

Abstract
Cerebral cavernous malformation (CCM) is a disease of vascular malformations known to be caused by mutations in one of three genes: CCM1, CCM2 or CCM3. Despite several studies, the mechanism of CCM lesion onset remains unclear. Using a Ccm1 knockout mouse model, we studied the morphogenesis of early lesion formation in the retina in order to provide insight into potential mechanisms. We demonstrate that lesions develop in a stereotypic location and pattern, preceded by endothelial hypersprouting as confirmed in a zebrafish model of disease. The vascular defects seen with loss of Ccm1 suggest a defect in endothelial flow response. Taken together, these results suggest new mechanisms of early CCM disease pathogenesis and provide a framework for further study.
INTRODUCTION
Cerebral cavernous malformation (CCM) is a disease characterized by the formation of dilated vascular lesions in the brain, retina and skin (1). CCM lesions lack the typical blood-tight barrier of healthy vessels, resulting in chronic leakiness and associated inflammation in the surrounding tissue and most commonly lead to headaches, seizures and strokes (2). The disease affects as much as 0.5% of the population (1,3,4) and can occur sporadically or be inherited in an autosomal dominant pattern. The inherited form of the disease is caused by the loss of function of one of three genes: CCM1, CCM2 or CCM3 (5).
Loss of CCM1, encoding the protein Krev-interaction trapped protein-1 (KRIT1), has been shown to disrupt several cellular processes in vitro and in vivo, including the appropriate organization of the junctional protein VE-Cadherin and endothelial cell polarity (6,7). Maintaining physiologic cell–cell junctions and cell polarity are critical to normal cell migration, flow response and appropriate signaling by various cascades (8). There are many other dysfunctional processes associated with loss of CCM1 in vitro, but how these interact in vivo to disrupt normal cellular physiology is unknown. Current models and systems have focused on established lesions, but the preceding events that initiate lesion formation have not been identified.
While models are useful for interrogation of the disease, later stage models can mislead when secondary downstream effects are interpreted as primary defects. Understanding the events that lead to interpretable signs of disease provides invaluable insight into the pathophysiology of CCM disease. Therefore we sought to analyze the developmental events that give rise to cerebral cavernous malformations associated with loss of CCM1 in hopes of identifying a morphological tipping point. The murine embryonic phenotype associated with homozygous deletion of Ccm1 consists of severe defects of the aorta and branchial arch arteries resulting in death, making the murine embryo a challenging system for studying the onset of CCM lesion formation (9). We therefore created an inducible mouse model of CCM disease where loss of a second CCM allele from the endothelium can be induced after birth in a heterozygous mouse (10,11), allowing normal developmental angiogenesis and vascular patterning to occur and mimicking the loss of heterozygosity thought to predispose to lesion formation. In our model, we find that retinal lesions develop early in life, adhere to a strict stereotypical pattern and form following hypersprouting of the retinal vasculature front. In addition, we find that cells deficient in CCM1 fail to align to flow in an in vitro model and that loss of ccm1 in the zebrafish phenocopies loss of blood flow, suggesting that failed endothelial flow response may be a contributing mechanism for CCM1 lesion development.
RESULTS
Generation of CCM1 mutant alleles
A homozygous germline knockout of Ccm1 results in vascular tissue defects as early as E8.5, including failure of the branchial arch artery and caudal portion of the dorsal aorta to form a lumen. These defects restrict and disrupt normal blood flow (9). In order to determine whether Ccm1 is required autonomously in endothelial cells, we created a conditional allele in which exons 4–8 of Ccm1 were flanked by loxP sites and Cre recombinase was expressed in specific cell types using different tissue-specific Cre drivers (Supplementary Material, Fig. S1). In embryos with a germline mutant Ccm1 allele and a second floxed allele of Ccm1, the endothelial loss of Ccm1 driven by co-expression of a tissue-specific Cre (Tie2-Cre) (12) resulted in mice with a homozygous deletion of Ccm1 restricted to the endothelium. With the loss of Ccm1 in the endothelium, embryos fail to survive beyond E12.5 due to failed vascular development. The branchial arch arteries fail to lumenize properly as seen by immunofluorescent staining of the vasculature (Fig. 1A–F). We examined the effects this impaired lumen formation could have on circulation by performing India ink microangiography, a technique used to assess vessel patency. The disrupted vessel formation prevented normal passage of ink through the branchial arch arteries (Fig. 1G and H). This failure in proper embryonic circulation could also be detected by in utero ultrasound (Fig. 1I and J). The development of the impaired lumen organization and failed circulation in the endothelial knockout is similar to that described previously in the germline knockout, a pattern akin to loss of Ccm2 but not Ccm3 (10,11).
Figure 1.
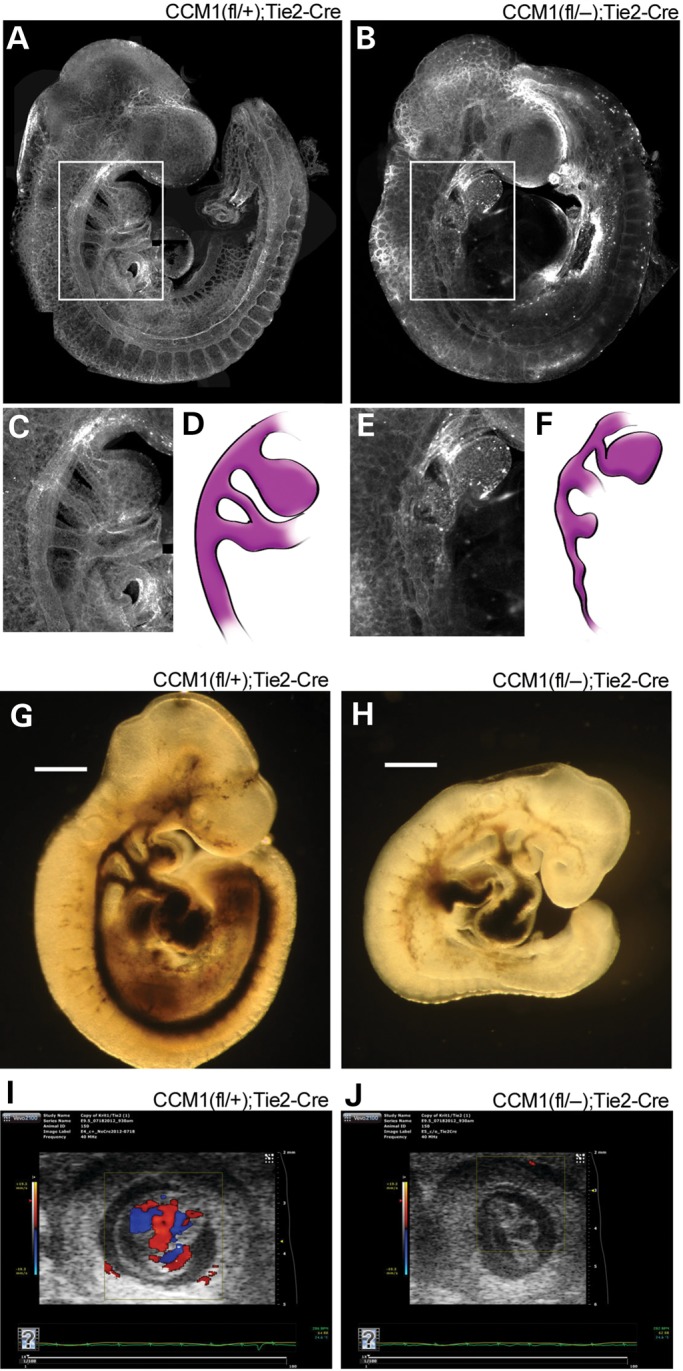
Endothelial loss of CCM1 results in abnormal embryonic vascular development. (A and B) Visualization of the vasculature, as indicated by lectin stain, or whole-mount embryos at E9.5. (C and E) Close-up image of the branchial arch artery morphology from the indicated white outlines in A and B. (D and F) Illustration of the branchial arch artery from the adjacent panel, included to highlight visualization. (G and H) Brightfield images of ink injected E9.5 embryos. Light blue arrows indicate areas where ink was able to flow through the vasculature. (I and J) In utero ultrasound of blood flow on E9.5 embryos. Embryos are located in the center of each image with the box indicating the area being monitored for flow. The heat map bar on the left indicates the directionality of the blood flow, red indicates blood flow towards the camera, blue indicates blood flow away from the camera. Absence of color demonstrates absence of flow.
Having observed that endothelial specific loss of Ccm1 generates defects that are nearly indistinguishable from the embryonic defects in germline knockouts (9), we inquired if post-natal induction of loss of a second allele of Ccm1 would produce a murine model that would more closely mimic human disease. Therefore, we utilized an inducible Cre, PDGFb-iCreERT2, the expression of which is also restricted primarily to the endothelium, but is activated only upon exposure to tamoxifen (13). To confirm the endothelial specificity, we crossed the PDGFb-CreERT2 mouse with the mTmG reporter mouse. Following tamoxifen administration the GFP expression was detected in the endothelium as confirmed with a co-stain with the endothelial marker lectin (Supplementary Material, Fig. S2). This system allowed us to knockout the second Ccm1 allele at any time point after birth, bypassing the lethal embryonic defects. Endothelial specific deletion of the floxed allele can be obtained by crossing the PDGF-iCreERT2 allele into the floxed Ccm1 background. This allele results in an induced endothelial knockout (ieKO) upon stimulation with tamoxifen. The loss of endothelial Ccm1 following tamoxifen treatment at birth leads to the formation of cavernous malformations in the brains of all animals. We tracked the development from simple, dilated telangiectasias to large multi-chambered caverns over time using live animal magnetic resonance imaging (MRI). Lesion pathology includes vascular leak, which results in the deposition of hemosiderin which is detected by the MRI and presents as dark masses within the brain. These lesions arise throughout the entire brain by 2 months and progress in severity with time (Fig. 2A–F). The presence of these lesions decreased the survival rate in mice, which correlated with increased lesion content (Fig. 2G and H). Further analysis showed that histologically these lesions phenocopy the hallmarks of human disease with dilated, thin-walled caverns surrounded by hemosiderin deposits (reflecting ongoing vascular leak), inflammation and fibrosis (Fig. 2I–O) (14). Lesions also consistently form in the retinas of all affected animals (Fig. 3). Because of its size and ease of access, the retina is well characterized with respect to normal vascular development (15) and is thus an ideal focal point for describing the early developmental mechanism of CCM lesion formation. Moreover, the retina is amenable to live-animal angiography, permitting us to ask questions of lesion perfusion as well as location.
Figure 2.

Post-natal endothelial loss of CCM1 phenocopies human disease. (A–F) Live animal magnetic resonance imaging (MRI) images of mice. Each row is from one mouse, kept alive, and imaged successively over time at the ages indicated in the panel. Light blue arrows indicate CCM lesions. (G) Kaplan–Meier survival curve, animals were given tamoxifen at birth and followed through life. Animals that survived were euthanized at 8 months of age. (H) Quantification of CCM brain lesion content over time. Lesion burden is determined as the total number of lesions observed in each tomographic view (slice) of the MRI per mouse. (I–N) Histological staining comparing samples from human CCM1 lesions and lesions from CCM1 knockout mice. H&E staining indicates tissue morphology of the lesion. Iron staining indicates leak into the brain tissue surrounding the lesion. Trichrome staining indicates fibrosis surrounding the lesion. Scale bar at 200 μm. (O) Whole-brain H&E histology of an 8-month old CCM1 knockout mouse. Black arrows indicate CCM lesions.
Figure 3.

Retinal CCMs develop in a stereotypical location. (A and B) Schematic indicating mouse retinal orientation, right in A and left in B, with an example angiograph showing the expected retinal vascular morphology. Adjacent to the example angiography are live angiographs taken from either control or CCM1 knockout mouse in the appropriate orientation. Blue arrows indicate lesion location. Scale bars are at 1000 μm. (C and D) Visualization of retinal vascular, as indicated by lectin staining, in whole-mount retinas from the mice shown in B (left eyes). (E and F) Schematic illustration highlighting lesion location and morphology. The superior and inferior veins are colored in purple with the major surrounding vessels in light gray.
In order to examine the location and perfusion of retinal cavernous malformations, we studied ieKO mice with in vivo fluorescein angiography. Using this method, we found that lesions stereotypically arise in the veins located both most superior and inferior within the retina (Fig. 3A and B). Extending out from the trunk of these veins is a tangled network of spherical vascular sacs, the hallmark of cavernous malformation anatomy (16). The individual caverns are confined to the periphery, vary in size and are interconnected by narrow vessels. These vessels also appear to be fully perfused as indicated by the ability of the fluorescein to fill the lesions. Visualization of these retinas via dissection and lectin staining confirmed our observations from the angiography (Fig. 3C–F).
We next examined the timing of lesion formation by looking at a series of retinas from mice at different ages. Retinas from ieKO remain indistinguishable from controls until postnatal Day 7 when the vascular network begins to exhibit vessel dilation and failure of plexus coalescence. Over time, the vascular dilations resolve into caverns and by p21 the lesions assume the well-defined morphology of a mature retinal CCM and recapitulate human retinal angiomas (Fig. 4A–J) (16). We found that this pattern of development held true for 100% of the retinas examined and that all of the Ccm1 knockouts developed retinal lesions.
Figure 4.

Retinal CCMs develop early and are limited to the superficial vascular plexus. (A–J) Timecourse of retinal vascular development in denoted genotypes. Retinas were dissected at the ages indicated, then lectin stained and imaged. Each image depicts a single leaflet of the dissected retina. Light blue arrows indicate the location of lesion development. Scale bars = 200 um. (K–R) Retinal depth images taken at 5 months of age from either a control or CCM1 knockout mouse. Each image was taken using confocal microscopy with the microscope focused in the retinal plane indicated in the panel (N,R represent merged image. Blue = deep layer. Red = intermediate layer. Green = superficial layer). K′–R′) Retinal depth images taken at 10 days of age from either a control or CCM1 knockout mouse. Each image was taken using confocal microscopy with the microscope focused in the retinal plane indicated in the panel (N,R represent merged image. Blue = deep layer. Red = intermediate layer. Green = superficial layer). (S–X) Cross sectional images from 5-month-old retinas from either control or CCM1 knockout mice. Each retina was frozen in OCT, sectioned and stained with lectin and DAPI to visualize both the vasculature and the surrounding tissue. (Y) Diagram depicting lesion growth across the retina. All scale bars set at 200 μm.
The normal growth of retinal vasculature begins at birth with endothelial cells expanding across the superficial layer (17). At P8 the formation of a vascular network in the deep retinal layer begins. Eventually the superficial and deep networks are joined together by the formation of an intermediate network. Once these layers are interconnected and the vascular network has reached the periphery of the retina, angiogenesis is complete and the retinal vasculature is mature. We found that CCM lesions arose from the superficial vascular plexus and remained superficial until later in disease progression. At P10 the dilations observed in early disease onset were limited to the superficial layer with no dilated caverns observed in the deep or intermediate layers (Fig. 4K′–R′). By 5 months the main body of the lesion remained superficial but some extension of caverns could be observed in the deeper layers as the mature lesion expanded in size (Fig. 4K–R). The superficial location of the lesions could also be seen using cross sectional analysis (Fig. 4S–X). To further expand on our observation that lesions were perfusable, we conducted ultrasound studies of the retinal vasculature. We found no difference in blood flow between ieKO and control mice, implying that retinal cavernous malformations are low flow lesions that do not increase the overall retinal blood flow (Fig. 5A–C).
Figure 5.

Retinal CCMs do not exhibit disrupted hemodynamics and are limited to a specific developmental window. (A and B) Ultrasound images of whole retinal vasculature from either control or knockout mice, orientation is en face with the optic nerve in the center of the eye. Red color indicates blood flow towards the camera, blue color indicates blood flow away from the camera. (C) Quantification of the volume time interval (VTI) of both left and right eyes as calculated by the Vevo2100 software. (D–F) Visualization of the retinal vasculature, as indicated by lectin stain, from a CCM1 knockout mouse that was induced at P1 (D imaged at P21), induced at P7 (E imaged at P21) or induced P21 (F imaged at 8 months). (G and H) Close-up of the vascular lesion from the insets in E and F. Scale bars set at 1000 μm.
The first 2 weeks of retinal development relies heavily on active angiogenesis, particularly as the superficial plexus expands (18,19). However, by P21, retinal angiogenesis is complete and the vascular structure is mature. To determine if lesion formation was dependent upon this early angiogenic process, we inactivated Ccm1 at varying time points after birth and followed the subsequent retinal development. We found that when Ccm1 is lost at the critical point of phenotype distinction, P7, lesions do form in the periphery of the retina by P21 and that these lesions demonstrate similar morphology as those of mice induced at birth, though the severity and size of the lesions are diminished (Fig. 5D and E). If we delay delivery of tamoxifen such that Ccm1 is not deleted until P21, after the retinal vasculature is mature, we find that lesions do not form (Fig. 5F). Therefore, our results in a distinct animal model confirm those of others that active angiogenesis is required for lesion formation (20).
Lack of CCM1 induces hypersprouting
Sprouting angiogenesis requires the coordinated effort of multiple endothelial cells and is found at the leading edge of the developing vascular plexus (18). It is dictated by the stochastic response of select endothelial cells responding to angiogenic stimuli. The responsive cells that form the sprouts (called tip cells) extend, migrate and fuse to develop a vascular plexus (19). The non-responsive cells that remain non-migratory are known as stalk cells. Therefore, it is possible that excessive sprouting and the subsequent anastomosis could contribute to lesion onset. Prior to vessel dilation, we find that retinas from ieKO mice induced at P1, exhibit endothelial hypersprouting (Fig. 6A–C). The sprouts observed showed a tip cell phenotype as indicated by the tip cell marker PDGFb as well as cellular morphology (Supplementary Material, Fig. S2). Although the retina allows better observation of the three-dimensional architecture of the cavernous malformation than the mouse brain, it is difficult to observe the dynamics of vascular development in the intact mouse eye in real time. Sprouting angiogenesis is a dynamic process, therefore we sought to explore this phenotype in a model more amenable to live imaging. Zebrafish have the advantage of ex vivo development and transparency that allow the real-time observation of sprouting angiogenesis. (21). Therefore, we sought to evaluate sprouting angiogenesis in a zebrafish model.
Figure 6.

Loss of CCM results in hypersprouting. (A and B) Images showing sprouting at the retinal vascular front at Day p7 as indicated by lectin stain. White asterisks denote sprouts. Scale bars set at 50 μm. (C) Quantification of sprouts per field at p7, P-value <0.05. (D and E) Endothelial sprouting of the zebrafish CVP at 28hpf. White asterisks denote sprouts. Scale bars set at 100 μm. (F and G) Zebrafish tail vasculature at 48 hpf. White box indicates area of inset. (H and I) Inset of tail vasculature. (J and K) Schematic illustration highlighting the tail morphology seen in F and G.
To examine early angiogenic sprouting, we injected a morpholino targeted against ccm1 (22) (or a scrambled control) at the single cell stage of embryos carrying the endothelial specific reporter kdrl;GFP. We found that the caudal vein plexus (CVP) development exhibited hypersprouting at 28 h post-fertilization (hpf) (Fig. 6D and E) and then went on to develop into a large vascular cavern, a defect similar to early retinal lesions (Fig. 6F–K). The dilation of the CVP is also reminiscent of the dilated caudal dorsal aorta in developing CCM1 knockout mouse embryos, which also demonstrated alterations in artery/vein specification. Loss of this morphological distinction between the caudal artery and the cadual vein in the zebrafish and the resulting dilated morphology, highlights overlap in the vascular pathology of both mouse and zebrafish. Under normal developmental circumstances the CVP arises from the process of ventral sprouting. Ventral sprouting initiates at 20 hpf when endothelial cells begins to sprout and migrate from the dorsal aorta (23). The cells then expand as a population and coalesce into the distinct vascular network of the CVP (Supplementary Material, Movie S1). In contrast, when ccm1 is reduced, the cells do not form a distinct vascular network and the CVP develops as a large vascular sac (Supplementary Material, Movie S2). Therefore these data suggest that the formation of CCMs could arise from a defect of population-wide endothelial hypersprouting and anastomosis, resulting in a malformed vascular cavern.
Lack of CCM1 impairs flow response
Flow can regulate vascular morphology during development (24) and shear stress can direct overall network architecture. Furthermore blood flow can attenuate endothelial sprouting (25). The effects of flow and the endothelial mechanism to detect flow may provide an explanation for the hypersprouting observed as well as why lesions form stereotypically.
The silent heart morpholino, which knocks down cardiac troponin, results in a non-beating heart resulting in a complete lack of blood flow (26). We find that at 28 hpf silent heart morphants exhibit the same hypersprouting defect as seen in the ccm1 morphants. In addition, the silent heart morphants go on to develop the CVP dilation as seen in the ccm1 morphants (Fig. 7A–G, Supplementary Material, Movie S3). However, flow is present in 37% of the ccm1 morphants and yet these morphants still develop the same defects (Supplementary Material, Movies S4 and S5), suggesting defects in blood flow or the ability to sense blood flow might be contributing to lesion formation.
Figure 7.

Loss of CCM results in impaired flow response. (A–C) Images of zebrafish tail morphology at 48 hpf. White box indicates area of inset. (D–F) Inset of tail vasculature. (G) Quantification of endothelial CVP sprouting at 28 hpf. P-value <0.05. (H) Quantification of I-L, P-value <0.0001. (I–L) Endothelial monolayers stained with phalloidin and exposed to either static (F and G) or flow (H and I) conditions. Scale bars at 40 μm.
We therefore hypothesized that CCM1 is required for the endothelial cell to sense or respond to flow. We predicted that cells deficient in CCM1 would exhibit hallmark defects, such as an inability to align to the direction of flow (27). To test endothelial response to shear stress, we suppressed CCM1 expression in monolayers of HUVEC cells using siRNA. These monolayers were then exposed to a period of laminar shear stress and their alignment was analyzed by measuring the major angle of an ellipse fitted to each cell in relation to the direction of flow. Cells depleted of CCM1 failed to align with the direction of blood flow, consistent with a role for flow in the formation of CCM lesions (Fig. 7H–L).
DISCUSSION
The current field of CCM study has focused primarily on late-stage disease, with read-outs being in mature disease states. Such an approach is limited in scope and can overlook the stimulus that initiates lesion formation. Understanding the developmental process of CCM formation is critical in identifying the key molecular events that lead to disease onset. By taking a developmental approach, our study represents a careful analysis of the early morphological events of CCM formation. We demonstrate that retinal lesions develop in a stereotypic pattern and location. In addition, we find that Ccm1 must be lost during a particular developmental window in order for lesions to form, consistent with the observations of Boulday et al. (20). Furthermore, we find that prior to any sort of vascular dilation phenotype endothelial cells exhibit a hypersprouting behavior. Hypersprouting in conjunction with the predictable development of the lesions from veins suggests that local cues may be a contributing factor to disease onset.
Previous work demonstrating that blood flow can regulate sprouting and organization suggested a potential mechanism for the development of CCM lesions. The difference in arterial and venous flow rates provides local developmental cues to endothelial cells via the process of flow response (24). A defect in flow response could explain why lesions develop in a stereotypic pattern and are restricted to the superior and inferior veins. Our observation that endothelial cells deficient in CCM1 do not align to flow and the ability of silent heart to phenocopy the sprouting and dilation phenotype seen with ccm1 knockdown support the hypothesis that response to flow may be disrupted. In addition, in a previous work by Hogan et al. (28) it was observed in vivo that loss of CCM1 results in a round cell shape that does not elongate in the direction of flow. This observation is consistent with our finding of failed alignment in vitro. If endothelial cells cannot sense or respond to the local flow appropriately, development of the local vascular architecture could be disrupted. Further study is needed in order to determine whether the defect is in sensing flow or in response to flow and how those processes may influence the development of the local vasculature. Taken together, our data demonstrate the predictable nature of retinal CCMs and provide a new approach to the study of this disease. Furthermore, our results illustrate the importance of both the physiology and environment in the etiology of vascular disease.
MATERIALS AND METHODS
Mice
Animals were housed at the University of Utah and all experiments were approved by the University of Utah Institutional Animal Care and Use Committee. The Ccm1 floxed allele was generated by inserting LoxP sites that flank exons 4–8 (Krit1tm1Kwhi, here referred to as Ccm1flox). A germline mutant allele (Krit1tm1.1Kwhi, here referred to as Ccm1-) was generated by crossing these mice against mice expressing Cre recombinase in the germline. See Supplementary Material, Figure S1. The mT/mG (B6.129 (Cg)-Gt (ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) was obtained from Jackson Labs.
Human samples
Human tissue samples were provided by Connie Lee and Amy Akers (Angioma Alliance, Norfolk, VA, USA) and Randy Jensen (University of Utah, Salt Lake City, UT, USA) and were obtained with written informed consent. Human experiments were approved by the Institutional Review Board of the University of Utah.
Zebrafish
Zebrafish were maintained under standard husbandry conditions in the University of Utah CZAR and were approved by IACUC. The transgenic lines used were Tg (kdrl:GFP) for endothelial visualization and Tg (gata1:dsRed) for red blood cell visualization. Morpholino injections were done using standard injection techniques and the previously described morpholinos for CCM1 (22) and Silent Heart (26).
Analysis of mouse embyos
Embryos were harvested at time indicated and confocal immunofluorescence or fetal ultrasound was performed as previously described (11). Ink injections were done as previously described (29)
MRI
MRI studies were conducted as previously described (10)
Antibodies
For endothelial visualization in retina immunofluorescence Alexa Fluor Isolectin GS-IB4 (Invitrogen) was used. For actin visualization Alexa Fluor phalloidin (Invitrogen) was used.
Histology
Adult mouse brains were removed after fixation in 4% formaldehyde for at least 3 days or zinc-buffered formalin overnight. The brain was then removed and sliced into three coronal sections and was embedded into paraffin by standard protocol. Histological staining for iron and trichrome were carried out using the Dako Artisan system according to manufacturer's instructions.
Microscopy
All fluorescent still images were taken using the Nikon AR1 confocal microscope using either a ×10, ×20 or ×40 objective. For live zebrafish imaging, the Nikon Eclipse Ti widefield was used in conjunction with temperature controlled stage. Fish were mounted in 1% low-melt agarose on a glass cover slip. The cover-slips were mounted onto the stage and E3 solution was used to prevent the agarose from drying. Temperature in the imaging chamber was set to 30°C. For brightfield histological imaging, the Zeiss Axioplan 2 microscope was used with either the ×10 or ×4 objective. For the whole-brain histology image (Fig. 2O) several individual panels were taken and then photo-merged into one single image.
Live animal angiography
Prior to anesthesia, 1% Tropicamide was administered to animals to facilitate eye dilation. Animals were then anesthetized using isoflurane, injected with 100 μl of fluorescein, and positioned on the imaging platform. Images were then taken using the Heidelberg SPECTRALIS Ultra-Widefield Angiography Module with the 102° lens while animals were held in consistent orientation.
Retinal processing
Eyes were enucleated and fixed overnight in 4% Paraformaldehyde (PFA) at 4°C. Retinas were then dissected and stained as previously described (19). After staining, retinas were placed into phosphate buffered saline (PBS), cut four times to section the cup into leaflets, and then flat-mounted onto slides using Dako Faramount Aqueous mounting media. For cross-sectional analysis, eyes were embedded into OCT for frozen sectioning per standard protocol. Sections were cut at 10 μm thickness and then stained with both lectin and DAPI, then mounted with Dako Faramount Aqueous mounting media for imaging.
Fluorescence in situ hybridization
Eyes were collected at Day 5 (P5), fixed with 4% PFA 4H and immediately dissected. Retinas were fixed ON with 4% PFA at 4C and transferred to METOH the day after and stored at −80°C Retinas were rehydrated by thorough washing steps with 75, 50 and 25% METOH–PBS–0.1%Tween at RT. Then retinas were treated with Proteinase K (Invitrogen 20 mg/ml. Cat. no. 25530-049) during 20 min, fixed with 4%PFA-0.1%Glutaraldehyde (Sigma, 25% glutaraldehyde solution. Cat. no. G5882) and pre-hybridize at 65°C during 2H with Hb4 hybridization buffer. Probe-hybridization was performed ON at 65°C using Hb4D5 buffer. Hybridization mix was prepared using the digoxigenin (DIG)-probe at concentration of 1 µg/ml. The day after, retinas were thorough washing steps using 50% formamide deionized (Sigma. Cat. no. F9037)-2XSSCT, 2XSSCT and 0.2XSSCT at 65°C, cool down on PBS–0.1%Tween and blocked with PBS–0.1%Tween–8%Sheep serum (Sigma. Cat. no. S2263) during 2H at RT. After that, retinas were incubated ON at 4°C with the sheep anti-DIG peroxidase (POD) antibody (1:500; Anti-DIG-POD Fab fragments. Roche. Cat. no. 11207733910). Next day, retinas were washed during 2H at intervals of 30 min with TBS–0.1%Tween and incubated during 20–30 min with fluorescein tyramide solution (TSA) diluted (1:500) on PBS–0.1%Tween, at RT. For TSA solution synthesis check the link, http://www.xenbase.org/xenwiki/index.php/Flourescin_Tyramide_Synthesis.
Posteriorly, 0.001% H2O2 was added to the TSA solution to activate TSA reaction. After 30–45 min TSA reaction was stopped washing quickly the retinas with PBS several times. Before proceeding with Isolectin IF, retinas were washed during 2 day at 4°C in slow agitation with PBS–0.1%Tween to reduce ISH unspecific signal and post-fixed with 4%PFA.
Ultrasound studies of the Eye
Mice were anesthetized with 1.5% isoflurane inhalational anesthetic and monitored for pulse and respiration. Images were acquired from the eye, including specific studies of the retinal artery and vein with B-mode, color and pulsed-wave Doppler modes using a Vevo 2100 high frequency ultrasound machine (VisualSonics).
Cell culture and flow
Human umbilical vein endothelial cells were obtained from Lonza and grown according to the manufacturer's instructions. Human CCM1 or non-targeting siRNA was obtained from Dharmacon. EC transfection with siRNAs was carried out in growth media with 1% serum with details of the siRNA transfection protocol as previously described (30). For flow experiments, cells were plated in IBIDI 8-well chamber slides (VI0.4) and exposed to a 16 dynes/cm2 flow rate (simulating arterial shear stress) or static conditions for 24 h. Cells were then fixed with 4% PFA and actin cytoskeleton was visualized using phalloidin.
Statistical analysis
For CCM penetrance and lesion content, we performed a two-tailed t-test. For quantification of sprouting, we performed a 2-tailed t-test. For analysis of cell alignment, we performed a two-way analysis of variance test. P-values are as indicated in the figure legends.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by NIH/NHLBI (grant R01HL065648).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Brent Bisgrove for the zebrafish Tg (kdrl:GFP) line; O. Abdullah and E. Hsu and the University of Utah Small Animal Imaging Facility; Christopher Rodesch, Mike Bridge and the University of Utah Cell Imaging/Fluorescence Facility; and Sheryl Tripp and the Immunohistochemistry Research and Development Lab at ARUP Laboratories. This work was further supported by the Small Animal Ultrasound core laboratory at the University of Utah. We would also like to thank Kirk Thomas, Shannon Odelberg and Chadwick Davis for their advice, discussions and contributions to the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Otten P., Pizzolato G.P., Rilliet B., Berney J. A Study of 131 Cases of Cavernomas of the Cns, discovered on Retrospective Analysis of 24,535 Autopsies. Neurochirurgie. 1989;35:82–83. [PubMed] [Google Scholar]
- 2.Clatterbuck R., Eberhart C., Crain B., Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood–brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry. 2001;71:188–192. doi: 10.1136/jnnp.71.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson J.R., Awad I.A., L1ti'le J.R. Natural history of the cavernous angioma. J. Neurosurg. 1991;75:5. doi: 10.3171/jns.1991.75.5.0709. [DOI] [PubMed] [Google Scholar]
- 4.Vernooij M.W., Arfan Ikram M., Tanghe H.L., Vincent A.J.P.E., Hofman A., Krestin G.P., Niessen W.J., Breteler M.M.B., van der Lugt A. Incidental findings on brain MRI in the general population. N. Engl. J. Med. 2007;357:7. doi: 10.1056/NEJMoa070972. [DOI] [PubMed] [Google Scholar]
- 5.Plummer N.W., Zawistowski J.S., Marchuk D.A. Genetics of cerebral cavernous malformations. Curr. Neurol. Neurosci. Rep. 2005;5:5. doi: 10.1007/s11910-005-0063-7. [DOI] [PubMed] [Google Scholar]
- 6.Lampugnani M.G., Orsenigo F., Rudini N., Maddaluno L., Boulday G., Chapon F., Dejana E. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J. Cell Sci. 2010;123:1073–1080. doi: 10.1242/jcs.059329. [DOI] [PubMed] [Google Scholar]
- 7.Glading A., Han J., Stockton R.A., Ginsberg M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007;179:247–254. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamalice L., Le Boeuf F., Huot J. Endothelial cell migration during angiogenesis. Circ. Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 9.Whitehead K.J., Plummer N.W., Adams J.A., Marchuk D.A., Li D.Y. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–1448. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 10.Chan A.C., Drakos S.G., Ruiz O.E., Smith A.C.H., Gibson C.C., Ling J., Passi S.F., Stratman A.N., Sacharidou A., Patricia Revelo M., et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Invest. 2011;122:5. doi: 10.1172/JCI44393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitehead K.J., Chan A.C., Navankasattusas S., Koh W., London N.R., Ling J., Mayo A.H., Drakos S.G., Jones C.A., Zhu W., et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009;15:177–184. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kisanuki Y.Y., Hammer R.E., Miyazaki J., Williams S.C., Richardson J.A., Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 13.Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K., Fruttiger M. Efficient, inducible Cre-Recombinase activation in vascular endothelium. Genesis. 2007;46:6. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 14.Liu H., Rigamonti D., Badr A., Zhang J. Ccm1 assures microvascular integrity during angiogenesis. Transl. Stroke Res. 2010;1:146–153. doi: 10.1007/s12975-010-0010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stahl A., Connor K.M., Sapieha P., Chen J., Dennison R.J., Krah N.M., Seaward M.R., Willett K.L., Aderman C.M., Guerin K.I., et al. The mouse retina as an angiogenesis model. Invest. Ophthal. Vis. Sci. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labauge P., Denier C., Bergametti F., Tournier-Lasserve E. Genetics of cavernous angiomas. Lancet Neurol. 2007;6:237–244. doi: 10.1016/S1474-4422(07)70053-4. [DOI] [PubMed] [Google Scholar]
- 17.Dorrell M.I., Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog. Retin Eye Res. 2006;25:277–295. doi: 10.1016/j.preteyeres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Bentley K., Mariggi G., Gerhardt H., Bates P.A. Tipping the balance: robustness of tip cell selection, migration and fusion in angiogenesis. PLoS Comput. Biol. 2009;5:e1000549. doi: 10.1371/journal.pcbi.1000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell. Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulday G., Rudini N., Maddaluno L., Blecon A., Arnould M., Gaudric A., Chapon F., Adams R.H., Dejana E., Tournier-Lasserve E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J. Exp. Med. 2011;208:1835–1847. doi: 10.1084/jem.20110571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gore A.V., Monzo K., Cha Y.R., Pan W., Weinstein B.M. Vascular development in the Zebrafish. Cold Spring Harb.Perspect. Med. 2012;2 doi: 10.1101/cshperspect.a006684. a006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mably J.D., Chuang L.P., Serluca F.C., Mohideen M.A., Chen J.N., Fishman M.C. Santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133:3139–3146. doi: 10.1242/dev.02469. [DOI] [PubMed] [Google Scholar]
- 23.Herbert S.P., Huisken J., Kim T.N., Feldman M.E., Houseman B.T., Wang R.A., Shokat K.M., Stainier D.Y. Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science. 2009;326:294–298. doi: 10.1126/science.1178577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.le Noble F., Fleury V., Pries A., Corvol P., Eichmann A., Reneman R.S. Control of arterial branching morphogenesis in embryogenesis: go with the flow. Cardiovasc. Res. 2005;65:619–628. doi: 10.1016/j.cardiores.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Munn J.W.S., Munn L.L. Fluid forces control endothelial sprouting. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sehnert A.J., Huq A., Weinstein B.M., Walker C., Fishman M., Stainier D.Y. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 27.Li Y.S., Haga J.H., Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 28.Hogan B.M., Bussmann J., Wolburg H., Schulte-Merker S. ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum. Mol. Genet. 2008;17:2424–2432. doi: 10.1093/hmg/ddn142. [DOI] [PubMed] [Google Scholar]
- 29.Urness L.D., Sorensen L.K., Li D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000;26:3. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 30.Saunders W.B., Bayless K.J., Davis G.E. MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J. Cell Sci. 2005;118:2325–2340. doi: 10.1242/jcs.02360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}