

Abstract
Signaling through the IL-7 receptor (IL-7R) is required for development and maintenance of the immune system. The receptor for IL-7 is heterodimeric, consisting of a common γ chain (γc, encoded by Il2rg) and an α subunit (IL-7Rα, encoded by Il7r). The Il7r gene is expressed specifically in the immune system in a developmental stage-specific manner. It is not known how the Il7r gene is transcriptionally regulated during B cell development. The goal of this study is to elucidate the function of the Il7r promoter region in developing B cells. Using a combination of 5′ rapid amplification of cDNA ends analysis, transient transfection assays, and DNase I hypersensitivity mapping, we identified the location of the Il7r promoter. Using a combination of electrophoretic mobility shift analysis, chromatin immunoprecipitation experiments, and RNA interference experiments, we found that the Ets transcription factors PU.1 and GA-binding protein (GABP) activate the Il7r promoter by interacting with a highly conserved Ets binding site. In committed B lineage cells, GABP can promote Il7r transcription in the absence of PU.1. However, the results of retroviral gene transfer experiments suggest that PU.1 is uniquely required to initiate transcription of the Il7r locus at the earliest stages of progenitor B cell generation. In summary, these results suggest that Il7r transcription is regulated by both PU.1 and GABP in developing B cells.
The cytokine interleukin-7 (IL-7)2 is essential for development and maintenance of the immune system (reviewed in Ref. (1)). IL-7 is produced constitutively by stromal cells of the bone marrow, fetal liver, thymus, and by epithelial cells (2). IL-7 promotes both proliferation and differentiation of developing B and T lymphocytes (3, 4). The receptor for IL-7 (IL-7R, encoded by Il7r) is heterodimeric, consisting of an IL-7-specific α chain (IL-7Rα) and a common γ chain (γc, encoded by Il2rg), which is shared with other subunits comprising the receptors for IL-2, IL-4, IL-9, IL-15, and IL-21. The Il2rg gene is expressed by all hematopoietic cells (5). Transcription of the Il7r gene is initiated in common lymphoid progenitor cells (6). IL-7Rα is expressed in developing B cells but is down-regulated upon B cell maturation (7, 8). In developing T cells, IL-7Rα is initially down-regulated at the CD4 and CD8 double negative 3 stage of thymic development (9). However, Il7r transcription is re-activated after the CD4 and CD8 double-positive stage and is expressed on peripheral CD4 or CD8 single-positive T cells (7). Therefore, the Il7r gene is expressed in a cell type and developmental stage-specific manner (7, 10). Targeted null mutation of either the Il7r (11, 12) or the Il2rg gene (13, 14) results in a profound block to early B and T cell development in mice. In human patients, mutations in the IL7RA gene cause a type of severe combined immunodeficiency in which the major deficiencies are in T cell development, whereas B and NK cells are relatively normal in number (15). Therefore, the IL-7Rα is critically required for the proper development and function of lymphoid cells. Dynamic regulation of IL-7Rα may be important for the generation of appropriate immune responses. Therefore it is important to elucidate the molecular mechanism by which Il7r transcription is regulated.
Little is known about how the Il7r gene is regulated during B cell development. The Ets transcription factor PU.1 (encoded by the gene Sfpi1) is required to activate Il7r transcription during murine fetal liver hematopoiesis (16). However, PU.1 is also required to generate B cell progenitors (17). Therefore it is unclear whether PU.1 is required to maintain Il7r transcription after cell commitment to the B cell lineage. Several recent studies demonstrate that conditional deletion of the Sfpi1 gene in committed B lineage cells does not result in loss of IL-7Rα expression (18–20). Furthermore, ectopic expression of the COE family (Collier/Olf/EBF) transcription factor EBF (Early B cell factor) can activate Il7r transcription and differentiation into pro-B cells in progenitor cells lacking PU.1 (17). The mechanism by which Il7r transcription is maintained in developing B cells in the absence of PU.1 is unknown.
The goal of this study was to elucidate the function of the Il7r promoter region in developing B cells. To accomplish this goal, we determined the location of the Il7r promoter using 5′ RACE, transient transfection analysis, and DNase I hypersensitivity analysis. Using a combination of EMSA, chromatin immunoprecipitation, and RNA interference experiments we found that a conserved Ets transcription factor binding site in the Il7r promoter region can be alternatively utilized by PU.1 or another Ets factor, GA-binding protein (GABP), in developing B cells. Activity of the Il7r promoter is dependent on the Ets site as well as on downstream initiator consensus sequences. In addition, we found that PU.1 is uniquely required to initiate transcription of the Il7r locus at the earliest stages of B cell progenitor cell generation. However, once the Il7r gene is activated, GABP can promote Il7r transcription in the absence of PU.1. These results suggest that Il7r transcription is regulated by both PU.1 and GABP in developing B cells.
EXPERIMENTAL PROCEDURES
5′ RACE Analysis
RNA was prepared from 38B9 pro-B cell lines, IL-7-dependent fetal liver-derived pro-B cells, and sorted peripheral T cell subsets including CD4+ naïve T cells, CD8+ naïve T cells, CD4+ memory T cells, and CD8+ memory T cells. 5′ RACE analysis was performed using a GENERACER kit (Invitrogen) according to the manufacturer’s instructions. The Il7r exon 1-specific reverse primer used was 5′-GCGAAAGCTCTACCCAGAGCCAT-3′. PCR products were gel-purified and cloned using the pCR-TOPO2.1 cloning kit (Invitrogen). At least 5 clones were sequenced from each cell type analyzed.
Reporter Vector Construction
All DNA constructs generated in the course of this study were constructed using standard molecular genetic approaches. Details of the construction of each vector are available upon request. Unless indicated otherwise, all reporter vectors utilized the pGL3-basic luciferase vector (Promega). In all approaches using PCR to clone DNA segments, LA-TAQ (TaKaRa, Otsu, Shiga, Japan) DNA polymerase was used to amplify regions of interest. Site-directed mutagenesis of the Il7r promoter was performed using the QuikChange XL kit (Stratagene) according to the manufacturer’s instructions. To mutate the Ets site core sequence from GGAA to CGAC, complementary oligonucleotides were synthesized and high pressure liquid chromatography purified (Integrated DNA Technologies, Coralville IA) as follows: forward 5′-CAATCCTTTTGCTCAGACGTCGTGTTTCTGGCACTTGC-3′, reverse 5′-GCAAGTGCCAGAAACACGACGTCTGAGCAAAAGGATTG-3′. The oligonucleotides used to mutate putative initiator sequences were: −120 (forward 5′-CTGTGGTTTAGCAGGGGCGGAGCTGGTTTGGGTC-3′, reverse 5′-GACCCAAACCAGCTCCGCCCCTGCTAAACCACAG-3′), and −90 (forward 5′-GGGTCTCCCTCTCTCTGGTGCACTTGCACATACAACCG-3′, reverse 5′-CGGTTGTATGTGCAAGTGCACCAGAGAGAGGGAGACCC-3′). All mutations were confirmed by DNA sequencing.
Transient Transfection Analysis
220–8, 38B9 (21), EL4, and RAW264.7 cell lines were maintained in complete medium containing RPMI 1640 (Invitrogen), 10% fetal bovine serum (HyClone, Logan UT), 100 units/ml penicillin/streptomycin, 2 mM L-glutamine, 5 × 10−5 M β-mercaptoethanol, and 0.5 mM HEPES buffer. For 38B9 and EL4 cells, 4 × 106 cells were electroporated with 10 μg of plasmid DNA and 200 ng of pRL-TK vector (encoding Renilla luciferase) using 220 V and 950 microfarad settings in a 4-mm gap electroporation cuvette (ISC Bioexpress). RAW264.7 cells (105/condition) were transfected using 200 ng of plasmid DNA and 4 ng of pRL-TK vector using Lipofectamine (Invitrogen). Luciferase activity was determined after 24 h of culture using a Dual Luciferase assay kit (Promega). All transient transfection data reported in this study are the mean ± S.D. or S.E. of at least three separate experiments performed in duplicate.
EMSA
Nuclear lysates were obtained from cultured cells using standard methods. In vitro translations were performed using a coupled transcription/translation kit (Promega). DNA binding reactions and acrylamide gel electrophoresis were performed as previously described (22). Antibody supershift analyses were performed by adding 1 μl of anti-PU.1 (Santa Cruz, Santa Cruz CA), anti-GABPα, or anti-GABPβ1 (gifts from Warren Leonard). 1 μl of normal rabbit serum was used as a control in antibody supershift experiments. Probes used were annealed fully complementary double-stranded oligonucleotides containing the wild type Il7r Ets site (5′-AAACAGGAAGTCTG3′), a mutant Il7r Ets site (5′-AAACACGACGTCTG-3′), or an E2A binding site (5′-TACATCTGTGCAGTTCGCTG-3′).
Chromatin Immunoprecipitation
38B9 or WEHI-231 cells in log phase growth were fixed for 10 min at room temperature by adding formaldehyde (1% final) with gentle rocking. Cross-linking was stopped by the addition of glycine to a final concentration of 0.125 M. Chromatin solutions with a mean DNA fragment size of 0.5–1.0 kb were prepared as previously described (16). Chromatin from 1 × 107 cells was incubated at 4 °C with 15 μl of anti-GABPα (Santa Cruz) or anti-actin (Sigma) antibodies for 16 h, followed by addition of Salmon Sperm DNA/Protein A-agarose gel slurry (Upstate, Charlottesville VA) for 1 h. Immunoprecipitates were collected by centrifugation, washed three times in RIPA buffer, and eluted with Elution Buffer (50 mM NaHCO3, 1% SDS). Cross-linking was reversed by adding NaCl (0.3 M final) for 4 h at 65 °C. The resulting solution was treated with RNase A for 20 min at 37 °C and Proteinase K for 16 h at 37 °C. DNA was purified using the Wizard SV Gel and PCR Purification System (Promega) and analyzed by semiquantitative PCR (30–35 cycles) with primer pairs identifying the Il7r and Hprt1 gene promoters (16).
RNA Interference Experiments
220–8 pro-B cells were electroporated as described above with 20 μg of pBS/U6 expression vector as a control, or 20 μg of pBS/U6 expression vectors encoding short hairpin RNAs directed against GABPα (23), as well as 1 μg of pEYFP-N1 to enable selection by cell sorting. EYFP-positive cells were isolated by cell sorting 60 h after transfection, and gene expression in these cells were assessed by real-time PCR as previously described (23). For inducible RNA interference experiments, an oligonucleotide containing a previously described small interfering RNA (5′-AGAAGACAGAAGTTCACCG-3′) (23) directed against the coding region of GABPα was adapted as a microRNA by ligation into the tetracycline-regulated retroviral vector SIN-TREmi30-PIG (TMP) (Open Biosystems, Huntsville, AL) according to the manufacturer’s instructions. Retrovirus was prepared as described below and used to infect 38B9 pro-B cells. Infected cells were enriched by cell sorting for GFP expression. To express the reverse tetracycline transactivator protein, 38B9 cells or sorted 38B9-TMP-GABPα cells were infected with pRevTet-ON retrovirus (Clontech) and selected by culture in 0.8 mg/ml G418 (Mediatech, Herndon, VA). To induce expression of the microRNA directed against GABPα, 38B9 + pRevTet-ON, or 38B9 + pRevTet-ON + TMP-GABPα cells were cultured with 2 μg/ml doxycycline (Sigma) for 72 h. At 72 h total RNA was prepared to measure GABPα expression, or flow cytometric analysis was performed to measure IL-7Rα expression.
DNase I Hypersensitive Site Mapping
DNase I treatment was performed as described in Ref. 24. Following DNA extraction, 10 μg of DNA for each sample was digested with EcoRI or ScaI to completion and separated by electrophoresis on an 0.8% agarose gel in TAE followed by Southern blot analysis. Probe 1 (+4569 bp to +4688 bp) and probe 2 (−6556 bp to 5992 bp) were used for Southern blot analysis.
Antibodies and Flow Cytometry
Single cell suspensions were stained with phycoerythrin or biotin-conjugated antibodies and streptavidin-phycoerythrin or streptavidin-allophycocyanin secondary reagents. Propidium iodide (Invitrogen) was used to exclude dead cells from analysis. Anti-mouse monoclonal antibodies utilized in this study were M1/70-biotin (CD11b, BD Pharmingen), 1D3-phycoerythrin (CD19, BD Pharmingen), and A7R34-biotin (IL-7Rα). Flow cytometric analysis was performed on a BD Immunocytometry systems FACScalibur dual-laser instrument. Cell sorting and green or yellow fluorescent protein (GFP or YFP) analysis was performed on a FACSVantage instrument with FACSDiva upgrade (BD Immunocytometry Systems).
Construction and Generation of Retroviruses
We have previously described the construction of the MSCV-IRES-GFP (MIG)-PU.1 retrovirus (16). MIG-GABPα was constructed by ligation of a FLAG-tagged GABPα cDNA (provided by Dr. Alan Rosmarin) into the MIGR1 retroviral vector EcoRI restriction site (25), and was confirmed by sequence analysis. GABPβ1 cDNA was amplified from EL-4 mRNA using RT-PCR with gene-specific primers, and cloned into the pCR-TOPO 2.1 vector (Invitrogen). After confirmation by DNA sequencing, GABPβ1 cDNA was ligated into the EcoRI restriction site of a retroviral vector (MIYR1) in which enhanced GFP (EGFP) had been replaced with EYFP (Clontech). Plat-E retroviral packaging cells (26) were maintained in complete Dulbecco’s modified Eagle’s medium (Invitrogen). Plat-E cells were transiently transfected with retroviral vectors using Lipofectamine reagent according to the manufacturer’s instructions (Invitrogen). Retrovirus-containing supernatants were collected 48 h after transfection by centrifugation.
To confirm protein expression, lysates of transfected Plat-E cells were prepared on the day of virus collection using Laemmli buffer according to standard protocols. Lysates were electophoresed on 10% polyacrylamide denaturing gels and transferred to nitrocellulose membranes using a Trans-Blot SD apparatus (Bio-Rad). Membranes were probed using anti-FLAG M2 (Sigma), or anti-GABPβ1 (gift of Dr. Warren Leonard) antibodies. Protein bands were visualized using a horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibodies (Pierce) and the Super-Signal West Pico kit (Pierce) before exposure to x-ray film.
Retroviral Transduction and Culture of Fetal Liver Progenitors
Lineage-depleted wild type or Sfpi1−/− fetal liver progenitor cells were generated by negative selection of day 14.5 fetal liver cells with immunomagnetic beads as previously described (27). For infections, 105 Lin− progenitors were mixed with 1 ml of retrovirus-containing supernatant, 1 ml of complete Iscove’s modified Dulbecco’s medium, and 16 μg of Polybrene, and then centrifuged in flat-bottom 24-well plates (Greiner Bio-One, Monroe, NC) for 2 h at 3100 × g. At the end of the centrifugation, infected progenitors were cultured in 20 ng/ml IL-3, 100 ng/ml SCF, and 10 ng/ml IL-6 in complete Iscove’s modified Dulbecco’s medium (Hyclone). After 48 h of culture, the infection frequency was measured by flow cytometric analysis of an aliquot of cells for GFP or YFP fluorescence. Infected progenitors were placed into culture with irradiated S17 stromal cells and IL-7-conditioned medium as previously described (16). Cultures were incubated for 9–11 days before counting and flow cytometric analysis for cells expressing CD11b, CD19, and IL-7Rα.
RESULTS
Functional Identification of the IL-7Rα Promoter
To investigate transcriptional regulation of the Il7r gene, it was necessary to clarify the promoter region that is functional in developing B cells. We previously reported that Il7r promoter constructs incorporating a published transcription start site (at position −946 relative to the translation start codon (28) had undetectable activity in transient transfection experiments (16). We therefore performed 5′ RACE with RNA from B cell lines that express high levels of IL-7Rα including 38B9 pro-B cells and IL-7-dependent fetal liver derived pro-B cell lines. In addition, we prepared 5′ RACE products from sorted naïve (TCRβ+ CD44−) or memory (TCRβ+ CD44hi, day 40 post-LCMV infection) peripheral T cells, from both CD4+ and CD8+ peripheral compartments (29). PCR was performed on RACE products using a forward RACE-specific primer and a reverse Il7r primer located in exon 1 of the cDNA (Fig. 1A). Two major groups of PCR products were detected in each tissue examined (Fig. 1B) that were gel purified, cloned, and sequenced to determine the transcription start site(s). Each cell type examined displayed an identical pattern of transcription start sites. Three sites were identified more frequently than others: a site at −120 (relative to the translation start site) was found in 39% (13/33) of clones, a site at −90 was found in 21% (7/33) of clones, and a site at −86 was found in 15% (5/33) clones (Fig. 1C). No PCR products were obtained using a gene-specific primer proximal to a previously described −946 bp transcription start site (28). These results suggest that the major transcription start sites in the Il7r gene are within 120 bp upstream of the open reading frame.
FIGURE 1. Identification of the Il7r promoter region.
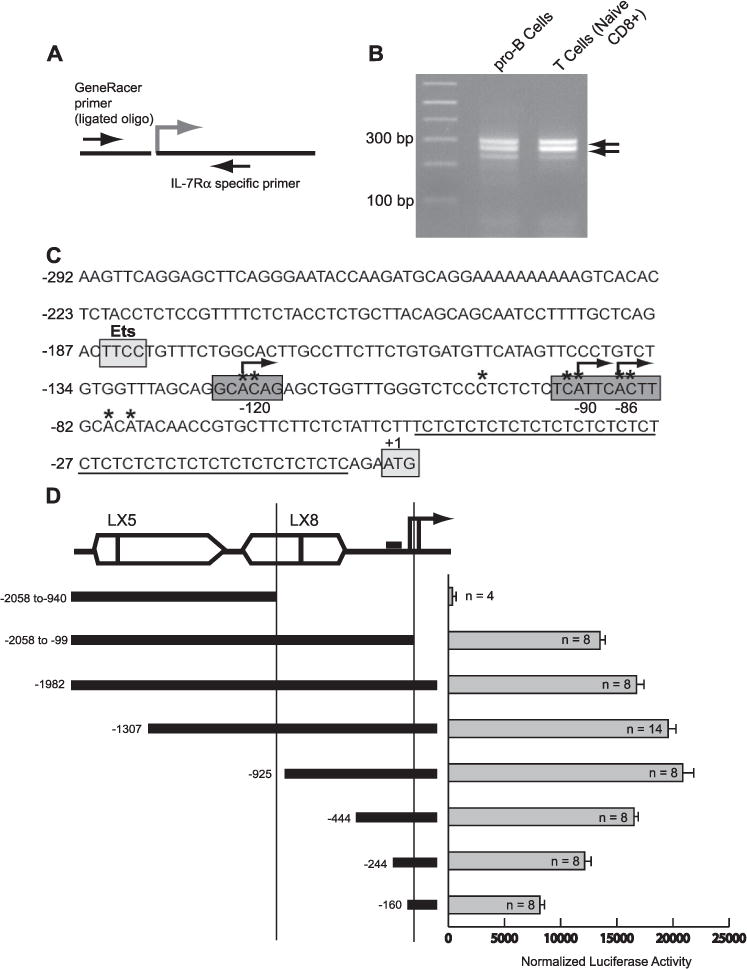
A, overview of the 5′ RACE method. B, amplification of 5′ RACE products from both B and T cells reveals major transcription start sites downstream from the Ets binding site. 5′ RACE PCR products from pro-B cells and naïve CD8+ T cells were visualized by gel electrophoresis and ethidium bromide staining. The products shown were cloned and sequenced to identify transcription start sites. Clones were sequenced from 5′ RACE products prepared from pro-B cell lines, IL-7-dependent pro-B cells, T cell lines, and purified naïve and memory CD4+ and CD8+ peripheral T cells. C, DNA sequence of the Il7r transcription initiation region. Transcription start sites mapped using 5′ RACE in B and T cells are marked with an asterisk. Frequently occurring transcription start sites are marked with an arrow. A(CT)n/(GA)n microsatellite sequence is underlined. Also indicated are the Ets binding site and the location of the translation start site. D, maximal IL-7Rα promoter activity is contained in the 925 bp upstream of the transcription start sites. Transient transfection analysis was performed by electroporation of 38B9 pro-B cells with equimolar amounts of the indicated luciferase reporter plasmids, along with equimolar amounts of a Renilla luciferase plasmid. A schematic of the Il7r promoter region is shown at the top left. Open boxes indicate the relative location of LX5 and LX8 LINE-1 elements. The solid lines with arrows represent known transcription start sites. The two vertical lines represent, from left to right, naturally occurring HincII and BsaI sites in the Il7r promoter region. The small filled rectangle represents the Ets site. Results (bar graph on right) are expressed as mean firefly luciferase activity normalized to Renilla luciferase activity. Error bars represent the S.E. of n experiments.
To determine which promoter region is functional in B cells we cloned various DNA segments into the pGL3 luciferase vector to perform transient transfection analysis. The 38B9 pro-B cell line (21) was chosen for transient transfection analysis because it expresses high levels of both PU.1 and IL-7Rα mRNA transcripts and protein (data not shown). Of the various constructs transfected into 38B9 cells, the −925 promoter had the highest activity (Fig. 1D). The activity of the promoter was significantly reduced by deletion of the conserved region (−160). Activity of the promoter was reduced but not eliminated by removing two of the major transcription start sites located at −86 and −90 (−2058 to −99); this was consistent with the evidence for multiple transcription start sites. Finally, a construct resembling one used in the original description of the Il7r promoter (−2058 to −940), which ends at a HincII restriction site (28), had activity below background levels, consistent with our lack of detection of transcription initiating in this area. In summary, transient transfection analysis suggests that the Il7r promoter is located in a region between −925 and the transcription start sites. The −925 promoter was therefore used in subsequent experiments.
An Intact Ets Site Is Essential for Activity of the Il7r Promoter in B and T Cells
To identify regulatory sites crucial for regulation of the Il7r promoter, we first performed sequence comparisons using the ClustalW algorithim. The Il7r gene has been annotated in genomes sequenced from human (Homo sapiens), chimp (Pan troglodytes) dog (Canis familiaris), rat (Rattus norvegicus), mouse (Mus musculus), pufferfish (Fugu rubripes), and green spotted puffer (Tetraodon nigroviridis). Alignment of the promoter region revealed that a ~108-bp block of sequence was highly conserved in every one of these species (Fig. 2A). This conserved block of sequence contains a previously identified Ets binding site (the sequence ACAGGAAGTC, which was 100% conserved in each species examined. This conserved sequence block appears at a similar position (~150 bp upstream of the open reading frame) in the Il7r gene from each species examined. There was no measurable sequence identity upstream or downstream of this region, including at the transcription start sites (data not shown). The high degree of sequence identity between species in this region suggests that it is likely to be functionally important across multiple species.
FIGURE 2. Activity of the Il7r promoter requires an intact Ets binding site and functional initiator sequences.
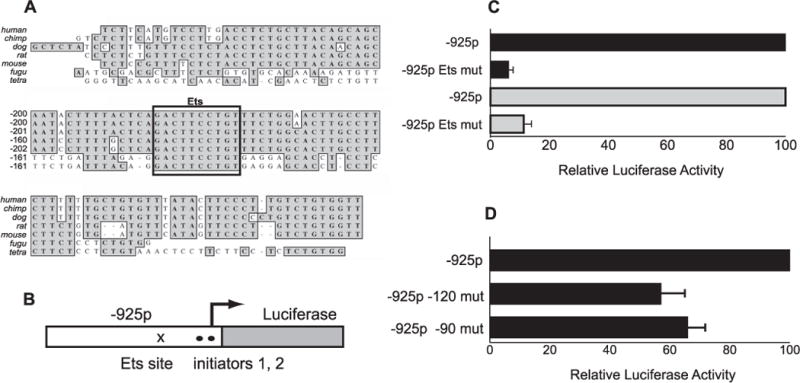
A, the Ets binding site is conserved across multiple species. A ClustalW alignment is shown using Il7r promoter sequences from H. sapiens (human), P. troglodytes (chimp), C. familiaris (dog), R. norvegicus (Rat), M. musculus (mouse), F. rubripes (fugu, i.e. pufferfish), and T. nigroviridis (tetra). The boxed sequence contains the Ets core binding site (TTCC) and conserved flanking sequence. B, overview of luciferase reporter plasmids. The pGL3-IL-7Rα −925 promoter plasmid was modified by site-directed mutagenesis of the Ets binding site (x) or by mutation of putative initiator sequences (●). C, mutation of the Ets site abolishes activity of the Il7r −925 promoter in B and T cells. Plasmids containing a wild type (−925p) or mutant (−925p Ets mut) Il7r −925 promoter were transiently transfected into 38B9 pro-B cells or EL-4 T cells, along with equimolar amounts of a Renilla luciferase plasmid. Results are expressed as % of full activity (normalized firefly luciferase activity of mutant Ets site plasmids/parent plasmids × 100). Error bars represent S.E. ± mean (n = 6). D, effect of initiator mutations on activity of the Il7r promoter. Site-directed mutagenesis of putative initiator sequences was performed as described under “Experimental Procedures.” Plasmids tested contained mutations in putative initiator sequences at positions −120 or −90, and were transiently transfected into 38B9 pro-B cells, along with equimolar amounts of a Renilla luciferase plasmid. Results are expressed as relative luciferase activity (mean normalized firefly luciferase activity of the mutation-containing plasmid divided by the mean normalized firefly luciferase activity of the parent plasmid). Error bars represent S.E. ± mean (n = 6).
We then aimed to test whether the Ets site is required for activity of the −925 Il7r promoter. The Ets site core sequence was mutated from GGAA to CGAC using site-directed mutagenesis. The activity of the mutant −925 construct was then tested by transient transfection analysis in 38B9 pro-B cells and in EL4 T cells (Fig. 2C). In 38B9 pro-B cells, mutation of the Ets site reduced activity of the −925 promoter by over 90% (Fig. 2C, top two bars). The Il7r −925 promoter construct also had significant activity when transiently transfected into the EL4 T cell line, which does not express PU.1 (data not shown). Mutation of the Ets site reduced activity of the −925 promoter by over 80% in EL4 T cells (Fig. 2C, bottom two bars). This result confirms previous findings that the Ets site is essential for activity of the Il7r promoter in T cells (23, 30). In summary, these results show that the Ets binding site is required for activity of the Il7r promoter in both B and T cells. These results suggest that an Ets family transcription factor other than PU.1 interacts with this site in T cells.
The three most frequent transcription start sites in the Il7r promoter initiate at CA dinucleotide sequences, which is a common feature of initiator sequences. In fact, the start site at −90 (Fig. 1C) has the initiator consensus sequence TCATTC (31). This raises the possibility that function of the Il7r promoter utilizes interactions between the Ets binding site and initiator sequences, a combination that has been previously described (32, 33). To test if initiator sequences are utilized in the Il7r promoter, we used site-directed mutagenesis to change the DNA sequence at −120 (see Fig. 1C) from GCACAG to GGGCGG, and the sequence at −90 from TCATTC to TGGTGC (34). Mutagenesis was performed on the −925 promoter reporter vector, and these constructs were tested using transient transfection analysis in 38B9 pro-B cells. Mutation of the sequences at −120 and −90 reduced activity of the promoter to 57 and 66% of the wild type promoter, respectively (Fig. 3D). Combined mutation of these two sequences did not reduce activity further (data not shown). Taken together, these results suggest that the Il7r promoter contains functional initiator sequences.
FIGURE 3. GABP interacts with the Il7r Ets site in EBF-rescued, Sfpi1−/−Spi-B−/− pro-B cells.
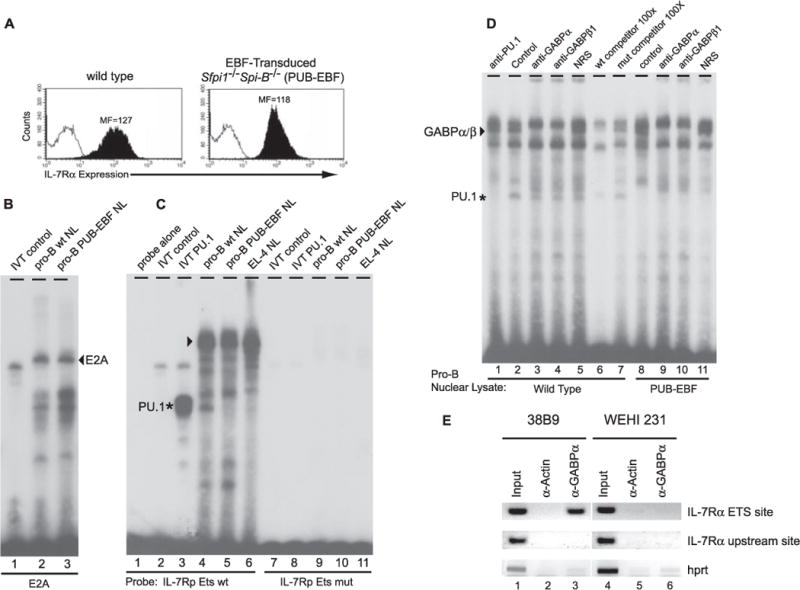
A, wild type and EBF-rescued, Sfpi1−/−Spi-B−/− (PUB-EBF) pro-B cells express equivalent levels of IL-7Rα. Flow cytometry was used to phenotype the indicated cell lines using biotinylated antibodies against CD11b (open histogram) or IL-7Rα (filled histogram). The mean fluorescence (MF) for IL-7Rα staining is indicated. B, EMSA for E2Ain wild type and PUB-EBF pro-B cells. EMSA was performed using a radiolabeled oligonucleotide probe containing a wild type E2A binding site. An E2A complex (arrow) was equally present in wild type and PUB-EBF pro-B cells. C, EMSA of Ets site using nuclear lysates from PUB-EBF pro-B cells. EMSA was performed using a radiolabeled oligonucleotide probe containing a wild type (lanes 1–6) or mutant (lanes 7–11) Il7r promoter Ets site. In vitro translated (IVT) PU.1 was used as a positive control (lane 3). APU.1 complex (asterisk) was evident in nuclear lysates from wild type pro-B cells (lane 4) but not PUB-EBF pro-B cells (lane 5). In addition, a slower migrating complex (arrow) was present in all nuclear lysate samples including the EL-4 T cell line (lane 6). D, the Il7r promoter utilizes GABP at the Ets site in PUB-EBF pro-B cells. EMSA was performed using nuclear lysates from wild type (lanes 1–7) or PUB-EBF (lanes 8–11) pro-B cells, and a radiolabeled oligonucleotide probe containing the wild type Il7r promoter Ets site. Antibody supershift analyses were performed using normal rabbit serum (NRS, lanes 5 and 11) as a negative control, or rabbit polyclonal antibodies recognizing PU.1 (lane 1), GABPα (lanes 3 and 9), or GABPβ1 (lanes 4 and 10). Competition experiments were carried out using 100-fold excess unlabeled wild type (lane 6) or mutant (lane 7) Il7r promoter Ets site oligonucleotide probes. E, GABP interacts with the Ets site in pro-B cells. Chromatin prepared from 38B9 pro-B cells or WEHI-231 B cells was immunoprecipitated using an anti-actin (α-actin) or anti-GABPα (α-GABPα) antibody. PCR was performed on DNA purified from input chromatin (Input) or immunoprecipitated chromatin, using primer pairs surrounding the Il7r Ets site, an upstream site in the Il7r promoter (IL-7Rα upstream site), or the promoter of the hprt1 gene (hprt). PCR products were visualized by agarose gel electrophoresis and ethidium bromide staining. wt, wild type.
GABP Interacts with the Ets Site in Pro-B Cells
As described in the Introduction, activation of Il7r transcription during fetal lymphocyte development requires PU.1 (16). However, once activated, Il7r transcription does not require continued PU.1 expression (17–20). Because we showed above that the Ets binding site is required for activity of the Il7r promoter, this raises the question of whether another Ets factor substitutes for PU.1 in Sfpi1 mutant B lineage cells. The Ets transcription factor GABP was recently found to be essential for Il7r transcription in T cells (23). GABP is a heterodimeric transcription factor consisting of a DNA-binding α subunit that is a member of the Ets family of transcription factors, and a β subunit containing an ankyrin domain that contains the activation domain (35). We therefore hypothesized that GABP might also interact with the Ets site in developing B cells.
As shown in Fig. 3A, IL-7Rα is expressed at high levels in cultured pro-B cells that are deficient in both PU.1 and the closely related Ets transcription factor Spi-B (17). We performed EMSA experiments using an Il7r promoter oligonucleotide probe and nuclear lysates from either wild type or PU.1/Spi-B-deficient (PUB-EBF) pro-B cell lines. Similar quality of nuclear lysates was confirmed by EMSA using oligonucleotide probes containing a binding site for E2A, which is expressed at high levels in all B cells (36) (Fig. 4B). Using the Il7r Ets site probe, in vitro translated PU.1 formed a monomeric complex as expected (Fig. 3C, lane 3). Nuclear lysates from wild type but not PUB-EBF pro-B cells contained a DNA binding activity corresponding to PU.1 (Fig. 3C, lanes 4 and 5). This activity was confirmed to be PU.1 by supershifting the complex with an anti-PU.1 antibody (Fig. 3D, lanes 1 and 2). Furthermore, PU.1 did not bind to an oligonucleotide probe containing a mutated Ets site (GGAA → CGAC, Fig. 3C, lanes 8 and 9) and could be competed by excess unlabeled oligonucleotide containing the wild type but not a mutated Ets site (Fig. 3D, lanes 6 and 7).
FIGURE 4. GABP regulates Il7r transcription in pro-B cells.
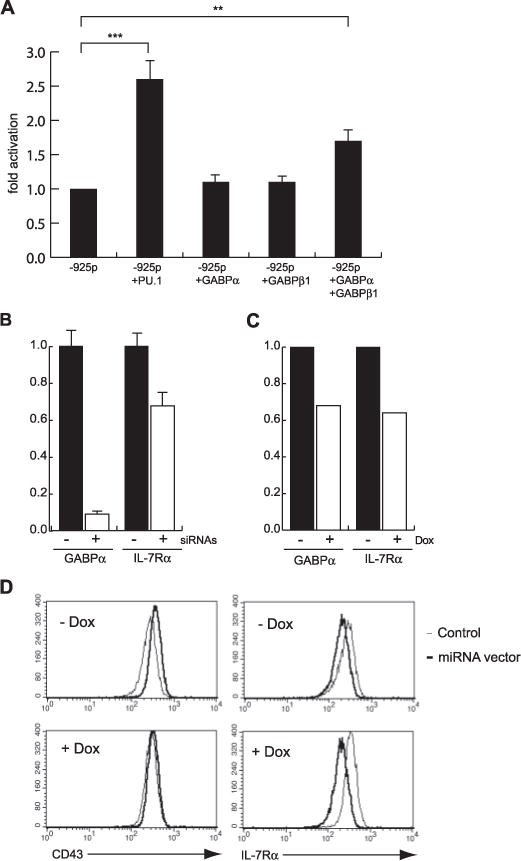
A, an Il7r reporter construct is activated by ectopic expression of PU.1 or GABP subunits. 38B9 pro-B cells were transiently transfected with equimolar amounts of DNA containing a pGL3-Basic vector containing the −925 Il7r promoter, as well as pCDNA3 expression vectors encoding PU.1, GABPα, GABPβ1, or both GABPα and GABPβ1. Empty pCDNA3 vector was used as a control and to equalize amounts of DNA. Transfections were normalized by transfecting equimolar amounts of a Renilla luciferase plasmid. Results are expressed as-fold activation above baseline reporter activity. Error bars represent standard deviation of the mean. B, RNA interference of GABPα transcription in pro-B cells. 220–8 pro-B cells were transiently transfected with empty expression vector (filled bars) or expression vector encoding short hairpin RNAs directed against GABPα (open bars). 60 h after sorting transfected cells, total RNA was prepared and real-time RT-PCR was performed to measure levels of mRNAs encoding either GABPα (left bars) or IL-7Rα (right bars). C, inducible RNA interference of GABPα transcription in pro-B cells. 38B9 cells infected with vectors encoding a reverse tetracycline-transactivator protein (rtTA, filled bars), or both rtTA and a tetracycline-regulatable vector expressing an anti-GABPα microRNA (open bars) were induced with doxycycline (Dox). After 72 h, total RNA was prepared and real time RT-PCR was performed to measure levels of mRNAs encoding either GABPα (left bars) or IL-7Rα (right bars). D, flow cytometric analysis of cells described in C.38B9 cells were analyzed for expression of cell-surface expression of CD43 or IL-7Rα.
Wild type, PUB-EBF, and EL4 T cell nuclear lysates contained DNA binding activities resulting in several slower-migrating complexes in the EMSA (Fig. 3C, lanes 4–6). Nuclear lysates from wild type pro-B cells contained proteins resulting in both slow and fast migrating complexes on the Il7r promoter Ets site probe (Fig. 3D, lane 2). One of several slower migrating complexes could be supershifted with either anti-GABPα or anti-GABPβ1 antisera (Fig. 3D, lanes 3 and 4), and could be competed with unlabeled excess oligonucleotide containing a wild type but not mutant Ets site (Fig. 3D, lanes 6 and 7). Therefore this complex contains a GABPα/β1 heterodimer. In addition to PU.1 and GABP complexes, several other DNA-binding complexes were evident. The identity of these DNA-binding proteins is unknown, but they likely do not represent Ets transcription factors because they were competed equally efficiently by unlabeled excess oligonucleotide containing either a wild type or mutant Ets site (Fig. 3D, compare lane 6 with lane 7). The GABPα/β1 heterodimeric complex (but not the PU.1 complex) was also present in PUB-EBF pro-B cells (Fig. 3D, lanes 8–11).
To confirm that GABP interacts with the Ets site in pro-B cells, we performed chromatin immunoprecipitation experiments. Chromatin was prepared from 38B9 pro-B cells or WEHI-231 B cells, which do or do not express the Il7r gene, respectively, and were immunoprecipitated with an anti-actin antibody as a negative control, or with an anti-GABPα antibody. PCR was performed on immunoprecipitated DNA after reversal of cross-linking, using primers specific for the Il7r Ets site and control primers specific for a site ~1800-bp upstream of the Ets site (“IL-7Rα site 1”) or for the promoter of an unrelated gene (hprt). As shown in Fig. 3E, the IL-7Rα Ets site was specifically immunoprecipitated with anti-GABPα antibody only in 38B9 cells. In summary, this data suggests that GABP associates directly with the Il7r Ets site in pro-B cells. The EMSA experiments suggest that in either wild type or Sfpi1−/− pro-B cells this complex contains a GABPα/β1 heterodimer.
GABP Trans-activates the Il7r Promoter in Pro-B Cells
We next investigated whether GABP regulates activity of the Il7r promoter in pro-B cells, using transient transfection assays as well as RNA interference experiments. As shown in Fig. 4A, co-transfection of a PU.1 expression vector into the 38B9 pro-B line increased activity of the Il7r −925 promoter by 2.7 ± 0.8-fold (n = 8). Co-transfection of expression vectors encoding GABPα or GABPβ1 separately did not significantly increase activity of the Il7r −925 promoter. However, co-transfection of both GABPα and GABPβ1 together increased activity of the Il7r −925 promoter by 1.7 ± 0.1-fold (n = 6) (Fig. 4A). This result is in agreement with data showing that GABP functions as an obligate heterodimer (37). These data suggest that GABP can activate transcription of the Il7r promoter in pro-B cells.
RT-PCR analysis showed that GABPα, GABPβ1, and GABPβ2 mRNA are expressed in IL-7-dependent pro-B cells, wild type total fetal liver cells, wild type fetal liver cells, and Sfpi1−/− fetal liver cells (data not shown). Therefore GABP is expressed in cell types in which IL-7Rα is also expressed. To test whether GABPα activates Il7r in pro-B cells, we performed several types of RNA interference experiments to “knockdown” GABPα mRNA in 220–8 or 38B9 pro-B cells. First, 220–8 pro-B cells were transiently transfected with a mixture of several small interfering RNAs directed against GABPα (23). As shown in Fig. 4B, GABPα mRNA was reduced to 9 ± 2% of normal levels when 220–8 pro-B cells were transfected with expression vectors encoding GABP short hairpin RNA oligonucleotides compared with empty vector. Reduction of GABPα mRNA led to reduction of IL-7Rα mRNA to 68 ± 11 of normal levels. To confirm and extend this result, we generated 38B9 pro-B cell lines stably transduced with a tetracycline-regulatable retroviral vector encoding a microRNA directed against GABPα (see “Experimental Procedures” for details). Such cells were also transduced with a retroviral vector encoding reverse tetracycline transactivator protein (rtTA). Induced expression of the anti-GABPα microRNA by culture in doxycycline for 72 h led to a substantial block to cell growth, suggesting that GABP might be involved in proliferation of pro-B cells (data not shown). Culture in doxycycline also caused a moderate reduction in GABP mRNA (68% of uninduced levels) as well as IL-7Rα mRNA (64% of uninduced levels) (Fig. 4C). Flow cytometric analysis revealed that culture in doxycycline led to reduced cell surface IL-7Rα expression relative to control cells (Fig. 4D, right panels). As a control for specificity, we found that CD43 expression was not affected by inducible GABPα knockdown (Fig. 4D, left panels). In summary, reduction of GABPα mRNA levels in pro-B cells using RNA interference leads to a reduction in IL-7Rα mRNA and cell surface expression. This result suggests that GABP directly regulates Il7r transcription in pro-B cells.
GABP Is Insufficient to Activate Il7r Transcription in PU.1−/− Fetal Liver Progenitor Cells
To test whether ectopic expression of GABP subunits is sufficient to activate Il7r transcription in Sfpi1−/− fetal liver progenitors, we constructed a MSCV-IRES-EGFP (MIG) retroviral vector encoding murine GABPα, and a MSCV-IRES-EYFP (MIY) retroviral vector encoding GABPβ1 (Fig. 5A). FLAG-tagged GABPα and untagged GABPβ1 proteins were both expressed as measured by Western blotting after transient transfection of Plat-E retroviral packaging cells with retroviral vectors (Fig. 5B). Cell-free MIGR1, MIG-PU.1, MIG-EBF, MIG-GABPα, or MIY-GABPβ1 retroviral supernatants were used to infect Sfpi1−/− progenitor cells. Infection was measured by flow cytometric analysis of green or yellow fluorescence (GFP or YFP) expression 24 h after transduction. As shown in Fig. 5C, analysis of GFP and YFP fluorescence confirmed that a significant fraction of Sfpi1−/− progenitor cells were infected with both GABPα and GABPβ1. Single or double-infected progenitor cells were placed in culture on S17 stromal cells in the presence of IL-7 as previously described (16). To normalize for different efficiencies of infection, an identical number of infected cells (or in the case of both GABPα and GABPβ1, double-infected cells) were placed into culture in each of three experiments. At the end of 9–11 days, proliferation was measured in each well by cell counting; and flow cytometric analysis was performed on single cell suspensions. As expected, transduction with a control (MIGR1) retrovirus failed to rescue IL-7-dependent proliferation, whereas transduction with either PU.1 or EBF resulted in robust IL-7-dependent growth (Fig. 5D). In contrast, transduction with GABPα, GABPβ1, or GABPα and GABPβ1 cDNAs did not rescue IL-7-dependent growth (Fig. 5D). Flow cytometric analysis of PU.1 or EBF-rescued cells revealed that most cells in the culture were pro-B cells expressing both CD19 and IL-7Rα (data not shown). In addition, cultured IL-7-dependent pro-B cell lines could be readily established from Sfpi1−/− progenitors infected with PU.1 or EBF, but not from progenitors infected with GABPα/β1 subunits. In summary, ectopic expression of GABP is not sufficient to activate Il7r transcription in Sfpi1−/− fetal liver progenitor cells.
FIGURE 5. GABPα is insufficient to rescue Il7r transcription in Sfpi1−/− fetal liver hematopoietic progenitors.
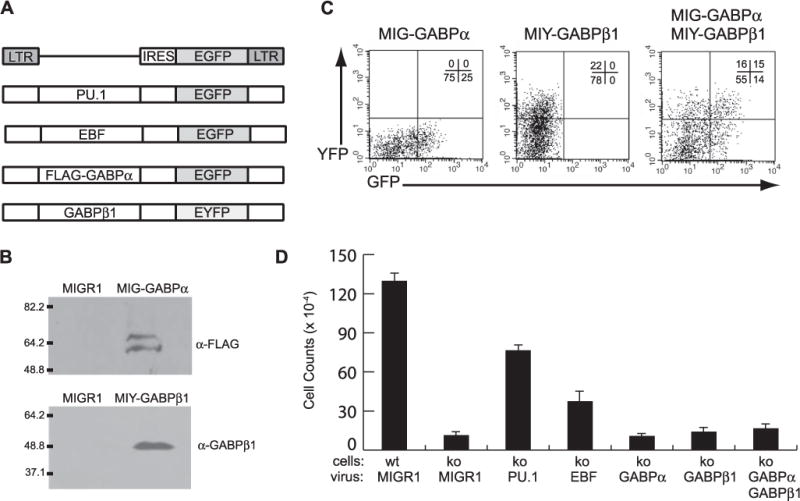
A, overview of retroviral vectors. Shown in schematic form are MIGR1 (first panel), MIG-PU.1 (second panel), MIG-EBF (third panel), MIG-GABPα (fourth panel), and MIY-GABPβ1 retroviral vectors (fifth panel). B, GABPα and GABPβ1 protein expression. Western blot analysis was used to demonstrate GABPα expression from retroviral packaging cells transiently transfected with the MIGR1 or MIG-GABPα retroviral plasmids, using an anti-FLAG antibody (top panel), or from MIY-GABPβ1 retroviral plasmids, using an anti-GABPβ1 antibody (bottom panel). C, flow cytometric analysis of Sfpi1−/− fetal liver progenitor cells infected with retroviral vectors. After 24 h of culture, Sfpi1−/− fetal liver progenitor cells infected with MIG-GABPα, MIY-GABPβ1, or both MIG-GABPα and MIY-GABPβ1 were analyzed for GFP fluorescence (x axis) and YFP fluorescence (y axis). The percent cells in each quadrant are shown in the upper right corner of each panel. D, PU.1 and EBF, but not GABP rescues IL-7-dependent proliferation of Sfpi1−/− progenitors. Sfpi1−/− progenitors were infected with MIGR1, MIG-PU.1, MIG-EBF, MIG-GABPα, MIY-GABPβ1, or both GABPα and GABPβ1 retroviruses and cultured on S17 stromal cells with IL-7 for 9 days. Proliferation was evaluated by cell counting. A summary of the results obtained from three independent experiments is shown. ko, knockout.
DNase I Hypersensitivity Mapping of the Il7r Gene
A major future direction of this work will be to identify tissue-specific regulatory regions of the Il7r gene. To identify potential regulatory regions in the Il7r promoter region, DNase I hypersensitivity analysis was performed to identify sites of open chromatin. Nuclei were prepared from NIH-3T3 cells as a negative control, as well as from RAW264.7 macrophage cells, SB+/+ pro-B cells, murine splenic B cells, thymic T cells, and splenic T cells. IL-7Rα is expressed at high levels in pro-B cells and in thymic or splenic T cells, but at low levels in macrophages and splenic B cells (data not shown). Altogether, 10 DNase I hypersensitive sites were identified (Fig. 6A, 6B). The transcription start site (promoter) was DNase I hypersensitive in every cell type except fibroblasts, which do not express the Il7r gene. In pro-B cells and T cells, DNase I hypersensitive sites were identified at −3.8, +1.5, +2.2, and +2.5 kb. Several additional sites were identified at −5.5, −4.6, and +2.6 kb that were highly hypersensitive only in splenic T cells. A site located at +1.5 kb was hypersensitive in B cells, T cells, and macrophages, but not in fibroblasts. In fibroblasts, RAW264 macrophages, and splenic B cells, additional sites were identified at +0.5 and +1.8 kb. Therefore these sites were observed only in cell types that do not express the IL-7Rα or express it at low levels. Interestingly, the DNase I hypersensitive site at −3.8 kb corresponds to a recently identified enhancer for the Il7r gene containing a glucocorticoid receptor binding site (30). The −3.8-kb hypersensitive site was most highly hypersensitive in peripheral T cells, and was not detected in cells that do not express the Il7r gene. These results suggest that the Il7r gene may be controlled by a combination of tissue-specific positive and negative regulatory elements.
FIGURE 6. The promoter region of the Il7r gene contains several groups of DNase I hypersensitive sites.
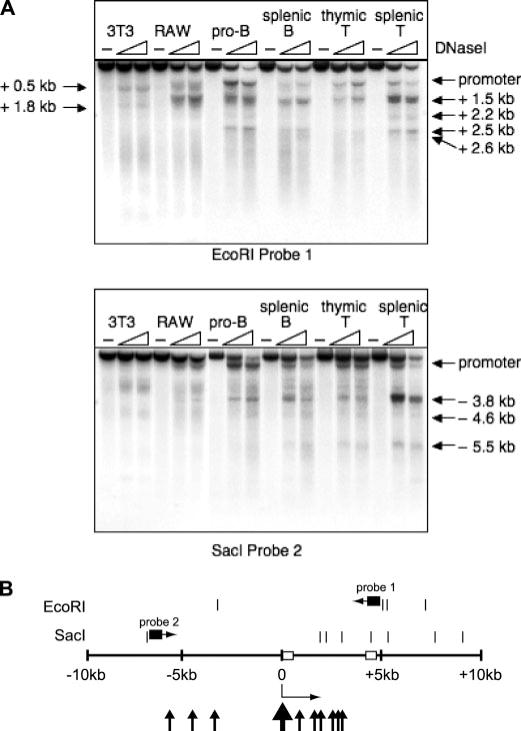
A, DNase I hypersensitivity analysis of the IL-7Rα gene. Nuclei were prepared from 3T3 fibroblasts (3T3), RAW264.7 macrophages (RAW), or SB+/+ pro-B cells (pro-B), splenic B cells, thymic T cells, or splenic T cells. Nuclei were either undigested (−) or digested with increasing concentrations of DNase I enzyme (slope). DNA prepared from these nuclei was digested with EcoRI (top panel) or SacI (lower panel) restriction enzymes, and Southern blotted with the indicated radiolabeled probes. B, summary of DNase I hypersensitivity analysis. Open boxes represent exons 1 and 2, respectively. The Il7r promoter is designated with a thick arrowhead. Thin arrowheads represent upstream and downstream DNase I hypersensitive sites.
Trans-activation of the Il7r Promoter by Potential Enhancer Sequences
To test if one or more of the DNase I hypersensitive sites identified above can function as an enhancer in transient transfection assays, we used PCR to clone the core region of the upstream (−3.8 kb) enhancer (30) as well as a 1-kb segment encompassing the +1.5-kb DNase I hypersensitive site. These DNA segments were ligated into the downstream SalI site of a pGL3 vector containing the Il7r −925 promoter. As a positive control for enhancer activity we used the full-length (~ 1 kb) Igh intronic enhancer (Eμ) (38). Transient transfection analysis revealed that the Eμ enhancer strongly activated transcription from the Il7r −925 promoter, increasing activity by 22-fold in 38B9 B cells, 5-fold in EL4 T cells, and ~2-fold in RAW264.7 macrophage cells (Fig. 7). The +1.5-kb DNA segment increased activity of the promoter 6.6-fold in RAW264 macrophages, but had no significant effect on the promoter in 38B9 cells or EL4 T cells. The −3.8-kb DNA segment increased activity of the promoter by a small but significant margin in T cells, had no effect in 38B9 pro-B cells, and significantly repressed activity in RAW264 macrophages (Fig. 7). Finally, we tested the activity of the upstream and downstream enhancers together in the upstream and downstream positions, respectively. In this configuration, the enhancers significantly increased activity of the Il7r promoter in EL4 T cells and RAW264 macrophages, but had no effect in 38B9 pro-B cells. These data suggest that the downstream DNA segment functions as an enhancer in macrophages, but not in T cells or pro-B cells. The upstream enhancer appears to function in T cells but not in macrophages or pro-B cells.
FIGURE 7. Function of cloned DNase I hypersensitive sites as enhancers.
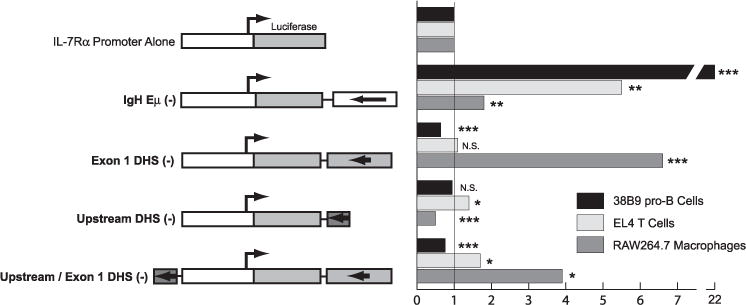
The full-length IgH intronic enhancer (Eμ), a ~300-bp DNA segment corresponding to the −3.5-kb DNase I hypersensitive site, or a ~ 1-kb DNA segment corresponding to the +1.5-kb DNase I hypersensitive site were ligated into the downstream (SalI) position in a pGL3 luciferase reporter vector containing the −925-bp Il7r promoter. Constructs were tested for activity by transient transfection of 38B9 pro-B cells (dark bars), EL4 T cells (light bars), or RAW264.7 macrophages (gray bars). Results are presented as -fold activation over vector containing the Il7r −925-bp promoter alone. N.S., not significant.
DISCUSSION
The goal of this study was to elucidate the function of the Il7r promoter region in developing B cells. First, we characterized the Il7r promoter region in detail, using a combination of 5′ RACE analysis, transient transfection analysis using cloned promoter constructs, and DNase I hypersensitivity analysis. 5′ RACE analysis identified a number of transcription start sites immediately upstream of exon 1. These start sites correspond to a DNase I hypersensitive site found at the same location. Using transient transfection assays, we determined that maximal promoter activity is contained in a 925-bp DNA segment upstream of the transcription start sites. This 925-bp promoter region is poorly conserved between species, with the exception of an Ets consensus binding site that was previously identified (16). Using transient transfection experiments, we showed that the intact Ets binding site is required for activity of the Il7r promoter. Activity of the promoter also requires initiator sequences downstream from the Ets binding site. Next, we showed using EMSA that the Ets family transcription factors GABP and PU.1 can both interact with the conserved binding site. RNA interference experiments demonstrated that GABP functions to activate Il7r transcription in pro-B cells. Finally, we performed retroviral gene transfer experiments to test if GABP can substitute for PU.1 in activating Il7r transcription in lymphoid progenitors. These experiments revealed that GABP cannot substitute for PU.1, suggesting that these factors have distinct functions during early lymphoid development, as is discussed below.
Little is yet known about GABP function in lymphocytes, because null mutation of the Gabpα gene results in pre-implantation embryonic lethality (39). The function of GABP within the immune system is best understood in myeloid cells (35). Recently, GABP was shown to interact with the conserved Ets binding site of the Il7r promoter region in T lymphocytes (23). This paper suggested that B cell versus T cell-specific regulation of the Il7r gene might be at least partially explained by differential recruitment of PU.1 versus GABP to the same site in the promoter. However, our results show that both PU.1 and GABP are expressed in developing B cells and can interact with the Il7r Ets site. Furthermore, in the absence of PU.1 (Sfpi1−/− pro-B cells) GABP is sufficient to maintain high-level transcription of the Il7r gene. These results suggest an explanation for why Il7r IL-7Rα levels are only moderately reduced when the Sfpi1 gene is deleted in an inducible manner in committed B lineage cells (18–20). We speculate that GABP might be capable of substituting for PU.1 for other target genes, as well as play additional important functions in the B cell lineage.
If both PU.1 and GABP function as activators at the Il7r promoter Ets site, then why is retroviral transduction of Sfpi1−/− fetal liver progenitor cells with GABP subunits insufficient to rescue IL-7-dependent proliferation? We note that forced expression of IL-7Rα cDNA is sufficient to rescue IL-7-dependent proliferation in this context, suggesting that activating Il7r transcription should be sufficient for rescue (16). There are several possible explanations. First, PU.1 and GABP might have different abilities to activate the Il7r gene from a closed chromatin structure. Second, PU.1 and GABP might have different abilities to activate downstream regulators that cooperate in activating transcription of the Il7r gene. One possible example of such a downstream regulator might be the EBF transcription factor. As we showed previously (17) and in the experiment shown in Fig. 5, ectopic expression of EBF is by itself sufficient to activate IL-7Rα transcription in Sfpi1−/− fetal liver progenitor cells. Interestingly, there are functional PU.1 binding sites in the Ebf1 gene, suggesting that PU.1 might directly activate EBF transcription during early lymphoid development (17). Therefore, we speculate that during early lymphoid development, PU.1, but not GABP, is capable of inducing expression of the Ebf1 gene. EBF would then work together with either PU.1 or GABP to activate Il7r transcription. EBF might be sufficient to activate Il7r transcription in Sfpi1 mutant progenitor cells because GABP is expressed in these cells (data not shown). Further work will be necessary to determine whether EBF acts on the Il7r gene directly or indirectly.
An important future direction of this work will be to identify regulatory regions that cooperate with the promoter to activate transcription in cell-type and developmental stage-specific manner. DNase I hypersensitivity analysis demonstrated that in pro-B cells and T cells the Il7r promoter region contains two groups of hypersensitive sites, located at −3.8 to −5.5 and at +1.5 to +2.6 kb within the first intron. Interestingly, several of these sites are also DNase hypersensitive in the macrophage cell line RAW264.7. Although expression of the Il7r gene is considered to be lymphoid-specific, and indeed is the hallmark of the “common lymphoid progenitor” (6), there are published studies demonstrating that macrophages can express the IL-7Rα and respond to IL-7 (40, 41). PU.1 is highly expressed in both B cells and macrophages, and indeed we previously found by chromatin immunoprecipitation analysis that PU.1 interacts with the Il7r promoter Ets site in both pro-B cells and in a macrophage cell line (16). Using transient transfection analysis, we found that the DNase I hypersensitive site(s) at +1.5 kb functions to activate the Il7r promoter in macrophages, but not in B or T cells. Conversely, the upstream DNase I hypersensitive site at −3.8-kb functions as an enhancer in T cells but not in B cells or macrophages. Interestingly, a DNase I hypersensitive site at + 0.5 kb was uniquely observed in 3T3 cells, RAW264 macrophages, and splenic B cells, which do not express IL-7Rα, or express it at low levels. Therefore it is possible that this DNase I hypersensitive site represents a negative regulatory element for the Il7r gene. No DNase I hypersensitive sites were identified in this study that functioned as an enhancer in pro-B cells. More work will be necessary to identify such enhancers.
In summary, our results suggest that PU.1 and GABP are key transcription factors responsible for regulating the Il7r promoter in developing B cells. We suggest that PU.1 is important for activating Il7r transcription at the earliest stages of B cell development, whereas GABP is important at later stages of B cell development. These experiments provide insight into the important issue of how the Il7r gene is dynamically regulated in the immune system.
Acknowledgments
We thank Sara Wojciechowski and the staff of the flow cytometry core facility at Cincinnati Children’s Hospital Research Foundation for expert assistance in cell sorting; Warren Leonard (National Institutes of Health) for anti-GABPα and GABPβ1 antisera; Hai-Hui Xue (University of Iowa) for performing RNA interference experiments and TaqMan real time RT-PCR analysis; Alan Rosmarin (Brown University) for GABPα cDNA; Harinder Singh (University of Chicago) for helpful discussions; and Iain Cartwright and Isaac Houston (University of Cincinnati College of Medicine) for critically reading the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants AI052175 (to R.P.D.) and AI057753 (to D.A.H.).
The abbreviations used are: IL, interleukin; RACE, rapid amplification of cDNA ends; EMSA, electrophoretic mobility shift assay; GABP, GA-binding protein; EYFP, enhanced yellow fluorescent protein; GFP, green fluorescent protein; RT, reverse transcriptase.
References
- 1.Ma A, Koka R, Burkett P. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 2.Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, Mosley B, March CJ, Urdal D, Gillis S. Nature. 1988;333:571–573. doi: 10.1038/333571a0. [DOI] [PubMed] [Google Scholar]
- 3.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dias S, Silva H, Jr, Cumano A, Vieira P. J Exp Med. 2005;201:971–979. doi: 10.1084/jem.20042393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo M, Ohashi Y, Tada K, Nakamura M, Sugamura K. Eur J Immunol. 1994;24:2026–2030. doi: 10.1002/eji.1830240914. [DOI] [PubMed] [Google Scholar]
- 6.Kondo M, Weissman IL, Akashi K. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 7.Sudo T, Nishikawa S, Ohno N, Akiyama N, Tamakoshi M, Yoshida H. Proc Natl Acad Sci U S A. 1993;90:9125–9129. doi: 10.1073/pnas.90.19.9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rolink A, Haasner D, Nishikawa S, Melchers F. Blood. 1993;81:2290–2300. [PubMed] [Google Scholar]
- 9.Van De Wiele CJ, Marino JH, Murray BW, Vo SS, Whetsell ME, Teague TK. J Immunol. 2004;172:4235–4244. doi: 10.4049/jimmunol.172.7.4235. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin RG, Friend D, Ziegler SF, Jerzy R, Falk BA, Gimpel S, Cosman D, Dower SK, March CJ, Namen AE, Park LS. Cell. 1990;60:941–951. doi: 10.1016/0092-8674(90)90342-c. [DOI] [PubMed] [Google Scholar]
- 11.Maki K, Sunaga S, Komagata Y, Kodaira Y, Mabuchi A, Karasuyama H, Yokomuro K, Miyazaki JI, Ikuta K. Proc Natl Acad Sci U S A. 1996;93:7172–7177. doi: 10.1073/pnas.93.14.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET, Paul WE, Katz SI, Love PE, Leonard WJ. Immunity. 1995;2:223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 14.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Proc Natl Acad Sci U S A. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giliani S, Mori L, de Saint Basile G, Le Deist F, Rodriguez-Perez C, Forino C, Mazzolari E, Dupuis S, Elhasid R, Kessel A, Galambrun C, Gil J, Fischer A, Etzioni A, Notarangelo LD. Immunol Rev. 2005;203:110–126. doi: 10.1111/j.0105-2896.2005.00234.x. [DOI] [PubMed] [Google Scholar]
- 16.DeKoter RP, Lee HJ, Singh H. Immunity. 2002;16:297–309. doi: 10.1016/s1074-7613(02)00269-8. [DOI] [PubMed] [Google Scholar]
- 17.Medina KL, Pongubala JM, Reddy KL, Lancki DW, Dekoter R, Kieslinger M, Grosschedl R, Singh H. Dev Cell. 2004;7:607–617. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Polli M, Dakic A, Light A, Wu L, Tarlinton DM, Nutt SL. Blood. 2005;106:2083–2090. doi: 10.1182/blood-2005-01-0283. [DOI] [PubMed] [Google Scholar]
- 19.Ye M, Ermakova O, Graf T. J Exp Med. 2005;202:1411–1422. doi: 10.1084/jem.20051089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, Arinobu Y, Geary K, Zhang P, Dayaram T, Fenyus ML, Elf S, Chan S, Kastner P, Huettner CS, Murray R, Tenen DG, Akashi K. Blood. 2005;106:1590–1600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alt F, Rosenberg N, Lewis S, Thomas E, Baltimore D. Cell. 1981;27:381–390. doi: 10.1016/0092-8674(81)90421-9. [DOI] [PubMed] [Google Scholar]
- 22.Schweitzer BL, Huang KJ, Kamath MB, Emelyanov AV, Birshtein BK, DeKoter RP. J Immunol. 2006;177:2195–2207. doi: 10.4049/jimmunol.177.4.2195. [DOI] [PubMed] [Google Scholar]
- 23.Xue HH, Bollenbacher J, Rovella V, Tripuraneni R, Du YB, Liu CY, Williams A, McCoy JP, Leonard WJ. Nat Immunol. 2004;5:1036–1044. doi: 10.1038/ni1117. [DOI] [PubMed] [Google Scholar]
- 24.Lefevre P, Lacroix C, Tagoh H, Hoogenkamp M, Melnik S, Ingram R, Bonifer C. J Biol Chem. 2005;280:27552–27560. doi: 10.1074/jbc.M502422200. [DOI] [PubMed] [Google Scholar]
- 25.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 26.Morita S, Kojima T, Kitamura T. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 27.DeKoter RP, Singh H. Science. 2000;288:1439–1441. doi: 10.1126/science.288.5470.1439. [DOI] [PubMed] [Google Scholar]
- 28.Pleiman CM, Gimpel SD, Park LS, Harada H, Taniguchi T, Ziegler SF. Mol Cell Biol. 1991;11:3052–3059. doi: 10.1128/mcb.11.6.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hildeman D, Yanez D, Pederson K, Havighurst T, Muller D. J Virol. 1997;71:9672–9678. doi: 10.1128/jvi.71.12.9672-9678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HC, Shibata H, Ogawa S, Maki K, Ikuta K. J Immunol. 2005;174:7800–7806. doi: 10.4049/jimmunol.174.12.7800. [DOI] [PubMed] [Google Scholar]
- 31.Smale ST, Kadonaga JT. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 32.Garraway IP, Semple K, Smale ST. Proc Natl Acad Sci U S A. 1996;93:4336–4341. doi: 10.1073/pnas.93.9.4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu M, Yang XY, Schmidt T, Chinenov Y, Wang R, Martin ME. J Biol Chem. 1997;272:29060–29067. doi: 10.1074/jbc.272.46.29060. [DOI] [PubMed] [Google Scholar]
- 34.Javahery R, Khachi A, Lo K, Zenzie-Gregory B, Smale ST. Mol Cell Biol. 1994;14:116–127. doi: 10.1128/mcb.14.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosmarin AG, Resendes KK, Yang Z, McMillan JN, Fleming SL. Blood Cells Mol Dis. 2004;32:143–154. doi: 10.1016/j.bcmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Murre C, McCaw P, Baltimore D. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 37.Chinenov Y, Henzl M, Martin ME. J Biol Chem. 2000;275:7749–7756. doi: 10.1074/jbc.275.11.7749. [DOI] [PubMed] [Google Scholar]
- 38.Picard D, Schaffner W. Nature. 1984;307:80–82. doi: 10.1038/307080a0. [DOI] [PubMed] [Google Scholar]
- 39.Ristevski S, O’Leary DA, Thornell AP, Owen MJ, Kola I, Hertzog PJ. Mol Cell Biol. 2004;24:5844–5849. doi: 10.1128/MCB.24.13.5844-5849.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park LS, Friend DJ, Schmierer AE, Dower SK, Namen AE. J Exp Med. 1990;171:1073–1089. doi: 10.1084/jem.171.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobsen FW, Veiby OP, Jacobsen SE. J Immunol. 1994;153:270–276. [PubMed] [Google Scholar]