

Abstract
We report a hybrid fragmentation method involving electron transfer dissociation (ETD) combined with ultraviolet photodissociation (UVPD) at 193 nm for analysis of intact proteins in an Orbitrap mass spectrometer. Integrating the two fragmentation methods resulted in an increase in the number of identified c- and z-type ions observed when compared to UVPD or ETD alone, as well as generating a more balanced distribution of a/x, b/y, and c/z ion types. Additionally, the method was shown to decrease spectral congestion via fragmentation of multiple (charge-reduced) precursors. This hybrid activation method was facilitated by performing both ETD and UVPD within the higher energy collisional dissociation (HCD) cell of the Orbitrap mass spectrometer, which afforded an increase in the total number of fragment ions in comparison to the analogous MS3 format in which ETD and UVPD were undertaken in separate segments of the mass spectrometer. The feasibility of the hybrid method for characterization of proteins on a liquid chromatography timescale characterization was demonstrated for intact ribosomal proteins.
Electron capture dissociation and electron transfer dissociation (ECD and ETD, respectively)1,2 have become landmark ion activation/dissociation methods in the field of proteomics due to their ability to maintain labile post-translational modifications (PTMs) while indiscriminately fragmenting the polypeptide backbone. Both ECD and ETD promote similar mechanisms of ion activation and fragmentation and have been used extensively for localization of PTMs in bottom-up peptide-based analysis and in top down mass spectrometry for characterization of intact proteins.3 A compelling feature of electron-based activation methods is the ability to generate charge-reduced ions, including ample abundances of odd electron (radical) precursors that may be isolated and further energized. In this way, the fragmentation of odd electron (radical) versus even electron (closed shell) peptides and proteins may be conveniently compared, not only shedding light on the fundamental impact of radical-mediated processes but also allowing access to a different, often complementary, type of fragmentation behavior with analytical merits (sequencing, localization of modifications, etc.). The intriguing opportunities afforded by production and analysis of radical-type ions have motivated several groups to explore hybrid methods that combine ETD with a second activation method. For example, Heck and co-workers have recently devised new approaches based on hybrid combinations of fragmentation methods for more complete peptide fragmentation.4 In one case, electron transfer dissociation followed by transmission of all resulting ions into a multipole for higher energy collision induced dissociation (so-called EThcD), was shown to provide an informative array of predominantly b-, c-, y-, and z-type ions.4 Although the greater number of fragmentation channels increased both the complexity of the product ion spectrum and the fragment ion search space for all candidate peptides that fell within the precursor mass tolerance, the net increase in information more than compensated for the decrease in confidence from a typical database search.4 Moreover, the hybrid EThcD method improved the localization scores obtained for identification of phosphorylation sites of peptides.5 We have evaluated the use of hybrid methods combining electron transfer reactions to generate radical cations, followed by collision-induced dissociation (CID), infrared multiphoton dissociation (IRMPD), or ultraviolet photodissociation (UVPD) for characterization of the sites of modification of nucleic acids.6 The most diverse array of fragment ions was obtained from the ETUVPD hybrid method, an outcome that proved particularly beneficial for specific localization of modifications for which fragmentation was suppressed for other activation methods.6 We have also explored the use of UVPD to characterize radical peptide cations produced by electron transfer reactions, finding that the location of very basic sites (like Arg) at the C- versus N-terminus influenced the resulting fragmentation behavior and the preference for radical-directed versus photoactivated cleavages.7
Now that available bioinformatic platforms can accommodate high throughput top down MS/MS analyses that result in a multitude of ion types, such as the diverse array of fragments that arise from UVPD,8 the potential for hybrid fragmentation of intact proteins is feasible even for complex mixtures. Recently, we have demonstrated the utility of 193 nm UVPD for intact protein characterization in both single protein infusion and high throughput type liquid chromatography mass spectrometry (LC-MS) experiments.8−10 Typically, product ion spectra following UV photoactivation are characterized by a large proportion of the total ion current residing in the surviving precursor ion and a complex distribution of fragment ions (a, b, c, x, y, z) in an array of charge states. For those proteins in higher charge states, the crowded spectra confound deconvolution algorithms and are artifactually enriched in fragment ions of low mass and lower (and more easily deconvoluted) charge.9 This spectral complexity is the result not only of closely spaced isotopic peaks due to high charge states but also from the multitude of ion types generated by UVPD. In general, previous studies of 193 nm UVPD for top down proteomics have reported product ion spectra that have large contributions from a- and a+1-type ions representing the N terminus (with much lower proportions of b- and c-type ions), and a mixture of x-, y-, y–1-, and z-type ions arising from the C terminus.8,10 Despite the increase in search space associated with accommodating all of these ion types in an unweighted search algorithm, the sheer number of identified fragment ions has been shown to allow nearly complete protein characterization (via backbone cleavages present at nearly every inter-residue position).8,10 Although the number of fragment ions is very high, the method could benefit from an increase in the number and abundances of complementary C-terminally derived ions, such as the radical containing z-type ions that result from ETD, as well as a decrease in ions that are duplicative for the same inter-residue position (a and a+1, for example). The recent strategy from the Heck group for performing ETD in a DC gradient-only multipole11 affords an opportunity to implement ETD and UVPD together in a high performance Orbitrap mass spectrometer,8,12 as described herein. Although ETUVPD can be readily implemented in an MS3 format (in which the ETD step in the LIT precedes UVPD in the HCD cell), the ability to perform ETD in the HCD cell, per the Heck concept,11 provides more flexibility. For example, ETD can precede or follow UVPD in the HCD cell or both activation processes can be undertaken simultaneously. Ultraviolet irradiation electron transfer dissociation (UVIETD), has the potential to alleviate one of the main drawbacks of UVPD when performed by itself. A high proportion of the total ion current in the product ion spectrum resides in and/or falls very close to the m/z of the unfragmented precursor. This high peak density results in crowded spectra that are difficult to deconvolute due to their high charge states and close proximity to one another. Because ET kinetics have strong charge state dependence,13 initiating the ETD reaction following UVPD favors the likelihood of preferential dissociation of the (more highly charged) unreacted precursor above the fragment ions also present in the cell. Here we report the analytical merits of hybridizing ETD and UVPD for top down proteomics, with emphasis on the ability to achieve a more balanced array of product ions as well as a more uniform distribution of the ion current across the available m/z landscape.
Materials and Methods
Model Protein Studies
Bovine ubiquitin, horse myoglobin, and bovine carbonic anhydrase were purchased from Sigma-Aldrich (St. Louis, MO) and intact ribosomes were purchased from New England Biolabs (Ipswich, MA). All other solvents and chemicals were purchased from Sigma-Aldrich. Proteins were suspended in 50/49/1 methanol/water/formic acid (v/v/v) at a final concentration of 10 μM. They were infused directly into an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) customized for implementation of UVPD.12 Ultraviolet irradiation was achieved via a single (unless otherwise noted) 5 ns laser pulse from a Coherent ExciStar (Santa Clara, CA) 193 nm excimer laser. Feasibility studies utilizing an MS3 mode were performed via electron transfer dissociation in either the linear ion trap (LIT) or the higher energy collision dissociation (HCD) cell, followed by UVPD of the resulting ETD product ions in the HCD cell. The m/z range of the ion isolation window was varied to accommodate solely the singly charge reduced radical precursor or to encompass as many of the product ions and charge-reduced precursors as possible (±300 m/z around the initial multiprotonated precursor). For intact protein studies, ubiquitin, myoglobin, and carbonic anhydrase were reconstituted at 10 μM in 50/49/1 acetonitrile/water/formic acid (v/v/v). Spectra were acquired using 75, 200, and 500 averaged scans, respectively. Both MS1 and product ion spectra were acquired at 240k resolving power (at m/z 400). For all analyses, the HCD cell pressure was reduced to ∼2 mTorr relative to the standard HCD cell operating pressure of 10 mTorr (a pressure difference of 0.04 × 10–10 Torr measured in the Orbitrap chamber) which enhanced the detection of low abundance and larger fragment ions in the Orbitrap mass analyzer.
ETUVPD
Custom changes to the ion trap control language (ITCL) were made to allow the ETD reaction to occur within the HCD cell and to accommodate laser triggering for ion irradiation in the HCD cell of the Orbitrap mass spectrometer.
Ribosomal LC-UVPD-MS/MS
Ribosomes were prepared as described elsewhere.10,14,15 Briefly, intact ribosomal protein was isolated via acid precipitation of rRNA. Ribosomes were mixed with acetic acid (1 M) to a final concentration of 60% (v/v). The nucleic acids were allowed to precipitate, and the samples were centrifuged. The protein containing supernatant was reduced and alkylated. Ribosomes were analyzed using an Eksigent nanoLC Ultra system coupled to the Orbitrap Elite mass spectrometer. MS1 and tandem mass spectrometry (MS/MS) spectra were acquired using 1 or 3 averaged scans, respectively.
Bioinformatics
Fragment ion matching for intact proteins was performed using a version of ProSightPC 3.0 (Thermo Fisher) that was customized to accommodate the fragment ion types encountered with 193 nm UVPD.8 All product ions were matched within 10 ppm of their theoretical masses.
Results and Discussion
To date, UVPD has yielded extremely rich fragmentation patterns of intact proteins, yielding high sequence coverages and exceptional capabilities for pinpointing modifications albeit at the expense of sensitivity due to the greater division of ion signal into many fragment ion channels.8−10 Optimizing the utility of the diverse fragmentation pathways for protein identification and characterization has required search algorithms to accommodate an array of fragment ion types (a, a+1, b, c, x, x+1, y, y–1, and z),8 and with this multiplicity comes a penalty due to the concomitant increase in fragment ion search space.10 Despite this trade-off, the amount of information obtained using UVPD outweighs the reduction in sensitivity and expanded search space.10 We have shown previously that the successful characterization of intact proteins such as ubiquitin, myoglobin, and carbonic anhydrase by UVPD arises in large part from the significant number of mostly a-type ions that span a high proportion of the protein backbone.8 By combining both ETD (which results in predominantly c- and z-type ions) and UVPD, we anticipated that the ion current might be more evenly distributed, especially balancing C-terminal fragment ions with N-terminal ions. This hypothesis holds true for some proteins (e.g., myoglobin, 22+; carbonic anhydrase, 34+) but is less notable for others (ubiquitin, 13+) by ETD, UVPD, and ETUVPD, as shown in Figure 1 for ubiquitin and in Figure S1 (Supporting Information) for myoglobin. As expected, c/z ions are more dominant in the ETD spectra, and the UVPD spectra display primarily a ions along with contributions from b, c, z, y, and z ions. The ETUVPD spectra show distributions that are intermediate between the ones observed for ETD and UVPD.
Figure 1.
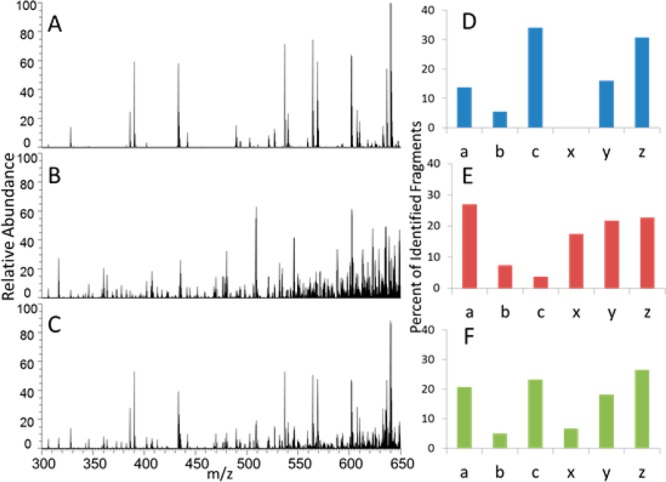
MS/MS spectra of ubiquitin (13+). (A) ETD (15 ms in HCD cell), (B) UVPD (one pulse, 2.5 mJ in HCD cell), (C) ETUVPD (15 ms ETD in HCD cell followed by UVPD using one pulse 2.5 mJ in HCD cell), and corresponding distribution of ion types in panels D, E, and F. All spectra are shown on the same scale.
An additional consequence of uniting ETD and UVPD is the ability to enhance the analysis of odd electron ions (such as charge-reduced precursors formed upon ETnoD and radical fragment ions initially formed upon ETD), ones that may dissociate by different, highly informative pathways and further enrich the resulting MS/MS spectra. Improved results have been reported for CID after electron transfer reactions by capitalizing on the instability of electron adducted precursors for peptide level proteomics in so-called “charge-reduced CID” (CRCID).16 The general idea of enhancing ETD fragmentation by supplemental activation has been termed “activated-ion ETD”,17−20 but as of yet, there have been no studies integrating ETD and UVPD at the protein level.
Additional feasibility experiments were conducted by comparing ETD in the linear ion trap (LIT) versus in the HCD cell prior to ultraviolet irradiation. The apparent decrease in efficiency of electron transfer from the reagent ion to the protein polycation was observed as expected11 when the reaction was performed in the HCD cell when compared to the reaction in the LIT. In the HCD cell, the reduced overlap between the reagent ion and analyte ion clouds leads to a decrease in the frequency of collisions between reagent anions and analyte cations. For this reason, hybrid ETUVPD experiments were undertaken to evaluate the overall dissociation efficiency when the ET step was undertaken in the LIT (8 ms ET reaction time) versus the HCD cell (15 ms reaction period), in each case with UVPD performed in the HCD cell. Similar distributions and types of product ions were observed for both hybrid variations, as illustrated for ubiquitin (13+) in Figure S2 (Supporting Information). For ETUVPD in which both ET and UVPD were undertaken in the HCD cell, a longer activation period was required to attain the same level of S/N due to the lower effectiveness of ET in the HCD cell, as mentioned above. The initial ETUVPD feasibility experiment provided evidence that there is little to no additional secondary fragmentation resulting in convoluting internal ions, an outcome consistent with prior results obtained using EThcD for peptides.4,5 Although internal ion formation can be used for diagnostic purposes in top down experiments with extensive a priori knowledge of the protein of interest,21 accommodating internal ions in a high throughput identification search strategy would cause a prohibitively large increase in fragment ion search space.
Performing MS/MS in the HCD cell allows trapping, activation, and analysis of a wider m/z range of product ions compared to MS/MS undertaken in the LIT. Specifically, performing ETD in the LIT allows isolation and transfer to the HCD cell of a range of product ions ±300 m/z units of the selected precursor ion. Undertaking ETD in the HCD cell and subsequent activation by UVPD does not require reisolation after ETD, and so all product ions may be simultaneously trapped, activated, and analyzed. This allows comparison of ETUVPD based on isolation of specific charge-reduced precursors from ET or broad populations of ions encompassing nearly the entire product ion spectrum resulting from ETD. For example, ubiquitin was infused and the z = 13 charge state was selected for ETD in the LIT, and the dominant product ions (as expected) were charge-reduced precursors (ETnoD). Subsequent photoirradiation of individually isolated singly, doubly, and triply charge-reduced species in the HCD cell resulted in mainly UVPD-type fragmentation, shown in Figure 2A,B,C. The abundance of the intact charge-reduced proteins decreased with each electron adduction, and for that reason, the signal-to-noise of the resulting fragment ions also decreased during the subsequent UVPD step, resulting in identification of only the most abundant fragment ions (Figure 2F). Also, because a large population of UVPD fragments have m/z values close to the precursor, fragment ions of higher m/z values are more likely to be identified upon photodissociation of more charge-reduced precursors (because the selected charge-reduced precursor and resulting fragment ion isotopes in lower charge states are less crowded and shifted higher in the m/z landscape) (Figure 2C). This latter benefit of ETUVPD is further illustrated in Figure S3 (Supporting Information) for which the number of fragment ions specific to each activation method and their respective distributions across the m/z range from m/z 300 to 950 are shown. These results for individual charge-reduced precursors showcase the potential benefits of combining ETUVPD results from several charge states or ideally via analysis of multiple precursor charge states at once (as is possible in the HCD cell).
Figure 2.
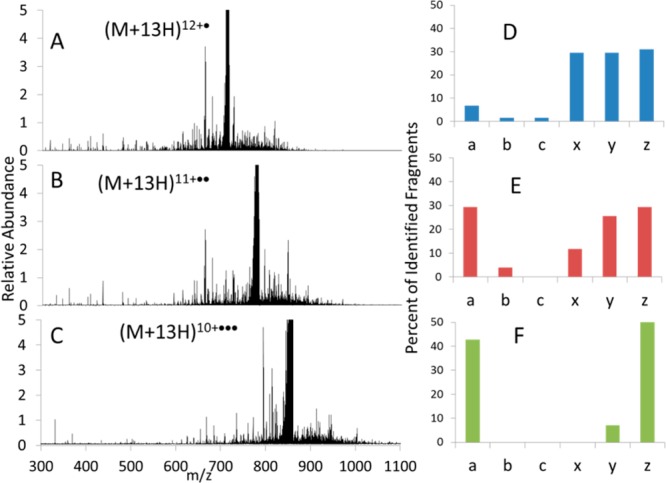
ETUVPD (8 ms ETD of ubiquitin (13+) in the LIT followed by one 1 mJ laser pulse in the HCD cell): (A) 12+·, (B) 11+··, (C) 10+···, and corresponding distribution of ion types in panels D, E, and F. All spectra are shown on the same scale.
This strategy of simultaneous UVPD of a broader range of precursors and product ions was implemented and evaluated via ETD of the z = 13 charge state of ubiquitin in the HCD cell followed by UVPD of the entire population of both charge-reduced and nonreduced precursors as well as product ions arising from ETD. This “broadband” ion activation by UVPD offers two potential benefits. First, the total ion population available for UVPD activation is increased relative to UVPD of a single charge-reduced species. Second, the potential for broad ion isolation in the HCD cell allows detection of a wider m/z range of product ions generated in the initial ETD reaction as well as the additional ones from UVPD. In the context of characterization of intact proteins, obtaining high sequence coverages and maximizing dissociation efficiencies are premium benefits, both of which are feasible with the broadband ETUVPD approach. To capitalize on these benefits, ETUVPD with broad ion isolation was undertaken for ubiquitin along with comparison to UVPD alone and ETUVPD with selected ion isolation (see Figure 3). Electron transfer activation in the HCD cell followed by a single 5 ns UV pulse (2.5 mJ) resulted in a fragment ion distribution that resulted from contributions from both ETD and UVPD (Figure 3C). Figure 3 shows expansions of the spectral region from m/z 720 to 780 for UVPD (12+), ETUVPD in which the charge-reduced 12+· ions generated by ETD in the LIT were isolated and subjected to UVPD in the HCD cell, and for ETUVPD in which all the products arising from ETD of the 13+ ions of ubiquitin in the HCD were subsequently subjected to UVPD. The shaded regions are unique fragments not seen upon standalone UVPD or UVPD after isolation of the charge-reduced 12+· ions in the selective ETD/UVPD spectrum and are only observed upon broadband ETUVPD. Although many of the fragment ions are the same in all three spectra, the new ones generated upon ETUVPD using broad precursor isolation provide additional sequence coverage. After demonstration of feasibility of ETUVPD and evaluation of initial metrics, all subsequent hybrid MS/MS experiments were performed via both ETD and UVPD in the HCD cell.
Figure 3.

(A) UVPD (one 2.5 mJ laser pulse) of ubiquitin 12+, (B) ETUVPD (8 ms ETD in LIT of ubiquitin 13+ followed by one 1.8 mJ laser pulse of ubiquitin 12+·) (MS3), (C) ETUVPD (15 ms ETD of 13+ ubiquitin in HCD cell followed by one 2.5 mJ laser pulse of all product ions). All spectra are shown on the same scale.
ETUVPD Decreases Spectral Congestion
Although UVPD of intact proteins provides the richest spectra of any MS/MS method due to fragmentation at nearly every inter-residue position, a resulting complication is the spectral congestion and overlapping isotopic envelope of the fragment ions, thus requiring high resolution of the mass analyzer. The resolving power of Fourier transform mass analyzers is proportional to acquisition time, and for the crowded spectra that are produced by UVPD, maximum resolution is required. Heck and co-workers have shown previously that the ETD reaction for intact proteins in the HCD multipole is a slow reaction that culminates largely in charge-reduced peaks of the unfragmented (intact) precursor.11 Although this outcome is not particularly beneficial for generating informative fragment ions, the result is quite advantageous for the hybrid ETUVPD method. The extent of spectral crowding and the difficulty associated with accurate deconvolution of the complex product ion spectra that result after UVPD have been reported previously using a set of green fluorescent protein (GFP) variants.9 The assignable product ions were routinely biased toward lower charge states, even for interrogation of higher charge state precursors, due to the difficulty associated with effectively deconvoluting the higher charge products in the crowded spectra.9 Taken together with visual inspection of the spectra and the total number of deconvoluted product ions (including those that were not matched to assignable fragment ions in the protein sequence), it is likely that the observed difficulty in deconvolution was the result of the combination of higher charge states (which have more closely spaced isotopic peaks) and product ions that overlap the same m/z region of the highly charged precursor.9 For the present study, intact proteins of varying sizes were infused and analyzed in an optimized method in which both activation events were performed in the HCD cell prior to detection in the Orbitrap analyzer (Figure 4). Ubiquitin (8.5 kDa), myoglobin (16.9 kDa), and carbonic anhydrase (29 kDa) were activated using all methods under investigation; UVPD, ETD, and ETUVPD. In all cases, the greatest total number of matched fragment ions resulted from ETUVPD (as exemplified by the results for myoglobin in Figure 4). Visual inspection of the spectra in Figure 4 clearly depicts how the ion current is distributed more effectively across the m/z landscape by combining the two activation methods. ETUVPD spectra from the z = 22 charge state of myoglobin showed not only a moderate increase in the number (and percentage) of matched fragment ions compared to UVPD or ETD alone, but also an increase in the total number of deconvoluted ions and percentage of the fragment ions matched to the protein sequence.
Figure 4.

Shown for each of the three fragmentation strategies are (A) the total number of matched fragment ions (with the number of redundant ones shown in parentheses), (B) the total number of deconvoluted fragment ions (matched plus unmatched), and (C) the percent of fragment ions matched to the protein sequence (calculated by dividing the number of matched fragment ions by the total number of deconvoluted fragments). All results correspond to the z = 22 charge state of myoglobin (16.9 kDa). On the right are product ion spectra resulting from (D) UVPD, (E) ETD, and (F) ETUVPD of myoglobin (22+). All activation events were performed in the HCD cell. All spectra are shown on the same scale.
The increase in the number of deconvoluted fragment ions was observed for both ETD and ETUVPD, an expected outcome owing to the ability of ETD to more effectively spread the ion current out across the m/z landscape via charge reduction. If this increase in the number of deconvoluted ions was accompanied by a substantial decrease in the percentage of those ions that were matched to the protein sequence, one could assume that the new ions were largely due to secondary or nonspecific fragmentation, but combining ETD with UVPD resulted in increases in the total number of deconvoluted fragments and in the percentage that could be matched to the protein sequence relative to UVPD alone. The increase in the number of deconvoluted fragment ions relative to UVPD alone corresponds to a more even distribution of “true positive” fragment ions, further confirmed by evaluation of the standard deviation of both the abundances and the m/z distribution of all identified ions. UVPD still gave the best overall sequence coverage (87%) relative to ETD (68%) or ETUVPD (79%) for myoglobin.
With respect to the product ion abundances for each of the MS/MS methods, the average abundance was 4.0 × 103 (±7.7 × 103) for UVPD, 1.0 × 104 (±1.0 × 104) for ETD, and 7.7 × 103 (±9.4 × 103) for ETUVPD. The distributions of all matched product ions were grouped in 100 Th bins for each method (Figure S4, Supporting Information). The matched product ions fall into a greater number of bins for ETD and ETUVPD, thus indicating a broader distribution of product channels and more product charge states. These metrics reflect the ability of ETUVPD to enhance protein characterization by apportioning product ion current more evenly for both the x (mass to charge ratio) and y (intensity) variables, resulting in more informative spectra.
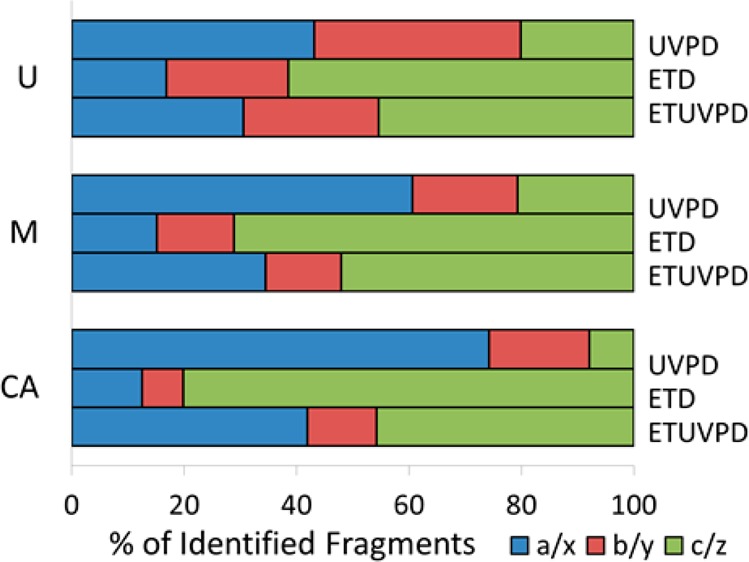
In this context, evaluation of the ion type distributions of the three model proteins reveals a trend toward more evenly distributed fragment ion pairs (i.e., a/x, b/y, c/z) for ETUVPD. Shown in Figure 5 are the ion type distributions obtained using UVPD, ETD, and ETUVPD for each protein, which are abbreviated as U, M, and CA for ubiquitin (averaging 13+, 12+, and 11+ precursor charge states), myoglobin (averaging 23+, 22+, and 21+ charge states), and carbonic anhydrase (34+), respectively. The distribution of fragments for UVPD is generally biased toward a- and x-type ions, especially as the protein mass increases. ETD resulted in a majority of c- and z-type ions, as expected. ETUVPD showed the most uniform distribution between a/x and c/z types, thus supporting the concept that ion types from both standalone UVPD and ETD are combined for the hybrid methods. Interestingly, the portion of N-terminal versus C-terminal product ions does not vary significantly from UVPD to ETD to ETUVPD (Figure S5). In terms of sequence coverage (calculated based on the number of interresidue cleavages relative to the total number of interresidue backbone bonds), the coverages obtained for myoglobin (22+) were 87% for UVPD, 68% for ETD, and 79% for ETUVPD. For carbonic anhydrase (34+), the sequence coverages were 68% for UVPD, 62% for ETD, and 73% for ETUVPD. The sequence maps for myoglobin and carbonic anhydrase are shown in Figures S6 and S7 (Supporting Information), respectively. Thus, although the distribution of ions changes for the hybrid method relative to UVPD or ETD alone, the net sequence coverage does not improve significantly.
Figure 5.
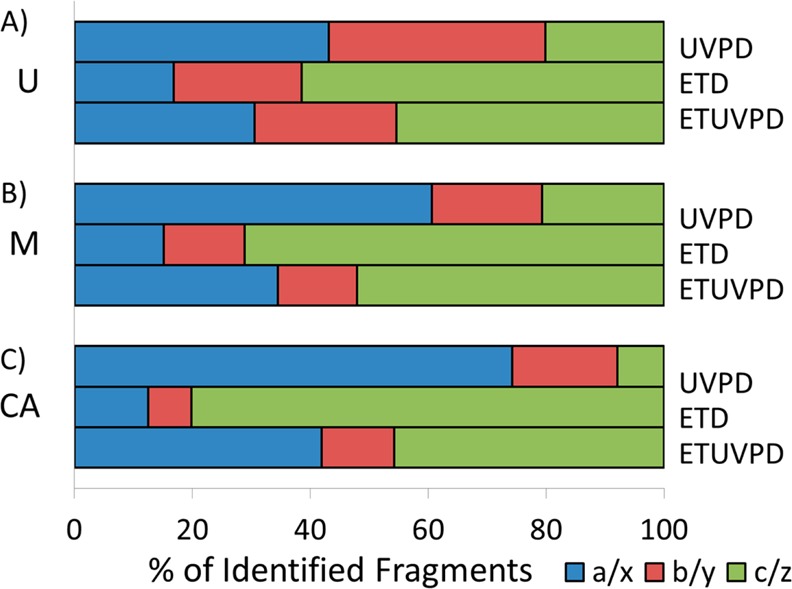
Shown are percentages of ion type pairs of total fragment ions identified by UVPD, ETD, and ETUVPD for (A) ubiquitin (U), (B) myoglobin (M), and (C) carbonic anhydrase (CA), with all activation events performed in the HCD cell (UVPD, one 2.5 mJ laser pulse; ETD, 15 ms; ETUVPD, 4 ms ETD, and one 1.0 mJ laser pulse).
LC-UVIETD and LC-ETUVPD
From the optimization and survey results for ubiquitin, myoglobin, and carbonic anhydrase, several sets of hybrid fragmentation conditions were chosen for analysis of the E. coli ribosome. The ribosomal proteome is composed of approximately 55 small and basic proteins. The positively charged Lys and Arg side chains interact with the rRNA phosphate backbone to maintain the ribosomal structure as a whole. This proteome is an ideal sample for evaluating the hybrid methods due to the likelihood of observing high charge states and the well-known positive correlation between precursor charge and ET reaction efficiency.13 For this phase of hybrid activation experiments, all fragment ions (and nondissociated and charge-reduced precursors) were simultaneously subjected to ETUVPD in the HCD cell. Additionally, further assessment of the impact of the ET reaction period on ETUVPD was undertaken for higher throughput LCMS applications. Using a digital delay generator, laser irradiation could be triggered either at the end of the electron transfer reaction period (termed ETUVPD, as described above) or at the beginning of the electron transfer reaction period (termed ultraviolet irradiation ETD or UVIETD), and the duration of the electron transfer reaction period could be varied. Two electron transfer reaction periods were chosen: 10 and 30 ms. A 10 ms period was chosen to maximize the production of intact charge-reduced proteins, and as such, distribute the ion current more effectively across the m/z landscape prior to UVPD. A 30 ms period was used to enhance the degree of radical-directed dissociation of the proteins in a manner complementary to the distinctive fragmentation promoted by UVPD.
Experiments utilizing all configurations (10 or 30 ms electron transfer period and preceded or followed by UVPD) were compared based on several metrics. The false discovery rate (FDR), average −log (E value) (where lower E values or higher −log (E values) reflect better matches), average number of fragments, and individual protein E values were compared to deduce which method was most ideally suited for combining both identification- and characterization-centric approaches. Interestingly, the FDR curves associated with all four iterations of the hybrid methods (ETUVPD 10 ms, ETUVPD 30 ms, UVIETD 10 ms, and UVIETD 30 ms) resulted in nearly identical curves (see Figure S8, Supporting Information). The nearly identical FDR curves as well as the earlier infusion studies that confirmed similar percentages of matched ions (32% for UVPD compared to 36% for ETUVPD, Figure 4) indicate that there is a low degree of additional internal fragmentation resulting from ETUVPD. Internal fragments cause a massive increase in product ion search space that is prohibitive for high throughput analysis, and one could expect that sequential activation using two methods is more likely to result in this undesirable outcome. Our results show that ETUVPD does not result in extensive internal fragmentation. We have previously shown that the laser energy required for efficient photodissociation is roughly inversely proportional to protein mass, meaning that fragmentation of larger proteins (or polypeptides) is achieved with less energy deposition than that required for efficient fragmentation of smaller proteins (or peptides).8 The lack of internal fragments observed from the hybrid formats can be explained by the propensity of each method alone to preferentially fragment larger (and more highly charged) polypeptides.
Among the proteins that were identified by all dissociation methods (n = 33 proteins, see Table S1, Supporting Information), UVPD resulted in the highest average −log (E value) at 60, followed closely by the ETUVPD for 30 ms (58) and then UVIETD for 10 ms (58) (where a higher −log (E value) indicates a better match). As for the protein complement identified in all analyses, UVPD by itself achieved the best fragmentation outcomes (as defined by the best E values) for 11 of the proteins, UVIETD for 10 ms was optimal for 9 of them, and ETUVPD for 30 ms was the best option for 7 of them. Both ETUVPD for 10 ms and UVIETD for 30 ms were only best for 3 of the 33 proteins each. This disparity in which method is optimal can be attributed to the vastly different ion type distributions achieved with each method. Shown in Figure 6 is the distribution of ion types for each method for the 33 protein identifications.
Figure 6.
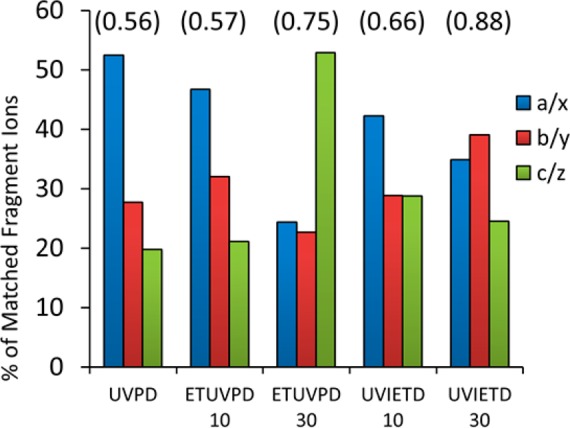
Histogram showing the distribution of ion types using UVPD and the hybrid methods for a ribosomal protein mixture. Shown above each set of bars in parentheses is the E value contribution per fragment ion.
From the fragment ion distributions shown in Figure 6, it is clear that hybridizing ETD with UVPD allows optimization of the ion population to suit the application. While one could argue that the total number of matched ions should be the only metric used to determine which method is best, changing the ion type distribution for higher confidence could be very useful for exploiting unweighted search algorithms. This is best exemplified by examining the average contribution of each matched fragment ion to the total E value for each identified protein (−log (E value)/number of identified fragments). For the UVPD data in Figure 6, the a- and x-type ions are dominant, typical of “canonical” 193 nm UVPD. UVPD, on average, produced more than 95 fragment ions per protein identification; however, a large proportion of the ion current resided in a- and x-type ions, which are sometimes duplicative for the same inter-residue position in the protein sequence. Comparing this to the results obtained by using UVIETD (10 ms), the average number of identified ions was lower (around 79), but it still gave the best score for 9 of the 33 proteins identified. The increase in the contribution of c- and z-type ions (which are not duplicative for the same inter-residue position) compensates for the overall lower total number of fragment ions and results in a positive impact on the E value per fragment ion. This simple calculation for these two methods revealed that the contribution per fragment to the E value using UVPD was 0.56 compared to 0.66 for UVIETD for 10 ms. In essence, this increase in the E value contribution per fragment underscores the possibility of achieving greater sensitivity via creating fewer fragment ions but ones with a more optimal distribution of ion types. UVPD alone of the Escherichia coli ribosome provides a high level of protein identification and characterization but is dominated by a- and x-type ions,10 as reconfirmed in this study. The primary utility of using hybrid ETUVPD activation is the ability to modulate the fragmentation distribution more evenly among identifiable ion types.
Conclusions
Presented is a new method that combines ETD and UVPD simultaneously performed in the HCD cell of an Orbitrap Elite mass spectrometer to provide a more balanced contribution from each complementary pair of ion types, and specifically, to increase the contribution of c- and z-type ions. Pilot studies on several benchmark proteins of varying size as well a mixture of intact ribosomal proteins demonstrated the utility of ETUVPD and UVIETD as compelling methods for enhancing top down protein characterization. The enhancement arises from the production of a more diverse set of ion types facilitating characterization via representation of overlapping sections of the protein sequence from both termini, especially when exploiting the advantage of the ultrahigh resolution available in single protein infusion experiments. Additionally, the ETUVPD and UVIETD approaches demonstrate successful hybridization of activation methods with the ability to modulate the activation time in the ETD step to achieve the preferred ion types characteristic of either ETD or UVPD.
Acknowledgments
Funding from the NSF (CHE-1402753), NIH 1K12GM102745 (fellowship to J.R.C.), and the Welch Foundation (F1155) are gratefully acknowledged. We thank Jens Griep-Raming and Dirk Nolting of Thermo Fisher Scientific for assistance in modification of the Orbitrap Elite mass spectrometer for photodissociation and for providing assistance with performing electron transfer dissociation in the HCD cell.
Supporting Information Available
Various MS/MS spectra of myoglobin (ETD, UVPD, ETUVPD, 22+) and ubiquitin (ETUVPD, 13+), the distributions of ion types and percentages of N- and C-terminal product ions, the m/z distribution of product ions of myoglobin (UVPD, ETD, ETUVPD, 22+), sequence coverage maps of myoglobin (ETD, UVPD, and ETUVPD, 22+) and carbonic anhydrase (34+), the false discovery rate curves for MS/MS of the E. coli ribosomal proteome, and summaries of the E (expectation) values of all E. coli ribosomal proteins identified by each MS/MS method. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† Both authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. J. Am. Chem. Soc. 1998, 120, 3265. [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Yu X.; Costello C. E.; Lin C.; O’Connor P. B. Anal. Chem. 2012, 84, 6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Anal. Chem. 2012, 84, 9668. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F. M.; Mechtler K.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2013, 12, 1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. I.; Brodbelt J. S. Anal. Chem. 2010, 83, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. A.; Cheng R. R.; Kaoud T. S.; Dalby K. N.; Makarov D. E.; Brodbelt J. S. Chem.—Eur. J. 2012, 18, 5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Li W.; Holden D. D.; Zhang Y.; Griep-Raming J.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. J. Am. Chem. Soc. 2013, 135, 12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Kluwe C.; Ellington A.; Brodbelt J. S. Proteomics 2014, 14, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Cammarata M. B.; Robotham S. A.; Cotham V. C.; Shaw J. B.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. Anal. Chem. 2014, 86, 2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C.; Nolting D.; Altelaar A. F. M.; Griep-Raming J.; Mohammed S.; Heck A. R. J. Am. Soc. Mass Spectrom. 2013, 24, 1663. [DOI] [PubMed] [Google Scholar]
- Vasicek L. A.; Ledvina A. R.; Shaw J.; Griep-Raming J.; Westphall M. S.; Coon J. J.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2011, 22, 1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. M.; Wirtala M.; McAlister G. C.; Coon J. J. Mol. Cell. Proteomics 2007, 6, 1942. [DOI] [PubMed] [Google Scholar]
- Hardy S. J. S.; Kurland C. G.; Voynow P.; Mora G. Biochemistry (Moscow) 1969, 8, 2897. [DOI] [PubMed] [Google Scholar]
- Cannon J.; Lohnes K.; Wynne C.; Wang Y.; Edwards N.; Fenselau C. J. Proteome Res. 2010, 9, 3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-L.; Hühmer A. F. R.; Hao Z.; Karger B. L. J. Proteome Res. 2007, 6, 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D. M.; Ge Y.; McLafferty F. W. Anal. Chem. 2000, 72, 4778. [DOI] [PubMed] [Google Scholar]
- Ledvina A. R.; Beauchene N. A.; McAlister G. C.; Syka J. E. P.; Schwartz J. C.; Griep-Raming J.; Westphall M. S.; Coon J. J. Anal. Chem. 2010, 82, 10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taouatas N.; Drugan M. M.; Heck A. J. R.; Mohammed S. Nat. Methods 2008, 5, 405. [DOI] [PubMed] [Google Scholar]
- Ledvina A. R.; McAlister G. C.; Gardner M. W.; Smith S. I.; Madsen J. A.; Schwartz J. C.; Stafford G. C.; Syka J. E. P.; Brodbelt J. S.; Coon J. J. Angew. Chem., Int. Ed. 2009, 48, 8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb J.; Easterling M.; Agar J. J. Am. Soc. Mass Spectrom. 2010, 21, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.