

Abstract

Clostridium difficile (C. difficile) infection (CDI) rates have exhibited a steady rise worldwide over the last two decades and the infection poses a global threat due to the emergence of antibiotic resistant strains. Interstrain antagonistic interactions across the host microbiome form an important strategy for controlling the emergence of CDI. The current diagnosis method for CDI, based on immunoassays for toxins produced by pathogenic C. difficile strains, is limited by false negatives due to rapid toxin degradation. Furthermore, simultaneous monitoring of nontoxigenic C. difficile strains is not possible, due to absence of these toxins, thereby limiting its application toward the control of CDI through optimizing antagonistic interstrain interactions. Herein, we demonstrate that morphological differences within the cell wall of particular C. difficile strains with differing S-layer proteins can induce systematic variations in their electrophysiology, due alterations in cell wall capacitance. As a result, dielectrophoretic frequency analysis can enable the independent fingerprinting and label-free separation of intact microbials of each strain type from mixed C. difficile samples. The sensitivity of this contact-less electrophysiological method is benchmarked against the immunoassay and microbial growth rate methods for detecting alterations within both, toxigenic and nontoxigenic C. difficile strains after vancomycin treatment. This microfluidic diagnostic platform can assist in the development of therapies for arresting clostridial infections by enabling the isolation of individual strains, optimization of antibiotic treatments and the monitoring of microbiomes.
A toxin-mediated intestinal disease, Clostridium difficile infection (CDI), is commonly attributed to exposure to pathogenic C. difficile strains, following the elimination of healthy microflora in the gut, due to administration of antibiotics.1 Prior studies within animal models strongly suggest that asymptomatic colonization with nontoxigenic Clostridium difficile (NTCD) strains can reduce the incidence of CDI from toxigenic Clostridium difficile (TCD) strains.2−5 The development of such preventive therapies against CDI requires means to monitor NTCD colonization during antibiotic and other therapeutic interventions, so that the antagonistic interactions between differing strains during coinfection can be characterized and optimized. However, there is no independent method to simultaneously monitor physiological alterations in both NTCD and TCD strains, especially during antibiotic and therapeutic interventions. The gold standard of CDI diagnosis is a culture of the bacteria from stool samples and testing for toxin production levels (cytotoxicity assay).6 Given the time-consuming nature of toxigenic C. difficile culture, rapid diagnosis of CDI is usually accomplished by enzyme immunoassays (EIA) that can directly monitor TCD strains through detecting the glutamate dehydrogenase (GDH) levels as well as that of toxin A (TcdA) and/or toxin B (TcdB) levels. However, this method is hampered by poor sensitivity due to rapid degradation of the toxins,6 thereby requiring its combined application with polymerase chain reduction (PCR) to reduce false-positives and false-negatives.6,7 Furthermore, colonization by NTCD strains cannot be monitored by EIAs due to absence of the toxins or by PCR-restriction fragment analysis of the pathogenicity locus (PaLoc)1,8 due to absence of the PaLoc within NTCD strains. Hence, there is a need for methods to enable the simultaneous monitoring of levels and physiological alterations of each strain-type within mixed C. difficile samples, preferably in a label-free, nondestructive, and real-time manner.
S(Surface)-layer glyco-proteins are part of the cell wall envelope in Gram-positive and Gram-negative bacteria. They are integral toward surface recognition, colonization, host–pathogen adhesion, and virulence.9,10 A number of studies have shown that the antigenic variations of S-layers between C. difficile strains11−13 can serve as a potential alternative to serotyping by PCR-restriction fragment length polymorphism analysis and nucleotide sequencing,14 but these methods have not been applied toward the recovery of intact microbials of each strain. S-layer deficient mutant strains can exhibit morphological differences, such as lower surface roughness versus the wild type strain, within various microbial samples.15 Hence, the correlation of S-layer induced morphological or functional variations to the cell electrophysiology can enable interstrain distinctions for the separation of intact C. difficile. Similar distinctions may also be possible within other Gram-positive and Gram-negative microbials that exhibit interstrain S-layer variations. For instance, Campylobacter fetus exhibits antigenically distinct S-layers due to DNA inversion and recombination of surface array A gene (sapA),16 while Geobacillus stearothermophilus has various S-layer gene expressions depending on the oxygen level,16,17 and strains of C. fetus(18) and Lactobacillus helveticus(19) have been identified based on the S-layer gene after PCR amplification.
Dielectrophoresis (DEP) causes the frequency-selective translation of polarized bioparticles in a spatially nonuniform electric field, either toward (by positive DEP or pDEP) or away (by negative DEP or nDEP) from the high field regions of a microfluidic device, depending on the polarizability of the bioparticle versus that of the medium.20,21 Hence, in spite of the heterogeneous nature of microbial samples, the frequency response of the DEP velocity of individual cells toward or away from localized regions of a microfluidic device can be used to quantify the alterations in electrophysiology of each cell type, while the frequency-selective DEP collection rate at these localized regions can be used to quantify the relative levels of each cell type.22,23 Prior work on DEP analysis and separation of live versus dead Gram-positive or Gram-negative bacteria24,25 has been accomplished using either ac fields (10 kHz–10 MHz) on electrode-based devices26,27 or using purely dc fields on electrode-less devices with insulating constrictions.24 While ac fields enable subtle distinction of cells based on their membrane or wall capacitance28 and cytoplasm electrophysiology, the high field at electrode edges can cause irreversible adhesion or damage of cells. Electrodeless techniques, on the other hand, enable contactless DEP manipulation at higher throughputs (due to field nonuniformities across the entire device depth), and they are especially significant within applications where functional microbials need to be collected for subsequent analysis. However, their application under dc field limits the distinctions to those based solely on cell wall permeability, rather than including distinctions based on capacitance and permittivity effects. In spite of this, recent work has demonstrated their ability toward distinguishing serotypes of Escherichia coli(29) and discriminating wild type versus surface protein isogenic mutant bacteria strains.30 Recently, we have developed instrumentation for electrodeless DEP over a wide frequency bandwidth31 and applied it toward discerning persistent versus susceptible subpopulations of Cryptosporidium parvum through sensitive and label-free measurement of the DEP trapping force on single microbial cells.32 In this current work, we apply these capabilities toward the label-free distinction of intact C. difficile strains with systematic differences in cell wall morphology that occur due to their constituting S-layer, as correlated by an adhesion assay. Differences in cell wall roughness are shown to cause systematic differences in their DEP crossover frequency due to alterations in the net wall capacitance. Furthermore, systematic differences correlated to their cytoplasm polarizability are apparent within the high frequency dispersion spectra (1–4 MHz) of each C. difficile strain, especially after vancomycin treatment. The sensitivity of the DEP method toward monitoring alterations after vancomycin treatment is benchmarked against the toxin immunoassay and microbial growth rate methods for toxigenic and nontoxigenic C. difficile strains, respectively. On the basis of this, we envision future work on applying DEP techniques toward clinical isolates for eventual application toward the independent monitoring and separation of particular C. difficile strains from mixed C. difficile samples, in a nondestructive and label-free manner.
Experimental Methods
C. difficile Sample Preparation
All experiments were conducted in a biosafety level 2 (BSL2) certified laboratory. The C. difficile samples (purchased from ATCC) were transferred into the microfluidic chip within the biosafety cabinet and sealed with platinum electrodes to prevent leakage. The dielectrophoretic motion of the respective C. difficile cells under the external field can then be observed under the microscope, outside of the biosafety cabinet, since the well-sealed device obviates exposure. Following imaging, the chip was disposed as per standard BSL2 procedures. In some cases, the trapped cells were collected for measurements of cell viability and the supernatant was collected to identify toxigenic versus nontoxigenic strains using the toxin immunoassay. The C. difficile strains were cultured in brain heart infusion (BHI) broth (BD BBL Brain Heart Infusion) at 37 °C overnight under anaerobic conditions, before further antibiotic treatment or dielectrophoresis experiments. The overnight cultured bacteria suspension (250 μL) and 750 μL of the BHI with vancomycin (Novaplus) or without vancomycin (for the control groups) were mixed in Eppendorf tubes and incubated at 37 °C for 4 or 24 h. The vancomycin concentration for VPI10463 (high-toxigenic, HTCD) was 2 and 1 mg/mL for ATCC630 (low-toxigenic, LTCD) and VPI11186 (nontoxigenic, NTCD). Prior to the dielectrophoresis experiments, the BHI broth was replaced with 8.8% sucrose water and readjusted with BHI broth to optimize the medium conductivity at 105 ± 5% mS/m for enabling DEP-based distinction of C. difficile strains from mixed samples. All three strains were confirmed to be viable within this altered BHI media over the time frame of the DEP experiments, as per the colony forming unit (CFU) assay (see the Supporting Information).
Adhesion Assay
On the basis of prior work,33,34 the human colon epithelial cell line, HCT-8 (purchased from ATCC), was used as the host cell in this assay. The HCT-8 cells were cultured in RPMI medium supplied with 10% horse serum, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a 5% CO2 incubator. Cells were grown as a confluent monolayer in 6-well plates prior to the assay. PBS and RPMI-serum free medium were prereduced for oxygen removal by overnight incubation in the anaerobic chamber. Before the adhesion assay, the cells were washed twice with PBS and replaced to RPMI-serum free medium. Overnight C. difficile cultures were pelleted and resuspended in fresh BHI medium to avoid any interference from proteins or toxins. All three C. difficile strains were adjusted to equal concentration by optical density measurement. An equal concentration of each C. difficile strain was added to each well, and the plates were incubated in the anaerobic chamber at 37 °C for 3 h. After 3 h, nonadhered C. difficile cells were eliminated by three wash steps with PBS. Following this, 1 mL of PBS was added to each well, and the cells were scraped, vortexed, serially diluted and plated to enumerate adherent C. difficile colony-forming units (CFU). Each experiment was performed in triplicate and repeated at least three times in entirety. All standard deviations (SD) in this study were obtained by using
| 1 |
Growth Measurement
Overnight cultured C. difficile suspension (250 μL) and 750 μL of the BHI broth with or without vancomycin were mixed in Eppendorf tubes. The optical density at a wavelength of 600 nm (OD600) of the mixed bacteria suspensions, as measured by spectrophotometry (Eppendorf Biophotometer) before incubation, was indexed as the “0 h” time point. The OD600 number at later incubation time points (4 and 24 h) for the respective strain at each condition was normalized to its 0 h point. Each experiment was performed in triplicate and repeated at least three times in entirety.
Toxicity Enzyme-Linked Immunosorbent Assay
Total toxin (A/B) production was measured using the C. difficile TOX A/B II kit (Tech-Lab) according to the manufacturer’s instructions. Culture supernatants were collected at 0, 4, and 24 h by centrifugation at 3500 rcf for 5 min and stored at −20 °C. The supernatants of the VPI10463 strain were diluted 1 to 20, while the supernatants of the VPI11186 strain were undiluted. Each specimen was run in duplicate. Total toxin levels were determined by measuring A450 under a 96 well plate spectrophotometer. The A450 number at each time point (4 and 24 h) for each strain at each condition was normalized to its 0 h time point. Each experiment was performed in triplicate and repeated at least three times in entirety.
Sample Preparation for Transmission Electron Microscope Imaging
C. difficile samples cultured overnight (1 mL) were pelleted and fixed in 2% glutaraldehyde and 2% paraformaldehyde in PBS for 4 h at room temperature. The samples were pelleted and washed 3 times in DI water before resuspension in 2% osmium tetroxide. After 30 min, the samples were pelleted and washed 2 times in DI water before the dehydration process. The samples were dehydrated through a serial gradient ethanol solution (50%, 70%, 95%, and 100%) for 10 min for each sample. The samples then resuspended in 1:1 EtOH/EPON (epoxy resin) overnight, followed by 1:2 EtOH/EPON for 2 h and 1:4 EtOH/EPON for 4 h and 100% EPON for overnight. After embedding the samples in fresh 100% EPON, the samples were baked in a 65 °C oven. The EPON hardened samples were sectioned to 75 nm, mounted onto 200 mesh copper grids, and contrast stained with 0.25% lead citrate and 2% uranyl acetate for TEM imaging (JEOL 1230) at 80 kV (SIA digital camera).
Dielectrophoretic Characterization of C. difficile
The experimental setup has been described in our prior work.32,35 Briefly, standard PDMS (polydimethyl-siloxane) micromolding methods were used to microfabricate channels with sharp lateral constrictions (1000–15 μm). This so-called “electrodeless DEP device” was bonded using oxygen plasma treatment to a standard coverslip for easy microscopic viewing of DEP behavior. Using Pt electrodes at the inlet and outlet, ac fields were applied over a wide-frequency range (50 kHz to 5 MHz) by utilizing a power amplifier for particle trapping toward or away from high field points at the constriction tips. The trajectory of the unlabeled C. difficile of each strain type was observed under this field, as high frame per second movies to quantify the DEP velocity. The data acquisition was automated to enable the capture of movies at each frequency within about 10 s, with the entire frequency spectrum completed within about 5 min. The same cells can be measured multiple times under the DEP field since we use the electrodeless DEP technique under high frequency ac fields, with applied fields less than 300 Vpp/cm, thereby obviating electro-permeabilization effects, which we confirmed through viability analysis on microbials under the DEP field. For experiments within mixed C. difficile samples, the trapped microbials were released and the supernatant was analyzed with the immunoassay to confirm toxigenicity. The DEP analysis was conducted on the microbials following the log phase stage of their culture period to ensure the insensitivity of DEP analysis to the temperature of the culture media and time for the culture. A full description of the simultaneous and automated dielectrophoretic tracking of single bioparticles can be found in our previous work.32
Results and Discussion
Morphological Differences between C. difficile Strains
We begin with an examination of the morphological differences between three particular C. difficile strains: the high-toxigenic VPI10463 strain (HTCD), the low-toxigenic strain ATCC630 (LTCD), and the nontoxigenic strain VPI11186 (NTCD). As per the transmission electron microscopy (TEM) images at 50k magnification in Figure 1a,c,e and at 100k magnification in Figure 1b,d,f, the three C. difficile strains show systematic variations in surface roughness in the cell wall region (see arrows), with the highest roughness apparent in the HTCD strain (Figure 1a,b), followed by LTCD (Figure 1c,d), and finally the NTCD strain (Figure 1e,f), which exhibits relatively smooth surface features. One of the chief differences between the respective strains is the S-layer on their cell wall, which exhibits the SlpA gene and Cwp gene sequence variations.7,11,13 On the basis of prior observations of a smoother cell surface for the S-layer deficient mutant Tannerella forsythia versus the wild type,15 we seek to correlate the interstrain morphological differences in C. difficile to their S-layer variations by using a standard adhesion assay. It has been shown that surface layer proteins are the chief determinant for the adherence of C. difficile to host cells34 and for binding to gastrointestinal tissues.36 Figure 1g shows a representative phase contrast image of the adherence of HTCD to the human colon epithelial host cells after three wash steps. As per Figure 1h, the HTCD strain shows the strongest adherence to the host cells, followed by LTCD and finally NTCD strains. This correlation of high cell wall roughness of the C. difficile strain-type to its enhanced host-cell adherence suggests an abundance of S-layer proteins within the HTCD strain, with successively lower S-layer protein levels within the LTCD and NTCD strains due to their relatively smoother features and poorer adhesion to the host cells. We also note that the average cell wall thickness of the HTCD strain (32.1 ± 3.8 nm) is lower than that of the NTCD strain (38.3 ± 5.2 nm), as averaged over 10 cells, as per the measurements in the Supporting Information, Figure S1.
Figure 1.
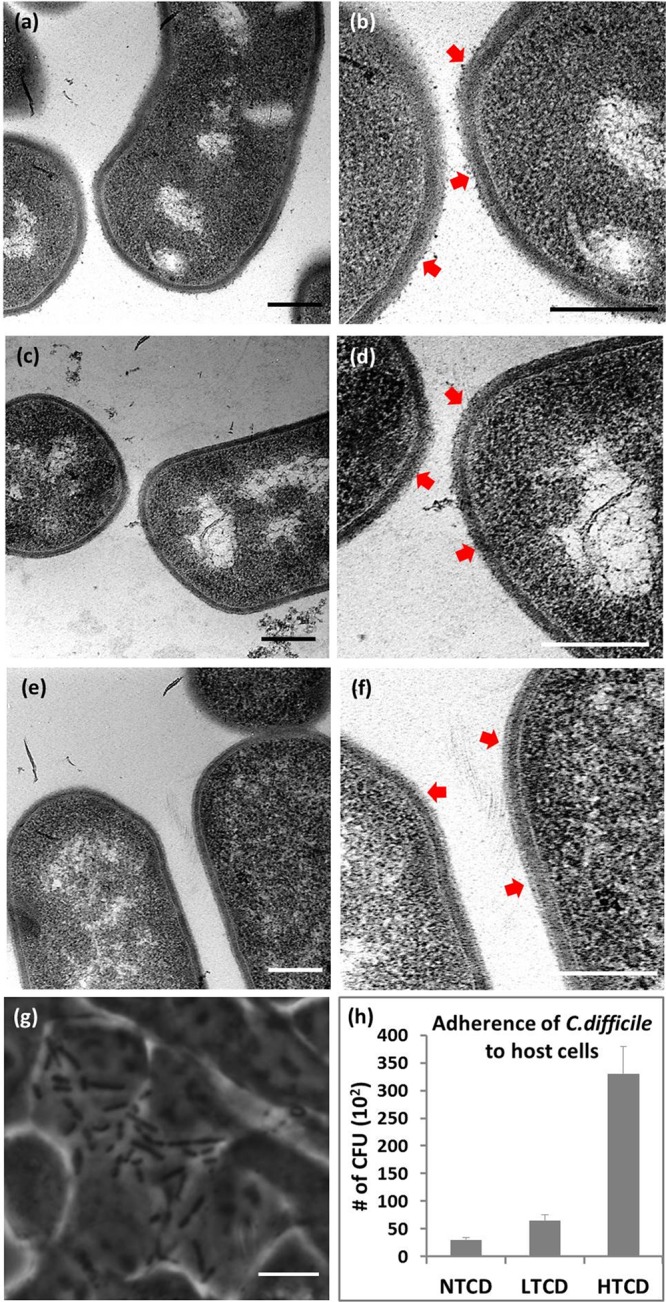
Transmission electron microscopy images of the C. difficile strains at 50k magnification (a, c, and e) and 100k magnification (b, d, and f). TEM scale bars are 0.2 μm and arrows indicate S-layer features. (a and b) HTCD (High-toxigenic C. difficile, VPI10463); (c and d) LTCD (Low-toxigenic C. difficile, ATCC630); and (e and f) NTCD (nontoxigenic C. difficile, VPI11186) strains. (g) A phase contrast image showing HTCD adherence to the human colon epithelial host cells. Scale bar: 5 μm. (h) Variations in adherence of each C. difficile strain to human colon epithelial cells by enumerating colony-forming units (CFU).
Independent Dielectrophoretic Monitoring of C. difficile Strains
Dielectrophoresis (DEP) of biological particles, such as C. difficile, can be characterized using a shell model.20,21 Herein, the net capacitance (C) due to the dielectric properties of the cell wall and membrane screens the electric field at low frequencies to cause negative DEP (nDEP), whereas the low resistance (R) due to conductive properties of the cytoplasm dominates at high frequencies to cause positive DEP (pDEP), with the crossover frequency (fxo) from nDEP to pDEP being determined by the inverse of the RC time constant due to the net resistance and capacitance of the system. On the basis of a parallel-plate model for the cell wall with spacing, d, and material permittivity, ε, its capacitance rises with surface area (A):
| 2 |
Changes in surface roughness and area of the cell wall would cause systematic differences in the net capacitance of each C. difficile strain. Hence, on the basis of the interstrain differences in surface roughness in Figure 1, HTCD strains should have the highest net capacitance, followed by the LTCD and then by the NTCD strains. The DEP crossover (fxo) for each C. difficile strain can be related to these differences in net cell wall capacitance (Cnet) at a given media conductivity (σm) as follows:28
| 3 |
Hence, we anticipate the lowest fxo for the HTCD strain, followed by that of the LTCD strain and finally the NTCD strain. However, in order to observe substantial differences in fxo between the strains, it is necessary to optimize the media conductivity (σm) using the strain-types that exhibit maximum differences in their cell wall roughness. Below a critical value of σm, the high resistance of the surrounding media will dominate the net RC time constant of the system, thereby driving the fxo to low values and making it insensitive to differences in wall capacitance between the three strains. On the other hand, above a critical σm value, pDEP cannot be effectively observed (pDEP requires particle conductivity to exceed media conductivity), thereby posing complications toward determining the fxo, due to lack of a clear crossover. As a result, DEP measurements need to be carried out within media of relatively high conductivity to enable the observation of significant differences in the fxo between the C. difficile strains. This is challenging due to the disruptive effects of electrolysis, electrothermal flow,37−39 and electropermeabilization of cells within electrode-based DEP devices at substantial σm. Hence, in this current work, the influence of these disruptive effects on DEP observations is reduced by the use of electrodeless microfluidic devices, wherein heat dissipation is enhanced by using channels of high surface to volume ratio and wherein cell trapping under DEP does not occur at the electrode but instead at or away from the tips of insulator constrictions that are designed to locally enhance electric fields.35,40,41 Figure 2a,b shows the electrodeless device with external electrodes (1 cm apart) to initiate localized microbial trapping in the constriction region. This electrodeless device geometry also enables facile and automated dielectrophoretic tracking due to the well-defined particle trajectories, either toward (by pDEP) or away (by nDEP) from highly localized constriction tips (Figure 2c), with a symmetric field profile across the device depth. In this manner, as per prior work,32 the translational velocity under the DEP trapping force is measured for ∼20 individual microbial cells to quantify the DEP spectra. Upon optimization of σm at 100 mS/m, well separated DEP spectra for each strain are apparent, as per Figure 2d. Example images in vicinity of the constriction region of the device after 30 s of ∼300 Vpp/cm field at the optimal frequency for nDEP and pDEP behavior are shown in Figure 2e for each strain, with arrows to denote the direction of translation, and the respective velocity values at each frequency are reported in Figure 2d. On the basis of this, while nDEP is highest at 100 kHz for all strains, the magnitude of the nDEP velocity is significantly lower for the HTCD strain versus the LTCD and NTCD strains. Furthermore, the crossover from nDEP to pDEP behavior occurs at successively lower values for the HTCD strain (300 ± 75 kHz) versus the LTCD (500 kHz) and NTCD (900 ± 75 kHz) strains, which is consistent with its progressively higher net wall capacitance due to higher surface area and lower cell wall thickness, as per the TEM images in Figure 1. It is noteworthy that the absolute fxo value of the HTCD strain is significantly lower than the other strains, not only due to its higher cell wall capacitance but also due to its significantly higher surface conductance, as judged by the lower magnitude of its nDEP velocity at low frequencies versus other strains. Also apparent is the successive reduction in the magnitude of maximum positive DEP force levels, from highest for HTCD to a lower level for LTCD and lowest for the NTCD strain. This indicates a gradual reduction in cytoplasm polarizability for the respective strains, since their sizes are identical. While the magnitude of highest pDEP occurs at 400 kHz for the HTCD strain, it occurs at 1 MHz for the LTCD and NTCD strains. Finally, it is apparent that in spite of the reduction in cytoplasm polarizability for the NTCD strain versus other strains, a discernible level of positive DEP can be observed up to ∼2 MHz with the NTCD strain, up to ∼3 MHz with the LTCD strain, and up to ∼1 MHz with the HTCD strain. These characteristic spectral features in the 0.05–5 MHz range; i.e., the fxo, the frequency and magnitude of maximum pDEP, and the frequency bandwidth for pDEP can offer the means to fingerprint each C. difficile strain and separate intact microbials of each strain-type from mixed C. difficile samples. More generally, since numerous other Gram-positive and Gram-negative microbials exhibit S-layer variations, these results with C. difficile suggest the broader applicability of frequency-resolved DEP spectra toward enabling interstrain distinctions.
Figure 2.

(a, b) Illustration of example electrodeless DEP device with platinum electrodes (1 cm apart) for localized microbial trapping in the constriction region. (c) Illustration of DEP trapping within a constriction device. (d) Well-separated DEP spectra (velocity under FDEP) for each strain: HTCD, LTCD, and NTCD; (e) DEP behavior of each C. difficile strain in the constriction region after 30 s of ac field, ∼300 Vpp/cm of nDEP (first column) at 100 kHz for all three strains or pDEP (second column) at 400 kHz, 1 MHz, and 2 MHz for HTCD, LTCD, and NTCD, respectively. Scale bar: 30 μm.
Alterations to Each C. difficile Strain upon Vancomycin Treatment
Alterations to the electrophysiology of cells upon antibiotic treatment, such as distinguishing the degree of cell wall permeabilization versus cytoplasm disruption, can be quantified by analyzing the dielectrophoretic frequency spectra of treated versus untreated cells.26,32,42 Herein, we utilize DEP to probe relative differences in the mechanism of microbial disruption for each C. difficile strain after vancomycin treatment, especially since similar measurements based on toxin production and growth rate can only indicate the overall alterations in cell viability, without providing information on the disruption mechanism. Furthermore, DEP spectra can offer information on the optimal frequencies for separating vancomycin treated cells from untreated cells of each C. difficile strain, thereby enabling a means for quantifying the efficacy of vancomycin treatment on each strain, especially within heterogeneous C. difficile samples. In general, all the three strains become less polarizable due to functionality alterations to the cell after 24 h of vancomycin treatment. However, the HTCD strain requires almost twice as much vancomycin levels than required for LTCD and NTCD strains to cause alterations to the DEP spectra. As a result of vancomycin treatment, while the DEP spectra for the HTCD strain (Figure 3a) is shifted toward a higher crossover frequency (300 kHz to 600 kHz), the spectra for the LTCD strain (Figure 3b) and the NTCD strain (Figure 3c) are shifted toward successively lower crossover frequencies (500 kHz to 300 kHz for LTCD and 900 kHz to 600 kHz for NTCD). To quantify the relative alterations after vancomycin treatment, we show the steady reduction in DEP velocity for each strain at 1 MHz (Figure 3d) and the changes in crossover frequencies (Figure 3e). It is likely that vancomycin treatment alters the permeability of the cell wall and membrane regions, so that the lowered inverse RC time constant of the system enables DEP crossover at earlier frequencies, as observed for the NTCD and LTCD strains. For the HTCD strain, on the other hand, the need for higher vancomycin levels to cause alterations and the up-shifting of the DEP crossover frequency after vancomycin treatment suggest a relatively sturdier cell wall and membrane that is not easily permeabilized, in comparison to the LTCD and NTCD strains. This is consistent with the trend of our measurements on minimum inhibitory concentration (0.5 mg/L for NTCD and LTCD and 1 mg/mL for HTCD), indicating the need for higher antibiotic levels to deactivate HTCD versus other strains.43
Figure 3.

(a–c) Modification of dielectrophoretic spectra (velocity under DEP) can be used to monitor alterations after vancomycin treatment of (a) HTCD, (b) LTCD, and (c) NTCD strains. Note that reported velocities are averaged over 20 cells, of which an overwhelming majority (95–100%) exhibits the reported velocities, except for vancomycin treated cells at frequencies close to the DEP crossover, wherein this value drops to a 50–65% majority of the analyzed cells. (d) Alteration in the magnitude of the DEP response at 1 MHz (velocity under DEP) after vancomycin treatment and (e) change in DEP crossover frequency after vancomycin treatment offer information on alterations in cell electrophysiology.
Benchmarking DEP Velocities to Toxin Production and Growth Rate
In order to evaluate the sensitivity of DEP methods versus the current state of the art, we benchmark the DEP velocity data for HTCD and NTCD C. difficile strains after various levels of antibiotic treatment versus conventional diagnostic measures for the loss of C. difficile functionality, such as toxin production and growth rate values. To compare DEP data across the C. difficile strains, we choose the 1 MHz frequency, since all untreated strains show an equal level of pDEP, and the respective vancomycin treated samples continue to show pDEP. For measuring alteration in toxin production level and growth rate of C. difficile strains after antibiotic treatment, it is necessary to culture the microbial cells with the antibiotic over a period of 4–24 h to enable sufficient sensitivity. Hence, these results on the untreated or antibiotic treated microbials are reported as a proportion of their respective value versus that after a “0 h culture time” (indexed as “1”). Furthermore, the results after antibiotic treatment for a particular period of time are compared against their respective values on untreated microbials for the same period of culture time (this control value for each treatment time is indicated as “Un-0”, “Un-4”, or “Un-24” in Figure 4c–e). On the other hand, since DEP velocity measurements do not require microbial culture to enhance sensitivity, the “control” measurement for DEP velocity of untreated C. difficile is invariant with antibiotic treatment time. Figure 4a shows the steady exponential rise in toxin production levels with culture time for the untreated HTCD strain, while the alterations upon vancomycin treatment lead to only a mild rise (1.05 times) after 4 h of treatment and a small reduction after 24 h treatment, due to degradation of residual toxin level. The data also shows that the NTCD strain cannot be quantified by this method due to absence of toxin production. The growth rate data in Figure 4b follows a similar trend, with the untreated samples showing a steady exponential rise over time, whereas the vancomycin treated HTCD sample shows only a mild rise to 1.51 and 1.61 times after 4 and 24 h, respectively, and the vancomycin treated NTCD sample shows only a minimal rise to 1.1 and 1.05 times, after 4 and 24 h, respectively. Next, the DEP velocity data after various treatment periods (4 and 24 h) is compared to the toxin production level for the HTCD strain (Figure 4c), the growth rate data for the HTCD strain (Figure 4d), and the growth rate data for the NTCD strain (Figure 4e) over the same treatment periods (4 and 24 h), with the respective value for the untreated sample at the same time period serving as the “control” (Un-0, Un-4, or Un-24). As per the toxin production levels in Figure 4c, while alterations to the HTCD strain are apparent after 24 h of vancomycin treatment; i.e., a difference of 7.5 versus the control (Un-24) as per red solid lines along the X-direction, the alteration is just barely apparent after 4 h of vancomycin treatment; i.e., a difference of just 0.55 versus the control (Un-4) as per green solid lines along the X-direction. On the other hand, the DEP data shows a significant reduction in velocity, from 30.7 μm/s to 7.6 μm/s (green dashed lines in the Y-direction), right from the first time point of 4 h of vancomycin treatment, with further reduction to 4 μm/s after 24 h of vancomycin treatment. Similarly, the growth rate reduction of the HTCD strain is clear only after 24 h of vancomycin treatment in Figure 4d, with a difference of 2.2 versus the control (Un-24), as per red solid lines in the X-direction. In comparison to the minimal growth rate reduction in the HTCD strain after 4 h of vancomycin treatment, the respective reduction in the DEP velocity is substantial for the same 4 h treatment time. For the NTCD strain, while the reduction in growth rate is apparent in Figure 4e after 24 h of vancomycin treatment; i.e., a difference of 3.3 versus the untreated sample (Un-24) as per red solid lines in the X-direction, the alteration is not easily distinguishable after 4 h of vancomycin treatment; i.e., a difference of 0.74 versus the untreated sample (Un-4), as per green solid lines in the X-direction. On the other hand, just as with the HTCD strain, reduction in the DEP velocity is substantial (16 μm/s to ∼7 μm/s) right from the first time point of 4 h of vancomycin treatment as per green dashed lines in the Y-direction. Hence, since the DEP measurement method eliminates the need for microbial culture, which is required within conventional diagnostic methods for enhancing their sensitivity toward viable versus nonviable C. difficile, the DEP velocity measurement method enables the quantification of microbial alterations at smaller antibiotic doses. Furthermore, the uncertainties are lowered with the DEP method, since comparisons are required against only a single control (i.e., against the DEP velocity of the untreated sample) rather than against multiple control samples, as required with toxin immunoassay and growth rate methods (i.e., against the respective values for untreated C. difficile after microbial culture over time periods equivalent to each antibiotic treatment time point). As a result, we envision that DEP methods can be applied more easily toward optimizing antibiotic dosage and discerning the mechanism of their action.
Figure 4.

(a) Relative toxin production and (b) relative growth rate for HTCD and NTCD strains before and after vancomycin treatment at 0, 4, and 24 h. The data points after 4 and 24 h of treatment are normalized to their respective value at the 0 h time point. The differences in microbial toxin production (part c for HTCD) and growth rate (part d for HTCD and part e for NTCD) after vancomycin treatment are compared versus the control (0 h treatment) as arrows in the X-direction, while the alterations in DEP response after each treatment are shown as arrows in the Y-direction. Note that the respective control value for the Untreated sample at various time points is shown as Un-0, Un-4, and Un-24 (controls are invariant over treatment time for DEP data but not so for toxin production and growth rate data).
Interstrain Separations from Mixed C. difficile Samples
The quantitative DEP response measurements in Figures 2d and 3 suggest that particular C. difficile strains may be separated from each other based on their characteristic electrophysiology, by identifying an appropriate frequency with maximum differences in the magnitude and direction of the DEP force. We choose the approach of accomplishing separations based on differences in direction of DEP force, since it can demonstrate the differential spatial localization of each strain-type within a few seconds. The heterogeneous samples we choose are (i) a minority HTCD subpopulation within a majority population of NTCD strains with 2-fold higher concentration and (ii) a HTCD sample after incomplete antibiotic treatment over just 20 min, including a subpopulation of viable HTCD along with deactivated HTCD. For (i), as per Figure 5a, we choose a frequency of 400 kHz, wherein the HTCD strain exhibits strong pDEP behavior, while the NTCD strain continues to exhibit substantial nDEP. It is apparent from Figure 5c, that such a separation can be accomplished in a facile manner, as confirmed by the DEP response and toxin levels measured from pDEP trapped C. difficile (see Supplementary Movie 1 versus Supplementary Movie 2 in the Supporting Information). Similarly, using a 400 kHz field as indicated in Figure 5b for the separation of sample (ii), HTCD samples after vancomycin treatment at early time periods (20 min) show the presence of a C. difficile subpopulation with some viability (red solid arrows), along with a deactivated majority population (red dotted arrows) (Figure 5d, also see Supplementary Movie 3 versus Supplementary Movie 4 in the Supporting Information), compared to vancomycin treated HTCD samples after 4 h (Supplementary Movie 4 in the Supporting Information), thereby presenting a methodology to quantify the effectiveness of antibiotic treatments based on the DEP collection rate.
Figure 5.
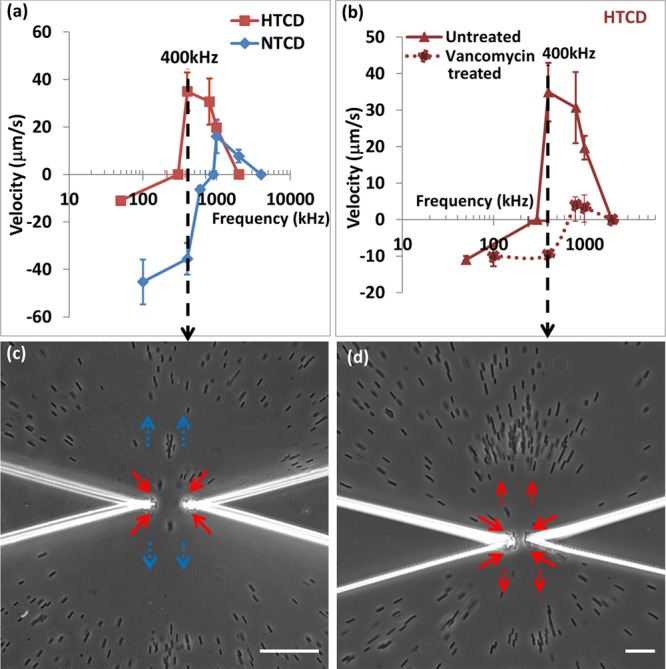
(a) Dielectrophoretic spectra of HTCD versus NTCD; (b) DEP spectra of HTCD after vancomycin treatment; (c) at 400 kHz, HTCD strain shows pDEP (red arrow) versus NTCD strain shows nDEP (blue arrow); (d) at 400 kHz, after 20 min vancomycin treatment, viable HTCD shows pDEP (red solid arrow) versus deactivated HTCD shows nDEP (red dotted arrow). Scale bar: 30 μm.
In summary, we demonstrate that morphological differences in the cell wall region of C. difficile strains, presumably due to differing S-layer glyco-protein levels as validated by their differing adhesion to a host cell, cause systematic variations in their crossover frequency for transition from negative to positive dielectrophoresis (DEP) behavior. As a result, the DEP spectra exhibit characteristic features that may be applied toward independently monitoring each C. difficile strain with differing S-layers as well as toward interstrain separation of intact cells from mixed C. difficile samples. Through benchmarking the DEP data against conventional measures of C. difficile activity, such as toxin production and growth rate, we demonstrate its superior sensitivity toward characterizing microbial alterations upon vancomycin treatment, thereby enabling the application of DEP methods toward the optimization of antibiotic treatments. Finally, through appropriate choice of frequency of the applied field, we demonstrate proof-of-concept separation of subpopulations of high-toxigenic C. difficile strains from a sample of nontoxigenic C. difficile, based on the direction of their dielectrophoresis behavior. In this manner, we present a methodology for isolation of individual strains from mixed C. difficile samples, quantification of antibiotic treatments, and the engineering of nutrient environments to control microbiomes. We envision that the highly sensitive features of DEP analysis, which enable the monitoring of antibiotic-induced C. difficile alterations at earlier times, can aid in development of antibiotic treatments with lower dosages. The ability of frequency-resolved DEP to selectively probe particular intracellular regions, such as the S-layer on the cell wall versus current methods based on overall microbial viability can aid monitoring needs for controlling infections through deactivating adhesion and colonization by toxigenic C. difficile. In this manner, DEP monitoring methods can aid in reducing the broader impacts of antibiotics to the microbiome.
Acknowledgments
This work is supported by University of Virginia’s Nanostar Seed Fund. The authors thank Dr. Glynis L. Kolling for helpful discussions. Dr. Guerrant and Dr. Warren were supported by NIH grant U19 AI109776.
Supporting Information Available
Table S1, viability of C. difficile after 3 h in microfluidic device; Supplementary Figure S1, representative measurements of cell wall thickness from TEM images of HTCD and NTCD strains; Supplementary Movie 1, dielectrophoretic separation (400 kHz) of the HTCD strain versus the NTCD strain from mixed samples; Supplementary Movie 2, positive dielectrophoretic response (400 kHz) of pure HTCD strain sample, as control; Supplementary Movie 3, dielectrophoretic separation (400 kHz) of viable versus deactivated HTCD after vancomycin treatment (20 min); and Supplementary Movie 4, negative dielectrophoretic response (400 kHz) of vancomycin treated HTCD sample (4 h) as control. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Yi-Hsuan Su: Experimental design, data analysis and interpretation, and manuscript preparation. Cirle Warren: Microbiology experimental design and data analysis. Richard L. Guerrant: Microbiology experimental design. Nathan Swami: Microfluidic device analysis, data interpretation, and manuscript preparation
Parts of this work were included as an oral presentation at the MicroTAS 2014 Conference.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Carroll K. C.; Bartlett J. G. Annu. Rev. Microbiol. 2011, 65, 501–521. [DOI] [PubMed] [Google Scholar]
- Natarajan M.; Walk S. T.; Young V. B.; Aronoff D. M. Anaerobe 2013, 22, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaro K. J.; Phillips S. T.; Cheknis A. K.; Sambol S. P.; Zukowski W. E.; Johnson S.; Gerding D. N. Antimicrob. Agents Chemother. 2013, 57, 5266–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villano S. A.; Seiberling M.; Tatarowicz W.; Monnot-Chase E.; Gerding D. N. Antimicrob. Agents Chemother. 2012, 56, 5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. W.; Curry S. R. Curr. Protoc. Microbiol. 2013, 309A31–39. [DOI] [PubMed] [Google Scholar]
- Cohen S. H.; Gerding D. N.; Johnson S.; Kelly C. P.; Loo V. G.; McDonald L. C.; Pepin J.; Wilcox M. H. Infect. Control Hosp. Epidemiol. 2010, 31, 431–455. [DOI] [PubMed] [Google Scholar]
- Hunt J. J.; Ballard J. D. Microbiol. Mol. Biol. Rev. 2013, 77, 567–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigaglia P.; Mastrantonio P. J. Clin. Microbiol. 2002, 40, 3470–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleytr U. B.; Schuster B.; Egelseer E. M.; Pum D. FEMS Microbiol. Rev. 2014, 38, 823–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan R. P.; Fairweather N. F. Nat. Rev. Microbiol. 2014, 12, 211–222. [DOI] [PubMed] [Google Scholar]
- Reynolds C. B.; Emerson J. E.; de la Riva L.; Fagan R. P.; Fairweather N. F. PLoS Pathog. 2011, 7, e1002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigaglia P.; Barketi-Klai A.; Collignon A.; Mastrantonio P.; Barbanti F.; Rupnik M.; Janezic S.; Kansau I. J. Med. Microbiol. 2013, 62, 1386–1393. [DOI] [PubMed] [Google Scholar]
- Mukherjee K.; Karlsson S.; Burman L. G.; Akerlund T. Microbiology 2002, 148, 2245–2253. [DOI] [PubMed] [Google Scholar]
- Karjalainen T.; Saumier N.; Barc M. C.; Delmee M.; Collignon A. J. Clin. Microbiol. 2002, 40, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekot G.; Posch G.; Messner P.; Matejka M.; Rausch-Fan X.; Andrukhov O.; Schaffer C. J. Dental Res. 2011, 90, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara M.; Sleytr U. B. J. Bacteriol. 2000, 182, 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara M.; Kuen B.; Mayer H. F.; Mandl F.; Schuster K. C.; Sleytr U. B. J. Bacteriol. 1996, 178, 2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casademont I.; Chevrier D.; Guesdon J. L. FEMS Immunol. Med. Microbiol. 1998, 21, 269–281. [DOI] [PubMed] [Google Scholar]
- Ventura M.; Callegari M. L.; Morelli L. FEMS Microbiol. Lett. 2000, 189, 275–279. [DOI] [PubMed] [Google Scholar]
- Jones T. B.Electromechanics of Particles; Cambridge University Press: Cambridge, U.K., 1995. [Google Scholar]
- Morgan H.; Green N. G.. AC Electrokinetics: Colloids and Nanoparticles; Research Studies Press: Philadelphia, PA, 2003. [Google Scholar]
- Pethig R. Biomicrofluidics 2010, 4, 022811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan H.; Sun T.; Holmes D.; Gawad S.; Green N. G. J. Phys. D: Appl. Phys. 2007, 40, 61–70. [Google Scholar]
- Lapizco-Encinas B. H.; Simmons B. A.; Cummings E. B.; Fintschenko Y. Anal. Chem. 2004, 76, 1571–1579. [DOI] [PubMed] [Google Scholar]
- Hawkins B. G.; Huang C.; Arasanipalai S.; Kirby B. J. Anal. Chem. 2011, 83, 3507–3515. [DOI] [PubMed] [Google Scholar]
- Hoettges K. F.; Hubner Y.; Broche L. M.; Ogin S. L.; Kass G. E.; Hughes M. P. Anal. Chem. 2008, 80, 2063–2068. [DOI] [PubMed] [Google Scholar]
- Rohani A.; Varhue W.; Su Y. H.; Swami N. S. Electrophoresis 2014, 35, 1795–1802. [DOI] [PubMed] [Google Scholar]
- Gascoyne P. R.; Shim S.; Noshari J.; Becker F. F.; Stemke-Hale K. Electrophoresis 2013, 34, 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. V.; DeMichele A. F.; Kemp L.; Hayes M. A. Anal. Bioanal. Chem. 2014, 406, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff W. A.; Willner D.; Hugenholtz P.; Rabaey K.; Buie C. R. PLoS One 2013, 8, e76751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmehini V.; Rohani A.; Su Y. H.; Swami N. S. Lab Chip 2014, 14, 4183–4187. [DOI] [PubMed] [Google Scholar]
- Su Y. H.; Tsegaye M.; Varhue W.; Liao K. T.; Abebe L. S.; Smith J. A.; Guerrant R. L.; Swami N. S. Analyst 2014, 139, 66–73. [DOI] [PubMed] [Google Scholar]
- Trejo F. M.; Minnaard J.; Perez P. F.; De Antoni G. L. Anaerobe 2006, 12, 186–193. [DOI] [PubMed] [Google Scholar]
- Merrigan M. M.; Venugopal A.; Roxas J. L.; Anwar F.; Mallozzi M. J.; Roxas B. A.; Gerding D. N.; Viswanathan V. K.; Vedantam G. PLoS One 2013, 8, e78404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurey V.; Rohani A.; Su Y. H.; Liao K. T.; Chou C. F.; Swami N. S. Electrophoresis 2013, 34, 1097–1104. [DOI] [PubMed] [Google Scholar]
- Calabi E.; Calabi F.; Phillips A. D.; Fairweather N. F. Infect. Immun. 2002, 70, 5770–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins B. G.; Kirby B. J. Electrophoresis 2010, 31, 3622–3633. [DOI] [PubMed] [Google Scholar]
- Xuan X. Electrophoresis 2008, 29, 33–43. [DOI] [PubMed] [Google Scholar]
- Nakano A.; Luo J.; Ros A. Anal. Chem. 2014, 86, 6516–6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanghavi B. J.; Varhue W.; Chavez J. L.; Chou C. F.; Swami N. S. Anal. Chem. 2014, 86, 4120–4125. [DOI] [PubMed] [Google Scholar]
- Liao K. T.; Tsegaye M.; Chaurey V.; Chou C. F.; Swami N. S. Electrophoresis 2012, 33, 1958–1966. [DOI] [PubMed] [Google Scholar]
- Hoettges K. F.; Dale J. W.; Hughes M. P. Phys. Med. Biol. 2007, 52, 6001–6009. [DOI] [PubMed] [Google Scholar]
- Gerber M.; Walch C.; Loffler B.; Tischendorf K.; Reischl U.; Ackermann G. J. Med. Microbiol. 2008, 57, 776–783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.