

Abstract
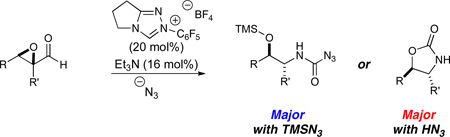
We report herein a nucleophilic carbene catalyzed redox azidation of epoxyaldehydes. The intermediate β-hydroxy acyl azides undergo thermal Curtius rearrangement followed by trapping with excess azide to form carbamoyl azides or, in a complementary sequence, by the hydroxy group to form oxazolidinones. Both products are formed in modest to good yields and diastereoselectivities. The use of an enantioenriched triazolium catalyst leads to modest asymmetric induction.
Introduction
The discovery of new reaction manifolds leading to improved selectivity and efficiency is an ongoing challenge. Specifically, reactions that manipulate standard organic functional groups by inducing a reversal of normal reactivity, termed umpolung,1 hold considerable promise due to a nonexistent background reaction. N-heterocyclic carbenes2 (NHC’s) have been shown to catalytically convert aldehydes into acyl anion equivalents under mild conditions in transformations such as the benzoin3,4 and Stetter5,6,7 reactions, along with a host of others.8
The desirability of catalytic formation of products in reagent efficient processes has increased the investigations of N-heterocyclic carbene reactivity. In concurrent seminal work, we9 and Bode10 demonstrated that α-reducible aldehydes lead to a novel reaction pathway under NHC catalysis in the presence of alcohols as nucleophiles. Upon formation of the acyl anion equivalent, redox reactivity occurs, resulting either in halide elimination,9 epoxide/aziridine opening,10 or “homoenolate/homoynolate ” protonation11 or electrophile trapping,12 with concomitant formation of an acyl azolium. The addition of an alcohol leads to catalyst turnover, giving the desired product. The aldehydes that have been successfully demonstrated to participate include α-halo aldehydes, epoxyaldehydes, aziridinoaldehydes, cyclopropanecarboxaldehydes,13 enals and ynals. Useful nucleophiles include alcohols, phenols and various amines, the latter only if coupled with a co-catalytic amount of typical acyl transfer agents such as 1-hydroxy-7-aza-benzotriazole (HOAt) or imidazole.14
We became interested in the formation of derivatized 1,2-amino alcohols, common pharmacophores, ligands, chiral auxiliaries and motifs in natural products.15 We considered that the use of azide as a nucleophile16 in this redox manifold would convert an epoxyaldehyde into a β-hydroxy acyl azide. Thermally induced Curtius rearrangement17 should proceed facilely to form a hydroxy isocyanate which would presumably cyclize to the oxazolidinone in the presence of non-nucleophilic solvent or to the hydroxycarbamate in an alcoholic medium. Importantly, the mechanism requires a net addition of HN3, or its silylated analog TMSN3, each of which is readily accessible and should have the desired reactivity to enable this sequence to occur (Scheme 2). We were attracted both by the facile access to amino alcohol derivatives but also by the potential to generate and control a newly formed carbinolamine stereocenter (II to III, Scheme 2) with the ultimate possibility of catalyst control in an asymmetric synthesis of branched carbinolamines.18,19
Scheme 2.

Plausible reaction pathway
Results and Discussion
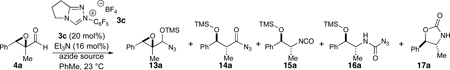
Initial investigations were focused on the reaction of epoxyaldehyde 4a in the presence of TMSN3 as the azidation agent. Preliminary results suggested a number of competing pathways were present, greatly complicating analysis of the reaction. Uncatalyzed (or weak nucleophile catalyzed) direct addition of TMSN3 to the aldehyde occurs under conditions which generate little or no active carbene providing silylated azidohydrin 13a. The productive reaction affords acyl azide 14a which undergoes slow Curtius rearrangement to isocyanate 15a. In the presence of excess TMSN3, isocyanate 15a readily affords carbamoyl azide 16a. Small amounts of oxazolidinone 17a were also formed under some of these reaction conditions. To complicate matters further, we noted a discernible difference in the rate of the Curtius rearrangement for the two diastereomers of the acyl azide as well as a difference in the rate of oxazolidinone formation for the corresponding isocyanates. The reaction analysis was thus complicated during initial screening phase and we sought to simplify the reaction in order to make useful conclusions. As such, the use of excess TMSN3 and long reaction times drives the formation of carbamoyl azide as the dominant product (entry 1 in Table 1), while the use of TESN3 affords no improvement. The addition of NaN3 upon consumption of epoxyaldehyde, which usually happens within 30 minutes of reaction initiation, provides shorter reaction times and slightly cleaner reaction mixtures (entry 3, Table 1).
Table 1.
Reaction optimization
 | |||||
|---|---|---|---|---|---|
| Entrya | Azide source | Time(h) | Product | drb | Product(%) |
| 1 | TMSN3 (2.5 equiv) | 20 | 16a | 5:1 | 82 |
| 2c | TESN3 (2.5 equiv) | 20 | 16a(TES) | 4:1 | 40 |
| 3c | TMSN3 (2.5 equiv)/ NaN3(1.0 equiv) | 12 | 16a | 4:1 | 78 |
| 4 | TMSN3/EtOH(1:1, 1 equiv) | 20 | 17a | 3.4:1 | 75 |
| 5 | TMSN3/EtOH/NaN3(1:1:1, 1 equiv "HN3") | 4 | 17a | 4:1 | 65 |
Catalyst 3c (20 mol %), NEt3 (16 mol %), PhMe (0.1 M), 23 °C.
Determined by 1H NMR.
Tert-butanol (5 equiv relative to substrate) added to help solubilize salts.
Complementary reactivity was observed from the in situ generation of hydrazoic acid by using equimolar ratios of TMSN3 and EtOH. Here, direct addition to form the azidohydrin is reversible and cyclization to the oxazolidinone is more rapid than the corresponding intermolecular addition of azide to form the carbamoyl azide (entry 4, Table 1). The use of NaN3, again leads to improved reactions through shorter reaction times and comparable yields and diastereoselectivities (entry 5, Table 1).
To further optimize the selectivity of the reaction by addressing the charge stabilization of the intermediates by solvation, various solvents were screened with toluene emerging optimal.20 A base screen revealed that DBU (pKa = 12), Et3N (pKa = 10.75), and imidazole (pKa = 6.95) each afford identical diastereoselectivities with nearly approximate yields, suggesting enol/enolate protonation does not occur via the ammonium.
Catalyst Effects on Selectivity
It has been demonstrated that the steric and electronic components of the azolium salt precursor, and particularly N-aryl substitution,8g affects the stability and reactivity of the carbene. The steric bulk of the azolium salt can provide significant steric influence on rate and selectivity of the reaction. As the basicity of the carbene can contribute to epimerization, various N-heterocyclic carbene precursors were investigated (Table 2). In order to ensure the best chance of generating an active catalyst from these electronically dissimilar azolium salts (of varying acidity) and given the insensitivity of the reaction to base structure, we chose to conduct this screen with the strong amine base DBU. Imidazolium 10 did not provide any reactivity under the reaction conditions (entry 1, Table 2), while the thiazolium catalyst 6 and the electron-rich mesityl triazolium 3b provide minimal selectivity (entries 2-3, Table 2). The electron-neutral triazolium 3a showed increased selectivity and a good yield (entry 4, Table 2). Finally, the electron-deficient triazolium 3c gave the best combination of selectivity and yield (entry 5, Table 2).
Table 2.
Catalyst Effect
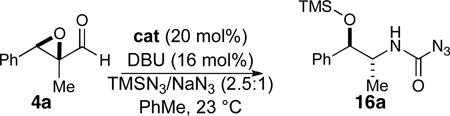
Catalyst (20 mol %), DBU (16 mol %), PhMe (0.1 M), 23 °C.
Determined by 1H NMR.
A variety of epoxyaldehydes were synthesized in an effort to determine substrate scope. Increasing the size of the alpha substituent to pentyl from methyl leads to improved diastereoselectivity under both TMSN3 and HN3 conditions (entries 2 and 10, Table 3). The use of the bulkier but more electron-withdrawing phenyl ring at the α-positions leads to poor selectivity. This is most likely due to the increased acidity of the α-proton at that position in the intermediate acyl azide. Varying the electronics and sterics of the β-phenyl group affords similar results (entries 4, 5, 12 and 13, Table 3). One exception to this is the use of 1-naphthyl which gives low yields and poor diastereoselectivities under TMSN3 conditions in a reaction complicated by competing formation of the oxazolidinone even under this protocol (entry 6, Table 3). Hydrazoic acid conditions with this substrate provide improved selectivities and yields (entry 14, Table 3). Aliphatic substitution at the beta position leads to depressed diastereoselectivities (entries 7 and 15, Table 3). Opening of the oxazolidinone ring to obtain protected and unprotected 1,2-amino alcohols is well precedented providing access to variety of structurally distinct motifs.21
Table 3.
Substrate Scope
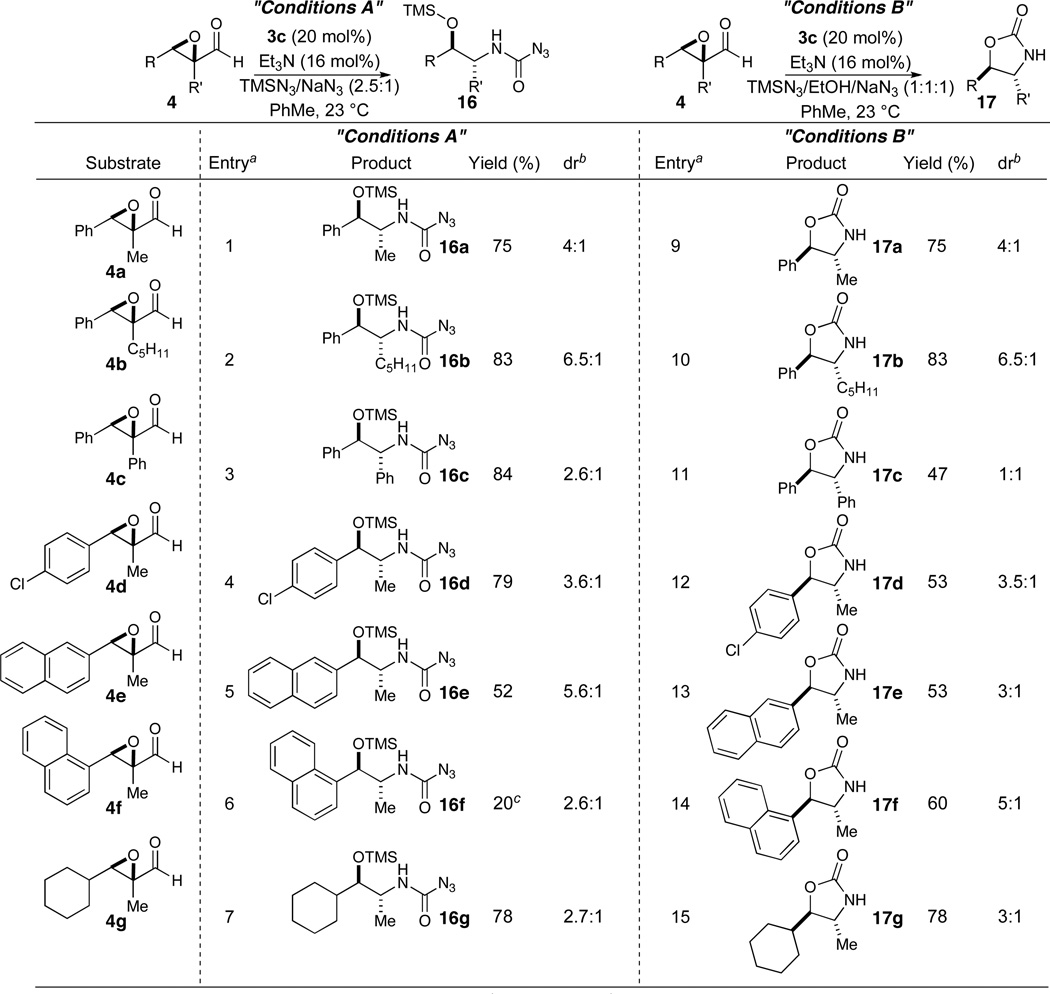 |
Catalyst 3c (20 mol %), NEt3 (16 mol %), PhMe (0.1 M), 23 °C.
Determined by 1H NMR.
Oxazolidinone 17f also formed in 56% yield and 5.6:1 dr.
Asymmetric Induction
The use of α-halo aldehydes as substrates allows us to investigate an enantioselective approach to α-branched carbinolamines relying on an asymmetric protonation event should a chiral azolium catalyst be used. In the event, haloaldehyde 18 when treated with catalyst 19 affords carbamoly azide 20 in 55% ee and modest yield (eq 5).
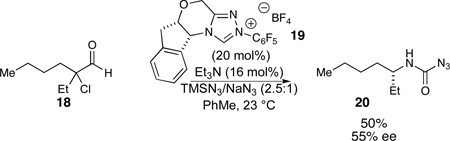 |
(5) |
Conclusion
We have shown that epoxyaldehydes afford 1,2-amino alcohol derivatives when treated with azidation agents in the presence of azolium salt and base. The overall process extends the utility of carbene-catalyzed redox processes of α-reducible aldehydes. Our results are consistent with the mechanism presented above although the discrepancy in selectivities between this process and redox using alcohol nucleophiles is not easily rationalized. Preliminary results further suggest that the use of chiral enantioenriched catalysts in this process lead to the potential for asymmetric synthesis of α-branched amines. Efforts at extending this reactivity and identifying improved catalysts for the asymmetric reaction are currently underway.
Experimental Section
General Procedure A: Synthesis of Carbamoyl Azides
A 5 ml flame dried round bottom flask was charged with a magnetic stir bar and azolium salt (0.027 g, 0.074 mmol, 0.20 equiv). To the flask under argon was added 2 ml of anhydrous toluene followed by triethylamine (0.006 g, 0.060 mmol, 0.16 equiv) and the mixture was allowed to stir for 20 min. tert-Butanol (0.137 g, 1.85 mmol, 5.0 equiv) was then added to the reaction vessel followed by azidotrimethylsilane (0.106 g, 0.924 mmol, 2.5 equiv). α, β-Epoxy aldehyde 4a (0.060 g, 0.370 mmol) was then dissolved in 1 ml of toluene (to obtain a final concentration of 0.1 M with respect to aldehyde), added to the reaction vessel and allowed to stir for 30 min. Upon consumption of starting material, sodium azide (0.024 g, 0.370 mmol, 1.0 equiv) was added to the reaction vessel under an argon atmosphere and the reaction was allowed to stir for 12 hours. The crude reaction mixture is taken up in a minimal amount of EtOAc, and filtered through a plug of silica eluting with 30 ml of EtOAc. The solution volume was reduced in vacuo and the crude oil was subjected to column chromatography, typically eluted with 95:5 hexanes:EtOAc to furnish the desired carbamoyl azide.
1,2-anti-1-phenyl-1-(trimethylsiloxy) propan-2-yl carbamoyl azide (16a)
Rf = 0.3 (95:5 Hex:EtOAc);1H NMR (400 MHz, CDCl3) δ 7.21-7.31 (m, 5H), 5.27 (br d, 1H), 4.65 (d, 1H, J = 2.80 Hz), 3.91-3.97 (m, 1H), 1.18 (d, 3H, J = 6.80 Hz), 0.04 (s, 9H); 13C NMR: (100 MHz, CDCl3) δ 155.9, 141.6, 128.3, 127.8, 126.2, 76.3, 53.4, 18.2, 13.2, 0.1; IR (NaCl, neat) 3317, 2958, 2444, 2139, 1689, 1491 cm−1; HRMS (FAB+) calcd for C13H21N4O2Si 292.13555. Found 292.13493.
General Procedure B: Synthesis of Oxazolidinones
A 5 ml flame dried round bottom flask was charged with magnetic stir bar and azolium salt (0.022 g, 0.061 mmol, 0.20 equiv). To the flask under argon was added 1 ml of anhydrous toluene and triethylamine (0.0048 g, 0.048 mmol, 0.16 equiv) and the mixture allowed to stir for 30 min under an argon atmosphere. Hydrazoic acid (see General Methods, Supplementary Information) is then added to the reaction mixture. α, β-Epoxy aldehyde 4a (0.060 g, 0.370 mmol, 1.0 equiv) is dissolved in 1 ml of toluene and added to the reaction vessel via syringe. Upon consumption of the substrate, sodium azide (0.023 g, 0.350 mmol, 1.0 equiv) is added to the reaction vessel under an argon atmosphere. The reaction was allowed to stir for two hours. The crude mixture was then filtered through a silica plug and eluted with 30 ml of ethyl acetate and concentrated in vacuo to yield a crude oil. The crude material was purified by column chromatography with typically 1:1 hexanes:EtOAc to furnish the desired compound. This compound (17a) matches literature spectral data.22
Supplementary Material
Scheme 1.

Carbene Catalyzed Redox Processes
ACKNOWLEDGMENT
This work is dedicated to our dear friend and colleague, the late Albert I. Meyers. We thank NIGMS (GM72586) and the Herman Frasch Foundation for support of this research. We acknowledge Eli Lilly, Johnson & Johnson and Boehringer-Ingelheim for additional support. We are extremely grateful to David M. Rubush and Philip A. Wheeler for experimental assistance. T. R. thanks the Monfort Family Foundation for a Monfort Professorship. We thank Donald Gauthier (Merck) for a generous gift of aminoindanol.
Footnotes
Supporting Information: Experimental procedures, characterization and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Seebach D, Corey EJ. J. Org. Chem. 1975;40:231–237. [Google Scholar]; (b) Seebach D. Angew. Chem. Int. Edit. Engl. 1979;18:239. [Google Scholar]
- 2. Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. Also see Igau A, Grutzmacher H, Baceiredo A, Bertrand G. J. Am. Chem. Soc. 1988;110:6463.
- 3.(a) Wöhler F, Liebig J. Ann. Pharm. 1832;17:249. [Google Scholar]; (b) Hassner A. The Benzoin and Related Acyl Anion Equivalent Reactions. In: Trost BM, Fleming I, editors. Comprehensive Organic Synthesis. Vol. 1. Oxford: Pergamon Press; 1991. p. 541. [Google Scholar]
- 4.(a) Enders D, Breuer K. Addition of acyl carbanion equivalents to carbonyl groups and enones. In: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I-III. Vol. 3. Berlin, Germany: Springer-Verlag; 1999. p. 1093. [Google Scholar]; (b) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Stetter H. Angew. Chem. Int. Edit. Engl. 1976;15:639. [Google Scholar]; (b) Stetter H, Kuhlmann H. Org. React. 1991;40:407. [Google Scholar]; (c) Christmann M. Angew. Chem. Int. Edit. 2005;44:2632. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]
- 6.(a) Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899. [Google Scholar]; (b) Christmann M. Angew. Chem. Int. Edit. 2005;44:2632. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]; (c) Mennen SM, Blank JT, Tran MB, Imbriglio JE, Miller SJ. Chem. Commun. 2005:195. doi: 10.1039/b414574g. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto Y, Tomioka K. Tetrahedron Lett. 2006;47:5843. [Google Scholar]
- 7.(a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (b) Kerr MS, Rovis T. Synlett. 2003:1934. [Google Scholar]; (c) Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;126:8876. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]; (d) Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]; (e) Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368. [Google Scholar]; (f) Moore JL, Kerr MS, Rovis T. Tetrahedron. 2006;62:11477. [Google Scholar]; (g) Liu Q, Rovis T. J. Am. Chem. Soc. 2006;128:2552. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Cullen SC, Rovis T. Org. Lett. 2008;10:3141. doi: 10.1021/ol801047k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Read de Alaniz J, Kerr MS, Moore JL, Rovis T. J. Org. Chem. 2008;73:2033. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Liu Q, Rovis T. Org. Proc. Res. Dev. 2007;11:598. doi: 10.1021/op600278f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Orellana A, Rovis T. Chem. Commun. 2008:730. doi: 10.1039/b716445a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Johnson JS. Angew. Chem. Int. Ed. 2004;43:1326. doi: 10.1002/anie.200301702. [DOI] [PubMed] [Google Scholar]; (c) Pohl M, Lingen B, Müller M. Chem. Eur. J. 2002;8:5288. doi: 10.1002/1521-3765(20021202)8:23<5288::AID-CHEM5288>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]; (d) Nair V, Bindu S, Sreekumar V. Angew. Chem. Int. Ed. 2004;43:5130. doi: 10.1002/anie.200301714. [DOI] [PubMed] [Google Scholar]; (e) Zeitler K. Angew. Chem. Int. Ed. 2005;44:7506. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]; (f) Webber P, Krische MJ. Chemtracts: Org. Chem. 2007;19:262. [Google Scholar]; (g) Rovis T. Chem. Lett. 2008;37:2. [Google Scholar]
- 9.(a) Reynolds NT, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2004;126:9518. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]; (b) Reynolds NT, Rovis T. J. Am. Chem. Soc. 2005;127:16406. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]
- 10.Chow KY-K, Bode JW. J. Am. Chem. Soc. 2004;126:8126. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]
- 11.(a) Sohn SS, Bode JW. Org. Lett. 2005;7:3873. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; (c) Zeitler K. Org. Lett. 2006;8:637. doi: 10.1021/ol052826h. [DOI] [PubMed] [Google Scholar]
- 12.(a) Burstein C, Glorius F. Angew. Chem. Int. Edit. 2004;43:6205. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (c) He M, Struble JR, Bode JW. J. Am. Chem. Soc. 2006;128:8418. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (d) He M, Uc GJ, Bode JW. J. Am. Chem. Soc. 2006;128:15088. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]
- 13.Sohn SS, Bode JW. Angew. Chem. Int. Edit. 2006;45:6021. doi: 10.1002/anie.200601919. [DOI] [PubMed] [Google Scholar]
- 14.(a) Vora HU, Rovis T. J. Am. Chem. Soc. 2007;129:13796. doi: 10.1021/ja0764052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sohn SS, Bode JW. J. Am. Chem. Soc. 2007;129:13798. doi: 10.1021/ja0768136. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ager DJ, Prakash I, Schaad DR. Chem. Rev. 1996;96:835. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]; (b) Bergmeier SC. Tetrahedron. 2000;56:2561. [Google Scholar]; (c) Fache F, Schulz E, Tommasino ML, Lemaire M. Chem. Rev. 2000;100:2159. doi: 10.1021/cr9902897. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi J, Ishibashi M. Heterocycles. 1996;42:943. [Google Scholar]; (e) Lee H-S, Kang SH. Synlett. 2004:1673. [Google Scholar]; (f) Meyers AI. J. Org. Chem. 2005;70:6137. doi: 10.1021/jo050470h. [DOI] [PubMed] [Google Scholar]
- 16.Scheidt specifically states that azide does not work as a nucleophile in his enal redox chemistry; see ref. 11b.
- 17.Smith PAS. Org. React. 1946;3:337. [Google Scholar]
- 18.Fu has recently reported asymmetric addition of hydrazoic acid to ketenes generating enantioenriched alpha-branched carbinolamines see: Dai X, Nakai T, Romero JAC, Fu GC. Angew. Chem. Int. Ed. 2007;46:4367. doi: 10.1002/anie.200700697.
- 19.It is well established that acylazides undergo Curtius rearrangement with retention of stereochemistry. Although the acylazide is somewhat sensitive to epimerization, Lebel has shown that the azidation/Curtius/trapping sequence proceeds with predominant retention of enantioselectivity; see: Lebel H, Leogane O, Huard K, Lectard S. Pure Appl. Chem. 2006;78:363.
- 20.Ether, hexanes and benzene give lower diastereoselectivity and decreased yields. THF and CH2Cl2 provide similar selectivity to that of ether and benzene, but with decreased yields. Acetonitrile provides only a minor amount of the desired product.
- 21.(a) Llebaria A, Triola G, Fabria’s G, Casas J. J. Org. Chem. 2003;68:9924. doi: 10.1021/jo030141u. [DOI] [PubMed] [Google Scholar]; (b) Tanaka N, Tamai T, Mukaiyama H, Hirabayashi A, Muranaka H, Ishikawa T, Kobayashi J, Akahane S, Akahane M. J. Med. Chem. 2003;46:105. doi: 10.1021/jm020177z. [DOI] [PubMed] [Google Scholar]; (c) Heathcock CH, Buse C, Kleschick WA, Pirrung MC, Sohn JE, Lampe J. J. Org. Chem. 1980;45:1066. [Google Scholar]; (d) Ohfune Y, Shimamoto K. Tet. Lett. 1988;29:5177. [Google Scholar]
- 22.Barta NS, Sidler DR, Somerville KB, Weissman SA, Larsen RD, Reider PJ. Org. Lett. 2000;2:2821. doi: 10.1021/ol006255z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.