

The distinction between plasmablastic lymphoma (PBL) and plasmacytoma may be confidently made on the basis of histopathological and clinical features. However, some cases can be challenging owing to a morphological overlap. The main significant differences include genetic aberrations [i.e. t(11;14); t(4;14), hyperdiploidy], and in particular the presence of Epstein-Barr virus (EBV)-encoded RNA (EBER), which is detectable in 75–100% of all PBL cases, in contrast to plasmacytoma (10% of cases).1 The exact role of EBV in the pathogenesis of PBL is still poorly understood.2 Although it has been suggested that PBL may occur as a high-grade transformation from a pre-existing plasmacytoma following EBV infection, this concept has not been well established and definitive proof is lacking.3,4
In December 2011, a 74-year old man presented for a right maxillary sinus solitary mass, infiltrating the oral cavity. Histological examination of the bioptic specimen showed a uniform proliferation of plasma cells with abundant cytoplasm, round eccentrically located nuclei and clock-face chromatin without nucleoli (Figure 1A). Neoplastic cells expressed CD38, CD138, CD79a, MUM-1; they were restricted for immunoglobulin heavy chain G (IgG) and light chain lambda (Igλ). CD20 and PAX-5 were negative. The proliferative index (Ki-67) was approximately 10%. A complete workup with computer tomography (CT) scan and bone marrow biopsy excluded other sites of involvement; therefore, the final diagnosis was extraosseous plasmacytoma. The patient was treated with one cycle of chemotherapy with: bortezomib 1.3 mg/m2 Days 1, 4, 8, 11; melphalan 9 mg/m2 Day 1–4; and prednisone 50 mg Days 1, 4, 8, 11 (VMP scheme), obtaining a 90% reduction of the mass. Subsequently, a 50 Gy of involved field radiotherapy treatment led to a complete remission, which was confirmed at CT/positron emission tomography. Ten months later the patient relapsed. The histological evaluation of the surgical specimen confirmed the previous diagnosis; however, an increase in the proliferative index was detected (Ki-67 approximately 40%). The patient was treated again with bortezomib associated with cyclophosphamide 500 mg Days 1, 8, and dexamethasone 20 mg Days 1, 4, 8, 11, achieving only a minimal response. Therefore, he was switched to a third-line therapy with lenalidomide 25 mg Days 1 and 21, cyclophosphamide 750 mg Days 1 and 8, and dexamethasone 20 mg every week (RCD scheme), obtaining an excellent but short response with a rapid growth of the mass in May 2013. The patient underwent surgical excision of the neoplastic lesion; laboratory tests showed hemoglobin 13.9 g/dL, white blood cell count 8.32 × 109/L, platelets 408 × 109/L. The histological examination of the specimen showed a diffuse and cohesive monomorphic proliferation of round- to oval-shaped large cells with scant cytoplasm, and prominent nucleoli (Figure 1D). Neoplastic cells were CD38, CD138 and MUM-1 positive, IgG/λ restricted, and CD20, CD79a, and PAX5 negative. The proliferative index (Ki-67) was approximately 95%. EBER was present in 90% of the neoplastic cells (Figure 1E). All of the neoplastic cells expressed the MYC protein. FISH analysis using dual-color break-apart probes did not identify any of the known translocations involving the MYC gene. To evaluate whether we were dealing with the transformation of the pre-exisisting plasmacytoma, CD56 and cyclin D1 positivity was checked in all the specimens.5 Both plasmacytomas and plasmablastic samples expressed CD56, whereas no cyclin D1 staining was identified. In addition, IGH-VDJ rearrangement was performed in all the specimens according to BIOMED-2 protocol by using framework regions 2 (FR2VH) and JH regions primers (VHFR2-JH).6 A productive IGHV rearrangement (VH3-23, DH3-22, JH4*02) with 93.27% identity of germ-line sequence was detected, thus confirming the clonal relationship of all the samples (Figure 1C and F). The final diagnosis was plasmablastic transformation of pre-existing plasmacytoma of the oral cavity. The patient was treated with liposomial doxorubicin 50 mg/m2 Day 1 and cytosine arabinoside 2 g Days 1–2, obtaining a minimal response. Thus, bendamustine 90 mg/m2 Days 1–2 for two cycles was administrated but with no response.
Figure 1.
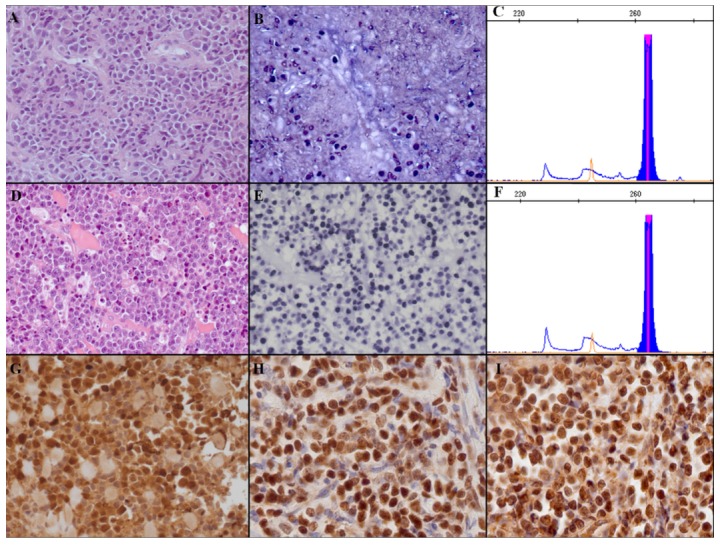
Morphological features of the samples. (A) A uniform population of plasma cells with abundant cytoplasm, round eccentrally located nuclei and clock-face chromatin without nucleoli is observed. (B) A small percentage of tumor cells are EBER-positive. (C) Neoplastic population is characterized by monoclonal IGHV rearrangement. (D) Histological specimen shows a diffuse and cohesive proliferation of round- to oval-shaped large cells with scant cytoplasm, and prominent nucleoli. (E) EBER positivity is observed in 90% of the neoplastic cells. (F) The same IGHV rearrangement of the plasmacytoma samples is shown. (G) EBNA1, and (H) Ea-D and (I) Ea-R expression in blastic cells of the plasmablastic lymphoma sample is present. [A, D: Haematoxylin and Eosin; B, E: EBER in situ hybridization; G: EBNA1 stain; H: Ea-D stain; I: Ea-R stain. A-B, D–E, G–I: original magnification: 20×].
Since EBV reactivation has been observed following bortezomib therapy,7 it was hypothesized that EBV was the trigger responsible for the transformation of plasmacytoma in PBL, determining the multiple rapid relapses of the disease. Therefore, we checked EBV also in the plasmacytoma samples. Interestingly, in these cases, only a small percentage of tumor cells with blastic appearance were EBER-positive, increasing from the first (10%) to the second (30%) biopsy (Figure 1B). In addition, the first sample was negative for MYC, whereas in the second specimen 30% of the neoplastic cells expressed the protein. Since the oncogenic role of EBV is controversial, and the type of viral program expressed by plasmablastic lymphoma is still a matter of debate, a complete genetic assessment of the virus latency and lytic genes by RT-qPCR and immunohistochemistry was performed. The expression of EBNA1 and the negativity for LMP-1 and EBNA-2 in all the three biopsies suggested a type I latency (Figure 1G). In the third biopsy, we detected expression of some genes/proteins involved in the lytic cycle (BZLF1, BMRF1, BHRF1 coding for the Zebra, Ea-D and Ea-R/p17 proteins, respectively) (Figure 1H–I). The fact that we did not find BLLF1- gp350/220 expression suggests an abortive lytic cycle in neoplastic cells.8 It is known that the reactivation of lytic cycle may play a role in EBV-driven lymphomagenesis by increasing the total number of latently infected cells.9 In addition, viral lytic genes, or cellular genes induced by viral lytic proteins could potentially encode paracrine factors that promote tumor growth.10 A number of physiological stimuli and chemical agents may favor the lytic form of EBV infection, including B-cell receptor stimulation, transforming growth factor-β, hypoxia and DNA damage.11 Since no response has been obtained by conventional therapy, and considering the high viral load in the blood (264,000 DNA copies/mL), the same therapy of post-transplant lymphoproliferative disorders (PTLD) was performed. Accordingly, rituximab 375 mg/m2 weekly for three doses and acyclovir were added to a continuous lenalidomide treatment Days 1–28 per month at 15 mg/day with the aim of: i) increasing potential response; ii) reducing the reservoir of latently infected cells; and iii) decreasing viral burden. Emerging data have suggested that rituximab may have therapeutic value also in patients with tumors which do not express CD20 by eliminating reactive B lymphocytes in the microenvironment that may support the growth and survival of neoplastic cells.12,13 Depleting reactive B cells may also enhance the anti-lymphoma immune response. As far as the rationale of using acyclovir is concerned, recent data on nasopharyngeal carcinoma and glioblastoma indicate that antiviral drugs may improve outcomes in viral-associated tumors by inhibiting lytic replication.14
In our case, the therapy induced a marked reduction of the mass, in parallel to a decrease in viral load (12,250 DNA copies/mL) (Figure 2A and B). At the last follow up (eleven months later) the patient was in complete remission.
Figure 2.
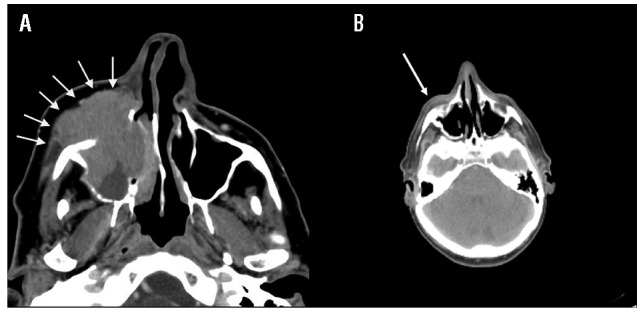
Head computed tomography findings. A reduction of the mass is observed between the first (A) and the second (B) head computed tomography (arrows).
In conclusion, the importance of our case consists in drawing attention to two main issues. First of all, it should be underlined that MYC protein expression can occur also in the absence of MYC translocation and may probably be due to alternative mechanisms, likely involving microRNA (miRNA) dysregulation. In our case, a downregulation of some miRNAs known to regulate MYC expression (i.e. hsa-miR34b, hsa-miR155, hsa-let7a, hsa-miR29a and -29b) has been detected, in comparison to normal lymph node and to MYC translocation-positive Burkitt lymphoma cases (Figure 3).15 This finding may explain the upregulation of MYC in the absence of the MYC translocation. On the other hand, the pathologist should consider the possibility of staining all plasmacytomas and probably all plasma cell myelomas for EBV in order to identify those patients at higher risk of transformation to PBL, which require a totally different and more aggressive therapeutic protocol and a closer follow up. In those patients in whom plasmacytoma de-differentiate to PBL, the therapeutic regimens used in PTLD to reduce the reservoir of latently infected cells and to inhibit lytic replication should be taken into account to improve the prognosis. Enhanced understanding of the multiple mechanisms regulating EBV reactivation will, hopefully, lead to the development of minimally toxic, highly specific therapies. The use of routine chemotherapy in combination with acyclovir appears to be a promising approach for treating patients with EBV-positive tumors.
Figure 3.
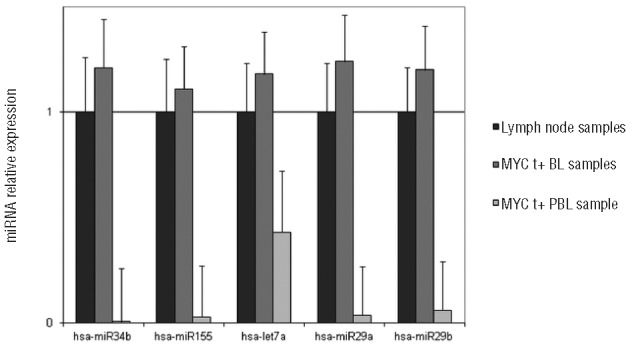
microRNAs relative expression level. In plasmablastic sample (PBL) a downregulation of hsa-miR34b, hsa-miR155, hsa-let7a, hsa-miR29a and -29b is detected, in comparison to normal lymph node and to MYC translocation-positive (MYC t+) Burkitt lymphoma (BL) samples.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.McKenna RW, Kyle RA, Kuehl WM, Gorgan TM, Harris NL, Coupland RW. Plasmablastic lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA. (eds). WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 4th edn. Lyon, France: IARC; 2008, pp 256–7. [Google Scholar]
- 2.Valera A, Balagué O, Colomo L, Martinez A, Delabie J, Taddesse-Heath L, et al. IG/MYC rearrangements are the main cytogenetic alteration in Plasmablastic lymphomas. Am J Surg Pathol. 2010;34(11):1686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saito M, Morioka M, Izumiyama K, Mori A, Irie T, Tanaka M, et al. Epstein-Barr virus-positive ileal extraosseous plasmacytoma containing Plasmablastic lymphoma component with CD20-positive lymph node involvement. Int J Gen Med. 2012;5:715–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qing X, Sun N, Chang E, French S, Ji P, Yue C. Plasmablastic lymphoma may occur as a high-grade transformation from plasmacytoma. Exp Mol Pathol. 2011;90(1):85–90. [DOI] [PubMed] [Google Scholar]
- 5.Colomo L, Loong F, Rives S, Pittaluga S, Martínez A, López-Guillermo A, et al. Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am J Surg Pathol. 2004;28(6):736–47. [DOI] [PubMed] [Google Scholar]
- 6.Van Dongen JJ, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003;17(12):2257–317. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Farre B, Rovira J, Martinez D, Valera A, Garcia-Herrera A, Marcos MA, et al. In vivo intratumoral Epstein-Barr virus replication is associated with XBP1 activation and early-onset post-transplant lymphoproliferative disorders with prognostic implications. Mod Pathol. 2014. April 25 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 8.Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. 2005; 79(22):13993–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma SH, Hedge S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. 2011;85(1):165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forte E, Luftig MA. The role of microRNAs in Epstein-Barr virus latency and lytic reactivation. Microbes and Infection. 2011;13(14–15):1156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenney SC, Mertz JE. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol. 2014. January 20 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Younes A, Oki Y, McLaughlin P, Copeland AR, Goy A, Pro B, et al. Phase 2 study of rituximab plus ABVD in patients with newly diagnosed classical Hodgkin lymphoma. Blood. 2012;119(18):4123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasamon YL, Jacene HA, Gocke CD, Swinnen LJ, Gladstone DE, Perkins B, et al. Phase 2 study of rituximab-ABVD in classical Hodgkin lymphoma. Blood. 2012;119(18):4129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soderberg-Nauclér C, Rahbar A, Stragliotto G. Survival in patients with Glioblastoma receiving valganciclovir. N Engl J Med. 2013; 369(10):985–6. [DOI] [PubMed] [Google Scholar]
- 15.Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C, et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008;216(4):440–50. [DOI] [PubMed] [Google Scholar]