

Abstract
RAS genes encode a family of 21 kDa proteins that are an essential hub for a number of survival, proliferation, differentiation and senescence pathways. Signaling of the RAS-GTPases through the RAF-MEK-ERK pathway, the first identified mitogen-associated protein kinase (MAPK) cascade is essential in development. A group of genetic syndromes, named “RASopathies”, had been identified which are caused by heterozygosity for germline mutations in genes that encode protein components of the RAS/MAPK pathway. Several of these clinically overlapping disorders, including Noonan syndrome, Noonan-like CBL syndrome, Costello syndrome, cardio-facio-cutaneous (CFC) syndrome, neurofibromatosis type I, and Legius syndrome, predispose to cancer and abnormal myelopoiesis in infancy. This review focuses on juvenile myelomonocytic leukemia (JMML), a malignancy of early childhood characterized by initiating germline and/or somatic mutations in five genes of the RAS/MAPK pathway: PTPN11, CBL, NF-1, KRAS and NRAS. Natural courses of these five subtypes differ, although hematopoietic stem cell transplantation remains the only curative therapy option for most children with JMML. With whole-exome sequencing studies revealing few secondary lesions it will be crucial to better understand the RAS/MAPK signaling network with its crosstalks and feed-back loops to carefully design early clinical trials with novel pharmacological agents in this still puzzling leukemia.
Introduction
Discoveries made about 50 years ago revealed the transforming power of the Harvey and Kirsten murine sarcoma retroviruses by a set of genes named RAS genes for their role in forming rat sarcomas. Subsequently, the contribution of RAS genes to cancer pathogenesis has been studied extensively. While the somatic dysregulation of RAS is a primary driver of cancer, there is a class of developmental disorders caused by germline mutations (as opposed to the somatic mutations found in cancer) in genes that encode components of the RAS signaling pathway. This chapter summarizes hematologic disturbances and cancer predisposition of these genetic disorders named “RASopathies”. It will focus on juvenile myelomonocytic leukemia (JMML), an infant leukemia with initiating germline and somatic mutations in genes of the RAS signal transduction pathway.
RAS and the RAS/mitogen-activated protein kinase signaling cascade
RAS turned out to be an essential cellular hub for a wide variety of signaling pathways including the three human RAS genes, KRAS, NRAS and HRAS (reviewed by Stephen et al.1). RAS proteins act as molecular switches by cycling between an active guanosine triphospate (GTP)-bound (RAS-GTP) and an inactive guanosine diphosphate (GDP)-bound (RAS-GDP) conformation (Figure 1).2 RAS-GTP concentrations are tightly regulated by the competitive action of guanosin exchange factors (GEFs) and GTPase activating proteins (GAPs).
Figure 1.

Genetic syndromes of the RAS-MAPK pathway: “Rasopathies” (modified from Tidyman et al.2).
RAS is activated by extracellular stimuli such as growth factor binding to receptor tyrosine kinases (RTKs) followed by RTK autophosphorylation and the creation of docking sites for adaptor molecules (e.g. GRB2). These molecules recruit and activate GEFs (e.g. SOS1) which displace GDP from RAS allowing RAS to bind to GTP. RAS-GTP binds to and activates a large number of effector pathways. The RAS-mediated mitogen-activated kinase (MAPK) pathway is one of several important downstream cascades. Activated RAS binds to RAF (RAF1, also known as CRAF, BRAF), the first MAPK of the signaling cascade. RAF phosphorylates MEK1 and/or MEK2, which in turn activate ERK1 and/or ERK2. Active ERKs serve as regulators of a large number of downstream processes both in the cytosol and nucleus.
Genetic syndromes of the RAS/mitogen-activated kinase pathway
In the last decade, a group of human genetic syndromes was discovered to result from germline mutations in genes of the RAS/MAPK pathway (Figure 1).2,3 These mutations induce the activation of the pathway, and thus, these disorders share common clinical features including reduced growth, facial dysmorphism, cardiac defects, ectodermal anomalies, variable cognitive deficits and susceptibility to certain malignancies. Based on the shared pathogenic mechanism with up-regulated RAS/MAPK signaling and the clinical overlap, these genetic diseases have been grouped as “neuro-cardio-facio cutaneous syndromes (NCFCS)” or “RASopathies” (Table 1).4–22
Table 1.
Summary of syndromes of the RAS/MAPK pathway, named RASopathies or neuro-cardio-facial cutaneous syndromes (adapted from Roberts et al.3 and Aoki et al.4).
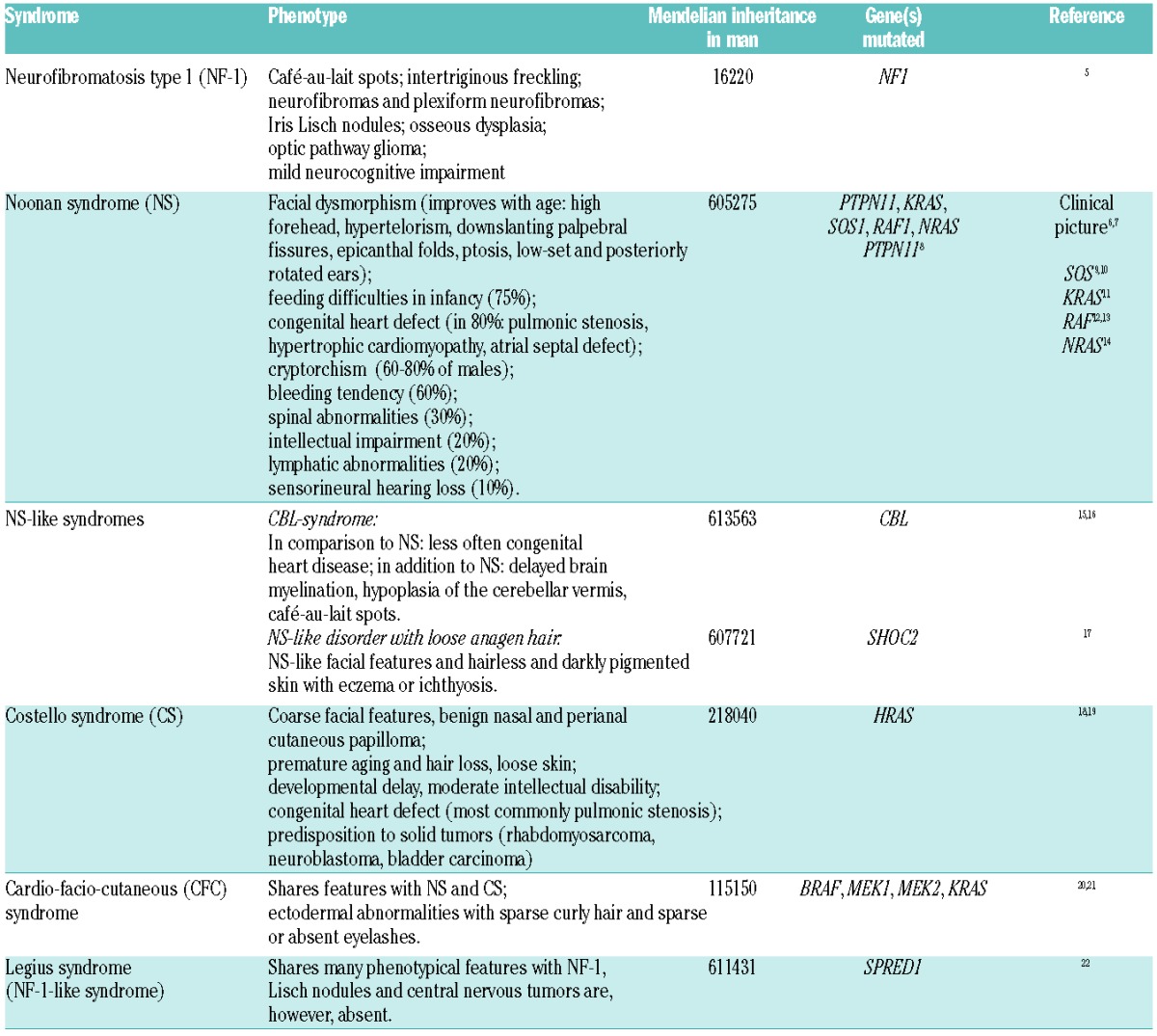
Noonan syndrome (NS) is the most common of these disorders occurring in 1 of 1000–2500 births (Table 1). It is characterized by a typical facial appearance, congenital heart defects and a variety of abnormalities in other organ systems. At present, heterozygous germline mutations in 6 genes are recognized in NS with PTPN11 mutations contributing for approximately half of the cases (Table 1). Other RASopathies consist in the two NS-like syndromes caused by mutations in CBL or SHOC2, Costello syndrome, cardio-facio-cutaneous syndrome (CFC), neurofibromatosis type 1 (NF-1) and Legius syndrome (Table 1).
Interestingly, germline mutation in genes involving the RAS/MAPK pathway may predispose to autoimmune disease as is known for the respective somatic mutations (see below). In a series of 42 patients with RASopathies, 52% were shown to have autoantibodies against various tissues.23 Other investigators suggested a predisposition to the development of systemic lupus erythematosus in NS.24
Malignancies in patients with germline mutations in RAS/MAPK pathway genes
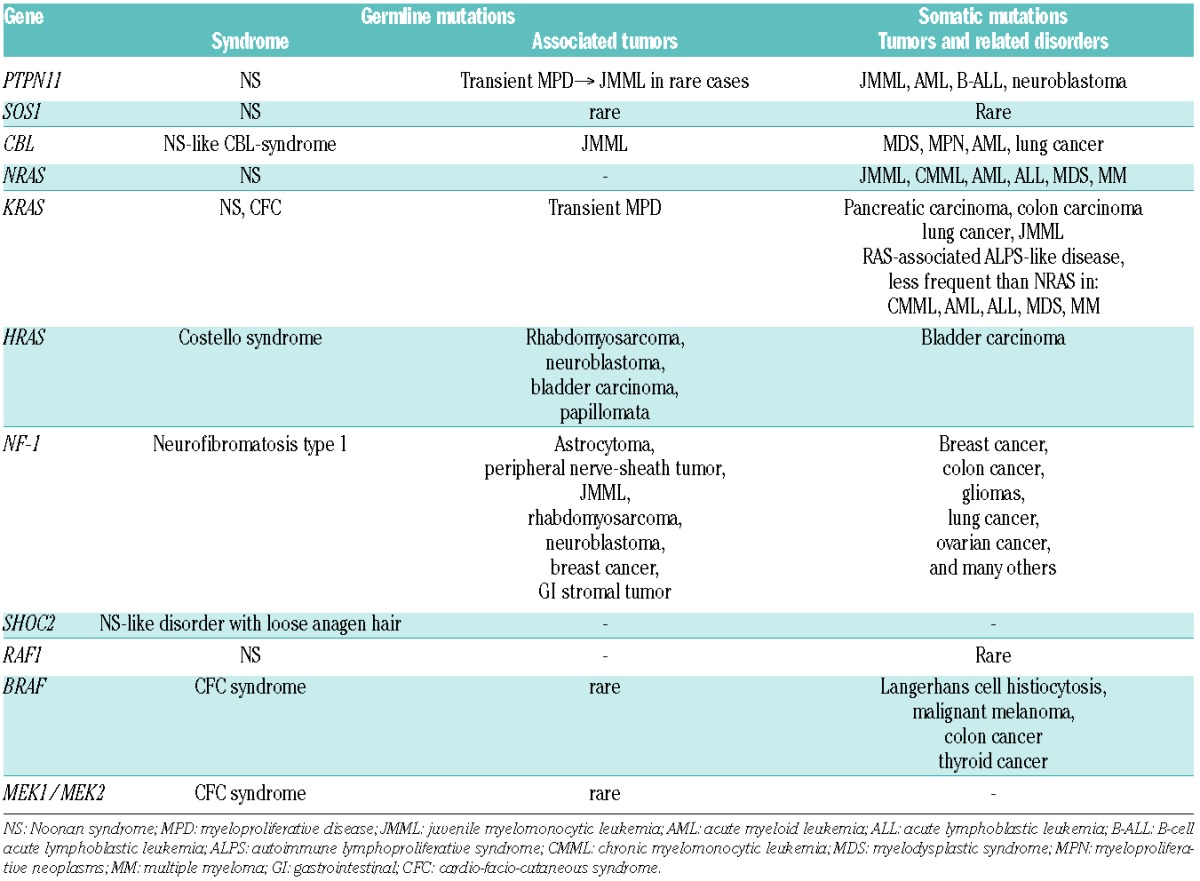
Besides NF-1, an extensively studied familial tumor syndrome, there is a clear correlation between germline mutations in the genes of the RAS/MAPK pathway and specific neoplasms (reviewed by Kratz et al.25). Since the RAS signaling pathway is a major contributor to tumorigenesis (Table 2) this is not surprising. From a literature review, the estimated rate of cancer was 11% for Costello syndrome, and 4% each for CFC syndrome and NS (excluding transient myeloproliferative disorder).25 In Costello syndrome, more than 70% of the malignancies are rhabdomyosarcomas.26 The patient age at the time of cancer diagnosis is comparable to that expected for sporadic cancers, thus, there is an early-childhood peak.
Table 2.
Disorders involving germline and somatic mutations in genes of the RAS/MAPK pathway (adapted from Aoki et al.4)
The most common hematopoietic disorder in NS is a transient myeloproliferative disorder (MPD)25–27 estimated to occur in up to 10% of all children with NS.28 While the abnormal myelopoiesis is benign in the vast majority of these cases, some children develop JMML. The following part of this chapter will focus on MPD in NS, and on JMML, a unique leukemia characterized by initiating genetic events in one of 5 genes of the RAS signal transduction pathway.
Transient myeloproliferative disorder in infants with Noonan syndrome
The transient MPD in Noonan syndrome25–27 is diagnosed in the neonatal period or early infancy. In contrast to JMML, NS/MPD is thought to be of polyclonal origin. It generally resolves spontaneously over months or years. However, leukocytosis and tissue invasion by monocytes and immature myeloid cells can have deleterious effects. Up to 30% of children with NS and severe MPD succumb to myeloproliferation in combination with their multiple other clinical problems (the EWOG-MDS, unpublished observation, 2014). To ameliorate the deleterious effects of abnormal myelopoiesis, mild cytoreductive therapy, such as treatment with 6-mercaptopurine, can be helpful. An estimated 10% of cases of NS/MPD acquire a cytogenetic abnormality and progress to JMML. Thus, NS/MPD is a tumor predisposition syndrome and affected patients should be followed closely. Nearly all patients with NS/MPD have mutations in PTPN11.29 Gain-of-function effect of these mutations on the protein product SHP2 is predicted to be intermediate between that expected in NS without myeloproliferation (lower gain of function) and somatic mutations in JMML (higher gain of function) (see below).30 In very rare cases, the underlying germline mutation in NS/MPD affects the KRAS gene.11
Juvenile myelomonocytic leukemia
Juvenile myelomonocytic leukemia is a clonal hematopoietic disorder of early childhood characterized by hepatosplenomegaly and organ infiltration due to excessive proliferation of cells of the monocytic and granulocytic lineages. It accounts for approximately 2.4% of all childhood hematologic malignancies.31
Clinical presentation
There is generally marked splenomegaly and hepatomegaly. Occasionally, spleen size is normal at diagnosis, but it rapidly increases thereafter. Dry cough, tachypnea and interstitial infiltrates on chest X-ray are signs of peribronchial and interstitial pulmonary infiltrates. Gut infiltrates may lead to diarrhea or gastrointestinal infections. Skin lesions other than petechiae are common and often present as eczematous eruptions, erythematous maculopapules or indurated raised lesions. In addition, multiple juvenile xanthogranulomas may be seen. Unlike acute monoblastic leukemia, JMML rarely involves the central nervous system.
Hematologic and laboratory features
Leukocytosis, anemia and thrombocytopenia are common findings in JMML. The median white blood cell count (WBC) varies from 25–33×109/L, and the WBC rarely exceeds100×109/L.32,33 Examination of the peripheral blood smear is the most important step in establishing the diagnosis. Almost all cases show a striking monocytosis, often with dysplastic cell forms. An absolute monocyte count exceeding 1×109/L is required for the diagnosis. Immature monocytes, along with myelocytes, metamyelocytes and nucleated red cells, are usually evident. The median blast cell percentage in blood smears is less than 2%. Thrombocytopenia may be severe and life threatening. Megakaryocytes are reduced or absent in approximately 75% of children.32 Bone marrow findings in JMML are not by themselves diagnostic but rather are consistent with the diagnosis.
A remarkable feature of many JMML cases is a markedly increased synthesis of hemoglobin F (HbF). Subtle immunological abnormalities like increased serum concentrations for IgG, IgM, and IgA or autoantibodies, such as antinuclear antibodies, antibodies against red cells giving rise to a positive antiglobin test are common in the absence of frank clinical autoimmune disease.32 Chromosomal studies of leukemic cells show monosomy 7 in approximately 25% of patients, other abnormalities in 10%, and a normal karyotype in 65%.32–34
Diagnosis
Approximately 90% of children with JMML have largely mutually exclusive mutations in one of 5 genes: PTPN11, NRAS, KRAS, NF-1 or CBL (Figure 2). Due to the size and complexity of the NF-1 gene, the diagnosis of NF-1 in children with JMML had in the past been based on the presence of 6 or more café-au-lait spots (and presence of an affected parent in approximately half of the patients). To identify abnormalities in the other genes, mutational screens have to be performed. Heterozygous point mutations noted in the PTPN11 and RAS genes may arise at a somatic or germline level. Consequently, genetic screens in leukemic cells need to be followed by studies in non-hematopoietic tissue such as fibroblasts, cells attached to nails, hair bulbs or epithelial buccal cells.
Figure 2.

The RAS signaling pathway and genes mutated in juvenile myelomonocytic leukemia (in black).
Viral infections, leukocyte adhesion deficiency variants,35 Wiskott Aldrich syndrome,36 infantile malignant osteopetrosis,35 and hemocytic lymphohistiocytosis (HLH)37 have to be excluded in the absence of a specific mutation. Of note, there are rare cases of myeloproliferative disorders other than JMML in young children who carry balanced translocations involving the GRAF gene,38 ALK gene,39 FIP1L1/RARA fusion gene,40 or the NUP98-HOXA9 fusion gene.41
Clinical course and prognostic factors
The natural course of JMML varies greatly depending on the genetic abnormality underlying the activation of the RAS/MAPK signaling pathway. However, in most series, the median survival time without hematopoietic stem cell transplantation (HSCT) is approximately one year.32 Low platelet count, age over two years at diagnosis and high HbF at diagnosis are the main predictors of short survival.32,34
Applying microarray analyses on archived material, Bresolin et al. demonstrated that JMML cases could be separated into a group with an acute myeloid leukemia (AML)-like and a group with a non-AML-like gene expression profile.42 There was a significant difference in the 10-year probability of survival after diagnosis between AML-like and non-AML-like patients (7% vs. 74%; P<0.001). Olk-Batz et al. studied the association of CpG island hypermethylation in JMML leukemic cells with known clinical risk factors.43 There was a strong association of interior prognosis with older age and elevated HbF at diagnosis. In multivariate models, DNA methylation retained an independent prognostic factor. These data may suggest that patients with a high-methylation phenotype may be good candidates for complementary approaches, e.g. pre-HSCT window therapies with DNA methyltransferase inhibitors.
Cell culture studies and induced pluripotent stem cells
The malignant transformation in JMML takes place at a stage of a committed stem cell that has the ability for myeloid, as well as early B-lymphoid, differentiation, while involvement of T-lymphoid precursors is generally absent.44,45 When cultured in semisolid media, JMML cells give rise to an excess number of monocyte-macrophage colonies in the absence of added growth factors. This so-called spontaneous proliferation depends on endogenous production of cytokines by monocytes. In addition, JMML myeloid progenitor cells were shown to display a striking hypersensitivity to GM-CSF.46 In fact, GM-CSF hypersensitivity became the hallmark of the disease, and represented an important diagnostic tool prior to the discovery of the molecular abnormalities of the RAS signaling pathway. Single cell profiling for changes in phosphoproteins demonstrated quite heterogeneous responses for readout of both RAS/MAPK and mTOR activation. However, for a subpopulation of CD33+CD14+CD38low JMML cells, a distinct pattern of STATA5 hyperphosphorylation in response to GM-CSF could be validated.47,48
Most recently, induced pluripotent stem cells (iPSCs) from malignant cells of 2 JMML patients with somatic heterozygous p.E76K missense mutations in PTPN11 have been generated.49 In vitro differentiation of iPSCs produced myeloid cells with increased proliferative capacity, constitutive activation of GM-CSF, and enhanced STAT5/ERK phosphorylation, similar to what has been seen in primary JMML cells. Furthermore, pharmacological inhibition of MEK kinase in iPSC-derived JMML cells reduced their GM-CSF independence. Thus, iPSCs offer the possibility of modeling the disease and exploring targeted therapy.
Juvenile myelomonocytic leukemia with neurofibromatosis type 1
The association between neurofibromatosis type 1 (NF-1) and JMML has long been established. A clinical diagnosis of NF-1 can be made in up to 12% of European32,34,50 and 4% of Japanese51 children with JMML. Approximately half of these cases have a parent affected with NF-1. In comparison to the other JMML subtypes, NF-1 is more common among JMML patients diagnosed after the age of five years.32 In the absence of HSCT, the clinical course of JMML in patients with NF-1 is invariably fatal.
Primary leukemic cells from children with NF-1 show a selective decrease in NF-1-like GAP activity and an increased percentage of RAS in the GTP-bound state.52 Finding of uniparental disomy (UPD) surrounding the NF-1 locus indicated that mitotic recombination in the leukemia-initiating cell is a crucial leukemic event.53,54 In a study on 15 children with NF-1 and JMML, two-thirds of patients had loss of heterozygosity (LOH) at the NF-1 locus, predominantly caused by segmental UPD disomy of large parts of chromosome arm 17q, and in a minority of cases by somatic interstitial deletions, while one-third had compound-heterozygous NF-1-inactivating mutations in leukemic cells.55 In a conditional murine model system, inactivation of somatic Nf-1 induces a myeloproliferative disorder with 100% penetrance and a subacute clinical course with progressive splenomegaly, a shift in hematopoiesis from the marrow to the spleen, and hypersensitivity of myeloid cells to GM-CSF.56
Juvenile myelomonocytic leukemia with somatic PTPN11 mutations
The discovery of heterozygous germline PTPN11 mutations in NS and in NS with transient myeloproliferation led to the prediction that children with ‘non-syndromic’ JMML may bear somatic PTPN11 mutations in their leukemic cells. Subsequently, several groups showed that PTPN11 is the most common mutated target in JMML with somatic PTPN11 mutations noted in approximately 35% of JMML cases (Figure 2).29,57 JMML with PTPN11 mutation is a rapidly fatal disorder unless the patient undergoes transplantation.
PTPN11 encodes SHP-2, a widely expressed cytoplasmic protein tyrosine phosphatase involved in multiple cell signaling processes. Despite its direct function in protein dephosphorylation, SHP-2 generally plays a positive role in transducing signals initiated from receptor and cytosolic kinases. SHP-2 contains two tandem SH-2 domains at the N-terminus and a catalytic PTP domain at the C-terminus. In its inactive state, PTPase activity is repressed by inhibition of the enzymatic cleft by the N-terminal SH-2 domain.58 Binding of the SH-2 domain to phosphorylated tyrosine residues induce a conformational shift that relieves the inhibitory interaction between the SH-2 domain and the catalytic PTP domain.
All PTPN11 mutations identified in JMML or in transient myeloproliferation in Noonan syndrome are missense mutations in the N-terminal SH-2 (exon 3) or PTP interacting surfaces (exon 13), while other exons are commonly mutated in the germline of patients with NS.30 Functionally, leukemia-associated SHP-2 mutants were shown to have significantly higher phospatase activity than mutants identified in NS,29,59 although this correlation did not hold true for all the mutant SHP-2 proteins examined.60 Approximately half of the mice reconstituted with hematopoietic progenitors transduced with leukemic Ptpn11 mutants succumbed to a severe MPD by seven months after transplantation,61 in contrast to a later-onset, non-lethal, chronic myelomonocytic hyperplasia observed in a mouse model bearing a common NS germline PTPN11 mutation.62
Juvenile myelomonocytic leukemia with somatic RAS mutations
The cancer-associated somatic heterozygous RAS mutations typically introduce amino acid substitutions at positions Gly12, Gly13 or Gln61 and lock Ras in the active GTP-bound state by diminishing the intrinsic Ras GTPas activity and/or by causing resistance to GAPs. In JMML, these somatic gain-of-function mutations occur in NRAS and KRAS and are found in leukemic cells at diagnosis in 20–25% of cases.44,50,51,63 Rarely, insertions are noted.64 For both, NRAS- and KRAS-associated disease, acquired loss of the wild-type RAS locus is followed by aggressive disease.65,66
Juvenile myelomonocytic leukemia with somatic KRAS mutations: children with JMML and heterozygous somatic KRAS mutation show a particularly aggressive form of JMML and generally have an urgent indication for HSCT. Similarly, mice expressing oncogenic KrasG12D in hematopoietic cells develop a fatal MPD with 100% penetrance that is characterized by leukocytosis, splenomegaly, and anemia, with death at approximately three months of age.67,68
There is a puzzling interface between KRAS-associated lymphoproliferative disease (RALD) and JMML with KRAS mutation.69,70 Individuals affected by RALD show overexpansion of lymphocytes with hepatosplenomegaly, lymphadenopathy and autoimmune phenomena like autoimmune hemolytic anemia, thrombocytopenia (Evan’s syndrome) and neutropenia. Lanzarotti et al. recently presented a case with RALD as initial presentation at five years of age and fatal JMML at 20 years of age.71 The authors speculate that RALD and JMML are not distinct entities but a continuum with additional genetic or epigenetic events contributing to the clinical phenotype and evolution.
Juvenile myelomonocytic leukemia with somatic NRAS mutations: like KRAS disease, JMML with NRAS mutation is a fatal disease in most instances unless HSCT is performed early in the course of the disease. Matsuda et al. suggested a mild clinical course for the G12S substitution in a number of children with NRAS disease.72 Other investigators agreed that, indeed, an occasional patient with JMML and NRAS mutations had spontaneous resolution of their myeloproliferative disorder despite persistence of the NRAS mutation in hematopoietic cells.44,70 However, there was no specific genotype-phenotype correlation for the different RAS codons. Doisaki et al. claimed that 2 of their patients with JMML, NRAS mutation and a mild and self-limiting course had somatic mosaicism of their oncogenic NRAS mutations.73 Future genetic and epigenetic studies will have to unravel the unusual clinical courses of these few patients with JMML and NRAS mutation. In our own experience, all children with spontaneous resolution of their disease had low HbF levels (see prognostic factors above).
Juvenile myelomonocytic leukemia with Noonan-like CBL syndrome
Based on the observation of UPD 11q in children affected by JMML, homozygous and rarely heterozygous CBL mutations were identified in up to 12% of JMML cases.74–77 Unexpectedly, a high percentage of children with JMML and CBL mutations had been noted to have developmental delay, cryptorchidism and impaired growth.15 In fact, and in contrast to adult MPN, children with JMML and CBL mutations were found to have a germline CBL missense mutation. Like in NF-1, leukemic cells displayed the germline mutation on one allele and acquired LOH on the other allele.15,16,76 Children with this germline CBL disorder display a NS-like phenotpye (Table 1).15,78
CBL, originally discovered as the cellular homolog of the v-Cbl oncogene (reviewed by Kales et al.79), is a member of a family of RING finger ubiquitin ligases that is responsible for the intracellular transport and degradation of a large number of receptor tyrosine kinases but also retains important adaptor functions. It contains a tyrosine kinase binding (TKB) and zinc binding RING finger domain that mediate the E3 ubiquitin ligase activity. These two highly conserved domains are separated by a short linker sequence crucial for ubiquitin ligase activity of CBL. CBL mutations reported in myeloid malignancies uniformly affect either the linker region or the RING finger domain. In JMML, mutations are located throughout the linker and RING finger domain (intron 7, exon 8, intron 8, and exon 9). Loss of ubiquitination of activated receptor tyrosine kinases is thought to contribute to the transforming potential of leukemia-associated mutant CBL proteins. Approximately 150 proteins are associated with or are regulated by CBL. Among these proteins is Grb2, an adaptor molecule that binds to CBL and prevents it from binding to SOS, a known guanine nucleotide exchange factor for RAS.
Many children with germline CBL mutations and JMML experience spontaneous regression of their myeloproliferation despite the persistence of LOH of the CBL locus in hematopoietic cells. Some of these children develop vasculitis-like optic neuritis later in life. Similarly, mice that lacked Cbl in T or B cells developed severe vascular lesions and autoimmunity.80,81 Of note, among children with JMML and CBL mutations treated with HSCT there was a high rate of partial rejection with stable mixed chimerism.15 Since none of these children are currently known to have vasculitis, it is conceivable that stable mixed chimerism is sufficient to improve T- and/or B-cell function. Further studies will have to clarify a possible role of HSCT in JMML with CBL mutations. Currently, most children with JMML and CBL germline mutations are followed closely in the absence HSCT, which is, however, indicated if chromosomal aberrations occur or the disease progresses rapidly.
Other genetic and epigenetic lesions in juvenile myelomonocytic leukemia
Whole exome sequencing in JMML is characterized by a paucity of gene mutations.82 The SETBP1 and JAK3 genes were the most frequently targeted genes mutated in up to 17% of JMML patients. Mutations were often subclonal and were involved in the progression of leukemia.82 Genetic alterations in SRFS2, TET2, RUNX1, JAK2 and ASXL1 do not play a significant role in JMML.51,75,83–86 Similarly, inactivation of the TP53 tumor suppressor gene and internal tandem duplication and activation of the Fms-like tyrosine kinase 3 (FLT3) gene are generally absent in JMML.87,88
With the paucity of genetic lesions, epigenetic changes such as abnormal DNA methylation pattern are of particular interest. Through studying CpG island hypermethylation it could be shown that a high-methylation phenotype characterized an aggressive biological variant of JMML and was an independent predictor of poor outcome in multivariate models.43
Hematopoietic stem cell transplantation
Allogeneic HSCT is still the only curative treatment for most children with JMML, specifically for all children with JMML and NF-1, somatic PTPN11 mutations, close to all children with somatic KRAS mutations, and the vast majority of children with somatic mutations in NRAS. HSCT results in a long-term survival of approximately half of the patients.89–91 Even with HSCT, the malignant JMML clone is difficult to eradicate, and the post-transplant relapse rate is high.
In a large prospective study of childhood JMML by EWOG-MDS, 100 patients were given unmanipulated HSCT after a preparative regimen with busulfan, cyclophosphamide and melphalan.89 The 5-year cumulative incidence of relapse was 35%, only age at diagnosis of over four years independently predicted an increased risk of relapse. The 5-year event-free survival rate over all patients was 52%, and did not differ significantly between patients treated with matched family or matched unrelated donor transplant. Japanese investigators chose a preparative regimen consisting of busulfan, fludarabine and melphalan in an attempt to decrease the mortality rate and reduce the risk of graft failure.92 In the current trial of the Children’s Oncology Group (COG), busulfan, cyclophosphamide, and melphalan is randomized against busulfan and fludarabine.93 Umbilical cord as stem cell source results in post-transplant survival similar to that observed with bone marrow or peripheral stem cells; disease-free survival after a single unmanipulated umbilical cord blood transplant was 43±5%.91
There is clear evidence that in JMML, graft-versus-leukemia (GvL) effect plays an important role.94 Re-emerging recipient cells and frank hematologic relapse have been successfully eradicated by immediate withdrawal of ongoing immunosuppressive therapy.95,96 However, donor lymphocyte infusion in JMML patients who relapsed following HSCT are largely unsuccessful.95,97 Despite the generally aggressive nature of re-emergence of the malignant clone after transplant, approximately30% of children have been cured by a second HSCT.98
Treatment other than stem cell transplantation
Conventional chemotherapy: intensive chemotherapy prior to HSCT does not reduce the relapse rate and nor does splenectomy.89,97 Clinical and hematologic responses in JMML have most consistently been attributed to mercaptopurine, administered either as single agent or in combination with low-dose cytarabine. For life-threatening disease, fludarabine in combination with high-dose cytarbine, or clofarabine may give temporary relief.
Epigenetic therapy: the first case report on the DNA-hypomethylating agent azacitidine in a child with JMML, monosomy 7 and KRAS mutation described an impressive response with clinical and genetic complete remission.99 A number of children have been treated off label since,100 and a phase II study was recently initiated in Europe.
Therapy targeting the RAS/MAPK pathway: initial attempts to develop new pharmacological agents targeted post-translational modifications of RAS proteins required for localization to the inner surface of the plasma membrane. The first obligatory step in this process is addition of a farnesyl moiety catalyzed by the enzyme farnesyl-protein transferase. Since NRAS and KRAS can be geranylgeranylated when farnesyltransferase is inhibited, the introduction of the farnesyltansferase inhibitor tipifarnib (R115777) in JMML was not successful.101
Since the 1990s, downstream drug targets have been studied. RAF-1 inhibitors like sorafenib have a selective sensitivity for most BRAF mutations in melanomas, but can otherwise paradoxically activate RAF kinase activity.102 Selective inhibition of MEK abrogated the myeloproliferative disease in Nf-1 and Kras mutant mice103,104 suggesting that MEK inhibitors might ameliorate hematologic abnormalities in JMML. Although therapeutic windows for pan inhibitors of the MAPK pathway may be limited,1 early clinical studies in JMML are needed to gain more insight into the specific signaling network of this rare disease.
Outlook for the future
There is the legitimate hope that our increasing knowledge on signaling transduction, feed-back loops, and JMML heterogeneity will soon translate into efficacious treatment with novel drugs. However, clinical studies in this rare disorder with genetically and clinically distinct subtypes will be a major challenge. While robust model systems may help to choose the most promising pharmacological agents for clinical evaluation, physicians need to make every effort to allow their patients to be enrolled in prospective clinical trials.
Acknowledgment
I am grateful to Brigitte Schlegelberger, M.D., Hannover, Germany, for critically reviewing the manuscript and to all members of the European Working Group of MDS in Childhood (EWOG-MDS) for their continuous collaboration.
Footnotes
This review article was originally published in the education book of the 19th congress of EHA (June 2014).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras Back in the Ring. Cancer Cell. 2014;25:272–81. [DOI] [PubMed] [Google Scholar]
- 2.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Current Opinion in Genetics & Development. 2009;19:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381: 333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aoki Y, Matsubara Y. Ras/MAPK syndromes and childhood hematooncological diseases. Int J Hematol. 2013;97:30–6. [DOI] [PubMed] [Google Scholar]
- 5.Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33. [DOI] [PubMed] [Google Scholar]
- 6.Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: a long-term follow-up study. Archives of Disease in Childhood. 2007; 92:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharland M, Burch M, Mckenna WM, Paton MA. A Clinical-Study of Noonan Syndrome. Archives of Disease in Childhood. 1992;67: 178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tartaglia M, Kalidas K, Shaw A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002;70:1555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–4. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–9. [DOI] [PubMed] [Google Scholar]
- 11.Schubbert S, Bollag G, Lyubynska N, et al. Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol. 2007;27:7765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12. [DOI] [PubMed] [Google Scholar]
- 13.Razaque MA, Nishizawa T, Komoike Y, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–7. [DOI] [PubMed] [Google Scholar]
- 14.Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause develop mental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuda K, Taira C, Sakashita K, et al. Long-term survival after nonintensive chemotherapy in some juvenile myelomonocytic leukemia patients with CBL mutations, and the possible presence of healthy persons with the mutations. Blood. 2010;115:5429–31. [DOI] [PubMed] [Google Scholar]
- 17.Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet. 2003;117C:42–8. [DOI] [PubMed] [Google Scholar]
- 19.Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. [DOI] [PubMed] [Google Scholar]
- 21.Niihori T, Aoki Y, Narumi Y, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–6. [DOI] [PubMed] [Google Scholar]
- 22.Brems H, Chmara M, Sahbatou M, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–6. [DOI] [PubMed] [Google Scholar]
- 23.Quaio CR, Carvalho JF, da Silva CA, et al. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med Genet A. 2012;158A:1077–82. [DOI] [PubMed] [Google Scholar]
- 24.Bader-Meunier B, Cave H, Jeremiah N, et al. Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Case report and systematic review of the literature. Semin Arthritis Rheum. 2013;43:217–9. [DOI] [PubMed] [Google Scholar]
- 25.Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157C:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasle H. Malignant diseases in Noonan syndrome and related disorders. Horm Res. 2009;72(Suppl 2):8–14. [DOI] [PubMed] [Google Scholar]
- 27.Bader-Meunier B, Tchernia G, Mielot F, et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130:885–9. [DOI] [PubMed] [Google Scholar]
- 28.Choong K, Freedman MH, Chitayat D, et al. Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol. 1999;21:523–7. [PubMed] [Google Scholar]
- 29.Tartaglia M, Niemeyer CM, Song X, et al. Somatic PTPN11 mutations in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. [DOI] [PubMed] [Google Scholar]
- 30.Kratz CP, Niemeyer CM, Castleberry RP, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106:2183–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasle H, Wadsworth LD, Massing BG, McBride M, Schultz KR. A population-based study of childhood myelodysplastic syndrome in British Columbia, Canada. Br J Haematol. 19991027–32. [DOI] [PubMed] [Google Scholar]
- 32.Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood. 1997;89:3534–43. [PubMed] [Google Scholar]
- 33.Luna-Fineman S, Shannon KM, Atwater SK, et al. Myelodysplastic and myeloproliferative disorders of childhood: a study of 167 patients. Blood. 1999;93:459–66. [PubMed] [Google Scholar]
- 34.Passmore SJ, Hann IM, Stiller CA, et al. Pediatric myelodysplasia: a study of 68 children and a new prognostic scoring system. Blood. 1995;85:1742–50. [PubMed] [Google Scholar]
- 35.Karow A, Baumann I, Niemeyer CM. Morphologic differential diagnosis of juvenile myelomonocytic leukemia--pitfalls apart from viral infection. J Pediatr Hematol. Oncol. 2009;31:380. [DOI] [PubMed] [Google Scholar]
- 36.Yoshimi A, Kamachi Y, Imai K, et al. Wiskott-Aldrich syndrome presenting with a clinical picture mimicking juvenile myelomonocytic leukaemia. Pediatr Blood Cancer. 2013;60:836–41. [DOI] [PubMed] [Google Scholar]
- 37.Unal S, Cetin M, Kutlay NY, et al. Hemophagocytosis associated with leukemia: a striking association with juvenile myelomonocytic leukemia. Ann Hematol. 2010;89:359–64. [DOI] [PubMed] [Google Scholar]
- 38.Borkhardt A, Bojesen S, Haas OA, et al. The human GRAF gene is fused to MLL in a unique t(5;11)(q31;q23) and both alleles are disrupted in three cases of myelodysplastic syndrome/acute myeloid leukemia with a deletion 5q. Proc Natl Acad Sci USA. 2000; 97:9168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rottgers S, Gombert M, Teigler-Schlegel A, et al. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia. 2010;24:1197–200. [DOI] [PubMed] [Google Scholar]
- 40.Buijs A, Bruin M. Fusion of FIP1L1 and RARA as a result of a novel t(4;17)(q12;q21) in a case of juvenile myelomonocytic leukemia. Leukemia. 2007;21:1104–18. [DOI] [PubMed] [Google Scholar]
- 41.Mizoguchi Y, Fujita N, Taki T, Hayashi Y, Hamamoto K. Juvenile myelomonocytic leukemia with t(7;11)(p15;p15) and NUP98-HOXA11 fusion. Am J Hematol. 2009;84: 295–7. [DOI] [PubMed] [Google Scholar]
- 42.Bresolin S, Zecca M, Flotho C, et al. Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol. 2010;28:1919–27. [DOI] [PubMed] [Google Scholar]
- 43.Olk-Batz C, Poetsch AR, Nollke P, et al. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood. 2011;117:4871–80. [DOI] [PubMed] [Google Scholar]
- 44.Flotho C, Valcamonica S, Mach-Pascual S, et al. Ras mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia. 1999;13:32–7. [DOI] [PubMed] [Google Scholar]
- 45.Busque L, Gilliland DG, Prchal JT, et al. Clonality in juvenile chronic myelogenous leukemia. Blood. 1995;85:21–30. [PubMed] [Google Scholar]
- 46.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925–9. [PubMed] [Google Scholar]
- 47.Kotecha N, Flores NJ, Irish JM, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hasegawa D, Bugarin C, Giordan M, et al. Validation of flow cytometric phospho-STAT5 as a diagnostic tool for juvenile myelomonocytic leukemia. Blood Cancer J. 2013;3:e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gandre-Babbe S, Paluru P, Aribeana C, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013;121:4925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez B, Kosmider O, Cassinat B, et al. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:460–8. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida N, Yagasaki H, Xu YY, et al. Correlation of Clinical Features With the Mutational Status of GM-CSF Signaling Pathway-Related Genes in Juvenile Myelomonocytic Leukemia. Pediatr Res. 2009;65:334–40. [DOI] [PubMed] [Google Scholar]
- 52.Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12: 144–8. [DOI] [PubMed] [Google Scholar]
- 53.Stephens K, Weaver M, Leppig KA, et al. Interstitial uniparental isodisomy at clustered breakpoint intervals is a frequent mechanism of NF1 inactivation in myeloid malignancies. Blood. 2006;108:1684–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flotho C, Steinemann D, Mullighan CG, et al. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene. 2007;26:5816–21. [DOI] [PubMed] [Google Scholar]
- 55.Steinemann D, Arning L, Praulich I, et al. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica. 2010; 95:320–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103:4243–50. [DOI] [PubMed] [Google Scholar]
- 57.Loh ML, Vattikuti S, Schubbert S, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–31. [DOI] [PubMed] [Google Scholar]
- 58.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. [DOI] [PubMed] [Google Scholar]
- 59.Fragale A, Tartaglia M, Wu J, Gelb BD. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat. 2004;23:267–77. [DOI] [PubMed] [Google Scholar]
- 60.Niihori T, Aoki Y, Ohashi H, et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J Hum Genet. 2005;50:192–202. [DOI] [PubMed] [Google Scholar]
- 61.Mohi MG, Williams IR, Dearolf CR, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–91. [DOI] [PubMed] [Google Scholar]
- 62.Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–57. [DOI] [PubMed] [Google Scholar]
- 63.Miyauchi J, Asada M, Sasaki M, et al. Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood. 1994;83:2248–54. [PubMed] [Google Scholar]
- 64.Reimann C, Arola M, Bierings M, et al. A novel somatic K-Ras mutation in juvenile myelomonocytic leukemia. Leukemia. 2006; 20:1637–8. [DOI] [PubMed] [Google Scholar]
- 65.Kato M, Yasui N, Seki M, et al. Aggressive transformation of juvenile myelomonocytic leukemia associated with duplication of oncogenic KRAS due to acquired uniparental disomy. J Pediatr. 2013;162(6):1285–8, 1288.e1. [DOI] [PubMed] [Google Scholar]
- 66.Matsuda K, Nakazawa Y, Sakashita K, et al. Acquisition of loss of the wild-type NRAS locus with aggressive disease progression in a patient with juvenile myelomonocytic leukemia and a heterozygous NRAS mutation. Haematologica. 2007;92:1576–8. [DOI] [PubMed] [Google Scholar]
- 67.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan IT, Kutok JL, Williams IR, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niemela JE, Lu L, Fleisher TA, et al. Somatic KRAS mutations associated with a human non-malignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117(10):2883–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takagi M, Shinoda K, Piao JH, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–90. [DOI] [PubMed] [Google Scholar]
- 71.Lanzarotti N, Bruneau J, Trinquand A, et al. RAS-associated lymphoproliferative disease evolves into severe juvenile myelomonocytic leukemia. Blood. 2014;123:1960–3. [DOI] [PubMed] [Google Scholar]
- 72.Matsuda K, Sakashita K, Taira C, et al. Quantitative assessment of PTPN11 or RAS mutations at the neonatal period and during the clinical course in patients with juvenile myelomonocytic leukaemia. Br J Haematol. 2010;148:593–9. [DOI] [PubMed] [Google Scholar]
- 73.Doisaki S, Muramatsu H, Hama A, et al. A favorable outcome in children with juvenile myelomonocytic leukemia (JMML) with RAS mutations [abstract]. Blood. 2010; 116:21. [Google Scholar]
- 74.Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009;114: 1859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muramatsu H, Makishima H, Jankowska AM, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010;115:1969–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perez B, Mechinaud F, Galambrun C, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet. 2010;47:686–91. [DOI] [PubMed] [Google Scholar]
- 77.Shiba N, Kato M, Park MJ, et al. CBL mutations in juvenile myelomonocytic leukemia and pediatric myelodysplastic syndrome. Leukemia. 2010;24:1090–2. [DOI] [PubMed] [Google Scholar]
- 78.Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet. 2010;87:250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kales SC, Ryan PE, Nau MM, Lipkowitz S. Cbl and human myeloid neoplasms: the Cbl oncogene comes of age. Cancer Res. 2010;70:4789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naramura M, Jang IK, Kole H, et al. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–9. [DOI] [PubMed] [Google Scholar]
- 81.Kitaura Y, Jang IK, Wang Y, et al. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases cbl and Cbl-b. Immunity. 2007;26:567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sakaguchi H, Okuno Y, Muramatsu H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45:937–41. [DOI] [PubMed] [Google Scholar]
- 83.Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119:e96–e99. [DOI] [PubMed] [Google Scholar]
- 84.Sugimoto Y, Muramatsu H, Makishima H, et al. Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br J Haematol. 2010;150: 83–7. [DOI] [PubMed] [Google Scholar]
- 85.Masunaga A, Mitsuya T, Kadofuku T, et al. Mutation analysis of AML1 gene in pediatric primary myelodysplastic syndrome and juvenile myelomonocytic leukemia. Leuk Res. 2008;32:995–7. [DOI] [PubMed] [Google Scholar]
- 86.Zecca M, Bergamaschi G, Kratz C, et al. JAK2 V617F mutation is a rare event in juvenile myelomonocytic leukemia. Leukemia. 2007;21:367–9. [DOI] [PubMed] [Google Scholar]
- 87.Xu F, Taki T, Yang HW, et al. Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children. Br J Haematol. 1999;105:155–62. [PubMed] [Google Scholar]
- 88.de Vries AC, Stam RW, Schneider P, et al. Role of mutation independent constitutive activation of FLT3 in juvenile myelomonocytic leukemia. Haematologica. 2007;92: 1557–60. [DOI] [PubMed] [Google Scholar]
- 89.Locatelli F, Nollke P, Zecca M, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105:410–9. [DOI] [PubMed] [Google Scholar]
- 90.Manabe A, Okamura J, Yumura-Yagi K, et al. Allogeneic hematopoietic stem cell transplantation for 27 children with juvenile myelomonocytic leukemia diagnosed based on the criteria of the International JMML Working Group. Leukemia. 2002;16:645–9. [DOI] [PubMed] [Google Scholar]
- 91.Locatelli F, Crotta A, Ruggeri A, et al. Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood. 2013;122:2135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yabe M, Sako M, Yabe H, et al. A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant. 2008;12:862–7. [DOI] [PubMed] [Google Scholar]
- 93.Dvorak CC, Loh ML. Juvenile Myelomonocytic Leukemia: Molecular Pathogenesis Informs Current Approaches to Therapy and Hematopoietic Cell Transplantation. Front Pediatr. 2014;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Locatelli F, Niemeyer C, Angelucci E, et al. Allogenic bone marrow transplantation for chronic myelomonocytic leukemia in childhood: A report from the European Working Group on Myelodysplastic Syndrome in Childhood. J Clin Oncol. 1997;15:566–73. [DOI] [PubMed] [Google Scholar]
- 95.Yoshimi A, Niemeyer CM, Bohmer V, et al. Chimaerism analyses and subsequent immunological intervention after stem cell transplantation in patients with juvenile myelomonocytic leukaemia. Br J Haematol. 2005;129:542–9. [DOI] [PubMed] [Google Scholar]
- 96.Tanoshima R, Goto H, Yanagimachi M, et al. Graft versus leukemia effect against juvenile myelomonocytic leukemia after unrelated cord blood transplantation. Pediatr Blood Cancer. 2008;50:665–7. [DOI] [PubMed] [Google Scholar]
- 97.Matthes-Martin S, Mann G, Peters C, et al. Allogeneic bone marrow transplantation for juvenile myelomonocytic leukaemia: a single centre experience and review of the literature. Bone Marrow Transplant. 2000;26: 377–82. [DOI] [PubMed] [Google Scholar]
- 98.Yoshimi A, Mohamed M, Bierings M, et al. Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia. 2007;21:556–60. [DOI] [PubMed] [Google Scholar]
- 99.Furlan I, Batz C, Flotho C, et al. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood. 2009;113:2867–8. [DOI] [PubMed] [Google Scholar]
- 100.Cseh A, Niemeyer CM, Catalá A, et al. Therapy with azacitidine in pediatric MDS and JMML A retrospective survey of the EWOG-MDS study. [abstract]. Haematologica 2012;97(s3):S6. [Google Scholar]
- 101.Castleberry R, Mignon L, Jayaprakash N, et al. Phase II Window study of the farnesyltransferase inhibitor R115777 (Zarnestra®) in untreated juvenile myelomonocytic leukemia (JMML): a children’s oncology group study. [abstract]. Blood. 2005;106(11): 2587. [Google Scholar]
- 102.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9. [DOI] [PubMed] [Google Scholar]
- 103.Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123:335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lyubynska N, Gorman MF, Lauchle JO, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3:76ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]