

Supplemental Digital Content is available in the text.
Key Words: leukemia, acute, lymphoblastic, maintenance therapy, 6-mercaptopurine, methotrexate, pharmacology, drug metabolism, pharmacokinetics, adherence
Abstract
The antileukemic mechanisms of 6-mercaptopurine (6MP) and methotrexate (MTX) maintenance therapy are poorly understood, but the benefits of several years of myelosuppressive maintenance therapy for acute lymphoblastic leukemia are well proven. Currently, there is no international consensus on drug dosing. Because of significant interindividual and intraindividual variations in drug disposition and pharmacodynamics, vigorous dose adjustments are needed to obtain a target degree of myelosuppression. As the normal white blood cell counts vary by patients’ ages and ethnicity, and also within age groups, identical white blood cell levels for 2 patients may not reflect the same treatment intensity. Measurements of intracellular levels of cytotoxic metabolites of 6MP and MTX can identify nonadherent patients, but therapeutic target levels remains to be established. A rise in serum aminotransferase levels during maintenance therapy is common and often related to high levels of methylated 6MP metabolites. However, except for episodes of hypoglycemia, serious liver dysfunction is rare, the risk of permanent liver damage is low, and aminotransferase levels usually normalize within a few weeks after discontinuation of therapy. 6MP and MTX dose increments should lead to either leukopenia or a rise in aminotransferases, and if neither is experienced, poor treatment adherence should be considered. The many genetic polymorphisms that determine 6MP and MTX disposition, efficacy, and toxicity have precluded implementation of pharmacogenomics into treatment, the sole exception being dramatic 6MP dose reductions in patients who are homozygous deficient for thiopurine methyltransferase, the enzyme that methylates 6MP and several of its metabolites. In conclusion, maintenance therapy is as important as the more intensive and toxic earlier treatment phases, and often more challenging. Ongoing research address the applicability of drug metabolite measurements for dose adjustments, extensive host genome profiling to understand diversity in treatment efficacy and toxicity, and alternative thiopurine dosing regimens to improve therapy for the individual patient.
The folate analog methotrexate (MTX) and the thio-substituted purine analog 6-mercaptopurine (6MP) became pioneering anticancer agents more than half a century ago, when first Farber et al1 and then Burchenal2,3 demonstrated that such drugs can induce temporary remissions in childhood leukemia. Soon cure became the goal,4,5 and through a series of trials it was shown that the chance of long-term remission of childhood acute lymphoblastic leukemia (ALL) was significantly improved, when patients received several years of postremission maintenance therapy with daily 6MP and weekly MTX.4–8 Today, ALL protocols include an induction regimen with 3 or 4 antileukemic drugs followed by several months of consolidation therapy.9–15 Then oral 6MP/MTX maintenance therapy is given until 2 to 3 years from diagnosis, the longer duration being reserved for boys due to their inferior prognosis with shorter therapy.16–20 Despite its long history, the antileukemic mechanisms of maintenance therapy remain to be revealed. Recent studies of NT5C2 mutations in relapsed ALL clones emerging during maintenance therapy21,22 support a direct antileukemic effect of maintenance therapy.23,24 ALL stem cells may be uniquely sensitive to inhibition of de novo pathways in nucleotide synthesis crucial for DNA repair, methylation, and mitotic duplication.25 In addition, maintenance therapy could modulate apoptotic pathways,26 or induce changes in the microenvironment of the leukemic stem cells,27–30 for example, by impeding antiangiogenesis.31,32
IS YEARS OF 6MP/MTX THERAPY NEEDED FOR ALL PATIENTS?
Maintenance therapy seems to be important for most ALL subsets, including T-cell ALL21 and other patients with hyperleukocytosis at diagnosis,33 adolescents,34 and Down syndrome patients with ALL.35 Observational studies support that 6MP/MTX maintenance therapy is superior to other drug combinations,33 and that poor physician compliance or poor patient adherence significantly increase the risk of relapse.36–39 As intensification of induction and consolidation therapy have improved cure rates of ALL, currently being >80%,9,15,40 the necessity of several years of the less toxic maintenance therapy has been questioned. A systematic review of 42 randomized studies with 12,000 childhood ALL cases indicated that longer maintenance therapy gave a slightly (although statistically significantly) lower risk of relapse, but with no difference in survival due to a higher risk of death in remission.19 Furthermore, longer duration of maintenance therapy as well as higher 6MP/MTX drug doses have in 3 recent studies been associated with an increased risk of second malignancies,41–43 but so far the subset of patients at risk of developing second cancers has not been identified. Shortening ALL therapy from 24 to 18 months significantly reduces the probability of event-free survival (pEFS),44 and if all chemotherapy is truncated at 52 weeks from diagnosis, the pEFS5y may be as low as 60%, even for non–high-risk ALL patients.45
The fact that some children with ALL are cured after only a few months of chemotherapy7,45 is not surprising, as monitoring of minimal residual disease (MRD) has shown that patients may have <10−5 leukemic cells in the bone marrow already at the end of induction therapy.46,47 Unfortunately, no randomized studies of therapy duration have included MRD monitoring.
DRUG DOSAGE
Maintenance therapy of childhood ALL is a challenging exception to the golden standard of body size–based drug dosing in cancer chemotherapy. Currently, there is no international consensus on administration of maintenance therapy, and protocols reflect tradition rather than empirical evidence.48 In most of continental Europe the starting dose of oral 6MP is 50 mg/m2/d, whereas in the United Kingdom, the Nordic countries, and most of the United States it is 75 mg/m2/d.48 For MTX, the starting dose varies from 20 to 40 mg/m2/d given orally, or sometimes parenterally to secure medication adherence and increase systemic drug exposure,48,49 although this route of administration has not been shown to be more efficacious and may even be more neurotoxic.48–51 In contrast, escalating intravenous MTX doses without leucovorin rescue during interim maintenance therapy seems to provide a better pEFS than oral MTX.52 However, without reliable pharmacological endpoints it is difficult to determine differences in the pharmacokinetic and pharmacodynamic aspects of these approaches.
The importance of 6MP/MTX dose intensity was first demonstrated in the 1960s, when children randomized to receive maintenance therapy with 6MP (50 mg/m2/d), MTX (20 mg/m2/wk), vincristine, and cyclophosphamide had longer remission duration than patients who only received half doses of these drugs to reduce toxicity.53
Because of large interindividual variations in 6MP/MTX bioavailability and cellular pharmacokinetics, patients receiving identical doses per body surface area may experience very different systemic and intracellular drug exposures.54–60 The doses of 6MP and MTX doses are poorly related to their AUC,60 and drug doses are not significantly related to relapse rates in multivariate analyses.61 Accordingly, the recommended 6MP and MTX doses in ALL protocols should only be regarded as starting doses that are to be individually adjusted according to myelotoxicity (Fig. 1, and Supplemental Fig. 1, Supplemental Digital Content 1, http://links.lww.com/JPHO/A72). The average 6MP and MTX doses prescribed during maintenance therapy and adjusted by WBC vary widely (Figs. 1, 2). Importantly, patients tolerant to the starting doses of 6MP and MTX, but for whom maintenance therapy is not intensified, have a poorer outcome compared both with patients who receive reduced drug doses due to leukopenia and with those who are upward dose adjusted to obtain target myelosuppression.36–38,62,63 Furthermore, recurrent unwarranted treatment interruptions due to a rise in aminotransferases are also an adverse factor for risk of relapse.37 In a more recent study, the cumulative duration of treatment interruptions, likely to be primarily determined by bone marrow toxicity and infections, did not seem to be related to an increased relapse rate in multivariate Cox regression analysis.61
FIGURE 1.
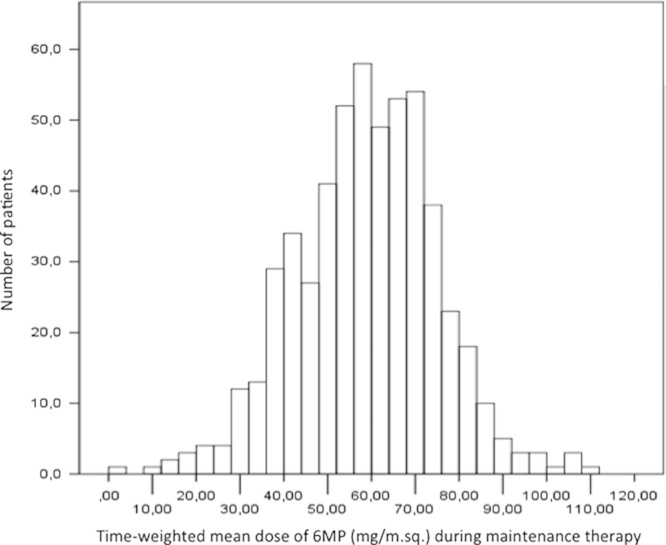
Distribution of mean prescribed 6-mercaptopurine (m6MP) doses during maintenance therapy (protocol starting oral dose: 75 mg/m2/d) for 538 patients included in the NOPHO ALL-92 maintenance therapy study.61 Means are based on a total of >28,000 registered drug doses and calculated by weighting each registered dose according to the time interval to the next measurement. The median m6MP dose for all patients is 59.4 mg/m2/d.
FIGURE 2.
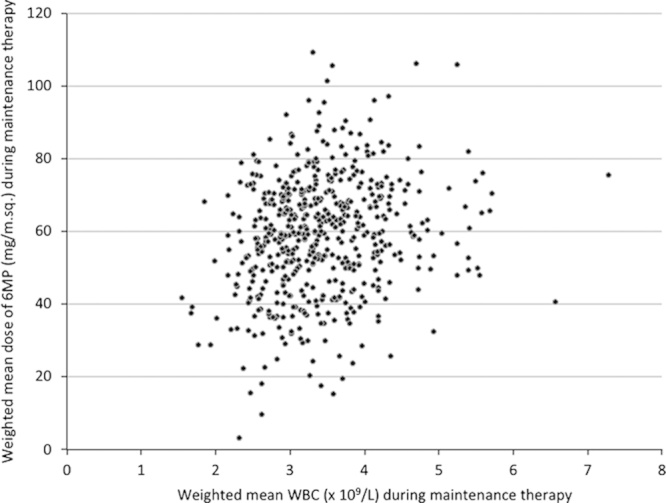
Distribution of mean white blood cell count (mWBC) and prescribed mean 6-mercaptopurine (m6MP) doses during maintenance therapy for 538 patients included in the NOPHO ALL-92 maintenance therapy study.61 Means are based on a total of >28,000 registered drug doses and blood counts and calculated by weighting each registration according to the time interval to the next registration. The median m6MP (59.4 mg/m2/d) and median mWBC (3.3×109/L) are significantly correlated (rS=0.20; P<0.001).
6MP AND MTX PHARMACOKINETICS AND PHARMACODYNAMICS
6MP
The absorption of 6MP is rapid, and the elimination half-life is short (1 to 2 h). Even though small studies (somewhat surprisingly) have been able to associate plasma 6MP concentrations to relapse rates,64,65 such measurements cannot be used for 6MP dose adjustments, due to very large (up to 70-fold) interindividual and intraindividual variations in bioavailability.54,60,66
6MP has 3 major metabolic pathways (Fig. 3). First, a substantial and highly varying fraction of 6MP is converted to inactive 6-thiouric acid by the enzyme xanthine oxidase in first pass metabolism.54,55,67 Xanthine oxidase and thiopurine methyltransferase (TPMT) can be inhibited by allopurinol,68–70 and coadministration of allopurinol thus requires 6MP dose reductions, as these interactions increases 6MP bioavailability and skews the metabolism of 6MP toward 6-thioguanine nucleotide (6TGN) production (Fig. 3). However, for childhood ALL this drug combination has not been explored beyond case reports.71
FIGURE 3.
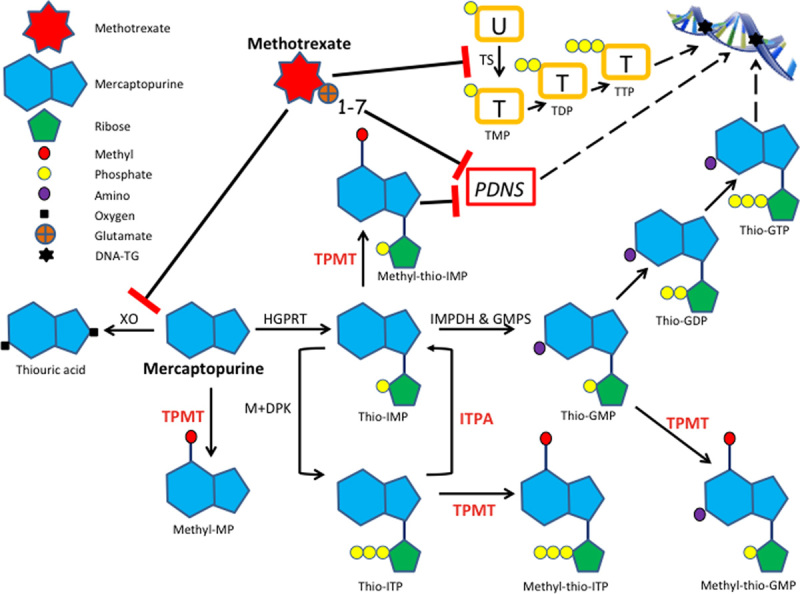
Simplified draft of 6-mercaptopurine (6MP) metabolism and methotrexate (MTX)-6MP interactions. DNA-TG indicates thioguanine nucleotides incorporated into DNA; GDP, guanosine diphosphate; GMP, guanosine monophosphate; GMPS, guanosine monophosphate synthetase; GTP, guanosine triphosphate: HGPRT, hypoxanthine guanine phosphoribosyl transferase; IMP, inosine monophosphate; IMPDH, inosine monophosphate dehydrogenase; ITP, inosine triphosphate; ITPA, inosine triphosphate pyrophosphatases; M+DPK, monophosphate and diphosphate kinases; MP, mercaptopurine; PDNS, purine de novo synthesis; TDP, thymidine diphosphate; TMP, thymidine monophosphate; TPMT, thiopurine methyltransferase; TTP, thymidine triphosphate; U, uridine monophosphate; XO, xanthine oxidase.
Second, 6MP is a prodrug that through a multistep process, involving hypoxanthine guanine phophoribosyl transferase mediated coupling of 6MP with phosphoribosyl pyrophosphate, base modification, and further phosphorylation to form 6TGN (Fig. 3). The deoxy form of 6TGN is then incorporated into DNA (DNA-TG) in nucleated cells,72–74 which may activate postreplication mismatch repair systems that lead to DNA strand breaks and apoptosis.24,75
A third metabolic pathway is thiomethylation of 6MP and some of its metabolites catalyzed by TPMT, thus reducing 6TGN formation76 (Fig. 3). Previously, methylated 6MP metabolites were considered largely insignificant for 6MP pharmacodynamics. However, some methylated metabolites, not least methylthioinosine monophosphates (MeMP), are strong inhibitors of purine de novo synthesis.77 As the purine salvage pathway is low in lymphoblasts that primarily depend on purine de novo synthesis,78 the reduced levels of endogenous nucleotides and the resulting enhanced DNA-TG incorporation in the presence of MeMP is likely to play a clinical role.74,79 Still, the impact of these pharmacodynamic interactions on relapse rates and toxicities remains undetermined, partly as a sufficiently sensitive and reliable assay for routine measurements of DNA-TG in nucleated blood has only recently become available.80
After a few weeks of oral 6MP therapy a steady state level in Ery-6TGN level is obtained.81,82 Early studies showed Ery-6TGN levels to be associated with both myelotoxicity and remission duration,83–85 even though the 6MP metabolite profiles differ widely between red blood cells and neutrophils.86 However, more recent studies have failed to confirm a significant association between Ery-6TGN and risk of relapse.38,61 Ery-6TGN levels reflect adherence to therapy87 and TPMT activity,63 but are only weakly, although statistically significantly, related to DNA-TG levels.74,79 Furthermore, 6MP dose increments to achieve higher Ery-6TGN levels primarily increase the methylated metabolite levels,88 which may enhance hepatotoxicity.89 There is a lack of large, prospective studies that explore whether monitoring of Ery-6TGN, Ery-MeMP, and DNA-TG adds dose adjustments advantages compared with adjustments by only myelotoxicity and hepatotoxicity.
As dose increments of 6MP increase the methylated metabolites and their associated toxicities far more than Ery-6TGN, several alternative treatment strategies have been efficacious in improving the 6TGN/MeMP ratio, including coadministration of allopurinol70,71,90 and splitting the daily 6MP dose in a morning and an evening dose,91,92 but it remains to be determined whether such approaches increase DNA-TG levels, ease 6MP dose adjustments to obtain target WBC/absolute neutrophil counts (ANC) levels, or reduce relapse rates of childhood ALL.
MTX
Although MTX disposition and pharmacodynamics have been well mapped in cancer cell lines,23,93,94 far less is known on how to implement such data into maintenance therapy strategies. The folate pathway gene expression profiles vary widely among subsets of ALL, which affects treatment efficacy of MTX.95 However, current guidelines for MTX dosing do not take into account the diversity of different leukemia subtypes’ sensitivity to MTX.
Bioavailability of low dose oral MTX is generally >90%, but is significantly reduced at doses >40 mg/m2.57 Similar to natural folates, MTX is converted into MTX polyglutamates (MTXPG, with 2 to 7 glutamyl residues) by the enzyme folylpolyglytamyl transferase, which enhances intracellular retention, inhibition of the target enzymes in purine and pyrimidine de novo synthesis, and treatment efficacy (Fig. 3).96 The propensity for MTX to undergo polyglutamation is higher for B-cell precursor ALL subtypes (not least the high-hyperdiploid cases) than for T-cell ALL.95,97,98 Accordingly, many groups offer high-dose MTX at doses of 5 g/m2/24 h during consolidation therapy to cure T-cell ALL. MTXPG bind tightly to and inhibit dihydrofolate reductase, the enzyme responsible for reducing folates to their bioactive tetrahydrofolate form.23 During weekly low-dose oral MTX therapy, MTXPG accumulates in red blood cell precursors in the bone marrow, and MTXPG with longer glutamyl chains are then retained in the erythrocytes (Ery-MTXPG) throughout their life span.99 Steady-state Ery-MTXPG is achieved after 4 to 8 weeks.100,101 High Ery-MTXPG levels have been associated with increased risk of myelotoxicity,100,102 but only a single Nordic study has found Ery-MTXPG levels significantly related to remission duration,102 and this association could not be confirmed in later studies,61,103 potentially due to more intensive use of intravenous MTX in these studies. No study has explored the impact on relapse rates of various Ery-MTX polyglutamate chain lengths. At steady state, Ery-MTXPG is both interindividually and intraindividually related to the dose of oral MTX and may thus be used for monitoring treatment adherence.34,100
PHARMACOGENETICS
Single nucleotide polymorphisms in genes that affect the disposition of anticancer agents influence the outcome of childhood ALL.104,105 However, so far only TPMT variants have influenced drug dosing,76,96 and it is poorly explored which host genome variants that ultimately determine the complex metabolism and efficacy of thiopurines and MTX, how this influences the toxicity profiles across ethnic groups,106–110 and how such data should be applied for dose adjustments.
TPMT
The normal substrate for TPMT is not known, and, in the absence of thiopurines, TPMT-deficient individuals are clinically and biochemically normal. In white individuals, the most common variants are *3A,*3B, and *3C all involving G460A and/or A719G and accounting for at least 90% of low-activity alleles among white individuals of North European decent.63,76 Approximately 5% to 10% are TPMT heterozygous carrying 1 wild-type and 1 low-activity allele, and 1 in 300 is TPMT deficient and at risk of life-threatening myelosuppression at standard 6MP doses.111,112 Although thiopurine dosing according to the TPMT genotype has been implemented by a few ALL study groups,48,79,113 the benefits of this strategy remain uncertain. Compared with TPMT wild-type patients, heterozygous patients experience higher intracellular 6TGN levels, more myelotoxicity, higher cure rates,63,113,114 but probably also a higher risk of second cancers.41,115,116 The German BFM group that administered lower starting doses of 6MP (50 mg/m2) failed to confirm the association with second cancers.117 It is noteworthy that, a recent study indicated that reduction of oral 6MP starting doses from 75 to 50 mg/m2/d, reflecting these BFM data, did reduce the risk of second cancers among TPMT heterozygous patients, but at the same time lead to an increased risk of relapse.43
Measuring TPMT activity in erythrocytes is an alternative to genotyping and may also identify rare low-activity variants missed by routine allele testing. However, as TPMT activity is inversely related to the erythrocyte age,118 the TPMT activity will in general be increased during maintenance therapy when the erythrocyte life span is shortened, and be low at diagnosis of ALL due to reduced erythropoiesis, hampering reliable discrimination of heterozygous and wild-type TPMT phenotypes.
Inosine Triphosphate Pyrophosphatase (ITPA)
Low-activity alleles of ITPA, the enzyme that dephosphorylates thioinosine triphosphate (Fig. 3), may increase methylated thiopurine metabolite levels,119,120 the risk of hepatotoxicity121,122 and of bone marrow toxicity123 with febrile neutropenia,119,124 and potentially also the risk of relapse.110 The frequency of ITPA low-activity alleles show wide interethnic variability being 1% to 2% among Hispanics, but almost 20% in Asian populations, which may influence tolerance to thiopurine therapy.125
Other 6MP Metabolizing Genes
Other 6MP metabolizing enzymes, such as xanthine oxidase and hypoxanthine guanine phosphoribosyl transferase (HGPRT) may vary among individuals,67,126 in part determined by genetic polymorphisms, and at least low HGPRT in B-cell precursor ALL has been associated with an inferior cure rate, although this association was not related to increased in vitro thiopurine resistance.127
MTX
Several groups have demonstrated that MTX treatment efficacy is associated with polymorphisms in dihydrofolate reductase,128 thymidylate synthetase,129,130 reduced folate carrier,131 5,10-methylenetetrahydrofolate reductase, and methylenetetrahydrofolate dehydrogenase132 (for reviews on childhood ALL and rheumatoid arthritis, see Davidsen and colleagues105,133–135). However, the results of these studies are often contradictory with some studies demonstrating improved cure rates for a specific genetic polymorphism, whereas others demonstrate the opposite, many of the studies are small, most only address 1 or a few of the many genetic polymorphisms involved in the disposition of MTX, and in general they address responses to high-dose MTX rather than low-dose MTX maintenance therapy. Furthermore, it is impossible to evaluate whether a specific polymorphism exert its modifying effect on relapse rate and/or toxicities directly through changed MTX disposition or indirectly by modifying endogenous folate levels. So far no groups have adjusted their MTX treatment strategies based on polymorphisms in the MTX/folate pathway.
TOXICITY AND RELAPSE RATE
Leukopenia
Dose adjustments guided by toxicity assumes that the individual variations in 6MP/MTX pharmacokinetics and/or pharmacodynamics affect leukemic and normal cells in parallel.136 For maintenance therapy, 6MP/MTX dosage is targeted to a preset degree of myelosuppression, generally a WBC of 1.5 to 3.0 (or 3.5)×109/L,48 but randomized studies demonstrating benefits hereof are lacking.137 Most observational studies have shown low WBC and/or ANC during maintenance therapy to be related to red blood cell levels of cytotoxic 6MP/MTX metabolites and/or to a reduced relapse rate.38,61,82,100,138–144 However, ANC correlates so closely with WBC, that it is virtually impossible to determine which of these 2 parameters is superior as guidance for dose adjustment (Fig. 4A). In the Nordic Society for Paediatric Haematology and Oncology (NOPHO) ALL92 maintenance therapy study,61 patients with an average ANC <2.0×109/L during maintenance therapy had a significantly better relapse-free survival than patients with higher ANC levels (Fig. 4B), and ANC was somewhat more strongly associated with relapse rates than WBC level, although the latter was the dose adjustment target in that protocol. Nevertheless, several factors challenge 6MP/MTX dose adjustments by the leukocyte counts.
FIGURE 4.
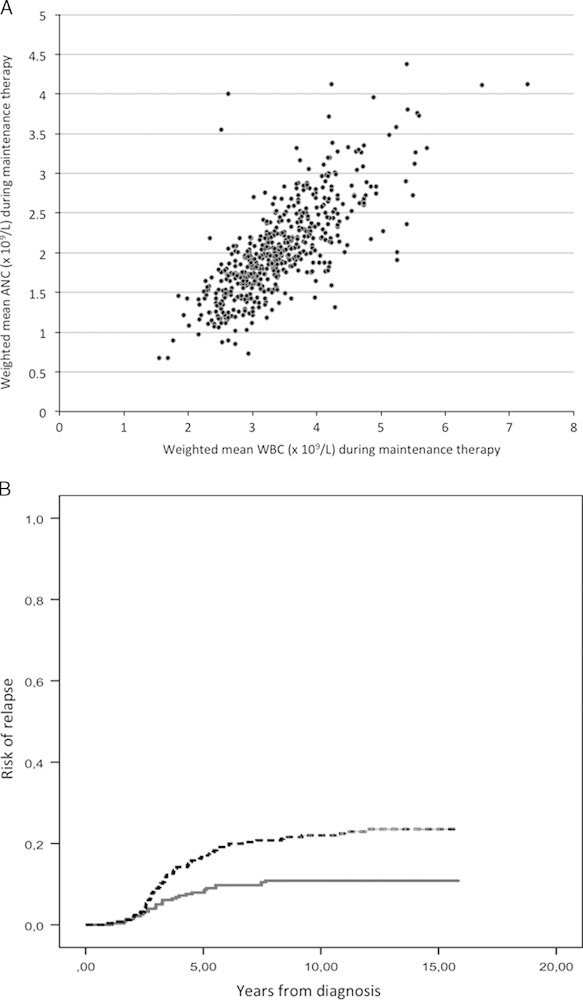
A, Mean white blood cell (mWBC) and absolute neutrophil counts (mANC) for 538 patients included in the NOPHO ALL-92 maintenance therapy study.61 Means are based on a total of >28,000 blood counts and calculated by weighting each measurement according to the time interval to the next measurement. Each dot represents 1 patient. mWBC and mANC are highly correlated (rS=0.77; P<0.001). The target range for WBC was 1.5 to 3.5×109/L in the NOPHO ALL92 protocol from which the data were retrieved. B, Kaplan-Meier relapse risk plots for patients with a mean absolute neutrophil count (mANC) at the end of maintenance therapy above or below 2.0×109/L=median mANC for all patients (upper curve, N=248, relapse risk 23.5%±2.7%; lower curve, N=280, relapse risk 10.9%±1.9%; P<0.001). Mean absolute neutrophil counts during maintenance therapy are calculated by weighting each measurement according to the time interval to the next measurement.
First, physicians may be more inclined to decrease 6MP/MTX drug doses in case of toxicity than to escalate doses in patients insufficiently myelosuppressed,36 requiring different strategies for 6MP dose adjustments, if the 6MP starting dose is 50 versus 75 mg/m2.
Second, the WBC levels reflect both treatment intensity and the child’s normal WBC level, which varies both between and within age groups, and by ethnicity. Thus, patients with lower WBC levels during therapy also have low WBC after cessation of therapy (rS=0.76; P<0.00001),145 and even more important, the best predictor of the rise in WBC after cessation of therapy is not the WBC level during maintenance therapy, but Ery-6TGN and Ery-MTXPG.102 Thus, an average WBC during therapy of 3.5×109/L could reflect more intensive treatment than an average WBC of 3.2×109/L, if the patients’ off-therapy WBC levels were 8.5 and 4.5×109/L, respectively. In support hereof, the red blood cell 6MP/MTX metabolite levels are overall higher in the former patient, and the rise in WBC following cessation of maintenance therapy is a stronger predictor of relapse than the average WBC level during maintenance therapy, associating a high rise in WBC with a reduced relapse rate after cessation of maintenance therapy.146
Third, often it is not possible to suppress WBC levels to a target range of 1.5 to 3.0×109/L by dose intensification without unacceptable extramedullary toxicity, including hepatotoxicity (see below).
Finally, an aggressive approach with higher 6MP doses and higher treatment intensity to achieve low WBC levels may be counteracted by treatment interruptions,61 or lead to an increased risk of second cancers.41,42 Other not yet understood mechanisms such as induction of dormant leukemic stem cells61 due to inhibition of purine de novo synthesis could also increase the risk of relapse for the dose-intensified patients.
Thrombocytopenia
Thrombocyte counts during and after cessation of maintenance therapy are significantly correlated (rS=0.74, P<0.0001),102 but thrombocytopenia is rarely a dose-limiting factor during 6MP/MTX maintenance therapy. However, if 6MP is substituted with the alternative thiopurine, thioguanine (6TG), Ery-6TGN levels become 7-fold higher and severe thrombocytopenia becomes 5- to 10-fold more common.147 Furthermore, 10% to 15% of 6TG-treated patients develop sinusoidal occlusive disease and/or portal hypertension, which often is accompanied by thrombocytopenia.148–150 Patients on 6MP with unexplained thrombocytopenia should be explored for hypersplenism and persistent Parvovirus B19 infection.151
Hepatotoxicity
6MP and MTX are hepatotoxic and 2-fold elevations or more of serum aminotransferases are frequent,100,146,152–156 but usually normalize within a few weeks after discontinuation of maintenance therapy.146,153 Hypoglycemic episodes during fasting157–160 have been associated with high levels of methylated 6MP metabolites.161 It can be counteracted by evening meals with slowly absorbed carbohydrates, by administration of rapidly absorbed carbohydrates (eg, apple juice) in case of symptoms, or by shifting to morning dosage. The latter reduces Ery-MeMP levels but the impact on relapse rate is unknown.161 Few patients develop symptoms of hypoglycemia such as severe nausea, itching, or malaise to a degree that requires dose reductions. A moderate rise in bilirubin or reduced levels of coagulation factors is common, but the risk of serious and/or permanent liver damage seems low.162–164 Accordingly, most study groups do not recommend dose reductions in case of high aminotransferase levels48 unless accompanied by biochemical evidence of severe hepatic dysfunction, that is, bilirubin 3 times above the upper normal limit and/or coagulation factor II-VII-X <0.50 IU/L. Such patients should be explored for other causes, including hepatotropic vira (eg, B or C virus153,154), veno-occlusive syndrome (VOD), or Gilbert syndrome with reduced glucuronyltransferase activity and elevated unconjugated bilirubin.
In accordance with the high incidence of hepatotoxicity seen with methylmercaptopurine riboside therapy,165,166 and the low rate of hepatotoxicity in patients with low TPMT activity,38,89 most cases of high aminotransferase levels can be related to high levels of methylated 6MP metabolites,89,167 but have also more rarely been proposed to be associated with high Ery-6TGN168 or Ery-MTXpg,100 or to accumulation of 6MP in the liver.169
A small Danish study from the 1980s linked increased aminotransferases levels during maintenance therapy with a reduced relapse rate.170 This could reflect reduced first-pass effect on oral 6MP with higher systemic 6MP exposure among developing hepatotoxicity, or higher levels of methylthioinosine monophosphate causing both liver toxicity and inhibition of purine de novo synthesis in leukemic cells,78,171 which could have increased the incorporation of 6TGN into DNA.74,79 Alternatively, elevated aminotransferases and reduced relapse risks could merely reflect that the patients were adherent to maintenance therapy. Importantly, patients who continued therapy despite an increase in aminotransferases had a lower relapse rates than patients with treatment interruptions due to hepatotoxicity.37
6MP AND MTX INTERACTION
MTX inhibits xanthine oxidase and thereby increases 6MP bioavailability (Fig. 3).172 In addition, inhibition of purine de novo synthesis can preserve phosphoribosyl pyrophosphate availability, a necessary substrate for 6TGN formation.171 Cellular depletion of reduced (activated) folates may also lower the availability of S-adenosyl methionine, the methyl donor in TPMT-mediated methylation, and thereby reduce 6MP metabolite methylation. For TPMT wild-type patients (but not for patients with TPMT low-activity alleles), Ery-6TGN and Ery-MTXPG correlate significantly during maintenance therapy (Supplemental Fig. 2, Supplemental Digital Content 2, http://links.lww.com/JPHO/A73), and the longer chained MTX polyglutamates seem most important for enhancing Ery-6TGN accumulation.173 Because of these synergisms, it is probably better to lower the doses of both 6MP and MTX than to withhold one drug and continue the other in case of dose-limiting toxicity. As the interindividual variation in bioavailability and pharmacokinetics is higher for 6MP than for MTX, above target WBC during maintenance should initially lead to upward dose adjustments in 6MP, followed by dose increments for MTX once the maximum tolerated dose of 6MP has been achieved.
6MP VERSUS 6TG FOR MAINTENANCE THERAPY
In 3 randomized studies by the US CCG, the German COALL, and the British UKALL groups that all compared 6MP with 6TG as the maintenance therapy thiopurine, only males below 10 years of age seemed to have reduced relapse rates with 6TG (OR=0.70; 95% confidence interval, 0.58-0.84), although with no significant difference in overall survival.174 The lack of MeMP and the associated inhibition of purine de novo synthesis for patients on 6TG may explain why this thiopurine failed to improve EFS, even though children receiving 6TG had several-fold higher Ery-6TGN levels.88,147,174,175 More worrying is that, 10% to 15% of patients on 6TG developed VOD, a few of which were sufficiently severe to require liver transplantation.148,176–178 It is noteworthy that, the German COALL study147 did not report 6TG-associated VOD, the only major difference from the other 2 studies being the absence of vincristine/glucocorticosteroid reinductions during maintenance therapy in the German trial. However, the biology behind this association remains uncertain.
PHYSICIAN COMPLIANCE AND PATIENT ADHERENCE TO MAINTENANCE THERAPY
With the complexity of multiple factors influencing therapy (physician compliance, patient adherence, drug disposition, toxicity), there are many potential layers of failure to optimize maintenance therapy. Toxicity-guided dosing relies heavily on physicians’ willingness to comply with the protocol guidelines, their experience with maintenance therapy, and their ability to explain the pharmacology, the biology of toxicities, and the importance of treatment adherence to patients and parents. Patient adherence will on the contrary reflect the patient’s/family’s willingness to accept burdensome toxicities of 6MP and MTX and frequent hospital visits to cure a disease that no longer can be detected. For very young children, not least infants, treatment adherence has been jeopardized by the commercially available 6MP tablets having been developed for adult-sized patients,179 and not until recently has a liquid formulation of 6MP been marketed (although still not formally tested) in children.180
Several groups have reported poor treatment adherence to maintenance therapy in a significant proportion of childhood ALL patients.39,87,181,182 The reasons for poor medication adherence can be biological, psychological, and social, and they vary across age groups and by ethnicity.183,184 Various approaches to address this challenge have been proposed, including routine measurements of Ery-6TGN/MeMP/MTXPG, but such analyses are only available in a few centers. If 6MP/MTX metabolite measurements are unavailable, nonadherence should be suspected in TPMT heterozygous patients with persistent WBC >3.0 to 3.5×109/L despite prescribed 6MP dose increments, and in patients with a TPMT wild-type genotype/phenotype, if dose increments do not lead to a rise in aminotransferases.
The randomized Brazilian ALL99 study indicated that intermittent oral high-dose 6MP with IV MTX 200 mg/m2/6 h not only improved adherence but also gave better pEFS than oral 6MP (50 mg/m2/d) with IM MTX 25 mg/m2/wk, although only for boys.185 However, the extent of patient adherence in the oral 6MP arm is difficult to assess, as 6MP/MTX metabolite measurements were not done, and it is also unclear whether the difference in 6MP and/or MTX dosing in the 2 treatment arms caused the difference in EFS.
CIRCADIAN SCHEDULE
The circadian schedule has a strong impact on efficacy and toxicity of a number of anticancer agents.186 Two maintenance therapy studies from the 1980s and 1990s found that the risk of relapse was several-fold higher for patients who reported taking 6MP and MTX in the morning compared with patients on evening schedule.187,188 It was speculated that differences in biological activity between malignant lymphoid cells and normal bone marrow cells determined these chronochemotherapeutical findings,189,190 but whatever the biological mechanism, changing patients from morning to evening schedule seemed a simple procedure to improve outcome, and this has become the general standard.48 However, a recent large study of 526 children on maintenance therapy with almost 10,000 E-6TGN/MTXpg measurements, found no association between relapse rates and the cumulative duration of evening dosage for the individual patient, when adjusting for 6MP and MTX doses, WBC levels during maintenance therapy, and Ery-6TGN and Ery-MTXPG levels.191
FOOD AND MAINTENANCE THERAPY
Several small studies,56,59,192,193 although not all,194–196 have demonstrated reduced bioavailability for both MTX and 6MP, when the drugs are administered together with food, and for 6MP specifically with milk due to its content of xanthine oxidase.197 Accordingly, nearly all study groups recommend 6MP and MTX to be taken without concomitant ingestion of food.48 Still, titrating the dose of MTX and 6MP by toxicity should counterbalance lower bioavailability, and restricting the individual patients’ choices of drug administration could reduce adherence. Only 1 large clinical study has explored the prognostic impact on administering 6MP/MTX with food, and this study demonstrated no significant influence of concomitant food ingestion on relapse rate, this also being the case within subgroups defined by their circadian schedule.188
COADMINISTRATION OF OTHER DRUGS
It is unproven that alternative or additive components of maintenance therapy such as intravenous 6MP,198,199 6TG, allopurinol, high-dose MTX,9 vincristine/glucocorticoid,200,201 or more intensive reinductions19 significantly reduce relapse rates with contemporary ALL therapy, although they can add to the burden of myelotoxicity202 and hepatotoxicity, which may necessitate 6MP and MTX dose reductions.155,203,204 Specifically, vincristine/glucocorticoid pulses during 6MP/MTX have been applied by many groups, but so far most, although not all,205 randomized studies have failed to demonstrate benefits of such pulses.200,201,206,207 Folate supplementation has been widely used to counteract MTX-induced toxicity without compromising efficacy in rheumatoid arthritis208 or posttransplantation.209 However, folate supplementation should probably be avoided during maintenance therapy, as it has been shown to influence both 6MP metabolism210 and myelotoxicity.211 Finally, trimethoprim-sulfamethoxazole given as Pneumocystis jiroveci pneumonia prophylaxis212 interferes with MTX213 and 6MP pharmacokinetics,214 and also enhances myelotoxicity leading to lower prescribed 6MP and MTX doses,215 but in spite hereof does not seem to increase relapse rates,215 and thus seems safe to prescribe to avoid this life-threatening infection.
RELAPSE DURING MAINTENANCE THERAPY
Several high-risk ALL subsets such as T-cell ALL,216 patients with hyperleukocytosis,217 and patients with t(1;19)[E21-PBX1], MLL rearrangements,218 or hypodiploidy219 nearly always relapse during maintenance therapy. In contrast, the majority of other B-cell precursor ALL relapses occurs within 2 to 3 years after cessation of treatment.220 Relapse during maintenance therapy has been associated with insufficient 6MP treatment intensity as indicated by low Ery-6TGN levels,61 or it may simply reflect poor treatment compliance/adherence.39,221 Two recent studies demonstrated activating mutations in the NT5C2 gene, which plays a role in nucleotide homeostasis, in approximately 15% to 20% of both B-cell precursor22 and T-cell ALL patients21 that relapse during 6MP/MTX maintenance therapy. It is noteworthy that, such mutations are rare among B-cell precursor ALL patients that relapse off therapy,22 indicating that such patients relapse rapidly when NT5C2 mutations emerge, or that the survival advantage for NT5C2-mutated clones at the cost of normal hematopoietic cells disappears once 6MP therapy is discontinued. In the future, targeted deep sequencing may allow routine screening for emerging clones with mutations that hamper the efficacy of thiopurines or MTX, which would allow modification of maintenance therapy to counteract such resistance mechanisms.
CONCLUSIONS AND FUTURE DIRECTIONS
During the last decades more attention has been paid to dose titration by myelotoxicity, and some groups even monitor 6MP and MTX metabolites to reveal poor treatment adherence. However, until it has been determined that such therapeutic drug monitoring eases dose adjustments, improves cure rates, and/or reduce toxicity, maintenance therapy should be adjusted according to the WBC, and lack of myelotoxicity and hepatotoxicity regarded as a surrogate marker for nonadherence. Future research should address the applicability of DNA-TG monitoring, extensive host single nucleotide polymorphism profiling, screening methods for resistant leukemic subclones, and alternative thiopurine dosing regimens to improve maintenance therapy for the individual patient.
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.jpho-online.com.
Supported by grants from the Danish Childhood Cancer Foundation and the Danish Cancer Society.
The authors declare no conflict of interest.
REFERENCES
- 1.Farber S, Diamond LK, Mercer RD, et al. Temporary remissions in acute leukemia in children produced by folic acid antagonist 4-aminopteroylglutamic acid (aminopterin). N Engl J Med. 1948;238:787–793. [DOI] [PubMed] [Google Scholar]
- 2.Burchenal JH, Murphy ML, Ellison RR, et al. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood. 1953;8:965–987. [PubMed] [Google Scholar]
- 3.Thomas A. Joe Burchenal and the birth of combination chemotherapy. Br J Haematol. 2006;133:493–503. [DOI] [PubMed] [Google Scholar]
- 4.Frei E, III, Caron M, Levin RH, et al. The effectiveness of combinations of antileukemia agents in inducing and maintaining remission in children with acute leukemia. Blood. 1965;26:642–656. [PubMed] [Google Scholar]
- 5.Simone JV. The treatment of acute lymphoblastic leukaemia. Br J Haematol. 1980;45:1–4. [DOI] [PubMed] [Google Scholar]
- 6.Holland JF, Glidewell O. Chemotherapy of acute lymphocytic leukemia of childhood. Cancer. 1972;30:1480–1487. [DOI] [PubMed] [Google Scholar]
- 7.Lonsdale D, Gehan EA, Fernbach DJ, et al. Interrupted vs. continued maintenance therapy in childhood acute leukemia. Cancer. 1975;36:341–352. [DOI] [PubMed] [Google Scholar]
- 8.Rivera GK, Pinkel D, Simone JV, et al. Treatment of acute lymphoblastic leukemia. 30 years’ experience at St. Jude Children’s Research Hospital. N Engl J Med. 1993;329:1289–1295. [DOI] [PubMed] [Google Scholar]
- 9.Schmiegelow K, Forestier E, Hellebostad M, et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–354. [DOI] [PubMed] [Google Scholar]
- 10.Conter V, Arico M, Basso G, et al. Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:255–264. [DOI] [PubMed] [Google Scholar]
- 11.Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children’s Oncology Group Report. Leukemia. 2010;24:285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamps WA, van der Pal-de Bruin KM, Veerman AJ, et al. Long-term results of Dutch Childhood Oncology Group studies for children with acute lymphoblastic leukemia from 1984 to 2004. Leukemia. 2010;24:309–319. [DOI] [PubMed] [Google Scholar]
- 13.Salzer WL, Devidas M, Carroll WL, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children’s oncology group. Leukemia. 2010;24:355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24:265–284. [DOI] [PubMed] [Google Scholar]
- 15.Schrappe M, Nachman J, Hunger S, et al. Educational symposium on long-term results of large prospective clinical trials for childhood acute lymphoblastic leukemia (1985-2000). Leukemia. 2010;24:253–254. [DOI] [PubMed] [Google Scholar]
- 16.Sather H, Miller D, Nesbit M, et al. Differences in prognosis for boys and girls with acute lymphoblastic leukaemia. Lancet. 1981;1:739–743. [DOI] [PubMed] [Google Scholar]
- 17.Riehm H, Feickert HJ, Schrappe M, et al. Therapy results in five ALL-BFM studies since 1970: implications of risk factors for prognosis. Hamatol Bluttransfus. 1987;30:139–146. [DOI] [PubMed] [Google Scholar]
- 18.Chessells JM, Richards SM, Bailey CC, et al. Gender and treatment outcome in childhood lymphoblastic leukaemia: report from the MRC UKALL trials. Br J Haematol. 1995;89:364–372. [DOI] [PubMed] [Google Scholar]
- 19.Childhood ALL Collaborative Group. Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Lancet. 1996;347:1783–1788. [DOI] [PubMed] [Google Scholar]
- 20.Pui CH, Boyett JM, Relling MV, et al. Sex differences in prognosis for children with acute lymphoblastic leukemia. J Clin Oncol. 1999;17:818–824. [DOI] [PubMed] [Google Scholar]
- 21.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chabner BA, Allegra CJ, Curt GA, et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest. 1985;76:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waters TR, Swann PF. Cytotoxic mechanism of 6-thioguanine: hMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry. 1997;36:2501–2506. [DOI] [PubMed] [Google Scholar]
- 25.Kamen BA. Serendipity-methotrexate and 6-mercaptopurine for continuation therapy for patients with acute lymphoblastic leukemia: the leukemic stem cell and beyond? J Pediatr Hematol Oncol. 2009;31:383–384. [DOI] [PubMed] [Google Scholar]
- 26.Gale RP, Butturini A. Maintenance chemotherapy and cure of childhood acute lymphoblastic leukaemia. Lancet. 1991;338:1315–1318. [DOI] [PubMed] [Google Scholar]
- 27.Kumagai M, Manabe A, Pui CH, et al. Stroma-supported culture in childhood B-lineage acute lymphoblastic leukemia cells predicts treatment outcome. J Clin Invest. 1996;97:755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campana D, Coustan-Smith E, Manabe A, et al. Human B-cell progenitors and bone marrow microenvironment. Hum Cell. 1996;9:317–322. [PubMed] [Google Scholar]
- 29.Mudry RE, Fortney JE, York T, et al. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96:1926–1932. [PubMed] [Google Scholar]
- 30.Narendran A, Ganjavi H, Morson N, et al. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp Hematol. 2003;31:693–701. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Atayde AR, Sallan SE, Tedrow U, et al. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150:815–821. [PMC free article] [PubMed] [Google Scholar]
- 32.Keyhani A, Jendiroba DB, Freireich EJ. Angiogenesis and leukemia. Leuk Res. 2001;25:639–645. [DOI] [PubMed] [Google Scholar]
- 33.Schmiegelow K, Heyman M, Kristinsson J, et al. Oral methotrexate/6-mercaptopurine may be superior to a multidrug LSA2L2 Maintenance therapy for higher risk childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. J Pediatr Hematol Oncol. 2009;31:385–392. [DOI] [PubMed] [Google Scholar]
- 34.Schmiegelow K, Heyman M, Gustafsson G, et al. The degree of myelosuppression during maintenance therapy of adolescents with B-lineage intermediate risk acute lymphoblastic leukemia predicts risk of relapse. Leukemia. 2010;24:715–720. [DOI] [PubMed] [Google Scholar]
- 35.Bohnstedt C, Levinsen M, Rosthoj S, et al. Physicians compliance during maintenance therapy in children with Down syndrome and acute lymphoblastic leukemia. Leukemia. 2013;27:866–870. [DOI] [PubMed] [Google Scholar]
- 36.Peeters M, Koren G, Jakubovicz D, et al. Physician compliance and relapse rates of acute lymphoblastic leukemia in children. Clin Pharmacol Ther. 1988;43:228–232. [DOI] [PubMed] [Google Scholar]
- 37.Schmiegelow K. Prognostic significance of methotrexate and 6-mercaptopurine dosage during maintenance chemotherapy for childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1991;8:301–312. [DOI] [PubMed] [Google Scholar]
- 38.Relling MV, Hancock ML, Boyett JM, et al. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817–2823. [PubMed] [Google Scholar]
- 39.Bhatia S, Landier W, Shangguan M, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children’s oncology group. J Clin Oncol. 2012;30:2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pui CH, Mullighan CG, Evans WE, et al. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmiegelow K, Al-Modhwahi I, Andersen MK, et al. Methotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia—results from the NOPHO ALL-92 study. Blood. 2009;113:6077–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmiegelow K, Levinsen MF, Attarbaschi A, et al. Second malignant neoplasms after treatment of childhood acute lymphoblastic leukemia. J Clin Oncol. 2013;31:2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levinsen M, Rotevatn EO, Rosthoj S, et al. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: Influence on cure rates and risk of second cancer. Pediatr Blood Cancer. 2014;61:797–802. [DOI] [PubMed] [Google Scholar]
- 44.Riehm H, Gadner H, Henze G, et al. Results and significance of six randomised trials in four consecutive ALL-BFM studies. Hematol Blood Transf. 1990;33:439–450. [DOI] [PubMed] [Google Scholar]
- 45.Toyoda Y, Manabe A, Tsuchida M, et al. Six months of maintenance chemotherapy after intensified treatment for acute lymphoblastic leukemia of childhood. J Clin Oncol. 2000;18:1508–1516. [DOI] [PubMed] [Google Scholar]
- 46.Nyvold C, Madsen HO, Ryder LP, et al. Precise quantification of minimal residual disease at day 29 allows identification of children with acute lymphoblastic leukemia and an excellent outcome. Blood. 2002;99:1253–1258. [DOI] [PubMed] [Google Scholar]
- 47.Lauten M, Moricke A, Beier R, et al. Prediction of outcome by early bone marrow response in childhood acute lymphoblastic leukemia treated in the ALL-BFM 95 trial: differential effects in precursor B-cell and T-cell leukemia. Haematologica. 2012;97:1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arico M, Baruchel A, Bertrand Y, et al. The seventh international childhood acute lymphoblastic leukemia workshop report: Palermo, Italy, January 29-30, 2005. Leukemia. 2005;19:1145–1152. [DOI] [PubMed] [Google Scholar]
- 49.Chessells JM, Leiper AD, Tiedemann K, et al. Oral methotrexate is as effective as intramuscular in maintenance therapy of acute lymphoblastic leukaemia. Arch Dis Child. 1987;62:172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balis FM, Mirro JJ, Reaman GH, et al. Pharmacokinetics of subcutaneous methotrexate. J Clin Oncol. 1988;6:1882–1886. [DOI] [PubMed] [Google Scholar]
- 51.Chessells JM, Cox TC, Kendall B, et al. Neurotoxicity in lymphoblastic leukaemia: comparison of oral and intramuscular methotrexate and two doses of radiation. Arch Dis Child. 1990;65:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matloub Y, Bostrom BC, Hunger SP, et al. Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinkel D, Hernandez K, Borella L, et al. Drug dosage and remission duration in childhood lymphocytic leukemia. Cancer. 1971;27:247–256. [DOI] [PubMed] [Google Scholar]
- 54.Zimm S, Collins JM, Riccardi R, et al. Variable bioavailability of oral mercaptopurine. Is maintenance chemotherapy in acute lymphoblastic leukemia being optimally delivered? N Engl J Med. 1983;308:1005–1009. [DOI] [PubMed] [Google Scholar]
- 55.Lafolie P, Hayder S, Bjork O, et al. Large interindividual variations in the pharmacokinetics of oral 6-mercaptopurine in maintenance therapy of children with acute leukaemia and non-Hodgkin lymphoma. Acta Paediatr Scand. 1986;75:797–803. [DOI] [PubMed] [Google Scholar]
- 56.Poplack DG, Balis FM, Zimm S. The pharmacology of orally administered chemotherapy. A reappraisal. Cancer. 1986;58:473–480. [DOI] [PubMed] [Google Scholar]
- 57.Teresi ME, Crom WR, Choi KE, et al. Methotrexate bioavailability after oral and intramuscular administration in children. J Pediatr. 1987;110:788–792. [DOI] [PubMed] [Google Scholar]
- 58.Koren G, Solh H, Klein J, et al. Disposition of oral methotrexate in children with acute lymphoblastic leukemia and its relation to 6-mercaptopurine pharmacokinetics. Med Pediatr Oncol. 1989;17:450–454. [DOI] [PubMed] [Google Scholar]
- 59.Dupuis LL, Koren G, Silverman ED, et al. Influence of food on the bioavailability of oral methotrexate in children. J Rheumatol. 1995;22:1570–1573. [PubMed] [Google Scholar]
- 60.Balis FM, Holcenberg JS, Poplack DG, et al. Pharmacokinetics and pharmacodynamics of oral methotrexate and mercaptopurine in children with lower risk acute lymphoblastic leukemia: a joint children’s cancer group and pediatric oncology branch study. Blood. 1998;92:3569–3577. [PubMed] [Google Scholar]
- 61.Schmiegelow K, Bjork O, Glomstein A, et al. Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21:1332–1339. [DOI] [PubMed] [Google Scholar]
- 62.Pearson AD, Amineddine HA, Yule M, et al. The influence of serum methotrexate concentrations and drug dosage on outcome in childhood acute lymphoblastic leukaemia. Br J Cancer. 1991;64:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmiegelow K, Forestier E, Kristinsson J, et al. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia. 2009;3:557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayder S, Lafolie P, Bjork O, et al. 6-mercaptopurine plasma levels in children with acute lymphoblastic leukemia: relation to relapse risk and myelotoxicity. Ther Drug Monit. 1989;11:617–622. [DOI] [PubMed] [Google Scholar]
- 65.Koren G, Ferrazini G, Sulh H, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukemia in children. N Engl J Med. 1990;323:17–21. [DOI] [PubMed] [Google Scholar]
- 66.Lafolie P, Bjork O, Hayder S, et al. Variability of 6-mercaptopurine pharmacokinetics during oral maintenance therapy of children with acute leukemia. Med Oncol Tumor Pharmacother. 1989;6:259–265. [DOI] [PubMed] [Google Scholar]
- 67.Relling MV, Lin JS, Ayers GD, et al. Racial and gender differences in N-acetyltransferase, xanthine oxidase, and CYP1A2 activities. Clin Pharmacol Ther. 1992;52:643–658. [DOI] [PubMed] [Google Scholar]
- 68.Zimm S, Collins JM, O’Neill D, et al. Inhibition of first-pass metabolism in cancer chemotherapy: interaction of 6-mercaptopurine and allopurinol. Clin Pharmacol Ther. 1983;34:810–817. [DOI] [PubMed] [Google Scholar]
- 69.Roberts RL, Gearry RB, Barclay ML. Allopurinol-thiopurine combination therapy in inflammatory bowel disease: are there genetic clues to this puzzle? Pharmacogenomics. 2010;11:1505–1508. [DOI] [PubMed] [Google Scholar]
- 70.Blaker PA, Arenas-Hernandez M, Smith MA, et al. Mechanism of allopurinol induced TPMT inhibition. Biochem Pharmacol. 2013;86:539–547. [DOI] [PubMed] [Google Scholar]
- 71.Brackett J, Schafer ES, Leung DH, et al. Use of allopurinol in children with acute lymphoblastic leukemia to reduce skewed thiopurine metabolism. Pediatr Blood Cancer. 2014;61:1114–1117. [DOI] [PubMed] [Google Scholar]
- 72.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. [DOI] [PubMed] [Google Scholar]
- 73.Swann PF, Waters TR, Moulton DC, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–1111. [DOI] [PubMed] [Google Scholar]
- 74.Hedeland RL, Hvidt K, Nersting J, et al. DNA incorporation of 6-thioguanine nucleotides during maintenance therapy of childhood acute lymphoblastic leukaemia and non-Hodgkin lymphoma. Cancer Chemother Pharmacol. 2010;66:485–491. [DOI] [PubMed] [Google Scholar]
- 75.Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8:24–36. [DOI] [PubMed] [Google Scholar]
- 76.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;93:324–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stet EH, De Abreu RA, Bokkerink JP, et al. Reversal of 6-mercaptopurine and 6-methylmercaptopurine ribonucleoside cytotoxicity by amidoimidazole carboxamide ribonucleoside in Molt F4 human malignant T-lymphoblasts. Biochem Pharmacol. 1993;46:547–550. [DOI] [PubMed] [Google Scholar]
- 78.Bokkerink JP, Stet EH, De Abreu RA, et al. 6-Mercaptopurine: cytotoxicity and biochemical pharmacology in human malignant T-lymphoblasts. Biochem Pharmacol. 1993;45:1455–1463. [DOI] [PubMed] [Google Scholar]
- 79.Ebbesen MS, Nersting J, Jacobsen JH, et al. Incorporation of 6-thioguanine nucleotides into DNA during maintenance therapy of childhood acute lymphoblastic leukemia-the influence of thiopurine methyltransferase genotypes. J Clin Pharmacol. 2013;53:670–674. [DOI] [PubMed] [Google Scholar]
- 80.Jacobsen JH, Schmiegelow K, Nersting J. Liquid chromatography-tandem mass spectrometry quantification of 6-thioguanine in DNA using endogenous guanine as internal standard. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;881-882:115–118. [DOI] [PubMed] [Google Scholar]
- 81.Erb N, Haverland U, Harms DO, et al. High-performance liquid chromatographic assay of metabolites of thioguanine and mercaptopurine in capillary blood. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;796:87–94. [DOI] [PubMed] [Google Scholar]
- 82.Schmiegelow K, Bruunshuus I. 6-Thioguanine nucleotide accumulation in red blood cells during maintenance chemotherapy for childhood acute lymphoblastic leukemia, and its relation to leukopenia. Cancer Chemother Pharmacol. 1990;26:288–292. [DOI] [PubMed] [Google Scholar]
- 83.Bostrom B, Erdmann G. Cellular pharmacology of 6-mercaptopurine in acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1993;15:80–86. [PubMed] [Google Scholar]
- 84.Lilleyman JS, Lennard L. Mercaptopurine metabolism and risk of relapse in childhood lymphoblastic leukaemia. Lancet. 1994;343:1188–1190. [DOI] [PubMed] [Google Scholar]
- 85.Schmiegelow K, Schroder H, Gustafsson G, et al. Risk of relapse in childhood acute lymphoblastic leukemia is related to RBC methotrexate and mercaptopurine metabolites during maintenance chemotherapy. Nordic Society for Pediatric Hematology and Oncology. J Clin Oncol. 1995;13:345–351. [DOI] [PubMed] [Google Scholar]
- 86.Bergan S, Bentdal O, Sodal G, et al. Patterns of azathioprine metabolites in neutrophils, lymphocytes, reticulocytes, and erythrocytes: relevance to toxicity and monitoring in recipients of renal allografts. Ther Drug Monit. 1997;19:502–509. [DOI] [PubMed] [Google Scholar]
- 87.Lancaster D, Lennard L, Lilleyman JS. Profile of non-compliance in lymphoblastic leukaemia. Arch Dis Child. 1997;76:365–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Erb N, Harms DO, Janka-Schaub G. Pharmacokinetics and metabolism of thiopurines in children with acute lymphoblastic leukemia receiving 6-thioguanine versus 6-mercaptopurine. Cancer Chemother Pharmacol. 1998;42:266–272. [DOI] [PubMed] [Google Scholar]
- 89.Nygaard U, Toft N, Schmiegelow K. Methylated metabolites of 6-mercaptopurine are associated with hepatotoxicity. Clin Pharmacol Ther. 2004;75:274–281. [DOI] [PubMed] [Google Scholar]
- 90.Smith MA, Blaker P, Marinaki AM, et al. Optimising outcome on thiopurines in inflammatory bowel disease by co-prescription of allopurinol. J Crohns Colitis. 2012;6:905–912. [DOI] [PubMed] [Google Scholar]
- 91.Shih DQ, Nguyen M, Zheng L, et al. Split-dose administration of thiopurine drugs: a novel and effective strategy for managing preferential 6-MMP metabolism. Aliment Pharmacol Ther. 2012;36:449–458. [DOI] [PubMed] [Google Scholar]
- 92.Bell BA, Brockway GN, Shuster JJ, et al. A comparison of red blood cell thiopurine metabolites in children with acute lymphoblastic leukemia who received oral mercaptopurine twice daily or once daily: a Pediatric Oncology Group study (now The Children’s Oncology Group). Pediatr Blood Cancer. 2004;43:105–109. [DOI] [PubMed] [Google Scholar]
- 93.Fotoohi AK, Albertioni F. Mechanisms of antifolate resistance and methotrexate efficacy in leukemia cells. Leuk Lymphoma. 2008;49:410–426. [DOI] [PubMed] [Google Scholar]
- 94.Sorich MJ, Pottier N, Pei D, et al. In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med. 2008;5:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kager L, Cheok M, Yang W, et al. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest. 2005;115:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schmiegelow K. Advances in individual prediction of methotrexate toxicity: a review. Br J Haematol. 2009;146:489–503. [DOI] [PubMed] [Google Scholar]
- 97.Belkov VM, Krynetski EY, Schuetz JD, et al. Reduced folate carrier expression in acute lymphoblastic leukemia: a mechanism for ploidy but not lineage differences in methotrexate accumulation. Blood. 1999;93:1643–1650. [PubMed] [Google Scholar]
- 98.Galpin AJ, Schuetz JD, Masson E, et al. Differences in folylpolyglutamate synthetase and dihydrofolate reductase expression in human B-lineage versus T-lineage leukemic lymphoblasts: mechanisms for lineage differences in methotrexate polyglutamylation and cytotoxicity. Mol Pharmacol. 1997;52:155–163. [DOI] [PubMed] [Google Scholar]
- 99.Schroder H, Fogh K, Herlin T. In vivo decline of methotrexate and methotrexate polyglutamates in age-fractionated erythrocytes. Cancer Chemother Pharmacol. 1988;21:150–155. [DOI] [PubMed] [Google Scholar]
- 100.Schmiegelow K, Schroder H, Pulczynska MK, et al. Maintenance chemotherapy for childhood acute lymphoblastic leukemia: relation of bone-marrow and hepatotoxicity to the concentration of methotrexate in erythrocytes. Cancer Chemother Pharmacol. 1989;25:65–69. [DOI] [PubMed] [Google Scholar]
- 101.Schroder H. In vivo methotrexate kinetics and metabolism in human hematopoietic cells. Clinical significance of methotrexate concentrations in erythrocytes. Dan Med Bull. 1990;37:22–40. [PubMed] [Google Scholar]
- 102.Schmiegelow K, Schroder H, Schmiegelow M. Methotrexate and 6-mercaptopurine maintenance therapy for childhood acute lymphoblastic leukemia: dose adjustments by white cell counts or by pharmacokinetic parameters? Cancer Chemother Pharmacol. 1994;34:209–215. [DOI] [PubMed] [Google Scholar]
- 103.Graham ML, Shuster JJ, Kamen BA, et al. Red blood cell methotrexate and folate levels in children with acute lymphoblastic leukemia undergoing therapy: a Pediatric Oncology Group pilot study. Cancer Chemother Pharmacol. 1992;31:217–222. [DOI] [PubMed] [Google Scholar]
- 104.Aplenc R, Lange B. Pharmacogenetic determinants of outcome in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:421–434. [DOI] [PubMed] [Google Scholar]
- 105.Davidsen ML, Dalhoff K, Schmiegelow K. Pharmacogenetics influence treatment efficacy in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30:831–849. [DOI] [PubMed] [Google Scholar]
- 106.Chang JG, Lee LS, Chen CM, et al. Molecular analysis of thiopurine S-methyltransferase alleles in South-east Asian populations. Pharmacogenetics. 2002;12:191–195. [DOI] [PubMed] [Google Scholar]
- 107.Lu HF, Shih MC, Hsueh SC, et al. Molecular analysis of the thiopurine S-methyltransferase alleles in Bolivians and Tibetans. J Clin Pharm Ther. 2005;30:491–496. [DOI] [PubMed] [Google Scholar]
- 108.Lu HF, Shih MC, Chang YS, et al. Molecular analysis of thiopurine S-methyltransferase alleles in Taiwan aborigines and Taiwanese. J Clin Pharm Ther. 2006;31:93–98. [DOI] [PubMed] [Google Scholar]
- 109.Toft N, Nygaard U, Gregers J, et al. Genetic analyses of thiopurine methyltransferase polymorphisms in Greenlandic and Danish populations. Acta Paediatr. 2006;95:1665–1667. [DOI] [PubMed] [Google Scholar]
- 110.Kim H, Kang HJ, Kim HJ, et al. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: a possible association between survival rate and ITPA polymorphism. PLoS One. 2012;7:e45558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lennard L, Lewis IJ, Michelagnoli M, et al. Thiopurine methyltransferase deficiency in childhood lymphoblastic leukaemia: 6-mercaptopurine dosage strategies. Med Pediatr Oncol. 1997;29:252–255. [DOI] [PubMed] [Google Scholar]
- 112.Andersen JB, Szumlanski C, Weinshilboum RM, et al. Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr. 1998;87:108–111. [DOI] [PubMed] [Google Scholar]
- 113.Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus [see comments]. J Natl Cancer Inst. 1999;91:2001–2008. [DOI] [PubMed] [Google Scholar]
- 114.Lennard L, Lilleyman JS, Van Loon J, et al. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. [DOI] [PubMed] [Google Scholar]
- 115.Relling MV, Rubnitz JE, Rivera GK, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet. 1999;354:34–39. [DOI] [PubMed] [Google Scholar]
- 116.Thomsen JB, Schroder H, Kristinsson J, et al. Possible carcinogenic effect of 6-mercaptopurine on bone marrow stem cells: relation to thiopurine metabolism. Cancer. 1999;86:1080–1086. [DOI] [PubMed] [Google Scholar]
- 117.Stanulla M, Schaeffeler E, Moricke A, et al. Thiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin-Frankfurt-Munster protocols. Blood. 2009;114:1314–1318. [DOI] [PubMed] [Google Scholar]
- 118.Lennard L, Chew TS, Lilleyman JS. Human thiopurine methyltransferase activity varies with red blood cell age. Br J Clin Pharmacol. 2001;52:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stocco G, Cheok MH, Crews KR, et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Adam-de BT, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol. 2012;68:1233–1242. [DOI] [PubMed] [Google Scholar]
- 121.Tanaka Y, Manabe A, Nakadate H, et al. The activity of the inosine triphosphate pyrophosphatase affects toxicity of 6-mercaptopurine during maintenance therapy for acute lymphoblastic leukemia in Japanese children. Leuk Res. 2012;36:560–564. [DOI] [PubMed] [Google Scholar]
- 122.Wan Rosalina WR, Teh LK, Mohamad N, et al. Polymorphism of ITPA 94C>A and risk of adverse effects among patients with acute lymphoblastic leukaemia treated with 6-mercaptopurine. J Clin Pharm Ther. 2012;37:237–241. [DOI] [PubMed] [Google Scholar]
- 123.Zelinkova Z, Derijks LJ, Stokkers PC, et al. Inosine triphosphate pyrophosphatase and thiopurine s-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin Gastroenterol Hepatol. 2006;4:44–49. [DOI] [PubMed] [Google Scholar]
- 124.Adam de BT, Fakhoury M, Medard Y, et al. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br J Clin Pharmacol. 2011;71:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marsh S, King CR, Ahluwalia R, et al. Distribution of ITPA P32T alleles in multiple world populations. J Hum Genet. 2004;49:579–581. [DOI] [PubMed] [Google Scholar]
- 126.Lennard L, Hale JP, Lilleyman JS. Red blood cell hypoxanthine phosphoribosyltransferase activity measured using 6-mercaptopurine as a substrate: a population study in children with acute lymphoblastic leukaemia. Br J Clin Pharmacol. 1993;36:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pieters R, Huismans DR, Loonen AH, et al. Hypoxanthine-guanine phosphoribosyl-transferase in childhood leukemia: relation with immunophenotype, in vitro drug resistance and clinical prognosis. Int J Cancer. 1992;51:213–217. [DOI] [PubMed] [Google Scholar]
- 128.Dulucq S, St-Onge G, Gagne V, et al. DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood. 2008;111:3692–3700. [DOI] [PubMed] [Google Scholar]
- 129.Krajinovic M, Costea I, Primeau M, et al. Combining several polymorphisms of thymidylate synthase gene for pharmacogenetic analysis. Pharmacogenomics J. 2005;5:374–380. [DOI] [PubMed] [Google Scholar]
- 130.Rocha JC, Cheng C, Liu W, et al. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood. 2005;105:4752–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gregers J, Christensen IJ, Dalhoff K, et al. The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood. 2010;115:4671–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al. Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2004;4:66–72. [DOI] [PubMed] [Google Scholar]
- 133.Krajinovic M, Moghrabi A. Pharmacogenetics of methotrexate. Pharmacogenomics. 2004;5:819–834. [DOI] [PubMed] [Google Scholar]
- 134.Kager L, Evans WE. Pharmacogenomics of acute lymphoblastic leukemia. Curr Opin Hematol. 2006;13:260–265. [DOI] [PubMed] [Google Scholar]
- 135.Ranganathan P. An update on methotrexate pharmacogenetics in rheumatoid arthritis. Pharmacogenomics. 2008;9:439–451. [DOI] [PubMed] [Google Scholar]
- 136.Schmiegelow K. Maintenance chemotherapy of acute lymphoblastic leukemia in children. Dan Med Bull. 1998;45:510–532. [PubMed] [Google Scholar]
- 137.van Eys J, Berry D, Crist W, et al. Treatment intensity and outcome for children with acute lymphocytic leukemia of standard risk. A Pediatric Oncology Group Study. Cancer. 1989;63:1466–1471. [DOI] [PubMed] [Google Scholar]
- 138.Lennard L, Rees CA, Lilleyman JS, et al. Childhood leukaemia: a relationship between intracellular 6-mercaptopurine metabolites and neutropenia. Br J Clin Pharmacol. 1983;16:359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmiegelow K, Pulczynska MK, Seip M. White cell count during maintenance chemotherapy for standard-risk childhood acute lymphoblastic leukemia: relation to relapse rate. Pediatr Hematol Oncol. 1988;5:259–267. [DOI] [PubMed] [Google Scholar]
- 140.Dolan G, Lilleyman JS, Richards SM. Prognostic importance of myelosuppression during maintenance treatment of lymphoblastic leukaemia. Leukaemia in Childhood Working Party of the Medical Research Council. Arch Dis Child. 1989;64:1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gobrecht O, Gobel U, Graubner U, et al. Effect of dose intensity and therapy-induced leukocytopenia in intensive therapy on the prognosis of acute lymphatic leukemia in childhood. Results in 213 patients of the COALL-85 study. Klin Padiatr. 1992;204:230–235. [DOI] [PubMed] [Google Scholar]
- 142.Hayder S, Bjork O, Nilsson B. Relapse factors during maintenance therapy of acute lymphoblastic leukemia in children. Pediatr Hematol Oncol. 1992;9:21–27. [DOI] [PubMed] [Google Scholar]
- 143.Lucas K, Gula MJ, Blatt J. Relapse in acute lymphoblastic leukemia as a function of white blood cell and absolute neutrophil counts during maintenance chemotherapy. Pediatr Hematol Oncol. 1992;9:91–97. [DOI] [PubMed] [Google Scholar]
- 144.Chessells JM, Harrison G, Lilleyman JS, et al. Continuing (maintenance) therapy in lymphoblastic leukaemia: lessons from MRC UKALL X. Medical Research Council Working Party in Childhood Leukaemia. Br J Haematol. 1997;98:945–951. [DOI] [PubMed] [Google Scholar]
- 145.Schmiegelow K, Pulczynska MK. White-cell counts in childhood acute lymphoblastic leukemia. Eur J Haematol. 1990;44:72–74. [DOI] [PubMed] [Google Scholar]
- 146.Schmiegelow K, Ifversen M. Myelotoxicity, pharmacokinetics, and relapse rate with methotrexate/6-mercaptopurine maintenance therapy of childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1996;13:433–441. [DOI] [PubMed] [Google Scholar]
- 147.Harms DO, Gobel U, Spaar HJ, et al. Thioguanine offers no advantage over mercaptopurine in maintenance treatment of childhood ALL: results of the randomized trial COALL-92. Blood. 2003;102:2736–2740. [DOI] [PubMed] [Google Scholar]
- 148.Satti MB, Weinbren K, Gordon-Smith EC. 6-thioguanine as a cause of toxic veno-occlusive disease of the liver. J Clin Pathol. 1982;35:1086–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Broxson EH, Dole M, Wong R, et al. Portal hypertension develops in a subset of children with standard risk acute lymphoblastic leukemia treated with oral 6-thioguanine during maintenance therapy. Pediatr Blood Cancer. 2005;44:226–231. [DOI] [PubMed] [Google Scholar]
- 150.Escherich G, Horstmann MA, Zimmermann M, et al. Cooperative study group for childhood acute lymphoblastic leukaemia (COALL): long-term results of trials 82,85,89,92 and 97. Leukemia. 2010;24:298–308. [DOI] [PubMed] [Google Scholar]
- 151.Heegaard ED, Kerndrup GB, Carlsen NT, et al. [Thrombocytopenia caused by Parvovirus B19 infection in a child with acute lymphatic leukemia]. Ugeskr Laeger. 1999;161:6501–6502. [PubMed] [Google Scholar]
- 152.Topley JM, Benson J, Squier MV, et al. Hepatotoxicity in the treatment of acute lymphoblastic leukaemia. Med Pediatr Oncol. 1979;7:393–399. [DOI] [PubMed] [Google Scholar]
- 153.Bessho F, Kinumaki H, Yokota S, et al. Liver function studies in children with acute lymphocytic leukemia after cessation of therapy. Med Pediatr Oncol. 1994;23:111–115. [DOI] [PubMed] [Google Scholar]
- 154.Farrow AC, Buchanan GR, Zwiener RJ, et al. Serum aminotransferase elevation during and following treatment of childhood acute lymphoblastic leukemia. J Clin Oncol. 1997;15:1560–1566. [DOI] [PubMed] [Google Scholar]
- 155.Schmiegelow K, Bretton-Meyer U. 6-mercaptopurine dosage and pharmacokinetics influence the degree of bone marrow toxicity following high-dose methotrexate in children with acute lymphoblastic leukemia. Leukemia. 2001;15:74–79. [DOI] [PubMed] [Google Scholar]
- 156.Halonen P, Mattila J, Makipernaa A, et al. Erythrocyte concentrations of metabolites or cumulative doses of 6-mercaptopurine and methotrexate do not predict liver changes in children treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2006;46:762–766. [DOI] [PubMed] [Google Scholar]
- 157.Halonen P, Salo MK, Makipernaa A. Fasting hypoglycemia is common during maintenance therapy for childhood acute lymphoblastic leukemia. J Pediatr. 2001;138:428–431. [DOI] [PubMed] [Google Scholar]
- 158.Bay A, Oner AF, Cesur Y, et al. Symptomatic hypoglycemia: an unusual side effect of oral purine analogues for treatment of ALL. Pediatr Blood Cancer. 2006;47:330–331. [DOI] [PubMed] [Google Scholar]
- 159.El-Bitar MK, Muwakkit SA, Dabbagh O. Severe hypoglycemic seizures in a child receiving 6-mercaptopurine. J Pediatr Hematol Oncol. 2011;33:e75–e76. [DOI] [PubMed] [Google Scholar]
- 160.Trelinska J, Fendler W, Szadkowska A, et al. Hypoglycemia and glycemic variability among children with acute lymphoblastic leukemia during maintenance therapy. Leuk Lymphoma. 2011;52:1704–1710. [DOI] [PubMed] [Google Scholar]
- 161.Melachuri S, Gandrud L, Bostrom B. The association between fasting hypoglycemia and methylated mercaptopurine metabolites in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2014;61:1003–1006. [DOI] [PubMed] [Google Scholar]
- 162.Halonen P, Mattila J, Suominen P, et al. Iron overload in children who are treated for acute lymphoblastic leukemia estimated by liver siderosis and serum iron parameters. Pediatrics. 2003;111:91–96. [DOI] [PubMed] [Google Scholar]
- 163.Halonen P, Mattila J, Ruuska T, et al. Liver histology after current intensified therapy for childhood acute lymphoblastic leukemia: microvesicular fatty change and siderosis are the main findings. Med Pediatr Oncol. 2003;40:148–154. [DOI] [PubMed] [Google Scholar]
- 164.Halonen P, Salo MK, Schmiegelow K, et al. Investigation of the mechanisms of therapy-related hypoglycaemia in children with acute lymphoblastic leukaemia. Acta Paediatr. 2003;92:37–42. [DOI] [PubMed] [Google Scholar]
- 165.Bodey GP, Brodovsky HS, Isassi AA, et al. Studies of combination 6-mercaptopurine (NSC-755) and 6-methylmercaptopurine riboside (NSC-40774) in patients with acute leukemia and metastatic cancer. Cancer Chemother Rep. 1968;52:315–320. [PubMed] [Google Scholar]
- 166.Hewlett JS, Bodey GP, Wilson HE, et al. Combination 6-mercaptopurine and 6-methylmercaptopurine riboside in the treatment of adult acute leukemia: a Southwest Oncology Group study. Cancer Treat Rep. 1979;63:156–158. [PubMed] [Google Scholar]
- 167.Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. [DOI] [PubMed] [Google Scholar]
- 168.Rulyak SJ, Saunders MD, Lee SD. Hepatotoxicity associated with 6-thioguanine therapy for Crohn’s disease. J Clin Gastroenterol. 2003;36:234–237. [DOI] [PubMed] [Google Scholar]
- 169.Berkovitch M, Matsui D, Zipursky A, et al. Hepatotoxicity of 6-mercaptopurine in childhood acute lymphocytic leukemia: pharmacokinetic characteristics. Med Pediatr Oncol. 1996;26:85–89. [DOI] [PubMed] [Google Scholar]
- 170.Schmiegelow K, Pulczynska M. Prognostic significance of hepatotoxicity during maintenance chemotherapy for childhood acute lymphoblastic leukaemia. Br J Cancer. 1990;61:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bokkerink JP, Damen FJ, Hulscher MW, et al. Biochemical evidence for synergistic combination treatment with methotrexate and 6-mercaptopurine in acute lymphoblastic leukemia. Haematol Blood Transfus. 1990;33:110–117. [DOI] [PubMed] [Google Scholar]
- 172.Balis FM, Holcenberg JS, Zimm S, et al. The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther. 1987;41:384–387. [DOI] [PubMed] [Google Scholar]
- 173.Dervieux T, Hancock M, Evans W, et al. Effect of methotrexate polyglutamates on thioguanine nucleotide concentrations during continuation therapy of acute lymphoblastic leukemia with mercaptopurine. Leukemia. 2002;16:209–212. [DOI] [PubMed] [Google Scholar]
- 174.Escherich G, Richards S, Stork LC, et al. Meta-analysis of randomised trials comparing thiopurines in childhood acute lymphoblastic leukaemia. Leukemia. 2011;25:953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lancaster DL, Lennard L, Rowland K, et al. Thioguanine versus mercaptopurine for therapy of childhood lymphoblastic leukaemia: a comparison of haematological toxicity and drug metabolite concentrations. Br J Haematol. 1998;102:439–443. [DOI] [PubMed] [Google Scholar]
- 176.Vora A, Mitchell CD, Lennard L, et al. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet. 2006;368:1339–1348. [DOI] [PubMed] [Google Scholar]
- 177.Lennard L, Richards S, Cartwright CS, et al. The thiopurine methyltransferase genetic polymorphism is associated with thioguanine-related veno-occlusive disease of the liver in children with acute lymphoblastic leukemia. Clin Pharmacol Ther. 2006;80:375–383. [DOI] [PubMed] [Google Scholar]
- 178.Jacobs SS, Stork LC, Bostrom BC, et al. Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children’s Oncology Group/National Cancer Institute pilot trial (CCG-1942). Pediatr Blood Cancer. 2007;49:250–255. [DOI] [PubMed] [Google Scholar]
- 179.Breitkreutz J, Buckman J, Fischer R, et al. Comparative in vitro studies on different 6-mercaptopurine formulations for use in children. Paediatr Perinat Drug Ther. 2007;8:31–39. [Google Scholar]
- 180.Mulla H, Leary A, White P, et al. A step toward more accurate dosing for mercaptopurine in childhood acute lymphoblastic leukemia. J Clin Pharmacol. 2012;52:161–163. [DOI] [PubMed] [Google Scholar]
- 181.Lennard L, Welch J, Lilleyman JS. Intracellular metabolites of mercaptopurine in children with lymphoblastic leukaemia: a possible indicator of non-compliance? Br J Cancer. 1995;72:1004–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lau RC, Matsui D, Greenberg M, et al. Electronic measurement of compliance with mercaptopurine in pediatric patients with acute lymphoblastic leukemia. Med Pediatr Oncol. 1998;30:85–90. [DOI] [PubMed] [Google Scholar]
- 183.Pritchard MT, Butow PN, Stevens MM, et al. Understanding medication adherence in pediatric acute lymphoblastic leukemia: a review. J Pediatr Hematol Oncol. 2006;28:816–823. [DOI] [PubMed] [Google Scholar]
- 184.Hanghoj S, Boisen KA. Self-reported barriers to medication adherence among chronically ill adolescents: a systematic review. J Adolesc Health. 2014;54:121–138. [DOI] [PubMed] [Google Scholar]
- 185.Brandalise SR, Pinheiro VR, Aguiar SS, et al. Benefits of the intermittent use of 6-mercaptopurine and methotrexate in maintenance treatment for low-risk acute lymphoblastic leukemia in children: randomized trial from the Brazilian Childhood Cooperative Group—protocol ALL-99. J Clin Oncol. 2010;28:1911–1918. [DOI] [PubMed] [Google Scholar]
- 186.Hrushesky WJ, Bjarnason GA. Circadian cancer therapy. J Clin Oncol. 1993;11:1403–1417. [DOI] [PubMed] [Google Scholar]
- 187.Rivard GE, Infante-Rivard C, Hoyoux C, et al. Maintenance chemotherapy for childhood acute lymphoblastic leukaemia: better in the evening. Lancet. 1985;2:1264–1266. [DOI] [PubMed] [Google Scholar]
- 188.Schmiegelow K, Glomstein A, Kristinsson J, et al. Impact of morning versus evening schedule for oral methotrexate and 6-mercaptopurine on relapse risk for children with acute lymphoblastic leukemia. Nordic Society for Pediatric Hematology and Oncology (NOPHO). J Pediatr Hematol Oncol. 1997;19:102–109. [DOI] [PubMed] [Google Scholar]
- 189.Ramot B, Brok-Simoni F, Chweidan E, et al. Blood leucocyte enzymes. III. Diurnal rhythm of activity in isolated lymphocytes of normal subjects and chronic lymphatic leukaemia patients. Br J Haematol. 1976;34:79–85. [DOI] [PubMed] [Google Scholar]
- 190.Sletvold O, Smaaland R, Laerum OD. Cytometry and time-dependent variations in peripheral blood and bone marrow cells: a literature review and relevance to the chronotherapy of cancer. Chronobiol Int. 1991;8:235–250. [DOI] [PubMed] [Google Scholar]
- 191.Clemmensen KK, Christensen RH, Shabaneh DN, et al. The circadian schedule for childhood acute lymphoblastic leukemia maintenance therapy does not influence event-free survival in the NOPHO ALL92 protocol. Pediatr Blood Cancer. 2013;61:653–658. [DOI] [PubMed] [Google Scholar]
- 192.Pinkerton CR, Welshman SG, Glasgow JF, et al. Can food influence the absorption of methotrexate in children with acute lymphoblastic leukaemia? Lancet. 1980;2:944–946. [DOI] [PubMed] [Google Scholar]
- 193.Riccardi R, Balis FM, Ferrara P, et al. Influence of food intake on bioavailability of oral 6-mercaptopurine in children with acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1986;3:319–324. [DOI] [PubMed] [Google Scholar]
- 194.Lonnerholm G, Kreuger A, Lindstrom B, et al. Oral mercaptopurine in childhood leukemia: influence of food intake on bioavailability. Pediatr Hematol Oncol. 1989;6:105–112. [DOI] [PubMed] [Google Scholar]
- 195.Kozloski GD, De Vito JM, Kisicki JC, et al. The effect of food on the absorption of methotrexate sodium tablets in healthy volunteers. Arthritis Rheum. 1992;35:761–764. [DOI] [PubMed] [Google Scholar]
- 196.Hamilton RA, Kremer JM. The effects of food on methotrexate absorption. J Rheumatol. 1995;22:630–632. [PubMed] [Google Scholar]
- 197.de Lemos ML, Hamata L, Jennings S, et al. Interaction between mercaptopurine and milk. J Oncol Pharm Pract. 2007;13:237–240. [DOI] [PubMed] [Google Scholar]
- 198.Pinkel D. Intravenous mercaptopurine: life begins at 40. J Clin Oncol. 1993;11:1826–1831. [DOI] [PubMed] [Google Scholar]
- 199.Bostrom BC, Sensel MR, Sather HN, et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood. 2003;101:3809–3817. [DOI] [PubMed] [Google Scholar]
- 200.Conter V, Valsecchi MG, Silvestri D, et al. Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet. 2007;369:123–131. [DOI] [PubMed] [Google Scholar]
- 201.Eden T, Pieters R, Richards S. Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia—an individual patient data meta-analysis involving 5659 children. Br J Haematol. 2010;149:722–733. [DOI] [PubMed] [Google Scholar]
- 202.Adam de Beaumais T, Dervieux T, Fakhoury M, et al. The impact of high-dose methotrexate on intracellular 6-mercaptopurine disposition during interval therapy of childhood acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2010;66:653–658. [DOI] [PubMed] [Google Scholar]
- 203.Nygaard U, Schmiegelow K. Dose reduction of coadministered 6-mercaptopurine decreases myelotoxicity following high-dose methotrexate in childhood leukemia. Leukemia. 2003;17:1344–1348. [DOI] [PubMed] [Google Scholar]
- 204.van Kooten Niekerk PB, Schmiegelow K, Schroeder H. Influence of methylene tetrahydrofolate reductase polymorphisms and coadministration of antimetabolites on toxicity after high dose methotrexate. Eur J Haematol. 2008;81:391–398. [DOI] [PubMed] [Google Scholar]
- 205.De MB, Suciu S, Bertrand Y, et al. Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood. 2010;116:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bleyer WA, Sather HN, Nickerson HJ, et al. Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia: a report of the CCG-161 study by the Childrens Cancer Study Group. J Clin Oncol. 1991;9:1012–1021. [DOI] [PubMed] [Google Scholar]
- 207.Felice MS, Rossi JG, Gallego MS, et al. No advantage of a rotational continuation phase in acute lymphoblastic leukemia in childhood treated with a BFM back-bone therapy. Pediatr Blood Cancer. 2011;57:47–55. [DOI] [PubMed] [Google Scholar]
- 208.Shea B, Swinden MV, Tanjong GE, et al. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst Rev. 2013;5:CD000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Robien K, Schubert MM, Yasui Y, et al. Folic acid supplementation during methotrexate immunosuppression is not associated with early toxicity, risk of acute graft-versus-host disease or relapse following hematopoietic transplantation. Bone Marrow Transplant. 2006;37:687–692. [DOI] [PubMed] [Google Scholar]
- 210.Lennard L, Lilleyman JS, Maddocks JL. The effect of folate supplements on 6-mercaptopurine remission maintenance therapy in childhood leukaemia. Br J Cancer. 1986;53:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schroder H, Clausen N, Ostergard E, et al. Folic acid supplements in vitamin tablets: a determinant of hematological drug tolerance in maintenance therapy of childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1986;3:241–247. [DOI] [PubMed] [Google Scholar]
- 212.Poulsen A, Demeny AK, Bang PC, et al. Pneumocystis carinii pneumonia during maintenance treatment of childhood acute lymphoblastic leukemia. Med Pediatr Oncol. 2001;37:20–23. [DOI] [PubMed] [Google Scholar]
- 213.Ferrazzini G, Klein J, Sulh H, et al. Interaction between trimethoprim-sulfamethoxazole and methotrexate in children with leukemia. J Pediatr. 1990;117:823–826. [DOI] [PubMed] [Google Scholar]
- 214.Rees CA, Lennard L, Lilleyman JS, et al. Disturbance of 6-mercaptopurine metabolism by cotrimoxazole in childhood lymphoblastic leukaemia. Cancer Chemother Pharmacol. 1984;12:87–89. [DOI] [PubMed] [Google Scholar]
- 215.Levinsen M, Shabaneh D, Bohnstedt C, et al. Pneumocystis jiroveci pneumonia prophylaxis during maintenance therapy influences methotrexate/6-mercaptopurine dosing but not event-free survival for childhood acute lymphoblastic leukemia. Eur J Haematol. 2012;88:78–86. [DOI] [PubMed] [Google Scholar]
- 216.Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–2084. [DOI] [PubMed] [Google Scholar]
- 217.Vaitkeviciene G, Heyman M, Jonsson OG, et al. Early morbidity and mortality in childhood acute lymphoblastic leukemia with very high white blood cell count. Leukemia. 2013;27:2259–2262. [DOI] [PubMed] [Google Scholar]
- 218.Rubnitz JE, Camitta BM, Mahmoud H, et al. Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol. 1999;17:191–196. [DOI] [PubMed] [Google Scholar]
- 219.Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Forestier E, Johansson B, Gustafsson G, et al. Prognostic impact of karyotypic findings in childhood acute lymphoblastic leukaemia: a Nordic series comparing two treatment periods. For the Nordic Society of Paediatric Haematology and Oncology (NOPHO) Leukaemia Cytogenetic Study Group. Br J Haematol. 2000;110:147–153. [DOI] [PubMed] [Google Scholar]
- 221.Schmiegelow K, Pulczynska MK. Maintenance chemotherapy for childhood acute lymphoblastic leukemia: should dosage be guided by white blood cell counts? Am J Pediatr Hematol Oncol. 1990;12:462–467. [DOI] [PubMed] [Google Scholar]