.4103.943. [DOI] [PubMed] [Google Scholar]
- 6.Cubells J.F., Price L.H., Meyers B.S., Anderson G.M., Zabetian C.P., Alexopoulos G.S., Nelson J.C., Sanacora G., Kirwin P., Carpenter L., et al. Genotype-controlled analysis of plasma dopamine beta-hydroxylase activity in psychotic unipolar major depression. Biol. Psychiatry. 2002;51:358–364. doi: 10.1016/s0006-3223(01)01349-x. [DOI] [PubMed] [Google Scholar]
- 7.Kopeckova M., Paclt I., Goetz P. Polymorphisms and low plasma activity of dopamine-beta-hydroxylase in ADHD children. Neuro. Endocrinol. Lett. 2006;27:748–754. [PubMed] [Google Scholar]
- 8.Segurado R., Bellgrove M.A., Manconi F., Gill M., Hawi Z. Epistasis between neurochemical gene polymorphisms and risk for ADHD. Eur. J. Hum. Genet. 2011;19:577–582. doi: 10.1038/ejhg.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Healy D.G., Abou-Sleiman P.M., Ozawa T., Lees A.J., Bhatia K., Ahmadi K.R., Wullner U., Berciano J., Moller J.C., Kamm C., et al. A functional polymorphism regulating dopamine beta-hydroxylase influences against Parkinson's disease. Ann. Neurol. 2004;55:443–446. doi: 10.1002/ana.20063. [DOI] [PubMed] [Google Scholar]
- 10.Combarros O., Warden D.R., Hammond N., Cortina-Borja M., Belbin O., Lehmann M.G., Wilcock G.K., Brown K., Kehoe P.G., Barber R., et al. The dopamine beta-hydroxylase-1021C/T polymorphism is associated with the risk of Alzheimer’s disease in the Epistasis Project. BMC Med. Genet. 2010;11:162. doi: 10.1186/1471-2350-11-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mustapic M., Presecki P., Pivac N., Mimica N., Hof P.R., Simic G., Folnegovic-Smalc V., Muck-Seler D. Genotype-independent decrease in plasma dopamine beta-hydroxylase activity in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;44:94–99. doi: 10.1016/j.pnpbp.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamner M.B., Gold P.B. Plasma dopamine beta-hydroxylase activity in psychotic and non-psychotic post-traumatic stress disorder. Psychiatry Res. 1998;77:175–181. doi: 10.1016/s0165-1781(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 13.Mustapic M., Pivac N., Kozaric-Kovacic D., Dezeljin M., Cubells J.F., Muck-Seler D. Dopamine beta-hydroxylase (DBH) activity and -1021C/T polymorphism of DBH gene in combat-related post-traumatic stress disorder. Am. J. Med. Genet. 2007;144B:1087–1089. doi: 10.1002/ajmg.b.30526. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y., Wen G., Rao F., Zhang K., Wang L., Rodriguez-Flores J.L., Sanchez A.P., Mahata M., Taupenot L., Sun P., et al. Human dopamine beta-hydroxylase (DBH) regulatory polymorphism that influences enzymatic activity, autonomic function, and blood pressure. J. Hypertens. 2010;28:76–86. doi: 10.1097/HJH.0b013e328332bc87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y., Zhang K., Wen G., Rao F., Sanchez A.P., Wang L., Rodriguez-Flores J.L., Mahata M., Mahata S.K., Waalen J., et al. Human dopamine beta-hydroxylase promoter variant alters transcription in chromaffin cells, enzyme secretion, and blood pressure. J. Hypertens. 2011;24:24–32. doi: 10.1038/ajh.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cubells J.F., Zabetian C.P. Human genetics of plasma dopamine beta-hydroxylase activity: applications to research in psychiatry and neurology. Psychopharmacology (Berl.) 2004;174:463–476. doi: 10.1007/s00213-004-1840-8. [DOI] [PubMed] [Google Scholar]
- 17.Ross S.B., Wetterberg L., Myrhed M. Genetic control of plasma dopamine-beta-hydroxylase. Life. Sci. 1973;12:529–532. doi: 10.1016/0024-3205(73)90056-8. [DOI] [PubMed] [Google Scholar]
- 18.Goldin L.R., Gershon E.S., Lake C.R., Murphy D.L., McGinniss M., Sparkes R.S. Segregation and linkage studies of plasma dopamine-beta-hydroxylase (DBH), erythrocyte catechol-O-methyltransferase (COMT), and platelet monoamine oxidase (MAO): possible linkage between the ABO locus and a gene controlling DBH activity. Am. J. Hum. Genet. 1982;34:250–262. [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson A.F., Elston R.C., Siervogel R.M., Tran L.D. Linkage of a gene regulating dopamine-beta-hydroxylase activity and the ABO blood group locus. Am. J. Hum. Genet. 1988;42:160–166. [PMC free article] [PubMed] [Google Scholar]
- 20.Cubells J.F., van Kammen D.P., Kelley M.E., Anderson G.M., O'Connor D.T., Price L.H., Malison R., Rao P.A., Kobayashi K., Nagatsu T., et al. Dopamine beta-hydroxylase: two polymorphisms in linkage disequilibrium at the structural gene DBH associate with biochemical phenotypic variation. Hum. Genet. 1998;102:533–540. doi: 10.1007/s004390050736. [DOI] [PubMed] [Google Scholar]
- 21.Wei J., Ramchand C.N., Hemmings G.P. Possible control of dopamine beta-hydroxylase via a codominant mechanism associated with the polymorphic (GT)n repeat at its gene locus in healthy individuals. Hum. Genet. 1997;99:52–55. doi: 10.1007/s004390050310. [DOI] [PubMed] [Google Scholar]
- 22.Zabetian C.P., Anderson G.M., Buxbaum S.G., Elston R.C., Ichinose H., Nagatsu T., Kim K.S., Kim C.H., Malison R.T., Gelernter J., et al. A quantitative-trait analysis of human plasma-dopamine beta-hydroxylase activity: evidence for a major functional polymorphism at the DBH locus. Am. J. Hum. Genet. 2001;68:515–522. doi: 10.1086/318198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zabetian C.P., Buxbaum S.G., Elston R.C., Kohnke M.D., Anderson G.M., Gelernter J., Cubells J.F. The structure of linkage disequilibrium at the DBH locus strongly influences the magnitude of association between diallelic markers and plasma dopamine beta-hydroxylase activity. Am. J. Hum. Genet. 2003;72:1389–1400. doi: 10.1086/375499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cubells J.F., Sun X., Li W., Bonsall R.W., McGrath J.A., Avramopoulos D., Lasseter V.K., Wolyniec P.S., Tang Y.L., Mercer K., et al. Linkage analysis of plasma dopamine beta-hydroxylase activity in families of patients with schizophrenia. Hum. Genet. 2011;130:635–643. doi: 10.1007/s00439-011-0989-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Connor D.T., Levine G.L., Frigon R.P. Homologous radio-immunoassay of human plasma dopamine-beta-hydroxylase: analysis of homospecific activity, circulating plasma pool and intergroup differences based on race, blood pressure and cardiac function. J. Hypertens. 1983;1:227–233. doi: 10.1097/00004872-198310000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Tang Y.L., Epstein M.P., Anderson G.M., Zabetian C.P., Cubells J.F. Genotypic and haplotypic associations of the DBH gene with plasma dopamine beta-hydroxylase activity in African Americans. Eur. J. Hum. Genet. 2007;15:878–883. doi: 10.1038/sj.ejhg.5201838. [DOI] [PubMed] [Google Scholar]
- 27.Ott J., Kamatani Y., Lathrop M. Family-based designs for genome-wide association studies. Nat. Rev. Genet. 2011;12:465–474. doi: 10.1038/nrg2989. [DOI] [PubMed] [Google Scholar]
- 28.Tang Y., Buxbaum S.G., Waldman I., Anderson G.M., Zabetian C.P., Kohnke M.D., Cubells J.F. A single nucleotide polymorphism at DBH, possibly associated with attention-deficit/hyperactivity disorder, associates with lower plasma dopamine beta-hydroxylase activity and is in linkage disequilibrium with two putative functional single nucleotide polymorphisms. Biol. Psychiatry. 2006;60:1034–1038. doi: 10.1016/j.biopsych.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Li M.X., Yeung J.M., Cherny S.S., Sham P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2012;131:747–756. doi: 10.1007/s00439-011-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lampinen K.H., Ronnback M., Groop P.H., Nicholls M.G., Yandle T.G., Kaaja R.J. Increased plasma norepinephrine levels in previously pre-eclamptic women. J. Hum. Hypertens. 2014;28:269–273. doi: 10.1038/jhh.2013.84. [DOI] [PubMed] [Google Scholar]
- 32.Anand A., Charney D.S. Norepinephrine dysfunction in depression. J. Clin. Psychiatry. 2000;61( 10):16–24. [PubMed] [Google Scholar]
- 33.Fitzgerald P.J. Is norepinephrine an etiological factor in some types of cancer? Int. J. Cancer. 2009;124:257–263. doi: 10.1002/ijc.24063. [DOI] [PubMed] [Google Scholar]
- 34.Lewitt P.A. Norepinephrine: the next therapeutics frontier for Parkinson’s disease. Transl. Neurodegener. 2012;1:4. doi: 10.1186/2047-9158-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heneka M.T., Nadrigny F., Regen T., Martinez-Hernandez A., Dumitrescu-Ozimek L., Terwel D., Jardanhazi-Kurutz D., Walter J., Kirchhoff F., Hanisch U.K., et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA. 2010;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pasha D.N., Davis J.T., Rao F., Chen Y., Wen G., Fung M.M., Mahata M., Zhang K., Trzebinska D., Mustapic M., et al. Heritable influence of DBH on adrenergic and renal function: twin and disease studies. PloS one. 2013;8:e82956. doi: 10.1371/journal.pone.0082956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almli L.M., Fani N., Smith A.K., Ressler K.J. Genetic approaches to understanding post-traumatic stress disorder. Int. J. Neuropsychopharmacol. 2014;17:355–370. doi: 10.1017/S1461145713001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solovieff N., Roberts A.L., Ratanatharathorn A., Haloosim M., De Vivo I., King A.P., Liberzon I., Aiello A., Uddin M., Wildman D.E., et al. Genetic Association Analysis of 300 Genes Identifies a Risk Haplotype in SLC18A2 for Post-traumatic Stress Disorder in Two Independent Samples. Neuropsychopharmacology. 2014;39:1872–1879. doi: 10.1038/npp.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker D.G., Nash W.P., Litz B.T., Geyer M.A., Risbrough V.B., Nievergelt C.M., O'Connor D.T., Larson G.E., Schork N.J., Vasterling J.J., et al. Predictors of risk and resilience for posttraumatic stress disorder among ground combat Marines: methods of the Marine Resiliency Study. Prev. Chronic Dis. 2012;9:E97. doi: 10.5888/pcd9.110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blake D.D., Weathers F.W., Nagy L.M., Kaloupek D.G., Gusman F.D., Charney D.S., Keane T.M. The development of a Clinician-Administered PTSD Scale. J. Traumatic Stress. 1995;8:75–90. doi: 10.1007/BF02105408. [DOI] [PubMed] [Google Scholar]
- 41.King D.W., Leskin G.A., King L.A., Weathers F.W. Confirmatory factor analysis of the Clinician-Administered PTSD Scale: Evidence for the dimensionality of posttraumatic stress disorder. Psychol Assess. 1998;10:90–96. [Google Scholar]
- 42.Weathers F.W., Keane T.M., Davidson J.R. Clinician-administered PTSD scale: a review of the first ten years of research. Depress. Anxiety. 2001;13:132–156. doi: 10.1002/da.1029. [DOI] [PubMed] [Google Scholar]
- 43.Weathers F.W., Ruscio A.M., Keane T.M. Psychometric properties of nine scoring rules for the clinician-administered posttraumatic stress disorder scale. Psychol Assess. 1999;11:124–133. [Google Scholar]
- 44.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nievergelt C.M., Maihofer A.X., Shekthman T., Libiger O., Wang X., Kidd K.K., Kidd J.R. Inference of human continental origin and admixture proportions using a highly discriminative ancestry informative 41-SNP panel. Investig. Genet. 2013;4:13. doi: 10.1186/2041-2223-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 48.Klimentidis Y.C., Miller G.F., Shriver M.D. Genetic admixture, self-reported ethnicity, self-estimated admixture, and skin pigmentation among Hispanics and Native Americans. Am. J. Phys. Anthropol. 2009;138:375–383. doi: 10.1002/ajpa.20945. [DOI] [PubMed] [Google Scholar]
- 49.Nievergelt C.M., Wineinger N.E., Libiger O., Pham P., Zhang G., Baker D.G., Schork N.J. Marine Resiliency Study, I. Chip-based direct genotyping of coding variants in genome wide association studies: utility, issues and prospects. Gene. 2014;540:104–109. doi: 10.1016/j.gene.2014.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 51.Nagatsu T., Udenfriend S. Photometric assay of dopamine- -hydroxylase activity in human blood. Clin. Chem. 1972;18:980–983. [PubMed] [Google Scholar]
- 52.Sandelin A., Wasserman W.W., Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–W252. doi: 10.1093/nar/gkh372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klepper K., Drablos F. MotifLab: a tools and data integration workbench for motif discovery and regulatory sequence analysis. BMC Bioinformatics. 2013;14:9. doi: 10.1186/1471-2105-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yurgil K.A., Barkauskas D.A., Vasterling J.J., Nievergelt C.M., Larson G.E., Schork N.J., Litz B.T., Nash W.P., Baker D.G. Marine Resiliency Study Team. Association between traumatic brain injury and risk of posttraumatic stress disorder in active-duty Marines. JAMA Psychiatry. 2014;71:149–157. doi: 10.1001/jamapsychiatry.2013.3080. [DOI] [PubMed] [Google Scholar]
- 56.Heckman J.J., Robb R. Alternative methods for evaluating the impact of interventions—an overview. J. Econometrics. 1985;30:239–267. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.