

Abstract
Huntington's disease (HD) is a neurodegenerative disorder caused by a CAG trinucleotide repeat expansion in the huntingtin (HTT) gene. Disease pathogenesis derives, at least in part, from the long polyglutamine tract encoded by mutant HTT. Therefore, considerable effort has been dedicated to the development of therapeutic strategies that significantly reduce the expression of the mutant HTT protein. Antisense oligonucleotides (ASOs) targeted to the CAG repeat region of HTT transcripts have been of particular interest due to their potential capacity to discriminate between normal and mutant HTT transcripts. Here, we focus on phosphorodiamidate morpholino oligomers (PMOs), ASOs that are especially stable, highly soluble and non-toxic. We designed three PMOs to selectively target expanded CAG repeat tracts (CTG22, CTG25 and CTG28), and two PMOs to selectively target sequences flanking the HTT CAG repeat (HTTex1a and HTTex1b). In HD patient–derived fibroblasts with expanded alleles containing 44, 77 or 109 CAG repeats, HTTex1a and HTTex1b were effective in suppressing the expression of mutant and non-mutant transcripts. CTGn PMOs also suppressed HTT expression, with the extent of suppression and the specificity for mutant transcripts dependent on the length of the targeted CAG repeat and on the CTG repeat length and concentration of the PMO. PMO CTG25 reduced HTT-induced cytotoxicity in vitro and suppressed mutant HTT expression in vivo in the N171-82Q transgenic mouse model. Finally, CTG28 reduced mutant HTT expression and improved the phenotype of HdhQ7/Q150 knock-in HD mice. These data demonstrate the potential of PMOs as an approach to suppressing the expression of mutant HTT.
INTRODUCTION
The expansion of CAG trinucleotide repeats leads to at least nine autosomal dominant neurodegenerative diseases (1,2). Among them, Huntington's disease (HD), characterized by progressive motor, cognitive and psychiatric abnormalities leading to death 15–20 years after clinical onset, is the most common (3–5). HD is caused by an expansion mutation of the CAG repeat in the first exon of the HTT gene (6); disease inevitably results in individuals with 40 or more triplets and may occur with as few as 36 triplets (and perhaps fewer) (7,8). The CAG repeat is translated into a polyglutamine tract (polyQ) within the huntingtin protein (HTT). Most investigators have concluded that toxicity of the expanded polyQ is the primary pathogenic mechanism in HD. Recently, it was shown that CAG repeats, including that at the HD locus, can be translated into other homopolymeric tracts, including polyalanine and polyserine, through repeat-associated non-ATG (RAN) translation (9). In addition, mutant HTT RNA itself may be toxic (10–12), suggesting that both RNA and protein gain-of-function contribute to the disease pathogenesis.
If both expanded HTT protein and transcript are neurotoxic, then the most direct therapeutic approach, aside from altering genomic DNA, is to use knockdown strategies to prevent protein expression and degrade expanded transcripts or block their toxicity. While suppression, ideally, is specific for the products of the expanded allele, the current consensus is that bi-allelic approaches may be successful as long as the level of normal HTT remains above the 30% threshold required for normal cell function (13–15). Multiple strategies are under investigation, such as zinc finger peptides targeting double-stranded DNA to prevent transcription, therefore reducing both protein and RNA expression (16), or peptides that bind to expanded polyglutamine to block its toxic function (17). However, most mutant HTT knockdown strategies are based on small interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs). Published evidence demonstrates the potential of these approaches to significantly decrease the expression of expanded HTT protein without completely blocking the expression of normal HTT, or at least to preserve a minimally needed amount of normal HTT (13,14,18–22). One strategy takes advantage of the heterozygosity of single-nucleotide polymorphisms (SNPs) in HTT; the mutant HTT gene contains specific SNPs in 75–85% of HD patients, and targeting these SNPs with one or a pool of siRNAs or ASOs could provide allele-specific knockdown of HTT expression (23–27). A second approach to allelic specificity employs siRNAs or ASOs that target the expanded CAG repeat. Indeed, CAG repeat–targeting siRNAs are effective at reducing the expression of mutant HTT mRNA with at least partial allelic selectivity in vitro (28–34). However, implementation of siRNA-based silencing in vivo faces several major obstacles, including the challenge of efficient delivery into the CNS, the relatively low stability of siRNAs, potential off-target effects and the risk of immune activation (35). Compared with siRNA, ASOs have a major advantage in flexibility (as modifications can enhance their stability), RNA affinity, cellular uptake and biodistribution. ASOs such as gapmers [chimeric ASOs consisting of a DNA sequence with flanking locked nucleic acids (LNA) or 2′-O-methoxyethyl (MOE) nucleic acids] can be used for RNase H-dependent degradation of targeted transcripts, an approach used to degrade CUG repeats in mouse models (36,37). However, a number of CAG repeat loci, including the HD locus, contain antisense transcripts spanning the repeat region (38–40), and it is possible that targeted degradation of sense transcripts may trigger an up-regulation of these CUG repeat-containing antisense transcripts with potential neurotoxic effects (41–44). An alternative is to use ASOs that sterically block RNA translation and, potentially, RNA-mediated neurotoxicity without leading to transcript degradation. Such ASOs include peptide nucleic acids (PNAs), LNAs, chemically modified single-stranded RNAs (ssRNAs), and phosphorodiamidate morpholino oligonucleotides (PMOs). Indeed, selective inhibition of the mutant HTT allele by PNA and LNA ASOs targeting the CAG repeat of the HTT transcript has been demonstrated (30,31,45–47). Chemically modified ssRNA has also been successfully applied to specifically silence expanded HTT expression both in vitro and in vivo (48).
Here, we examine the potential of PMOs in HD therapeutics. PMOs have advantages over other ASOs in therapeutic applications: the absence of an electrical charge, the lack of dependence on the activity of RNase H or other catalytic proteins (49–51), water solubility, stability, lack of non-specific toxicity even at high concentrations and the capacity for extensive modifications. PMOs have shown great promise when injected peripherally, including preventing the sequestration of muscleblind-like protein 1 (MBNL1) in myotonic dystrophy 1 (DM1) (52) and inducing targeted exon skipping in Duchenne muscular dystrophy (DMD) (53–58). PMOs also can alter brain processes, such as reducing the anxiolytic effect of estrogen following direct delivery into the rat dorsal raphe nucleus (59), increasing the survival of postnatal spinal muscular atrophy mice (60–62), and modulating feeding behavior in rats (63).
In this study, we designed multiple PMOs targeted to the HTT CAG repeat region and to the region flanking the HTT repeat. The effectiveness and allelic selectivity of the PMOs were examined in HD patient–derived fibroblast lines with repeat expansions of 44, 77 or 109 CAG triplets and in two HD mouse models, N171-82Q transgenic and HdhQ7/Q150 knock-in mice. The effectiveness in ameliorating neurotoxicity was examined in a neuroblastoma cell line. Our data demonstrate that PMOs can significantly decrease HTT protein expression without decreasing transcript level, with target selectivity and knock-down efficacy determined by PMO sequence and concentration, and by target CAG repeat length.
RESULTS
The specificity and effectiveness of a PMO depend on its sequence and concentration, as well as on the length of the targeted CAG repeat
Based on the use of PMOs to ameliorate the toxicity of the CUG repeat expansion within the DMPK transcript associated with DM1 (52), we synthesized three CAG repeat–targeting PMOs, CTG22, CTG25 and CTG28, respectively (Fig. 1). The optimal length for a PMO is 25 bases and the longest PMO that could be synthesized was 30 bases. The high transfection efficiency of ∼90% was confirmed by delivering fluorescein isothyocyanate (FITC)-labeled standard control (Ctrl) PMO into multiple cell lines (Supplementary Material, Fig. S1) and visualizing FITC distribution through an EGFP filter. We evaluated the PMOs' specificity and effectiveness in reducing mutant HTT protein levels in three HD patient–derived fibroblast cell lines, HD 18/44, HD 20/77 and HD 19/109 (Supplementary Material, Table S1; numbers indicate CAG repeat sizes within normal and mutant HTT alleles). First, 1–20 µm of CTG25 PMO was applied to all three HD patient–derived fibroblast cell lines; 20 µm of a standard control PMO (Ctrl) served as a baseline. 48 h after the treatment, HTT protein levels were assessed by western blot. An antibody against full-length HTT (MAB2166) was used to assess the total level of endogenous HTT in line HD 18/44, as allelic separation was not possible on the gel. The same antibody successfully resolved the normal allele (lower band) from the mutant (upper band) in cell lines HD 20/77 and HD 19/109. In addition, the expression of mutant HTT was examined using 1C2 antibody (64). As shown in Figure 2A and C, CTG25 reduced total HTT expression in all three HD fibroblast lines. Blotting with 1C2 antibody demonstrated suppression of mutant HTT (Fig. 2B and C). In cell lines HD 20/77 and HD 19/109, a non-significant trend for allelic selectivity of CTG25 was observed, and 10 and 20 µm reduced total HTT levels in the HD 18/44 line to ∼40%, a potentially therapeutically significant level. We next hypothesized that increasing the CTG repeat length of the PMO may increase allelic selectivity due to increased cumulative binding of PMOs to the expanded CAG repeat (45). We observed that in cell line HD 19/109, treatment with 10 µm CTG28 reduced the mutant HTT level to ∼65% that of control PMO-treated, whereas the level of normal HTT levels remained at 90% that of control PMO-treated. Increasing the concentration of CTG28 to 20 µm further decreased mutant HTT to ∼50% while normal HTT expression remained at 75% that of control PMO-treated (Fig. 3A and C). Specific staining for mutant HTT confirmed this finding (Fig. 3B and C). However, this PMO did not significantly reduce mutant HTT protein levels in HD 18/44 or HD 20/77 (Fig. 3), indicating that increasing the length of CTG PMOs increases their selectivity for long repeats.
Figure 1.

Schematic representation of the PMO sequences and their target regions within human HTT mRNA.
Figure 2.

PMO CTG25 inhibits HTT expression. Cell lines HD 18/44, HD 20/77 and HD 19/109 were each treated for 48 h with 1, 5, 10 and 20 µm CTG25 or 20 µm standard control PMO (Ctrl). (A) Analysis of total HTT levels after CTG25 treatment. Antibody MAB2166 was used to discriminate between normal HTT (lower band) and mutant HTT (upper band) in cell lines HD 20/77 and HD 19/109. Since it is not possible to resolve normal and mutant HTT in the HD 18/44 cell line, total protein levels were analyzed. HD 18/44, one-way ANOVA, F = 2.960, P = 0.0747, n = 3; HD 20/77, two-way ANOVA, F(Allele) = 0.51, P(Allele) = 0.4821, F(PMO) = 63.07, P(PMO) < 0.0001, n = 3; HD 19/109, two-way ANOVA, F(Allele) = 1.35, P(Allele) = 0.2571, F(PMO) = 4.38, P(PMO) = 0.0057, n = 3; post hoc test *P < 0.05, **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group. (B) In addition, 1C2 immunoblotting was performed to assess mutant HTT level only. All HD cell lines, one-way ANOVA, n = 3. HD 18/44, F = 5.237, P = 0.0154; HD 20/77, F = 21.56, P < 0.0001; HD 19/109, F = 5.210, P = 0.0039; post hoc test *P < 0.05, **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group. (C) Representative western blot data. HTT antibodies are shown in parentheses. All experiments were performed three times.
Figure 3.

PMO CTG28 preferentially reduces mutant HTT expression. Cell lines HD 18/44, HD 20/77 and HD 19/109 were each treated with 1, 5, 10 and 20 µm CTG28 or 20 µm standard control PMO (Ctrl). (A) Analysis of the normal versus expanded HTT level. HD 18/44, one-way ANOVA, F = 1.015, P = 0.4449, n = 3; HD 20/77, two-way ANOVA, F(Allele) = 1.11, P(Allele) = 0.3053, F(PMO) = 3.27, P(PMO) = 0.0323, n = 3; HD 19/109, two-way ANOVA, F(Allele) = 9.56, P(Allele) = 0.0058, F(PMO) = 14.35, P(PMO) < 0.0001, n = 3; post hoc test **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group; #P < 0.05 versus normal HTT. (B) The levels of mutant HTT following CTG28 treatment. All HD cell lines, one-way ANOVA, n = 3. HD 18/44, F = 2.256, P = 0.1356; HD 20/77, F = 0.7091, P = 0.6039; HD 19/109, F = 9.021, P = 0.0024; post hoc test **P < 0.01 versus Ctrl PMO–treated group. (C) Representative western blot data. HTT antibodies are shown in parentheses. All experiments were performed three times.
During the course of this study, it was reported that a PMO consisting of seven CTG triplets selectively decreased the expression of mutant HTT in HD patient–derived fibroblasts (30). We therefore examined the effect of a similar PMO, CTG22 (seven CTG triplets and an additional C) in more detail. We found that, like with CTG25, total (Fig. 4A and C) and mutant (Fig. 4B and C) levels of HTT in all three HD cell lines were significantly reduced by CTG22 treatment; however, no significant discrimination between normal and mutant HTT was observed in any of the lines. These results demonstrate that as CTG repeat length decreases, suppression of total HTT expression increases but allelic selectivity decreases.
Figure 4.

Concentration- and targeted CAG repeat length-dependent reduction of HTT expression following treatment with PMO CTG22. Cell lines HD 18/44, HD 20/77 and HD 19/109 were each treated with 1, 5, 10 and 20 µm CTG22 or 20 µm standard control PMO (Ctrl) for 48 h. (A) Analysis of the normal versus expanded HTT level after CTG22 treatment. HD 18/44, one-way ANOVA, F = 4.380, P = 0.0265, n = 3; HD 20/77, two-way ANOVA, F(Allele) = 0.03, P(Allele) = 0.8677, F(PMO) = 5.51, P(PMO) = 0.0037, n = 3; HD 19/109, two-way ANOVA, F(Allele) = 0.83, P(Allele) = 0.3740, F(PMO) = 20.04, P(PMO) < 0.0001, n = 3; post hoc test *P < 0.05, **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group. (B) Mutant HTT levels after CTG22 treatment. All HD cell lines, one-way ANOVA, n = 3. HD 18/44, F = 5.637, P = 0.0122; HD 20/77, F = 8.637, P = 0.0028; HD 19/109, F = 6.499, P = 0.0076; post hoc test *P < 0.05, **P < 0.01 versus Ctrl PMO–treated group. (C) Representative western blot data. HTT antibodies are shown in parentheses. All experiments were performed three times.
Taken together, we conclude that the allelic specificity and effectiveness of a repeat-targeting PMO depends on the length of the PMO repeat, the concentration of the PMO, and the length of the repeat in the targeted gene.
The off-target effect of CAG repeat–targeting PMOs depends on the PMO sequence and concentration
Off-target effect is an important criterion in evaluating the therapeutic potential of repeat-targeting ASO approaches. Besides HTT, there are at least 40 human genes with seven or more consecutive CAG triplets (65). To assess the off-target effect of repeat-targeting PMOs in this study, we chose five control genes that normally contain long CAG repeats: ataxin-2 (ATXN2), ataxin-3 (ATXN3), TATA box-binding protein (TBP), androgen receptor (AR) and retinoic acid-induced 1 (RAI1). The repeat sizes of ATXN2, ATXN3 and TBP in the three HD cell lines examined are shown in Supplementary Material, Table S1. Treatment with CTG22 significantly reduced the levels of endogenous ATXN3 in both HD 20/77 and HD 19/109 fibroblasts (Fig. 5B). Reduction of endogenous ATXN2 by PMO CTG22 in cell line HD 20/77 was not statistically significant (Fig. 5A). A high concentration of PMO CTG25 also inhibited endogenous ATXN3 expression to ∼40% that of control PMO–treated cells from line HD 19/109 (Fig. 5B). PMO CTG28 had no inhibitory effect on endogenous ATXN2 and ATXN3 in any of the HD cell lines (Fig. 5A and B). None of the three CAG repeat–targeting PMOs had an inhibitory effect on the expression of TBP in nuclear protein extracts of cell line HD 19/109 (Fig. 5C). Since AR and RAI1 are expressed at low levels in HD patient–derived fibroblasts, we tested the effect of these CAG repeat–targeting PMOs on the endogenous levels of both proteins in the SH-SY5Y neuroblastoma cell line, and we observed no significant reduction (Fig. 5D). Representative western blot images for Figure 5 are shown in Supplementary Material, Figure S2. The data suggest that the off-target effect of repeat-targeting PMOs on endogenous genes is dependent on the sequence of the PMO and possibly on the sequence flanking the repeat and the subsequent tertiary structure of each repeat locus, as the length of the repeat in the endogenous control genes examined is not directly correlated to the off-target effect of the PMO.
Figure 5.

PMO off-target effects. Cell lines HD 18/44, HD 20/77, HD 19/109 and SH-SY5Y neuroblastoma cell line were each treated with PMOs CTG22, CTG25 and CTG28, and the expression of ATXN2 (A), ATXN3 (B), TBP (C), AR and RAI1 (D) were assessed 48 h after treatment. (A) ATXN2 expression was not significantly decreased following all PMO treatments. (B) The concentration range of 5–20 µm CTG22 significantly reduced the levels of ATXN3 in cell lines HD 20/77 and HD 19/109. CTG25 significantly inhibited ATXN3 expression in line HD 19/109. PMO CTG28 did not affect ATXN3 expression in any of the HD lines tested. (C) A high concentration (20 µm) of repeat-targeting PMOs had no significant effect on the level of TBP in line HD 19/109. (D) AR and RAI expression in the SH-SY5Y cell line were not decreased by a high concentration (20 µm) of repeat-targeting PMOs. All treatments, one-way ANOVA, n = 3. ATXN3 in HD 20/77 with CTG22, F = 3.505, P = 0.0490; ATXN3 in HD 19/109 with CTG22, F = 4.050, P = 0.0331; ATXN3 in HD 19/109 with CTG25, F = 29.15, P < 0.0001; post hoc test *P < 0.05, **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group. All experiments were performed three times.
Non-CAG repeat–targeting PMOs reduce HTT levels with high target selectivity
In addition to targeting the CAG repeat region in HTT RNA, targeting other regions may also be a feasible therapeutic approach if levels of normal HTT remain sufficient to support normal cell function (13,18). We therefore designed two non-CAG repeat–targeting PMOs, HTTex1a and HTTex1b (Fig. 1), and examined their effectiveness in suppressing HTT expression in cell line HD 19/109. HTTex1a binds to the first 25 bases downstream of the start codon in HTT RNA, and HTTex1b binds to the region immediately upstream of the CAG repeat. As expected, HTTex1a and HTTex1b modestly decreased both normal and mutant HTT levels without allelic selectivity (Fig. 6A, B and D). No effect on the expression of endogenous ATXN2 or ATXN3 was detected with either of the PMOs (Fig. 6C and D), as expected given the HTT specificity of these PMOs. The relatively small effect of these PMOs on HTT expression compared with repeat-targeting PMOs is presumably due to only one PMO being able to bind to each transcript. Nonetheless, the comparative absence of off-target effects suggests the potential value of these PMOs.
Figure 6.

Non-CAG repeat-targeting PMOs inhibit HTT expression in a non–allele-specific manner and without off-target effect. Cell line HD 19/109 was treated with non–repeat-targeting PMOs HTTex1a and HTTex1b. (A) Analysis of normal versus mutant HTT level. PMO HTTex1a, two-way ANOVA, F(Allele) = 1.35, P(Allele) = 0.2593, F(PMO) = 10.39, P(PMO) = 0.0001, n = 3; PMO HTTex1b, two-way ANOVA, F(Allele) = 0.03, P(Allele) = 0.8706, F(PMO) = 12.34, P(PMO) < 0.0001, n = 3; post hoc test *P < 0.05, **P < 0.01, ***P < 0.001 versus Ctrl PMO–treated group. (B) Mutant HTT level after HTTex1a and HTTex1b treatment. Both PMOs, one-way ANOVA, n = 3. PMO HTTex1a, F = 2.573, P = 0.1028; PMO HTTex1b, F = 8.018, P = 0.0037; post hoc test **P < 0.01 versus Ctrl PMO–treated group. (C) As expected, PMOs HTTex1a and HTTex1b did not have an effect on the expression of endogenous ATXN2 and ATXN3 control proteins. Both PMOs, one-way ANOVA, n = 3. (D) Representative western blot data. HTT antibodies are shown in parentheses. All experiments were performed three times.
RNA levels of HTT, HTTAS_v1 and control genes are not affected by PMOs
Since PMOs are designed to sterically block RNA translation through complementary binding with target RNA, PMO activity should not lead to the degradation of targeted transcripts (49). To confirm this, we analyzed HTT RNA levels, as well as the RNA levels of four endogenous control genes including ataxin-1 (ATXN1), ATXN3, TBP and atrophin-1 (ATN1) after treatment with each of the five PMOs. As expected, no significant effect on the levels of transcripts was observed (Fig. 7A). Since CTG22 was the most effective at reducing overall HTT levels (Fig. 4), we used this PMO to determine whether PMO-HTT transcript hybridization affects levels of the antisense transcript at the HD locus, HTTAS_v1 (38). Our results indicate that neither the levels of sense HTT nor HTTAS_v1 are significantly changed following introduction of the PMO (Fig. 7B). In addition, we observed that modulating the levels of the endoribonuclease dicer (DICER1) in HD cell line 19/109 transfected with PMO CTG22 has no significant effect on the levels of HTT (Supplementary Material, Fig. S3). Taken together, our experiments further confirm that PMO suppression of target expression does not involve degradation of either the targeted transcript or endogenous control genes.
Figure 7.

PMOs exhibit no significant effect on the RNA levels of HTT, HTTAS_v1 and endogenous control genes. (A) Cell line HD 19/109 was treated with 20 µm Ctrl, CTG22, CTG25, CTG28, HTTex1a and HTTex1b PMO for 48 h, and the RNA levels of HTT and endogenous control genes (ATXN1, ATXN3, TBP and ATN1) were examined using TaqMan real-time PCR. All genes, one-way ANOVA, n = 3. (B) Cell line HD 18/44 was treated with 20 µm either Ctrl and or CTG22 PMO for 48 h, and the RNA level of HTTAS_v1 was examined using TaqMan real-time PCR. Student's t-test, n = 3. Experiment was performed three times in triplicate.
PMOs show minimal cytotoxicity
Any ASO-based strategy to lower HTT expression must be evaluated for non-specific cell toxicity. Unlike other ASOs, PMOs typically do not have cytotoxic effects. We confirmed that this general rule extended to our PMOs, as we were unable to detect toxicity in HD cell lines treated with a high concentration (20 µm) of each of the PMOs (data not shown). We next determined the toxicity of PMOs in normal HEK293 cells, as these cells tend to be more sensitive to toxins than fibroblasts. As shown in Figure 8A, most of the PMOs were not toxic to HEK293 cells even at the highest concentrations, with the exception of CTG22, which was significantly toxic even at 5 µm. Since CTG22 induced the most off-target effect in HD cell lines (Fig. 5), we speculate that this may reflect the capacity of PMOs with short repeats to target a number of endogenous genes containing CAG repeats. The data also demonstrate that off-target effects of PMOs, and hence potentially other ASOs, differ by cell type, and this phenomenon should be taken into consideration when evaluating ASOs as potential therapeutics.
Figure 8.

PMOs show minimal cell toxicity and can protect cells against mutant HTT-associated neurotoxicity. (A) HEK293 cells were treated with each of the PMOs at the indicated concentrations. With the exception of CTG22, which showed high concentration-dependent toxicity in HEK293 cells, PMOs show no or minimal cytotoxicity even at the highest levels. All PMO concentrations, one-way ANOVA, n = 4; post hoc test *P < 0.05, ***P < 0.001 versus Ctrl PMO–treated group. (B) Mouse neuronal cell lines STHdh Q7/Q7 and STHdh Q111/Q111 were treated with the indicated concentrations of L-glutamate (L-Glu) for 72 h and a caspase 3/7 assay was used to identify the best l-Glu concentration to induce mutant HTT-related cytotoxicity for the subsequent studies. Two-way ANOVA, F(polyQ) = 112.50, P(polyQ) < 0.0001, F(Concentration) = 97.06, P(Concentration) < 0.0001, n = 4; post hoc test **P < 0.01, ***P < 0.001 versus STHdh Q7/Q7. (C) 48-h pre-treatment of PMO CTG25 protected STHdh Q111/Q111 cells from HTT neurotoxicity. Two-way ANOVA, F(polyQ) = 103.35, P(polyQ) < 0.0001, F(Treatment) = 11.45, P(Treatment) < 0.0001, n = 4; post hoc test ***P < 0.001, NS = no significance versus STHdh Q7/Q7. All experiments were performed three times in quadruplicate with similar results.
PMOs protect cells from mutant HTT-induced toxicity
Since CTG25 suppresses mutant HTT with minimal toxicity, we examined whether this PMO can protect neurons against mutant HTT-related toxicity. We used the STHdh cell model of HD, in which 5 mm sodium l-glutamate induces cytotoxicity in cell line STHdh Q111/Q111 expressing mutant HTT, but not in the control cell line STHdh Q7/Q7 expressing normal HTT (66). Using a modified protocol, we confirmed that incubation of 2 mm l-glutamate for 72 h was toxic to STHdh Q111/Q111, but not to STHdh Q7/Q7 cells (Fig. 8B). We then observed that pre-treatment of STHdh Q111/Q111 cells for 48 h with CTG25 protected the cells from glutamate-mediated mutant HTT-related neurotoxicity (Fig. 8C). This result indicates that PMO CTG25 can both suppress mutant HTT expression in patient-derived fibroblasts and reduce HTT neurotoxicity in a cell model of HD.
A PMO's ability to suppress HTT in an allele-selective manner may be cell type–specific
We next asked whether PMOs can suppress HTT expression in vivo. Since PMOs are transported into cells through endocytosis, and following release from the endosomes, freely diffuse into the cytoplasm and nucleus (67), we first examined to what extent PMOs can diffuse into the cytoplasm and the nucleus in neurons. We transfected a FITC-labeled control (Ctrl) PMO into mouse primary cortical neurons. Interestingly, we detected a much higher level of PMO release from neuronal endosomes than from fibroblast endosomes (Fig. 9). This finding confirms the cell type–specific effect of PMOs and provides a potential mechanistic explanation for cell type specificity.
Figure 9.
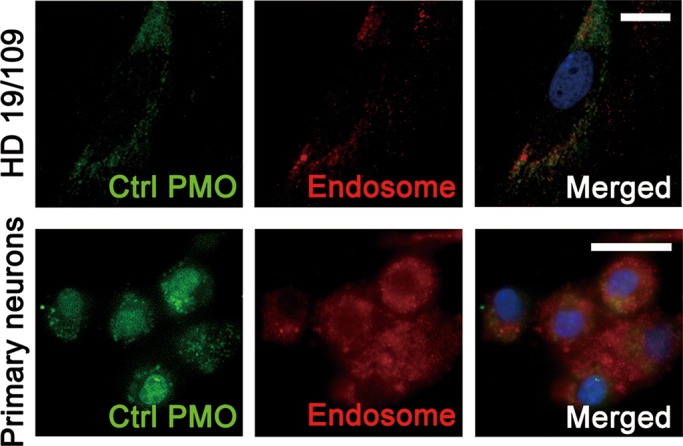
PMO intracellular diffusion is cell type–specific. HD fibroblast cell line 19/109 and wild-type mouse primary cortical neurons were treated with 20 µm fluorescein-labeled standard control PMO (Ctrl) for 48 h. In the HD fibroblasts, the majority of PMO remains within endosomes. PMO transfected into cultured primary cortical neurons is released from endosomes and diffuses in the cytoplasm and nucleus. Green, fluorescein-labeled Ctrl PMO; Red, Rab7 as an endosome marker; Blue, 4′,6-diamidino-2-phenylindole (DAPI) as a nucleus marker. Scale bar, 20 µm.
CTG25 and CTG28 inhibit mutant HTT expression in vivo
Since CTG25 reduced mutant HTT expression and rescued mutant HTT toxicity in vitro, we next examined the effect of CTG25 on HTT expression in vivo. We selected the N171-82Q transgenic HD mouse model for this experiment (68), as the repeat size in the model resembles that in HD 20/77 fibroblasts, in which PMO CTG25 was the most effective (Fig. 2A). A single low dose of (100 µg) standard control (Ctrl) or CTG25 PMO was injected into the right lateral ventricle of 6-week-old N171-82Q mice. The expression of the mutant HTT transgenic fragment in the frontal cortex was assessed 2 weeks post-injection. CTG25 was effective in reducing N171-82Q transgenic protein expression in the frontal cortex (Fig. 10A) without a significant effect on the levels of endogenous normal HTT and control proteins containing polyglutamine repeats (Fig. 10B). The decrease of the N171-82Q protein level in both the ipsilateral and the contralateral cortex suggests an efficient distribution of the PMO in vivo.
Figure 10.

CTG25 and CTG28 preferentially inhibit mutant HTT expression in vivo. Intracerebroventricular injection was used to deliver 100 µg of standard control PMO (Ctrl) or CTG25 PMO into the right lateral ventricle of six-week-old N171-82Q transgenic HD mice. Two weeks following the injection, the mice were sacrificed and 1C2 immunoblotting was used to assess the levels of HTT N171-82Q protein (A). CTG25 PMO successfully reduced HTT N171-82Q expression to 35–40% that of the Ctrl-injected without any significant effect on the levels of endogenous HTT, as well as on control proteins ATXN2, ATXN3, AR and TBP (B). Student's t-test, n = 3; *P < 0.05, ***P < 0.001 versus Ctrl PMO–injected group. The same ICV injections were also used to deliver Ctrl PMO or CTG28 PMO to the right lateral ventricle of 6-month-old HdhQ7/Q150 knock-in HD mice. Totally, three 100-µg ICV injections were administered bimonthly. Two months following the last injection, HdhQ7/Q150 knock-in HD mice were first assessed with the tail suspension test and then sacrificed to examine total HTT levels via MAB2166 immunoblotting. (C) CTG28 preferentially inhibited mutant HTT (upper mouse HTT band) to 15–45% that of Ctrl-injected in multiple brain regions (cortex, striatum and cerebellum), with minimal or no effect on normal HTT (lower mouse HTT band). Cortex, two-way ANOVA, F(Allele) = 180.9, P(Allele) < 0.001, F(PMO) = 115.9, P(PMO) < 0.001, n = 4; striatum, two-way ANOVA, F(Allele) = 190.6, P(Allele) < 0.001, F(PMO) = 7.84, P(PMO) = 0.0161, n = 4; cerebellum, two-way ANOVA, F(Allele) = 155.2, P(Allele) < 0.001, F(PMO) = 27.41, P(PMO) < 0.0001, n = 4; post hoc test *P < 0.05, ***P < 0.001 versus Ctrl PMO–injected group; #P < 0.05 versus normal HTT. (D) CTG28 had no effect on the expression of control proteins ATXN2, ATXN3 and TBP. Student's t-test, n = 4. (E) CTG28 injections decreased immobility time and increased latency to immobility and escape attempts of HdhQ7/Q150 knock-in HD mice in the tail suspension test. Two-way repeated-measure ANOVA; post hoc test *P < 0.05 versus Ctrl PMO–treated group; n = 4.
To further investigate the long-term efficacy and safety of PMOs, we also tested the effect of a repeat-targeting PMO in the HdhQ7/Q150 knock-in HD mouse model, which has both normal and mutant HTT expression at endogenous levels (69–71). We chose to use PMO CTG28 in this model based on its strong selectivity for expanded alleles in HD fibroblasts harboring 109 CAG repeats. Three successive, low-dose (100 µg) treatments of either Ctrl or CTG28 PMO were injected into the right lateral ventricle of 6-month-old HdhQ7/Q150 knock-in mice at 6, 8 and 10 months of age. At the age of 12 months, mice were assessed in the tail suspension test, which has been used to measure depressive-like behavior of YAC128 HD mice (72), and the expression of HTT in various brain regions associated with the disease pathology was also assessed. No loss of body weight was observed in mice with CTG28 injections, suggesting that PMO CTG28 does not induce systemic deleterious effect (Supplementary Material, Fig. S4). CTG28 was effective in inhibiting the expression of mutant HTT (Fig. 10C, upper mouse HTT band) in the ipsilateral frontal cortex, ipsilateral striatum and cerebellum without affecting the levels of normal HTT (Fig. 10C, lower mouse HTT band) or control proteins containing polyglutamine repeats (Fig. 10D). These data suggest that CTG28 exhibits efficient distribution in the brain, good efficacy in reducing mutant HTT and little off-target effect in vivo. Representative western blot images for the data quantified in Figure 10 are shown in Supplementary Material, Figure S2. Promisingly, preliminary evidence also suggests that CTG28 improves the behavioral phenotype of HdhQ7/Q150 HD mice (Fig. 10E). Mice injected with CTG28 showed decreased immobility, increased latency to immobility and increased escape attempts compared with mice with Ctrl injections in the tail suspension test, an assay generally interpreted as a test of depressive-like phenotype (72).
DISCUSSION
Our results demonstrate that PMOs can block the expression of mutant HTT, with the effectiveness, allelic specificity and off-target effects of this activity a function of PMO sequence, target repeat length and PMO concentration. PMO CTG22, with the fewest CTG triplets, was the most effective of the CTGn PMOs in inhibiting HTT expression; however, this PMO did not show allelic selectivity and triggered significant off-target effects. In contrast, the longest PMO, CTG28, showed the lowest effectiveness yet the highest allelic selectivity, primarily evident in the HD fibroblast line with 109 CAG repeats. Our first important finding is that PMO CTG25, which shows marginal allelic selectivity, is able to reduce the total level of HTT to ∼40–50% that of untreated cells in HD fibroblasts with an expanded allele containing 44 CAG triplets, a repeat expansion length in the range typical for HD patients. The effects on the normal and mutant alleles were similar, as demonstrated by the 1C2 antibody, which is specific for expanded polyglutamine repeats.
Of note, we observed a discrepancy between data obtained with the MAB2166 and 1C2 antibodies. For example 1 µm of CTG25 reduced total HTT in line HD 20/77 to 50% based on western blotting with MAB2166, but no significant reduction of the expanded allele was observed with 1C2. In contrast, while 10 µm of CTG25 reduced total HTT levels in this line to ∼10% as determined by MAB2166, 1C2 indicated a 50% reduction of expanded HTT. This suggests that MAB2166 differentially detects the normal and mutant alleles. Therefore, future studies of the effect of knockdown strategies should use multiple antibodies to measure normal and mutant HTT levels.
Yu et al. (48) recently described a repeat-targeting ASO capable of allele-specific HTT inhibition in vivo. While achieving allelic specificity is considered to be the ultimate goal in ASO-based HD therapy, work from Kordasiewicz et al. (37) demonstrates the potential therapeutic role of non–allele-specific ASO-based HD therapy. While, as expected, non-CAG repeat-targeting PMOs that we examined did not exhibit allelic specificity, these PMOs have the advantage of specificity to HTT and therefore are less likely to exhibit significant off-target effect than a PMO (or other ASO) targeting the repeat.
In addition to allelic selectivity and effectiveness, an ideal ASO therapy should exhibit no off-target effects. While the repeat-targeting ASO-based HD therapies reported so far are very promising (20), their off-target effects remain to be fully explored. As our data demonstrate, off-target effects are very likely to occur, especially with shorter repeat-targeting ASOs. In addition, our data indicate that off-target effects will increase with higher concentrations of ASOs; we speculate that prolonged administration of ASOs would have a similar effect. For instance, CTG22 and CTG25 both decreased ATXN3 expression in the HD 20/77 and HD 19/109 cell lines (Fig. 5B). Therefore, the length of the PMO CTG repeat, the length of the repeat in off-target genes and the structure of the repeat in the off-target gene as determined by flanking sequence all influence off-target effects. Since ATXN3 levels appear to be particularly susceptible to CAG repeat-targeting siRNAs (32) and PMOs, ATXN3 levels may provide a good marker for off-target effects of repeat-targeting siRNAs and ASOs. Our results also suggest that the differential tissue expression of CAG repeat-containing genes and the differential intracellular distribution of ASOs need to be considered when examining off-target effects.
The ability of PMOs to reduce target expression depends on their ability to enter cells through endocytosis and to be efficiently released from endosomes/lysosomes (73). Our results show that PMOs have a wider intracellular distribution in neurons, and therefore their selectivity and effectiveness may be higher when delivered to neuronal cells than to fibroblasts (Fig. 9). This suggests that, while the endogenous levels of normal and expanded HTT in fibroblasts serve as a useful model for an initial evaluation of an ASO (74), they may not be fully informative when it comes to the specificity and effectiveness of an ASO in neurons.
Our second and most important finding is that repeat-targeting PMOs are capable of significantly and selectively reducing mutant HTT expression in two different HD mouse models and that a safe reduction in one of the models is also accompanied by a preliminary improvement in phenotype. A single low-dose intracerebroventricular (ICV) injection of CTG25 in N171-82Q transgenic HD mice selectively and significantly reduced the levels of mutant HTT in the frontal cortex without a significant effect on endogenous HTT levels or the levels of a selection of other polyglutamine-containing proteins (Fig. 10A and B). However, we are aware of the limitations of this transgenic model, in which a human HTT fragment is expressed under the PrP promoter. Therefore, to further examine PMO pharmacokinetics, possible effects on behavior, and off-target effects, we examined the effect of three successive low-dose ICV injections of CTG28 over the course of 6 months on the HdhQ7/Q150 knock-in HD mouse model (Fig. 10C). CTG28 was effective in selectively inhibiting mutant HTT expression in widely distributed brain regions 2 months after the last injection. We observed that CTG28 has a higher efficacy in the cortex than in the striatum, perhaps reflecting differential PMO penetration into the two regions, or cell type selectivity of response to CTG28. We also determined that CTG28 treatment attenuated the abnormal behavior displayed by HdhQ7/Q150 knock-in mice on the tail suspension test. Unfortunately, only a limited behavioral analysis can be performed on HdhQ7/Q150 mice, as they exhibit very little phenotype at 12 months of age (69,70). While we did not detect significant off-target effects of CTG28 (Fig. 10D), it is important to note that the repeat length of repeat-containing mouse genes is almost universally shorter than those in the orthologous human genes (65), so any repeat-targeting strategy will require careful exploration of off-target effect in human tissue. The absence of loss of body weight relative to Ctrl PMO–injected mice [a simple assay of systemic health commonly used to assess PMOs (75–77)] following CTG28 injection provides additional evidence of the in vivo safety of PMOs (Fig. 10E).
PMOs are uniquely amenable to modifications, and a range of changes could address issues such as the need to enhance their delivery across the blood-brain barrier (BBB) using less invasive delivery routes than ICV or brain parenchyma injections, such as intravenous (IV) or intranasal (IN) delivery (75). At least two PMO modifications with increased effect in vivo have been reported: Vivo-Morpholinos (eight guanidinium groups on a dendrimeric scaffold linked to a PMO) and PPMOs, in which an arginine-rich cell-penetrating peptide is linked to a PMO. Direct intracranial administration of Vivo-Morpholinos provided an effective means to suppress protein expression of xCT, GLT1 and orexin in the brain for up to 7 days (76). However, Vivo-Morpholinos were reported to have low brain penetration when injected peripherally (51,77). Alternatively, peptide conjugation leads to the delivery of PPMOs to both muscle (78,79) and brain via the BBB (80) following tail vein injections. Recently, a systemic delivery of PPMO reduced CUG RNA neurotoxicity and resulted in corrected splicing in a mouse model of DM1 (81). However, despite the superior efficacy of Vivo-Morpholinos and PPMOs, reports of their toxicity will need to be taken into careful consideration as their therapeutic potential is explored (82,83).
ASO-based therapies may require targeting specific neuronal subpopulations to maximize effectiveness while minimizing toxicity and side effects. PMOs linked to nanoparticles have successfully been targeted to glioma cells in vivo (84), and a similar approach may serve to target PMOs to medium spiny neurons in HD. Both the repeat-targeting and non–repeat-targeting PMOs examined here may also be combined with other successful strategies, as a combination of multiple knockdown techniques may lead to the most effective treatment (15). Moreover, since PMOs have good diffusion into neuronal nuclei, additional PMOs could be designed to correct missplicing in HTT, similar to the strategy described for DMD (85).
In summary, published applications of PNAs, LNAs, chemically modified ssRNAs and other ASOs toward the selective inhibition of mutant HTT are encouraging. Our results support PMOs as an additional viable therapeutic tool for HD.
MATERIALS AND METHODS
Reagents and antibodies
Standard control PMO, custom-made PMOs and Endo-Porter (in DMSO solution) were obtained from Gene Tools, LLC (Philomath, OR, USA). The sequence for each of the PMOs used in this study is given in Figure 1. TaqMan Assay IDs for real-time PCR purchased from Applied Biosystems (Carlsbad, CA, USA) were as follows: HTT, Hs00918174_m1; ATXN1, Hs00165656_m1; ATXN3, Hs01026440_g1; TBP, Hs00427620_m1; ATN1, Hs00157312_m1; GUSB, Hs00939627_m1; HTTAS_v1, described by Chung et al. (38). For toxicity assays in cells, the Caspase-Glo 3/7 assay kit was used (G8091, Promega, Madison, WI, USA). Control and DICER1 siRNAs were purchased from Ambion (Carlsbad, CA, USA). The antibodies and concentrations used for immunoblotting and immunofluorescence were as follows: anti-HTT (MAB2166, 1:1000), anti-polyglutamine, 1C2 (MAB1574, 1:2000), anti–ataxin-3 (MAB5360, 1:500) anti-Androgen Receptor (06–680, 1:200), all from EMD Millipore (Billerica, MA, USA); anti-TBP (sc-273, 1:200), anti-histone H1 (sc-8030, 1:200) and anti–Retinoic Acid-Induced 1 (sc-365065, 1:100), all from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti–β-actin (ab8224, 1:5000) from Abcam (Cambridge, MA, USA); anti-DICER1 (SAB4200087, 1:200) from Sigma-Aldrich; anti-Rab7 (9367, 1:100) from Cell Signaling Technology (Danvers, MA, USA). The secondary antibodies were Cy3-conjugated goat-anti-rabbit IgG, HRP-conjugated goat-anti-rabbit IgG and HRP-conjugated goat anti-mouse IgG antibodies (Life Technologies, Grand Island, NY, USA).
Cell culture, transfection and PMO treatment
HD patient–derived fibroblast cell lines HD 18/44, HD 20/77 and HD 19/109 were obtained from the Baltimore Huntington's Disease Center. The fibroblasts were cultured in Minimal Essential Medium Eagle (MEM) supplemented with heat-inactivated 10% fetal bovine serum (FBS) and 1% MEM non-essential amino acid solution. HEK293 cells (CRL-1573; ATCC, Manassas, VA, USA) and neuroblastoma cell line SH-SY5Y (CRL-2266, ATCC) were cultured in Dulbecco's Modified Eagle Medium with 4.5 g/l glucose and supplemented with 10% FBS. Mouse primary cortical neurons were prepared as previously described (86) and cultured in neurobasal medium supplemented with 2% B-27 supplements. All media and reagents used for cell culture were obtained from Life Technologies. All cells were maintained at 37°C with 5% CO2 and plated either in 6-well plates (100 000 cells/well) or in 96-well plates (5000 cells/well) 1 day prior to siRNA transfection or PMO treatment. For knockdown of DICER1, 100 pmol of siRNA was transfected into HD 19/109 fibroblasts using Lipofectamine 2000 reagents, according to the manufacturer's manual (Life Technologies). For all PMO treatments, PMOs (at respective concentrations) and Endo-Porter (6 μm) were pre-mixed for 30 min and then added to the culture medium for 48 h before collection for analysis.
Western blot
Following PMO treatment, cells were washed with PBS, and total protein was extracted using RIPA buffer (Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer's protocol. For nuclear protein extraction, the NE-PER Nuclear Protein Extraction Kit (Pierce, Rockford, IL, USA) was used according to the manufacturer's protocol. Total protein extracts from mouse brains were prepared following tissue homogenization in RIPA buffer. Protein samples were subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE) using 3–8% tris-acetate gel (constant voltage of 120 V, for immunoblotting of HTT) and 4–12% bis-tris gel (constant voltage of 100 V, for immunoblotting of other proteins; Life Technologies). Proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) at a constant voltage of 20 V overnight and blocked with 5% milk in tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature (RT). Membranes were incubated with primary antibodies overnight at 4°C. After washing with TBS-T, membranes were incubated with the corresponding secondary antibodies and protein bands were detected using the ECL Prime Western Blotting System (GE Healthcare, UK). Images were analyzed with the ImageJ Software (NIH, http://rsbweb.nih.gov/ij/ 8 July 2014 last accessed date). Relative protein levels of TBP were obtained by normalizing TBP levels to that of histone H1, while β-actin was used as an internal control for other proteins.
RNA isolation and real-time PCR
Total RNA was isolated using TRIZOL reagent (Sigma-Aldrich), according to the manufacturer's manual. Total RNA was reverse-transcribed using random hexamer primers and SuperScript III reverse transcriptase (Life Technologies). The ABI 7900HT detection system (Applied Biosystems) was used for real-time PCR. Gene-specific TaqMan primers and probes (Applied Biosystems) were used to monitor the replication of PCR products. Human GUSB was selected as an internal control. Quantities of transcripts were obtained using the standard curve method and normalized to GUSB RNA levels. RNA expression levels were represented as a ratio of the gene of interest to GUSB.
Caspase-3/7 activity assay
Cells were treated with PMOs in 96-well plates and incubated with 100 µl/well of Caspase-Glo 3/7 reagent for 1 h at 37°C. A Fluoroskan Ascent Microplate Fluorometer (Thermo Scientific, Waltham, MA, USA) was used to quantify luminescence. Relative luminescence unit measurements of all treated groups were normalized to that of the non-treated group.
LDH assay
Released LDH activity was measured according to the manufacturer's protocol (Roche, Berlin, Germany). Briefly, 50 µl of conditioned medium from cell cultures were incubated with 50 µl of LDH assay buffer at RT for 20 min in the dark. The reaction mixture was stopped and the absorbance was read at 492 nm. Fresh medium was also assayed as background and subtracted from each group to get relative LDH activity. Relative LDH activity of the non-treated control group was defined as 100, and all other groups were normalized to this value.
Immunofluorescence
Cells were seeded onto Poly d-lysine-coated cover slips in a 24-well plate at 1 × 105 cells/well and incubated with culture medium containing 20 μm fluorescein-labeled standard control PMO. Cells were then washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min at RT. For endosome staining, HD 19/109 cells were permeabilized in PBS with 10% BSA and 0.1% Triton X-100 for 30 min and incubated in a primary antibody against Rab7 at 4°C overnight, followed by washing and 1-h incubation with a secondary antibody. All images were acquired using a Nikon Eclipse E400 epifluorescence microscope (Nikon, Tokyo, Japan) and MetaVue 4.6 software (Molecular Devices, Sunnyvale, CA, USA).
ICV injection in HD mouse
The breeding and subsequent use of N171-82Q transgenic mice and HdhQ7/Q150 knock-in mice were approved by the ACUC of Johns Hopkins University, Baltimore, MD. Mice were housed at the East Baltimore campus rodent vivarium and maintained on a standard circadian cycle with free access to water and standard chow. Six-week-old N171-82Q mice were anesthetized via intraperitoneal injection of a mixture of 80 mg/kg ketamine and 1 mg/kg dexmedetomidine, diluted in 0.9% sterile saline. Mice were then immobilized in a stereotactic device and injected in the right lateral ventricle (1.0 mm posterior to Bregma, 1.3 mm right, 2.0 mm deep); 100 µg of previously freeze-dried PMO were resuspended in 2 µl saline to create a 6 mm injection solution, which was injected using a 2 µl Hamilton syringe over 1 min. After injection, the needle was withdrawn and skin was closed with cyanoacrylate glue. Post-surgical health was monitored until sufficient recovery was attained in respiration and mobility levels. Fourteen days after ICV injection, mice were killed by rapid decapitation and frozen brain region punches were taken in a cryostat. Total lysate of the frontal cortex was prepared from each mouse and used in western blot analysis. Six-month-old HdhQ7/Q150 knock-in mice were subjected to the same injection protocol for three successive injections at 6, 8 and 10 months of age. The mice's body weight was measured weekly from the first injection. Two months after the last ICV injection, the mice were examined in the tail suspension test and then killed. The ipsilateral hemisphere of each mouse brain was dissected into the frontal cortex, striatum and cerebellum for western blot analysis.
Tail suspension test
The tail suspension test is a standard mouse behavioral phenotyping procedure that is used to assess depressive-like phenotype: mice administered monoamine oxidase inhibitors (MAOIs) and other pharmaceutical antidepressants display decreased immobility and increased latency to immobility (89, 90). Mice were allowed to acclimatize to individual housing in an isolated behavioral suite for 1 day prior to testing. Each mouse was hung by the distal 2 cm of tail to a shelf positioned ∼18 inches above the bench top. The mice were oriented such that the ventral aspect faced the observer. Each trial lasted 60 s and each mouse completed five trials separated by ∼15 min. Each trial was recorded by a digital camera and later scored by two observers (one blinded) for time spent attempting escape, time spent immobile and latency to immobility.
Statistical analysis
Experiments were repeated at least three times. Data were presented as mean ± SD. The results were analyzed using Student's t-test, one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test, or two-way ANOVA followed by Bonferroni's post hoc test as indicated. Statistical significance was set at P-value < 0.05.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (NS064138 to D.D.R.) and CHDI Foundation (Early Discovery Initiative to D.D.R.).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank HD patients and their families for their support, tissue donation and inspiration.
Conflict of Interest statement: None declared.
REFERENCES
- 1.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Riley B.E., Orr H.T. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev. 2006;20:2183–2192. doi: 10.1101/gad.1436506. [DOI] [PubMed] [Google Scholar]
- 3.Roos R.A. Huntington's disease: a clinical review. Orphanet J. Rare Dis. 2010;5:40. doi: 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peavy G.M., Jacobson M.W., Goldstein J.L., Hamilton J.M., Kane A., Gamst A.C., Lessig S.L., Lee J.C., Corey-Bloom J. Cognitive and functional decline in Huntington's disease: dementia criteria revisited. Mov. Disord. 2010;25:1163–1169. doi: 10.1002/mds.22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rickards H., De Souza J., van Walsem M., van Duijn E., Simpson S.A., Squitieri F., Landwehrmeyer B. Factor analysis of behavioural symptoms in Huntington's disease. J. Neurol. Neurosurg. Psychiatry. 2011;82:411–412. doi: 10.1136/jnnp.2009.181149. [DOI] [PubMed] [Google Scholar]
- 6.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 7.ACMG/ASHG statement. Laboratory guidelines for Huntington disease genetic testing. The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. Am. J. Hum. Genet. 1998;62:1243–1247. [PMC free article] [PubMed] [Google Scholar]
- 8.Semaka A., Creighton S., Warby S., Hayden M.R. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin. Genet. 2006;70:283–294. doi: 10.1111/j.1399-0004.2006.00668.x. [DOI] [PubMed] [Google Scholar]
- 9.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mykowska A., Sobczak K., Wojciechowska M., Kozlowski P., Krzyzosiak W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39:8938–8951. doi: 10.1093/nar/gkr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banez-Coronel M., Porta S., Kagerbauer B., Mateu-Huertas E., Pantano L., Ferrer I., Guzman M., Estivill X., Marti E. A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krauss S., Griesche N., Jastrzebska E., Chen C., Rutschow D., Achmuller C., Dorn S., Boesch S.M., Lalowski M., Wanker E., et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 2013;4:1511. doi: 10.1038/ncomms2514. [DOI] [PubMed] [Google Scholar]
- 13.Boudreau R.L., McBride J.L., Martins I., Shen S., Xing Y., Carter B.J., Davidson B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol. Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grondin R., Kaytor M.D., Ai Y., Nelson P.T., Thakker D.R., Heisel J., Weatherspoon M.R., Blum J.L., Burright E.N., Zhang Z., et al. Six-month partial suppression of huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135:1197–1209. doi: 10.1093/brain/awr333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aronin N., Moore M. Hunting down huntingtin. N. Engl. J. Med. 2012;367:1753–1754. doi: 10.1056/NEJMcibr1209595. [DOI] [PubMed] [Google Scholar]
- 16.Garriga-Canut M., Agustin-Pavon C., Herrmann F., Sanchez A., Dierssen M., Fillat C., Isalan M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl Acad. Sci. USA. 2012;109:E3136–E3145. doi: 10.1073/pnas.1206506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer P.O., Goswami A., Wong H.K., Okuno M., Kurosawa M., Yamada M., Miyazaki H., Matsumoto G., Kino Y., Nagai Y., et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010;28:256–263. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- 18.Drouet V., Perrin V., Hassig R., Dufour N., Auregan G., Alves S., Bonvento G., Brouillet E., Luthi-Carter R., Hantraye P., et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009;65:276–285. doi: 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
- 19.McBride J.L., Pitzer M.R., Boudreau R.L., Dufour B., Hobbs T., Ojeda S.R., Davidson B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol. Ther. 2011;19:2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsui M., Corey D.R. Allele-selective inhibition of trinucleotide repeat genes. Drug Discov. Today. 2012;17:443–450. doi: 10.1016/j.drudis.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sah D.W., Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Invest. 2011;121:500–507. doi: 10.1172/JCI45130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Engelman J., Friedlander R.M. Allele-specific silencing of mutant Huntington's disease gene. J. Neurochem. 2009;108:82–90. doi: 10.1111/j.1471-4159.2008.05734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfister E.L., Kennington L., Straubhaar J., Wagh S., Liu W., DiFiglia M., Landwehrmeyer B., Vonsattel J.P., Zamore P.D., Aronin N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr. Biol. 2009;19:774–778. doi: 10.1016/j.cub.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardi M.S., Jaspers L., Spronkmans C., Gellera C., Taroni F., Di Maria E., Donato S.D., Kaemmerer W.F. A majority of Huntington's disease patients may be treatable by individualized allele-specific RNA interference. Exp. Neurol. 2009;217:312–319. doi: 10.1016/j.expneurol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz D.S., Ding H., Kennington L., Moore J.T., Schelter J., Burchard J., Linsley P.S., Aronin N., Xu Z., Zamore P.D. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006;2:e140. doi: 10.1371/journal.pgen.0020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Bilsen P.H., Jaspers L., Lombardi M.S., Odekerken J.C., Burright E.N., Kaemmerer W.F. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington's disease patient-derived fibroblasts. Hum. Gene Ther. 2008;19:710–719. doi: 10.1089/hum.2007.116. [DOI] [PubMed] [Google Scholar]
- 27.Carroll J.B., Warby S.C., Southwell A.L., Doty C.N., Greenlee S., Skotte N., Hung G., Bennett C.F., Freier S.M., Hayden M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 2011;19:2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J., Liu J., Corey D.R. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 2010;17:1183–1188. doi: 10.1016/j.chembiol.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Mezer M., Wojciechowska M., Napierala M., Sobczak K., Krzyzosiak W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39:3852–3863. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiszer A., Olejniczak M., Switonski P.M., Wroblewska J.P., Wisniewska-Kruk J., Mykowska A., Krzyzosiak W.J. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol. Biol. 2012;13:6. doi: 10.1186/1471-2199-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J., Matsui M., Corey D.R. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann. N. Y. Acad. Sci. 2009;1175:24–31. doi: 10.1111/j.1749-6632.2009.04975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiszer A., Mykowska A., Krzyzosiak W.J. Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 2011;39:5578–5585. doi: 10.1093/nar/gkr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiszer A., Olejniczak M., Galka-Marciniak P., Mykowska A., Krzyzosiak W.J. Self-duplexing CUG repeats selectively inhibit mutant huntingtin expression. Nucleic Acids Res. 2013;41:10426–10437. doi: 10.1093/nar/gkt825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J., Liu J., Yu D., Chu Y., Corey D.R. Mechanism of allele-selective inhibition of huntingtin expression by duplex RNAs that target CAG repeats: function through the RNAi pathway. Nucleic Acids Res. 2012;40:11270–11280. doi: 10.1093/nar/gks907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L., Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu. Rev. Biomed. Eng. 2011;13:507–530. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 36.Lee J.E., Bennett C.F., Cooper T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl Acad. Sci. USA. 2012;109:4221–4226. doi: 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., Mazur C., McAlonis M.M., Pytel K.A., Artates J.W., Weiss A., Cheng S.H., Shihabuddin L.S., et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chung D.W., Rudnicki D.D., Yu L., Margolis R.L. A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum. Mol. Genet. 2011;20:3467–3477. doi: 10.1093/hmg/ddr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilburn B., Rudnicki D.D., Zhao J., Weitz T.M., Cheng Y., Gu X., Greiner E., Park C.S., Wang N., Sopher B.L., et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moseley M.L., Zu T., Ikeda Y., Gao W., Mosemiller A.K., Daughters R.S., Chen G., Weatherspoon M.R., Clark H.B., Ebner T.J., et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 41.Rudnicki D.D., Holmes S.E., Lin M.W., Thornton C.A., Ross C.A., Margolis R.L. Huntington's disease—like 2 is associated with CUG repeat-containing RNA foci. Ann. Neurol. 2007;61:272–282. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- 42.Wheeler T.M., Thornton C.A. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 43.Day J.W., Ranum L.P. RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., Thornton C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 45.Gagnon K.T., Pendergraff H.M., Deleavey G.F., Swayze E.E., Potier P., Randolph J., Roesch E.B., Chattopadhyaya J., Damha M.J., Bennett C.F., et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. 2010;49:10166–10178. doi: 10.1021/bi101208k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J., Matsui M., Gagnon K.T., Schwartz J.C., Gabillet S., Arar K., Wu J., Bezprozvanny I., Corey D.R. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 2009;27:478–484. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu J., Dodd D.W., Hudson R.H., Corey D.R. Cellular localization and allele-selective inhibition of mutant huntingtin protein by peptide nucleic acid oligomers containing the fluorescent nucleobase [bis-o-(aminoethoxy)phenyl]pyrrolocytosine. Bioorg. Med. Chem. Lett. 2009;19:6181–6184. doi: 10.1016/j.bmcl.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu D., Pendergraff H., Liu J., Kordasiewicz H.B., Cleveland D.W., Swayze E.E., Lima W.F., Crooke S.T., Prakash T.P., Corey D.R. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell. 2012;150:895–908. doi: 10.1016/j.cell.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 50.Summerton J.E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 2007;7:651–660. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- 51.Moulton J.D., Jiang S. Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules. 2009;14:1304–1323. doi: 10.3390/molecules14031304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wheeler T.M., Sobczak K., Lueck J.D., Osborne R.J., Lin X., Dirksen R.T., Thornton C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J., et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kinali M., Arechavala-Gomeza V., Feng L., Cirak S., Hunt D., Adkin C., Guglieri M., Ashton E., Abbs S., Nihoyannopoulos P., et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yokota T., Lu Q.L., Partridge T., Kobayashi M., Nakamura A., Takeda S., Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 57.Anthony K., Feng L., Arechavala-Gomeza V., Guglieri M., Straub V., Bushby K., Cirak S., Morgan J., Muntoni F. Exon skipping quantification by quantitative reverse-transcription polymerase chain reaction in Duchenne muscular dystrophy patients treated with the antisense oligomer eteplirsen. Hum. Gene Ther. Methods. 2012;23:336–345. doi: 10.1089/hgtb.2012.117. [DOI] [PubMed] [Google Scholar]
- 58.Malerba A., Kang J.K., McClorey G., Saleh A.F., Popplewell L., Gait M.J., Wood M.J., Dickson G. Dual myostatin and dystrophin exon skipping by morpholino nucleic acid oligomers conjugated to a cell-penetrating peptide is a promising therapeutic strategy for the treatment of Duchenne muscular dystrophy. Mol. Ther. Nucleic Acids. 2012;1:e62. doi: 10.1038/mtna.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hiroi R., McDevitt R.A., Morcos P.A., Clark M.S., Neumaier J.F. Overexpression or knockdown of rat tryptophan hyroxylase-2 has opposing effects on anxiety behavior in an estrogen-dependent manner. Neuroscience. 2011;176:120–131. doi: 10.1016/j.neuroscience.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porensky P.N., Mitrpant C., McGovern V.L., Bevan A.K., Foust K.D., Kaspar B.K., Wilton S.D., Burghes A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H., Janghra N., Mitrpant C., Dickinson R.L., Anthony K., Price L., Eperon I.C., Wilton S.D., Morgan J., Muntoni F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 2013;24:331–342. doi: 10.1089/hum.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitrpant C., Porensky P., Zhou H., Price L., Muntoni F., Fletcher S., Wilton S.D., Burghes A.H. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: towards a treatment for spinal muscular atrophy. PLoS One. 2013;8:e62114. doi: 10.1371/journal.pone.0062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oh I.S., Shimizu H., Satoh T., Okada S., Adachi S., Inoue K., Eguchi H., Yamamoto M., Imaki T., Hashimoto K., et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature. 2006;443:709–712. doi: 10.1038/nature05162. [DOI] [PubMed] [Google Scholar]
- 64.Trottier Y., Lutz Y., Stevanin G., Imbert G., Devys D., Cancel G., Saudou F., Weber C., David G., Tora L., et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 65.Butland S.L., Devon R.S., Huang Y., Mead C.L., Meynert A.M., Neal S.J., Lee S.S., Wilkinson A., Yang G.S., Yuen M.M., et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genomics. 2007;8:126. doi: 10.1186/1471-2164-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruiz C., Casarejos M.J., Gomez A., Solano R., de Yebenes J.G., Mena M.A. Protection by glia-conditioned medium in a cell model of Huntington disease. PLoS Curr. 2012:e4fbca54a2028b. doi: 10.1371/4fbca54a2028b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Summerton J.E. Endo-Porter: a novel reagent for safe, effective delivery of substances into cells. Ann. N. Y. Acad. Sci. 2005;1058:62–75. doi: 10.1196/annals.1359.012. [DOI] [PubMed] [Google Scholar]
- 68.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 69.Heng M.Y., Tallaksen-Greene S.J., Detloff P.J., Albin R.L. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J. Neurosci. 2007;27:8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 71.Woodman B., Butler R., Landles C., Lupton M.K., Tse J., Hockly E., Moffitt H., Sathasivam K., Bates G.P. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007;72:83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 72.Chiu C.T., Liu G., Leeds P., Chuang D.M. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington's disease. Neuropsychopharmacology. 2011;36:2406–2421. doi: 10.1038/npp.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abes R., Moulton H.M., Clair P., Yang S.T., Abes S., Melikov K., Prevot P., Youngblood D.S., Iversen P.L., Chernomordik L.V., et al. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies. Nucleic Acids Res. 2008;36:6343–6354. doi: 10.1093/nar/gkn541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakano S., Ozasa S., Yoshioka K., Fujii I., Mitsui K., Nomura K., Kosuge H., Endo F., Matsukura M., Kimura S. Exon-skipping events in candidates for clinical trials of morpholino. Pediatr. Int. 2011;53:524–529. doi: 10.1111/j.1442-200X.2011.03330.x. [DOI] [PubMed] [Google Scholar]
- 75.Dhuria S.V., Hanson L.R., Frey W.H., 2nd. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 76.Reissner K.J., Sartor G.C., Vazey E.M., Dunn T.E., Aston-Jones G., Kalivas P.W. Use of vivo-morpholinos for control of protein expression in the adult rat brain. J. Neurosci. Methods. 2012;203:354–360. doi: 10.1016/j.jneumeth.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morcos P.A., Li Y., Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. Biotechniques. 2008;45:613–614. doi: 10.2144/000113005. , 616, 618 passim. [DOI] [PubMed] [Google Scholar]
- 78.Leger A.J., Mosquea L.M., Clayton N.P., Wu I.H., Weeden T., Nelson C.A., Phillips L., Roberts E., Piepenhagen P.A., Cheng S.H., et al. Systemic delivery of a peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013;23:109–117. doi: 10.1089/nat.2012.0404. [DOI] [PubMed] [Google Scholar]
- 79.Betts C., Saleh A.F., Arzumanov A.A., Hammond S.M., Godfrey C., Coursindel T., Gait M.J., Wood M.J. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids. 2012;1:e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Du L., Kayali R., Bertoni C., Fike F., Hu H., Iversen P.L., Gatti R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011;20:3151–3160. doi: 10.1093/hmg/ddr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leger A.J., Mosquea L.M., Clayton N.P., Wu I.H., Weeden T., Nelson C.A., Phillips L., Roberts E., Piepenhagen P.A., Cheng S.H., et al. Systemic delivery of a peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013;23:109–117. doi: 10.1089/nat.2012.0404. [DOI] [PubMed] [Google Scholar]
- 82.Ferguson D.P., Dangott L.J., Lightfoot J.T. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques. 2014;56:251–256. doi: 10.2144/000114167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amantana A., Moulton H.M., Cate M.L., Reddy M.T., Whitehead T., Hassinger J.N., Youngblood D.S., Iversen P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007;18:1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 84.Ljubimova J.Y., Fujita M., Khazenzon N.M., Lee B.S., Wachsmann-Hogiu S., Farkas D.L., Black K.L., Holler E. Nanoconjugate based on polymalic acid for tumor targeting. Chem. Biol. Interact. 2008;171:195–203. doi: 10.1016/j.cbi.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D., et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl Acad. Sci. USA. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sciarretta C., Minichiello L. The preparation of primary cortical neuron cultures and a practical application using immunofluorescent cytochemistry. Methods Mol. Biol. 2010;633:221–231. doi: 10.1007/978-1-59745-019-5_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.