

Abstract
Inflammatory conditions are associated with the extracellular release of nucleotides, particularly ATP. In the extracellular compartment, ATP predominantly functions as a signalling molecule through the activation of purinergic P2 receptors. Metabotropic P2Y receptors are G-protein-coupled, whereas ionotropic P2X receptors are ATP-gated ion channels. Here we discuss how signalling events through P2 receptors alter the outcomes of inflammatory or infectious diseases. Recent studies implicate a role for P2X/P2Ysignalling in mounting appropriate inflammatory responses critical for host defence against invading pathogens or tumours. Conversely, P2X/P2Y signalling can promote chronic inflammation during ischaemia and reperfusion injury, inflammatory bowel disease or acute and chronic diseases of the lungs. Although nucleotide signalling has been used clinically in patients before, research indicates an expanding field of opportunities for specifically targeting individual P2 receptors for the treatment of inflammatory or infectious diseases.
Nucleotides—particularly ATP—are well known for their function as a universal energy currency1. Interestingly, ATP has a completely different role in the extracellular compartment, where it functions as a signalling molecule through the activation of nucleotide receptors1. These receptors are referred to as purinergic P2 receptors. In contrast to P1 receptors, which are activated by the ATP metabolite adenosine, P2 receptors are activated by ATP and/or other nucleotides (for example, UTP). On the basis of their signalling properties, P2 receptors can be further subdivided into metabotropic P2Y receptors (P2YRs) that are G-protein-coupled, and ionotropic P2X receptors (P2XRs) that are nucleotide-gated ion channels2. Although P2 receptors were originally described on the basis of their functional role in the central nervous system3,4, more recent studies demonstrate their widespread expression throughout different tissues (Supplementary Table 1) and implicate them in innate or adaptive immune responses2,5.
Cellular ATP release during inflammatory conditions
During certain conditions—for example inflammatory, ischaemic and hypoxic—several cell types release ATP from intracellular storage pools into the extracellular compartment2,5,6. Although ATP release can occur in an uncontrolled fashion (for example, during necrosis), many studies have examined molecular pathways that control extracellular ATP release5. For example, inflammatory cells can release ATP via pannexins or connexin hemichannels2,7. Pannexins—transmembrane protein channels that connect the intracellular with the extracellular space—have been implicated in the release of ATP from apoptotic cells8, and other studies have implicated connexins in extracellular nucleotide release6,7. Connexins were originally described as gap junction proteins consisting of two hemichannels. However, isolated hemichannels (connexons) can function as conduits between the cytoplasm and the extracellular space, thereby controlling ATP release, for example from inflammatory cells7 or vascular endothelia9. Other studies found that the release of uridine nucleotides such as UTP, UDP and UDP-glucose are increased during cystic fibrosis10. Together, these studies indicate that inflammatory disease conditions are associated with the extracellular release of nucleotides.
Molecular structure and signalling cascade of P2YR
P2YR belongs to the G-protein-coupled receptor (GPCR) family and contains an extracellular amino terminus, an intracellular carboxy terminus and seven transmembrane-spanning motifs (Fig. 1). At present, eight distinct mammalian P2YRs have been cloned and characterized (P2Y1/2/4/6/11/12/13/14R). The missing numbers represent either non-mammalian receptors (P2Y3R is the chicken orthologue of human P2Y6R) or other GPCRs that share some sequence homology with P2YRs but cannot be activated by nucleotides (for example, lysophosphatidic acid is a P2Y9R agonist)11. According to their phylogenetic and sequence divergence, two distinct P2YR subgroups have been proposed. The first group includes the P2Y1/2/4/6/11R subtypes, with a sequence homology of 35–52% in amino acid composition and the presence of a Y-Q/K-X-X-R defining motif in the transmembrane α-helix 7, thus affecting ligand-binding characteristics. The second group contains P2Y12/13/14R, with members sharing a sequence homology of 47–48% and the presence of the K-E-X-X-L motif in transmembrane α-helix 7 (ref. 11). There is some evidence suggesting that the two P2YR subgroups also differ in their primary coupling to G proteins: the P2Y1/2/4/6/11R group is coupled to Gq/G11 (leading to calcium release via phospholipase C/inositol-1,4,5-triphosphate activation). By contrast, P2Y12/13/14R bind to Gi/0 proteins, which inhibit adenylate cyclase and modulate flow through ion channels11,12. However, there are several instances in which other signalling pathways have been identified. For example, the discrepancy between structural-group affiliation and functional characteristics is highlighted by P2Y12R. Despite having only 20–25% sequence homology with P2Y1/2/4/6R12, there is considerable functional similarity. Indeed, P2Y12 and P2Y2R can both activate monomeric G-proteins (such as Rac and/or RhoA)13, and are the only P2YR subtypes that exhibit agonist-induced desensitization through GPCR kinases14. These studies indicate that despite some sequence homology among P2YRs, there are marked differences between individual members of the P2YR family regarding their intracellular signalling cascades.
Figure 1. Extracellular nucleotide release and signalling during inflammation.
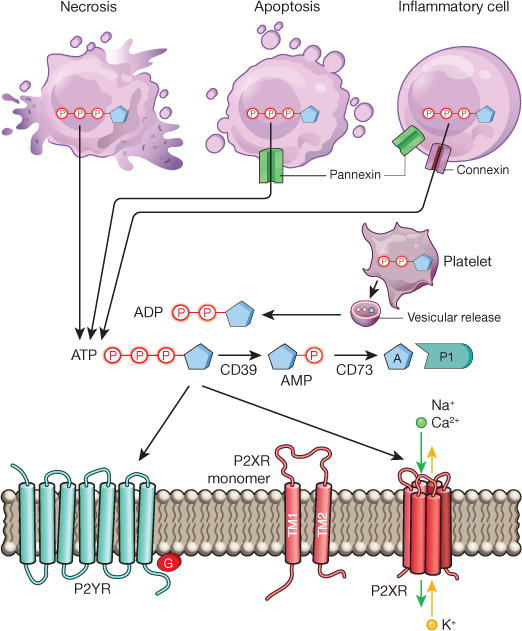
During inflammation, multiple cell types release nucleotides, for example ATP or ADP, from their intracellular compartments into the extracellular space. Nucleotides can be released during mechanical injury, necrosis, apoptosis or inflammatory cell activation. Several molecular pathways have been implicated in this process, such as vesicular ADP release from platelets, pannexin-mediated ATP release during apoptosis, and connexin-or pannexin-mediated ATP release from inflammatory cells, such as neutrophils. Extracellular nucleotides function as signalling molecules through the activation of purinergic P2 receptors. These receptors can be grouped into metabotropic P2Y receptors (P2YRs; GPCRs with seven transmembrane-spanning motifs) or ionotropic P2X receptors (P2XRs), which are nucleotide-gated ion channels. Each P2XR is formed by three subunits (P2XR monomers), each of which consists of two transmembrane regions, TM1 and TM2. Binding of three molecules of ATP to the assembled P2X channel causes opening of a central pore. These conformational changes allow for flux of ions such as sodium (Na+), calcium (Ca2+) and potassium (K+) across the membrane. ATP signalling is terminated by the enzymatic conversion of ATP to adenosine through the ectonucleoside triphosphate diphosphohydrolase CD39 (conversion of ATP/ADP to AMP) and the ecto-5′-nucleotidase CD73 (conversion of AMP to adenosine). Similar to ATP, adenosine (A) functions as an extracellular signalling molecule through the activation of purinergic P1 adenosine receptors.
Endogenous ligands for P2YR
The most abundant and best-characterized endogenous ligand for P2YR is the nucleotide ATP. ATP binds to all P2YRs except P2Y6R and P2Y14R12. Its binding characteristics exemplify the complexity of P2YR signalling: at low concentrations it is the only native agonist for P2Y11R, but at higher concentrations it functions as a partial agonist for P2Y1R and P2Y13R, or as an antagonist for human P2Y4R or P2Y12R11,12,15. Other nucleotides, such as ADP, UTP, UDP or UDP-glucose, exhibit more specificity for individual P2YRs. For example, ADP activates P2Y1R, P2Y12R and P2Y13R, whereas UTP primarily binds to P2Y2R and P2Y4R, and to a lesser extent to P2Y6R, for which UDP is its preferred native ligand. P2Y14R is predominantly activated by UDP-glucose and other UDP-sugars, and to a lesser degree by UDP11,12,15,16. Indeed, the capacity of different nucleotides to bind specifically to individual P2YRs, or to act as either agonists or antagonists, highlights the complexity of the P2Y system and suggests non-redundant signalling pathways.
Pharmacological compounds that act on P2YR
Owing to the fact that P2YRs have crucial roles in regulating immune responses, they became an obvious pharmacological target for the treatment of inflammatory or infectious diseases. Interestingly the parasiticide suramin, which was widely used in the 1920s for the treatment of human onchocerciasis and trypanosomiasis17,18, was later found to be a nonspecific inhibitor of P2YR and P2XR11,15. Although many P2YR-subtype-specific agonists or antagonists have been characterized in in vitro assays or animal studies of inflammatory disorders11,12,15, presently only two types of P2YR-specific compounds are used in patients: antithrombotic P2Y12R antagonists (for example, clopidrogel) and the P2Y2R agonist denufosol, which was examined for the treatment of cystic fibrosis, but eventually failed in clinical trials19. One of the important future challenges for targeting PYR signalling in patients will include the development of highly selective P2YR antagonists, or specific combined P2R antagonists (for example, a P2Y2/P2Y6/P2X7R antagonist), which could be used for the treatment of chronic inflammatory disorders.
Functional roles of P2YR in unchallenged mice
Mice with genetic deletions for human P2YR homologous genes have been generated and characterized, with the exception of P2RY11, which is not expressed in mice12. Despite their widespread expression and their functional involvement in many diseases, mice with global deletions for individual P2YRs display only mild phenotypical alterations when maintained unchallenged in a germ-free environment. For instance, P2ry2−/− mice have slightly lower plasma concentrations of aldosterone, renin and potassium20, whereas global deletion of P2ry4 is associated with lower exercise capacity and reduced myocardial hypertrophy during a swimming exercise21. These findings indicate the likelihood of some redundancy in the signalling system, or compensatory mechanisms following global P2ry gene deletion.
P2YR signalling during inflammatory disease states
Several studies over the past decade have highlighted fundamental roles for P2YRs during inflammatory and infectious diseases. Particularly, signalling events through P2Y2/6/12R have shaped an ambivalent view of their function as either friend or foe during inflammation.
P2Y2R
An early attempt at targeting P2Y signalling for the treatment of inflammatory disorders came from studies of P2Y2R agonists for the treatment of cystic fibrosis22,23. Cystic fibrosis is a life-shortening disease that affects over 30,000 children and adults in the United States24. The airways of patients with cystic fibrosis are susceptible to infection, characterized by neutrophilic inflammation. Although neutrophil proteases are critical for killing engulfed bacteria, neutrophil elastase accumulates in the airways of cystic fibrosis patients, impairing ciliary function, crippling bacterial clearance and degrading structural proteins24. From a molecular perspective, cystic fibrosis is characterized by a defect in the cystic fibrosis transmembrane conductance regulator gene, causing hyperabsorption of sodium leading to thickening of mucus, reduced mucociliary clearance and concomitant increases in susceptibility to bacterial infection22,23. Several studies have indicated that P2Y2R agonists can induce chloride secretion through inhibition of the epithelial sodium channel ENaC, activation of calcium-dependent chloride channels25, stimulation of mucin production, surfactant secretion and ciliary beating (Fig. 2)25. These observations were followed by the development of the P2Y2R agonist denufosol for the treatment of patients suffering from cystic fibrosis26. In 2005 denufosol entered clinical trials, and a 28-day intervention study in a small cohort indicated a potential benefit in lung function of cystic fibrosis patients27. Unfortunately, long-term follow-up (48 weeks) in 466 patients was not associated with improved pulmonary function or reduction of pulmonary exacerbations19. These disappointing findings may be related to an inflammatory role for P2Y2R signalling (see below)2.
Figure 2. P2Y2R signalling during injury resolution and chronic inflammation.
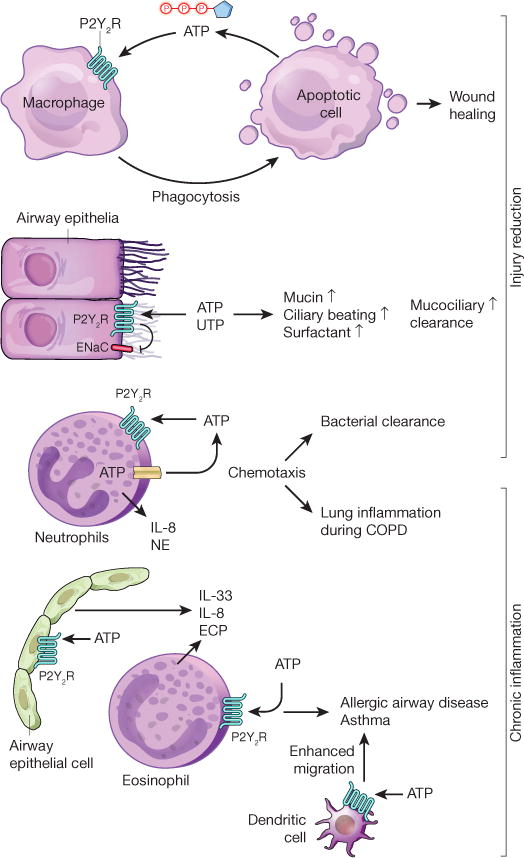
P2Y2R signalling on phagocytes, such as macrophages, contributes to the clearance of apoptotic cells, which release the P2Y2R agonist ATP as a ‘find-me’ signal. P2Y2R-mediated clearance of apoptotic cells and debris contributes to wound healing. Activation of P2Y2R by UTP or ATP promotes mucociliary clearance in the airways via inhibition of the epithelial sodium channel (ENaC), which is associated with concomitant increases in mucin production, surfactant-secretion and ciliary beating. Neutrophil-dependent ATP release and autocrine activation of P2Y2R contributes to purinergic chemotaxis, thereby enhancing bacterial clearance during pneumonia. On the other hand, P2Y2R-mediated release of IL-8 and neutrophil elastase (NE) from neutrophils contributes to the pathogenesis of chronic obstructive lung disease (COPD). ATP-elicited P2Y2R signalling on alveolar epithelial cells or eosinophils causes production of pro-allergic mediators (for example, IL-33, IL-8, eosinophil cationic protein) during allergic airway disease. Similarly, P2Y2R signalling on dendritic cells has a role during the induction and self-perpetuation of asthma.
In addition to a means for enhancing mucociliary clearance, P2Y2R agonists have been implicated in the promotion of wound healing28. In this context, P2Y2R signalling mediates the recruitment of leukocytes to the site of tissue damage as well as differentiation and proliferation of structural cells. Moreover, ATP release and concomitant P2Y2R signalling has been identified as a ‘find-me’ signal for leukocytes, promoting phagocytic clearance of apoptotic cells or bacteria by macrophages29 and neutrophils30,31, thereby contributing to the resolution of inflammation (Fig. 2).
Other studies have indicated that P2Y2R signalling contributes to fundamental leukocyte functions such as migration and mediator production by neutrophils, eosinophils, dendritic cells (DCs) and macrophages32,33. For example, migrating neutrophils can release ATP from their leading edge to amplify chemotactic signals and direct cell orientation by feedback signalling involving P2Y2R (Fig. 2)31. In addition, danger signals such as uric acid, complement factor 5a, Toll-like receptor ligands and interleukin (IL)-8 are stimulated by an autocrine ATP–P2Y2R loop to modulate migration and cytokine production of neutrophils or eosinophils2,31,34–36. Several studies point towards an ambivalent function of P2Y2R in neutrophilic inflammation: although P2ry2−/− mice are less capable of containing bacterial infections30,37, inappropriate activation of P2Y2R is associated with neutrophil-induced hyperinflammation and tissue damage during sepsis, chronic lung disease and hepatitis (Fig. 2)38–41. For example, neutrophils from patients with chronic obstructive pulmonary disease express higher levels of P2Y2R, which is associated with higher elastolytic activity and migration capacity upon ATP stimulation compared to healthy controls40. In the context of P2Y2R agonists for the treatment of cystic fibrosis, these findings could explain why the long-term use of inhaled denufosol in cystic fibrosis patients failed to improve clinical outcomes, as it may have been associated with enhanced neutrophil activation and increased lung inflammation, thus overcoming the beneficial effects of improved mucociliary clearance.
Studies in murine models of asthma or contact hypersensitivity demonstrate a contribution of P2Y2R to the induction and self-perpetuation of allergic diseases. Allergen provocation leads to ATP release and concomitant signalling through P2Y2R, thus favouring the recruitment of immature myeloid DCs and eosinophils to the site of allergen exposures42–44. This is associated with the production of pro-allergic mediators (for example, IL-33, IL-8, eosinophil cationic protein) from different cellular sources (Fig. 2)33,45,46. Similarly, studies in humans indicate that P2Y2R-induced migration and production of reactive oxygen species are enhanced in immature monocyte-derived DCs and eosinophils from allergic donors in the context of concomitant increases in P2Y2R expression43.
Taken together, these findings suggest that ATP-elicited activation of P2Y2R can function as a ‘friend’ in the defence against bacterial infections, promotion of wound healing or in enhancement of mucociliary clearance mechanisms. By contrast, it can also lead to uncontrolled inflammation, attenuated resolution, promotion of chronic inflammatory disease states and fibrotic remodelling47. Indeed, P2Y2R antagonists—as opposed to P2Y2R agonists—could evolve as useful drugs for the treatment for chronic inflammatory diseases, such as chronic obstructive pulmonary disease (Fig. 2)40,41.
P2Y6R
P2Y6R is highly expressed on stromal cells and can be activated by UDP48–50. The ambivalent behaviour (‘friend or foe’) of P2Y2R signalling during inflammatory diseases also applies to P2Y6R signalling. Recent in vivo studies demonstrate a role of P2Y6R in innate immune responses against bacterial infection51. P2Y6R activation triggers chemokine release from monocytes, DCs, eosinophils and endothelial cells, thus promoting recruitment of inflammatory cells towards the site of inflammation or infection11,48,51–53. Similarly, one study demonstrated that injured neurons release UTP and UDP, causing the upregulation of P2Y6R expression on microglia, and concomitant enhancement of their phagocytic capacity for dying cells54. UDP signalling through P2Y6R can therefore function as an ‘eat-me’ signal for microglia, thereby initiating the clearance of dying cells or debris in the central nervous system.
By contrast, P2Y6R signalling is detrimental in models of endothelial50 or epithelial49 inflammation. Mucosal P2Y6R expression is increased during experimentally induced intestinal inflammation such as occurs during inflammatory bowel disease (IBD)49. IBD is a heterogeneous group of disorders characterized by intestinal inflammation, including Crohn’s disease and ulcerative colitis. Here, pharmacological inhibition or genetic deletion of P2ry6 in murine models of intestinal inflammation is associated with improved disease outcomes55. Similarly, a functional role for P2Y6R signalling in promoting detrimental inflammation has been reported for chronic forms of lung disease, such as asthma48. Indeed, its functional role in promoting pathological airway inflammation during chronic lung disease is an important concern regarding potential considerations for the use of P2Y6R agonists for the treatment of cystic fibrosis as a means towards enhancing mucociliary clearance25. The idea that UDP-elicited P2Y6R activation can lead to self-perpetuating chronic inflammatory disorders is further supported by recent in vivo findings suggesting a functional role of P2Y6R in promoting atherosclerotic disease in murine models56. Taken together, these studies indicate that although activation of P2Y6R is important in initiating innate immune responses after infection, inappropriate P2Y6R signalling, predominantly on stromal cells, can drive detrimental immune responses in chronic inflammatory disorders such as atherosclerosis, chronic lung disease or IBD.
P2Y12R
P2Y12R, which is highly expressed on platelets, has fundamental roles in platelet activation and aggregation. Stimulation of P2Y12R inhibits adenylyl cyclase activity and increases phosphatidylinositol-3 kinase activity, resulting in the activation of the fibrinogen receptor (integrin αIIb β3), which is critical for platelet aggregation5. P2Y12R antagonists have been used successfully for antithrombotic therapy in patients5. Because platelets are a key source of inflammatory mediators, P2Y12R signalling has also been implicated in modulating inflammatory responses57. The fact that P2Y12R agonists trigger mediator release from platelets implicates inflammatory alternations in patients taking P2Y12R antagonists. Importantly, P2Y12R antagonists such as clopidogrel or ticagrelor are clinically used in patients as platelet inhibitors. Indeed, reduced levels of circulating inflammatory mediators (for example, tumour-necrosis factor-α, C-reactive protein, P-selectin) were found in patients receiving clopidogrel58. Preclinical studies confirm the proinflammatory role of PY12R signalling in models of vascular inflammation and asthma. For instance P2ry12−/− mice are protected in models of atherosclerosis59,60. Moreover, a surprising crosstalk involving leukotrienes and P2Y12R has been described during asthma. In brief, murine studies demonstrate that P2Y12R signalling on platelets is required for the pro-asthmatic action of leukotriene LTE4 (ref. 61). Furthermore, platelet-independent P2Y12R signalling events contribute to asthma, as P2Y12R antagonists can directly block cysteinyl leukotriene-induced release of eosinophil cationic protein from human eosinophils62, and ADP-elicited P2Y12R activation enhances the capacity of DCs to activate allergen-specific T cells63. The clinical observation that single nucleotide polymorphisms of the P2RY12 gene are associated with altered lung function in a cohort of asthmatic children provides additional evidence for a role of P2Y12R signalling in human asthma64. Taken together, these studies implicate P2Y12R signalling in promoting chronic inflammatory disorders such as asthma and atherosclerosis. However, additional clinical trials addressing the clinical efficacy of P2Y12R antagonists (such as clopidogrel or ticagrelor) for the treatment or prevention of chronic inflammation will be critical in establishing their clinical usefulness beyond current indications.
P2XR signalling during inflammation
Molecular structure and signalling cascade of P2XR
P2XRs are plasma membrane channels selective for monovalent and divalent cations (Na+, K+, Ca2+) which are directly activated by extracellular ATP (Fig. 1)65. Seven different subunits have been identified so far (P2X1–7R)65. The primary sequence of P2XRs has no important sequence homology with other ligand-gated ion channels, ATP-binding proteins or other known proteins66. P2XRs share a common topology with two transmembrane domains (TM1 and TM2), a large extracellular loop responsible for ligand binding, and an intracellular N and a longer C terminus67. The extracellular ‘loop’ starts in proximity of position 52, and ends near proline 329, therefore most of the P2XR protein protrudes from the plasma membrane. Evidence suggests that functional P2XRs are trimers, with three peptide subunits arranged around an ion-permeable channel pore, where ATP binding promotes subunit rearrangement and ion channel opening68. Three molecules of ATP seem to bind to the extracellular portions of P2XR67. Channel opening induces transmembrane ion fluxes, that is, Na+ and Ca2+ influx and K+ efflux, leading to plasma membrane depolarization, and—due to the increase of intracellular Ca2+ levels—activation of Ca2+ signalling cascades, such as p38 MAPK or phospholipase A2 activation67. Interestingly, P2X7R is capable of activating the NOD-like-receptor-mediated inflammasome assembly with pro-caspase 1 proteolytic activation and subsequent pro-IL-1 and pro-IL-18 cleavage and release of their biologically active forms65. The long C-terminal ‘tail’ of P2X7R allows it to undergo a conformational change resulting in the so called ‘permeability transition’; that is, P2X7R changes from a cationic channel to a wider pore, allowing transmembrane fluxes of small hydrophilic molecules (including ATP) with a molecular mass of approximately 900 (refs 69, 70).
In addition, some P2XRs, for example P2X1R, can form heterotrimers with P2X2R, P2X4R and P2X5R subunits, whereas P2X2R forms heterooligomers with P2X3R66. Together these findings indicate that despite the fact that all P2XRs are activated by ATP and share significant sequence homology with each other, highly distinct functions are unique for individual members of the P2XR family.
Endogenous and pharmacological ligands for P2XR
In contrast to P2YR signalling, for which more than one native agonist exists, human P2XRs share ATP as their main endogenous agonist71. Owing to the central role of P2X7R during inflammatory disorders, great efforts have been made to develop selective antagonists. For example, the P2X7R antagonist AZ9056 was used in patients with rheumatoid arthritis. Although initial studies were promising, Phase IIb/III studies with different P2X7R antagonists failed to improve long-term clinical outcomes72. In addition, there are ongoing PhaseII studies with P2X7R and P2X3R antagonists in chronic pain and chronic cough (http://www.clinicaltrials.gov/)72, highlighting the potential for P2XRs as drugable targets in the treatment of inflammatory disorders.
Functional roles of P2XR in unchallenged mice
Physiological roles of P2XRs are indicated by phenotypic manifestations in mice lacking individual P2XR subtypes. Although all single P2rx knockout mice are viable and survive to adulthood, some of them reveal unexpected phenotypes, suggesting that functional roles of individual receptor subunits cannot be compensated for by others. For instance, P2rx1−/− mice show reduced vas deferens contraction and are infertile73, whereas P2rx2−/− mice develop severe progressive hearing loss74, and P2rx3−/− mice experience urinary bladder hyporeflexia75. Mice lacking both P2rx2 and P2rx3 have enlarged spleens and increased numbers of immune cells76. These findings suggest P2XR-subtype-specific signalling functions under physiological conditions, including immune functions.
P2X7R signalling during inflammatory disease
Compelling evidence implicates P2XR during inflammation and immune response against microbes72,78. Although several other P2XRs are functional during inflammation (for example, P2X4R), P2X7R in particular has been shown to affect the outcomes of inflammatory or infectious diseases. This may be due to the fact that P2X7Rs are predominantly expressed on immune cells such as mast cells, macrophages, microglia and DCs (s)2. Indeed, functional studies implicate P2X7R in immune responses against bacterial and parasitic infection. For example, activation of P2X7R is involved in the formation of macrophage multinucleated giant cells, an important step for the control of tuberculosis (Fig. 3)77. Other studies implicate P2X7R signalling in the elimination of intracellular microbes— such as Mycobacterium tuberculosis, Chlamydia psittaci, Leishmania amazonensis or Toxoplasma gondii—by either killing of the pathogen or by inducing cell death of infected macrophages (Fig. 3)78. Human studies indicate that loss-of-function mutations in the P2RX7 gene are associated with increased susceptibility to tuberculosis or toxoplasmosis78. Owing to its proinflammatory role via activation of the inflammasome, and its direct cytotoxic or pro-apoptotic function, many reports implicate a role for the ATP–P2X7R axis in tumour suppression. P2X7R expression is lower in some types of cancer and loss-of-function mutations in the P2RX7 gene have been linked to the pathogenesis of chronic lymphatic leukaemia79. Additional evidence comes from studies demonstrating that loss-of-function mutations of the P2RX7 gene in patients with breast cancer are associated with an increased risk of progression to metastatic disease states80. This study specifically implicated DC–P2X7R signalling in resistance to chemotherapy (Fig. 3). DCs present antigens from dying cancer cells to prime tumour-specific interferon-γ (IFN-γ)-producing T cells. Dying tumour cells release ATP, which activates P2X7R expressed on DCs, which in turn causes inflammasome assembly and subsequent secretion of IL-1β (Fig. 3). Accordingly, anticancer chemotherapy was shown to be inefficient against tumours established in purinergic receptor P2rx7−/− hosts80. Together, such findings implicate ATP signalling through the P2X7R in host-defence mechanisms against intracellular pathogens and cancers.
Figure 3. P2X7R signalling during infection and inflammation.
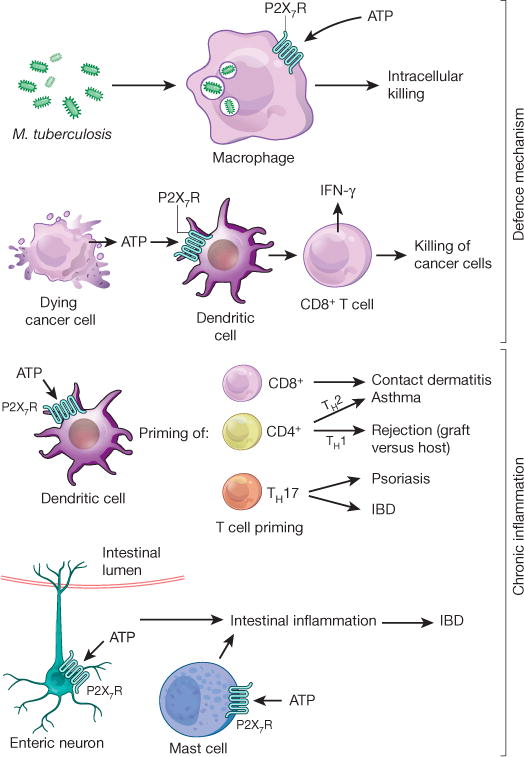
P2X7R is required for mounting an appropriate inflammatory response to defend against invading pathogens, for example during intracellular killing of Mycobacterium tuberculosis by macrophages. Dying tumour cells release ATP, which activates P2X7R expressed on DCs, which in turn promotes the priming of IFN-γ-producing cytotoxic CD8+ T cells that kill cancer cells. On the other hand, P2X7R signalling on DCs and concomitant T-cell priming contributes to allergic disease states, such as CD8+ T-cell-elicited contact dermatitis. DC-mediated T-cell priming under the control of P2X7R signalling has also been shown to promote TH1 responses that are implicated in graft-versus-host disease, which contributes to the rejection of a transplanted organ. Similarly, P2X7R-mediated T-cell priming towards a TH2 response promotes allergic airway disease during asthma. Priming of TH17 cells is critical during psoriasis and contributes to intestinal inflammation as occurs during IBD. P2X7R signalling on enteric neurons or mast cells has been implicated in promoting intestinal inflammation during IBD.
It is particularly interesting that P2X7R signalling on DCs can have very different effects on T-cell priming, depending on the specific context, including CD8+ and CD4+ T-cell differentiation (Fig. 3). In contrast to its essential role in immune priming for response against tumours or pathogens, the involvement of P2X7R in the polarization of antigen-specific effector T cells by DCs contributes to the induction and maintenance of chronic inflammation. P2X7R signalling on DCs is involved in the sensitization phase of allergic disorders such as contact hypersensitivity (CD8+ T-cell priming)44 and asthma (CD4+ T-cells, TH2 response)81, and contributes to transplant rejection (CD4+ T cells, TH1 response; Fig. 3). For example, recent studies implicate P2X7R signalling in graft-versus-host disease, a common complication following an allogeneic tissue transplant, in which immune cells in the tissue recognize the recipient as ‘foreign’, leading to an immunological reaction of transplanted immune cells against the host. Indeed, P2X7R inhibition or deficiency on DCs is associated with reduced severity of graft-versus-host disease82. As such, P2X7R signalling of antigen-presenting DCs led to an increased expression of CD80 and CD86 in vitro and in vivo and activated a cascade of proinflammatory events, including signal transducer and activator of transcription 1 (STAT1) phosphorylation, IFN-γ production and donor T-cell expansion82. Again, other studies report that P2X7R-mediated priming can contribute to TH17-driven autoimmune diseases, such as psoriasis (Fig. 3)83. By triggering the production of pro-allergic mediators from eosinophils, mast cells, macrophages and DCs, P2X7R signalling also contributes to the effector phase and chronification of allergic disorders42,81. Furthermore, increased expression of P2X7R can be found on eosinophils and macrophages in asthma (Fig. 3)81, and loss-of-function mutations in the P2rx7 gene have been associated with attenuated risk of allergen sensitization and asthma84.
In line with these findings, other studies report a detrimental role of P2X7R in promoting excessive inflammation during IBD, by showing that ATP derived from commensal bacteria activates a unique subset of lamina propria cells (CD70high CD11clow cells), leading to TH17 cell differentiation (Fig. 3)85. Indeed, germ-free mice exhibit lower concentrations of luminal ATP, accompanied by fewer lamina propria TH17 cells85. P2X7R also participates in IBD pathogenesis by mediating enteric neural death (Fig. 3)85,86. Finally, mast-cell-dependent mechanisms of intestinal inflammation are under the control of P2X7R, as increased P2X7R expression can be found in mast cells from Crohn’s patients and inhibition of P2X7R on mast cells dampens intestinal inflammation (Fig. 3)87.
Together, these findings expose P2X7R signalling during inflammation as a double-edged sword: these receptors have a critical role in mediating appropriate inflammatory and immunological responses against invading pathogens orcancer cells, respectively, but contribute to chronic inflammatory disease states in a wide range of inflammatory disorders, such as chronic lung disease40,88, asthma81,84 or IBD86,87, when activated inappropriately.
Termination of ATP signalling
Termination of P2R signalling involves the conversion of ATP/ADP to adenosine within the extracellular compartment by the activity of ectonucleotidases. The four main groups of ectonucleotidases are the ectonucleoside triphosphate diphosphohydrolases (NTPDases), ecto-5′-nucleotidase (CD73), ectonucleotide pyrophosphatase/phosphodiesterases and alkaline phosphatases89. NTPDases represent a family of ubiquitously expressed membrane-bound enzymes. The catalytic sites of plasma membrane-expressed NTPDases 1–3 and 8 are oriented towards the extracellular milieu90. Owing to its high expression in many tissues and its ability to catalyse the conversion of ATP (and ADP) down to AMP, many studies have found a functional role for NTPDase1 (CD39) in the termination of P2R signalling90,91. Next, extracellular AMP is converted to adenosine by CD73 (Fig. 1)5. Therefore, termination of ATP signalling is closely linked to the generation of extracellular adenosine. In many instances, adenosine-elicited P1R signalling dampens acute inflammation and tissue injury92,93, thus opposing inflammatory functions of P2Rs (Fig. 1)94,95.
Consistent with a protective role for the CD39/CD73 pathway in terminating inflammatory P2R signalling, and concomitantly increasing extracellular adenosine levels and signalling events, several studies show that Cd39−/− or Cd73−/− mice are prone to tissue injury during inflammatory conditions such as acute lung injury or intestinal inflammation96,97. For example, patients with a single nucleotide polymorphism associated with low levels of CD39 expression have increased susceptibility to Crohn’s disease, suggesting that deficiency in CD39 could be associated with IBD in humans96. Other reports suggest that CD39 exerts a protective thromboregulatory function in stroke by preventing P2R-mediated thrombosis91. Moreover, several studies implicate the CD39/CD73 pathway in the immunosuppressive roles of regulatory T cells (Treg). These are a group of CD4+ lymphocytes that suppress T-cell responses against a variety of pathogens and control inappropriate immune activation, thus limiting collateral tissue damage but allowing pathogen persistence98. As such, Treg cells from Cd39−/− mice demonstrate attenuated suppressive functions in vitro and fail to block rejection of allografts in vivo99. Similarly, Cd73−/− mice fail to resolve lung injury induced by lipopolysaccharide inhalation due to impairment of Treg functions100.
The ambivalence of CD39/CD73-mediated control of Tregs is further exemplified during infections with human immunodeficiency virus (HIV), the retrovirus known to cause AIDS in humans. HIV infections are characterized by a progressive CD4 lymphopenia in conjunction with defective HIV-specific CD8 responses that are critical for the control of viral replication98. As such, the consequences of Treg expansion, as seen during HIV infection, could have either a beneficial function by suppressing generalized T-cell activation, or could be fatal owing to attenuated HIV-specific responses and thus promoting viral persistence98. For example, studies demonstrate that HIV-1-positive patients have an increase of Treg-associated expression of CD39. These findings indicate that the CD39/CD73 pathway is involved in Treg suppression in HIV infection101. A genetic association study demonstrated that a polymorphism in the CD39 gene is associated with attenuated CD39 expression and slower progression to AIDS in HIV-infected patients101. Thus, it can be speculated that CD39+ Tregs are the most potent Treg subset to inhibit HIV-specific T-cell responses. This could at least in part account for their association with disease progression. Other examples for a detrimental role of CD39-dependent ATP breakdown come from studies of autoimmune hepatitis in which natural killer T cell dysfunction in Cd39−/− mice protects against concanavalin A-induced hepatitis. Heightened levels of apoptosis of Cd39−/− natural killer T cells in vivo and in vitro appear to be driven by unimpeded activation of P2X7R102. Similarly, enzymatic removal of ATP by apyrase (conversion of ATP/ADP to AMP) or ectopic CD39 expression attenuates clearance of apoptotic cells, indicating a detrimental role for CD39-dependent ATP phosphohydrolysis in dampening efficient corpse clearance via P2Y2R signalling29. Other studies demonstrate the existence of bacterial ectotriphosphate diphosphohydrolases—similar to human CD39—which are critical for the intracellular multiplication of Legionella pneumophila by preventing P2R-elicited immune responses. As such, these findings implicate bacterial ectotriphosphate diphosphohydrolases in virulence103. Together these studies exemplify an ambivalent role for the termination of P2R signalling via enzymatic phosphohydrolysis. Although this pathway is critical in preventing excessive P2R-dependent inflammation in a sterile environment104, CD39 function can become detrimental for the appropriate clearance of apoptotic debris29, inflammation directed against bacterial infections103 or by generating an immunosuppressive environment, which promotes the development or progression of cancer5.
Functional role of P1 signalling during inflammation
Extracellular AMP generated by phosphohydrolysis of precursor nucleotides (for example, ATP or ADP) has no clearly characterized signalling function (for example, through specific AMP receptors). However, extracellular AMP serves as the metabolic substrate for the extracellular generation of adenosine via CD73 (Fig. 1)5. Once generated within the extracellular compartment, adenosine can function via activation of four distinct P1 receptors: ADORA1, ADORA2A, ADORA2B or ADORA3. Adenosine signalling is terminated via uptake of adenosine from the extracellular towards the intracellular compartment through equilibrative nucleoside transporters and is metabolized to inosine via adenosine deaminase105, or to AMP via adenosine kinase106. Several studies implicate adenosine signalling in dampening excessive inflammation92. For example, Adora2a−/− mice experience increased inflammation including extensive tissue damage, more prolonged and higher levels of proinflammatory cytokines, and mortality when exposed to sub-threshold doses of inflammatory stimuli93. Other studies demonstrate that ADORA2B signalling dampens excessive inflammation during acute lung injury107, promotes ischaemia tolerance and improves anaerobic carbohydrate metabolism108,109. Similarly, genetic deletion or pharmacological blockade of equilibrative nucleoside transporters is associated with increased adenosine levels and improved outcomes during inflammatory disease states110,111. In most instances, the anti-inflammatory signalling effects of adenosine are associated with improved outcomes during inflammatory diseases such as IBD112, or during sepsis induced by caecal ligation and puncture113. However, other studies indicate that the anti-inflammatory effects of adenosine signalling can be detrimental in containing an infection with live bacteria. For example, a recent study demonstrates that antagonism of P1 receptors (for example, ADORA2B) can be useful in enhancing macrophage-mediated bacterial phagocytosis and improving polymicrobial sepsis survival in mice114. Together, these studies highlight that P1 and P2 receptors frequently have opposing effects in biological systems, and that shifting the balance from purinergic P2YR and P2XR signalling towards adenosine-mediated P1 signalling is an important therapeutic concept in efforts to dampen pathological inflammation and promote healing5.
Conclusions
The field of extracellular nucleotide signalling and metabolism is a dynamic area of research with important opportunities for novel treatments for inflammatory or infectious diseases. On the one hand, P2R signalling functions to coordinate appropriate immune responses against invading pathogens or tumours. Indeed, pharmacological approaches that amplify extracellular ATP signalling hold promise as therapies for the treatment of cancer or during uncontrolled infections with live pathogens. Such strategies could include inhibition of ATP breakdown (for example, via nucleotidase inhibitors) or treatment with P2 receptor agonists. Conversely, inadequate P2R signalling has been associated with excessive inflammation, chronification and inappropriate resolution and fibrosis in a wide range of inflammatory diseases. In this context, treatment strategies that block P2R signalling, promote extracellular conversion of ATP to adenosine and activate adenosine receptors have been implicated in the treatment of acute or chronic inflammatory diseases. We therefore anticipate that compounds targeting these pathways will be further exploited in the treatment of inflammatory conditions in human patients in the near future.
Note added in proof
Two reports appeared online regarding the atomic structure of the P2Y12R while the current review was in press. The first report provides a 2.6 Å resolution crystal structure of the human P2Y12R in complex with the non-nucleotide antagonist AZD1283 (ref. 115), thus providing important insights for the development of P2Y12R ligands and allosteric modulators as drug candidates. The second report provides the structures of the human P2Y12R in complex with a full agonist (2-methylthio-adenosine-5′-diphosphate) at a resolution of 2.5 Å, and the corresponding ATP derivative 2-methylthio-adenosine-5′-triphosphate at 3.1 Å resolution116. The agonist-bound P2Y12R structure answers ambiguities surrounding P2Y12R-agonist recognition, and suggests unexpected interactions with several residues.
Supplementary Material
Acknowledgments
We acknowledge S. A. Eltzschig for help with artwork during manuscript preparation. The present research is supported by Deutsche Forschungsgemeinschaft grant ID7/3-1 ID7/4-1 and a grant by the Boehringer-Ingelheim Foundation to I.D., as well as National Institutes of Health grants R01-DK097075, R01-HL0921, R01-DK083385, R01-HL098294, POIHL114457-01 and a grant by the Crohn’s and Colitis Foundation of America to H.K.E.
Footnotes
Author Contributions M.I., D.F. and H.K.E. all contributed to the writing of this paper.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Supplementary Information is available in the online version of the paper.
References
- 1.Khakh BS, Burnstock G. The double life of ATP. Sci Am. 2009;301:84–92. doi: 10.1038/scientificamerican1209-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nature Rev Immunol. 2011;11:201–212. doi: 10.1038/nri2938. Provides a comprehensive overview of the functional roles of purinergic signalling events on cells of the adaptive and innate immune systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fredholm B, Verkhratsky A. Purines — 80 years and very much alive. Acta Physiol. 2010;199:91–92. doi: 10.1111/j.1748-1716.2010.02113.x. [DOI] [PubMed] [Google Scholar]
- 4.Burnstock G. Purinergic signalling and disorders of the central nervous system. Nature Rev Drug Discov. 2008;7:575–590. doi: 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- 5.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eltzschig HK, Macmanus CF, Colgan SP. Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc Med. 2008;18:103–107. doi: 10.1016/j.tcm.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eltzschig HK, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 8.Chekeni FB, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. Identifies a functional role for pannexin-mediated ATP release from cells undergoing apoptosis as a ‘find-me’ signal for phagocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faigle M, Seessle J, Zug S, El Kasmi KC, Eltzschig HK. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE. 2008;3:e2801. doi: 10.1371/journal.pone.0002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8:359–373. doi: 10.1007/s11302-012-9304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abbracchio MP, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG. G protein-coupled adenosine (P1) and P2Y receptors: ligand design and receptor interactions. Purinergic Signal. 2012;8:419–436. doi: 10.1007/s11302-012-9294-7. Provides an update on the medicinal chemistry and pharmacology of the different subtypes of adenosine receptors and P2Y receptors, including recent advances in the identification and characterization of selective ligands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soulet C, et al. Gi-dependent and -independent mechanisms downstream of the P2Y12 ADP-receptor. J Thromb Haemost. 2004;2:135–146. doi: 10.1111/j.1538-7836.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 14.Hardy AR, et al. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood. 2005;105:3552–3560. doi: 10.1182/blood-2004-07-2893. [DOI] [PubMed] [Google Scholar]
- 15.von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110:415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Harden TK, Sesma JI, Fricks IP, Lazarowski ER. Signalling and pharmacological properties of the P2Y receptor. Acta Physiol. 2010;199:149–160. doi: 10.1111/j.1748-1716.2010.02116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawking F. Suramin: with special reference to onchocerciasis. Adv Pharmacol Chemother. 1978;15:289–322. doi: 10.1016/s1054-3589(08)60486-x. [DOI] [PubMed] [Google Scholar]
- 18.Voogd TE, Vansterkenburg EL, Wilting J, Janssen LH. Recent research on the biological activity of suramin. Pharmacol Rev. 1993;45:177–203. [PubMed] [Google Scholar]
- 19.Ratjen F, et al. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J Cyst Fibros. 2012;11:539–549. doi: 10.1016/j.jcf.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Rieg T, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 21.Horckmans M, et al. Gene deletion of P2Y4receptor lowers exercise capacity and reduces myocardial hypertrophy with swimming exercise. Am J Physiol Heart Circ Physiol. 2012;303:H835–H843. doi: 10.1152/ajpheart.00256.2012. [DOI] [PubMed] [Google Scholar]
- 22.Knowles MR, Clarke LL, Boucher RC. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N Engl J Med. 1991;325:533–538. doi: 10.1056/NEJM199108223250802. [DOI] [PubMed] [Google Scholar]
- 23.Parr CE, et al. Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proc Natl Acad Sci USA. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis SD, Ferkol T. Identifying the origins of cystic fibrosis lung disease. N Engl J Med. 2013;368:2026–2028. doi: 10.1056/NEJMe1303487. [DOI] [PubMed] [Google Scholar]
- 25.Burnstock G, Brouns I, Adriaensen D, Timmermans JP. Purinergic signaling in the airways. Pharmacol Rev. 2012;64:834–868. doi: 10.1124/pr.111.005389. [DOI] [PubMed] [Google Scholar]
- 26.Kellerman D, et al. Denufosol: a review of studies with inhaled P2Y2 agonists that led to Phase 3. Pulm Pharmacol Ther. 2008;21:600–607. doi: 10.1016/j.pupt.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Deterding RR, et al. Phase 2 randomized safety and efficacy trial of nebulized denufosol tetrasodium in cystic fibrosis. Am J Respir Crit Care Med. 2007;176:362–369. doi: 10.1164/rccm.200608-1238OC. [DOI] [PubMed] [Google Scholar]
- 28.Gendaszewska-Darmach E, Kucharska M. Nucleotide receptors as targets in the pharmacological enhancement of dermal wound healing. Purinergic Signal. 2011;7:193–206. doi: 10.1007/s11302-011-9233-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, et al. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3:ra45. doi: 10.1126/scisignal.2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. Shows that human neutrophils release ATP from the leading edge of the cell surface to amplify chemotactic signals and direct cell orientation by feedback through P2Y2R as a mechanism of purinergic chemotaxis. [DOI] [PubMed] [Google Scholar]
- 32.Myrtek D, Idzko M. Chemotactic activity of extracellular nucleotideson human immune cells. Purinergic Signal. 2007;3:5–11. doi: 10.1007/s11302-006-9032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrari D, et al. Activation of human eosinophils via P2 receptors: novel findings and future perspectives. J Leukoc Biol. 2006;79:7–15. doi: 10.1189/jlb.0505286. [DOI] [PubMed] [Google Scholar]
- 34.Kronlage M, et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci Signal. 2010;3:ra55. doi: 10.1126/scisignal.2000588. [DOI] [PubMed] [Google Scholar]
- 35.Ben Yebdri F, Kukulski F, Tremblay A, Sevigny J. Concomitant activation of P2Y2 and P2Y6 receptors on monocytes is required for TLR1/2-induced neutrophil migration by regulating IL-8 secretion. Eur J Immunol. 2009;39:2885–2894. doi: 10.1002/eji.200939347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi T, Kouzaki H, Kita H. Human eosinophils recognize endogenous danger signal crystalline uric acid and produce proinflammatory cytokines mediated by autocrine ATP. J Immunol. 2010;184:6350–6358. doi: 10.4049/jimmunol.0902673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geary C, et al. Increased susceptibility of purinergic receptor-deficient mice to lung infection with Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. 2005;289:L890–L895. doi: 10.1152/ajplung.00428.2004. [DOI] [PubMed] [Google Scholar]
- 38.Inoue Y, Chen Y, Hirsh MI, Yip L, Junger WG. A3 and P2Y2 receptors control the recruitment of neutrophils to the lungs in a mouse model of sepsis. Shock. 2008;30:173–177. doi: 10.1097/shk.0b013e318160dad4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayata CK, et al. Purinergic P2Y2 receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology. 2012;143:1620–1629. doi: 10.1053/j.gastro.2012.08.049. [DOI] [PubMed] [Google Scholar]
- 40.Lommatzsch M, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928–934. doi: 10.1164/rccm.200910-1506OC. [DOI] [PubMed] [Google Scholar]
- 41.Cicko S, et al. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185:688–697. doi: 10.4049/jimmunol.0904042. [DOI] [PubMed] [Google Scholar]
- 42.Idzko M, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nature Med. 2007;13:913–919. doi: 10.1038/nm1617. Shows that allergen challenge causes acute accumulation of ATP in the airways of asthmatic subjects or in mice with experimentally induced asthma, and further implicates purinergic signalling as a key mediator in allergen-driven lung inflammation. [DOI] [PubMed] [Google Scholar]
- 43.Muller T, et al. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy. 2010;65:1545–1553. doi: 10.1111/j.1398-9995.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- 44.Weber FC, et al. Lack of the purinergic receptor P2X7results in resistance to contact hypersensitivity. J Exp Med. 2010;207:2609–2619. doi: 10.1084/jem.20092489. Identifies P2X7R as a crucial receptor for extracellular ATP released in skin in response to contact allergens, and triggering of contact hypersensitivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Idzko M, et al. Stimulation of P2 purinergic receptors induces the release of eosinophil cationic protein and interleukin-8 from human eosinophils. Br J Pharmacol. 2003;138:1244–1250. doi: 10.1038/sj.bjp.0705145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunzli BM, et al. Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G223–G230. doi: 10.1152/ajpgi.00259.2006. [DOI] [PubMed] [Google Scholar]
- 48.Vieira RP, et al. Purinergic receptor type 6 contributes to airway inflammation and remodeling in experimental allergic airway inflammation. Am J Respir Crit Care Med. 2011;184:215–223. doi: 10.1164/rccm.201011-1762OC. [DOI] [PubMed] [Google Scholar]
- 49.Grbic DM, Degagne E, Langlois C, Dupuis AA, Gendron FP. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J Immunol. 2008;180:2659–2668. doi: 10.4049/jimmunol.180.4.2659. [DOI] [PubMed] [Google Scholar]
- 50.Riegel AK, et al. Selective induction of endothelial P2Y6 nucleotide receptor promotes vascular inflammation. Blood. 2011;117:2548–2555. doi: 10.1182/blood-2010-10-313957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z, et al. P2Y6 agonist uridine 5′-diphosphate promotes host defense against bacterial infection via monocyte chemoattractant protein-1-mediated monocytes/macrophages recruitment. J Immunol. 2011;186:5376–5387. doi: 10.4049/jimmunol.1002946. [DOI] [PubMed] [Google Scholar]
- 52.Idzko M, et al. Characterization of the biological activities of uridine diphosphate in human dendritic cells: influence on chemotaxis and CXCL8 release. J Cell Physiol. 2004;201:286–293. doi: 10.1002/jcp.20070. [DOI] [PubMed] [Google Scholar]
- 53.Ferrari D, et al. P2 purinergic receptors of human eosinophils: characterization and coupling to oxygen radical production. FEBS Lett. 2000;486:217–224. doi: 10.1016/s0014-5793(00)02306-1. [DOI] [PubMed] [Google Scholar]
- 54.Koizumi S, et al. UDP acting at P2Y6receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. Demonstrates that P2Y6R is upregulated when neurons are damaged, and implicates its signalling function as a sensor for phagocytosis by sensing diffusible UDP signals, a novel pathway mediating microglial phagocytosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grbic DM, et al. P2Y6 receptor contributes to neutrophil recruitment to inflamed intestinal mucosa by increasing CXC chemokine ligand 8 expression in an AP-1-dependent manner in epithelial cells. Inflamm Bowel Dis. 2012;18:1456–1469. doi: 10.1002/ibd.21931. [DOI] [PubMed] [Google Scholar]
- 56.Guns PJ, Hendrickx J, Van Assche T, Fransen P, Bult H. P2Y receptors and atherosclerosis in apolipoprotein E-deficient mice. Br J Pharmacol. 2010;159:326–336. doi: 10.1111/j.1476-5381.2009.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nature Rev Immunol. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 58.Muhlestein JB. Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb Haemost. 2010;103:71–82. doi: 10.1160/TH09-03-0177. [DOI] [PubMed] [Google Scholar]
- 59.Li D, et al. Roles of purinergic receptor P2Y, G protein-coupled 12 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:e81–e89. doi: 10.1161/ATVBAHA.111.239095. [DOI] [PubMed] [Google Scholar]
- 60.Yashiro K, et al. Involvement of platelet activation by P2Y12 receptor in the development of transplant arteriosclerosis in mice. Transplantation. 2009;87:660–667. doi: 10.1097/TP.0b013e318196305a. [DOI] [PubMed] [Google Scholar]
- 61.Paruchuri S, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206:2543–2555. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neves JS, Radke AL, Weller PF. Cysteinyl leukotrienes acting via granule membrane-expressed receptors elicit secretion from within cell-free human eosinophil granules. J Allergy Clin Immunol. 2010;125:477–482. doi: 10.1016/j.jaci.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ben Addi A, Cammarata D, Conley PB, Boeynaems JM, Robaye B. Role of the P2Y12 receptor in the modulation of murine dendritic cell function by ADP. J Immunol. 2010;185:5900–5906. doi: 10.4049/jimmunol.0901799. [DOI] [PubMed] [Google Scholar]
- 64.Bunyavanich S, Boyce JA, Raby BA, Weiss ST. Gene-by-environment effect of house dust mite on purinergic receptor P2Y12 (P2RY12) and lung function in children with asthma. Clin Exp Allergy. 2012;42:229–237. doi: 10.1111/j.1365-2222.2011.03874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- 66.Khakh BS, North RA. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron. 2012;76:51–69. doi: 10.1016/j.neuron.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442:527–532. doi: 10.1038/nature04886. Describes P2X signalling beyond its function in the autonomic nervous system, with particular focus on its key roles in afferent signalling, chronic pain and autocrine loops of endothelial and epithelial cells. [DOI] [PubMed] [Google Scholar]
- 68.Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X4ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. Presents the crystal structure of zebrafish P2X4R in its closed, resting state, including definition of the locations of three non-canonical, intersubunit ATP-binding sites, and evidence suggesting that ATP binding promotes subunit rearrangement and ion channel opening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alberto AV, et al. Is pannexin the pore associated with the P2X7 receptor? Naunyn Schmiedebergs Arch Pharmacol. 2013;386:775–787. doi: 10.1007/s00210-013-0868-x. [DOI] [PubMed] [Google Scholar]
- 70.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 71.Schwarz N, et al. Alternative splicing of the N-terminal cytosolic and transmembrane domains of P2X7 controls gating of the ion channel by ADP-ribosylation. PLoS One. 2012;7:e41269. doi: 10.1371/journal.pone.0041269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.North RA, Jarvis MF. P2X receptors as drug targets. Mol Pharmacol. 2013;83:759–769. doi: 10.1124/mol.112.083758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mulryan K, et al. Reduced vas deferens contraction and male infertility in mice lacking P2X1 receptors. Nature. 2000;403:86–89. doi: 10.1038/47495. [DOI] [PubMed] [Google Scholar]
- 74.Yan D, et al. Mutation of the ATP-gated P2X2 receptor leads to progressive hearing loss and increased susceptibility to noise. Proc Natl Acad Sci USA. 2013;110:2228–2233. doi: 10.1073/pnas.1222285110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cockayne DA, et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- 76.Coutinho-Silva R, Knight GE, Burnstock G. Impairment of the splenic immune system in P2X2/P2X3 knockout mice. Immunobiology. 2005;209:661–668. doi: 10.1016/j.imbio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 77.Falzoni S, et al. The purinergic P2Z receptor of human macrophage cells. Characterization and possible physiological role J Clin Invest. 1995;95:1207–1216. doi: 10.1172/JCI117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coutinho-Silva R, Ojcius DM. Role of extracellular nucleotides in the immune response against intracellular bacteria and protozoan parasites. Microbes Infect. 2012;14:1271–1277. doi: 10.1016/j.micinf.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 80.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nature Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 81.Muller T, et al. A potential role for P2X7R in allergic airway inflammation in mice and humans. Am J Respir Cell Mol Biol. 2011;44:456–464. doi: 10.1165/rcmb.2010-0129OC. [DOI] [PubMed] [Google Scholar]
- 82.Wilhelm K, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nature Med. 2010;16:1434–1438. doi: 10.1038/nm.2242. Reveals the relevance of ATP-induced activation of P2X7R for graft-versus-host disease development, indicating that the physiological metabolite ATP must be recognized as an endogenous danger signal that has detrimental effects when released into the extracellular space after tissue damage through the activation of recipient antigen-presenting cells. [DOI] [PubMed] [Google Scholar]
- 83.Killeen ME, Ferris L, Kupetsky EA, Falo L, Jr, Mathers AR. Signaling through purinergic receptors for ATP induces human cutaneous innate and adaptive Th17 responses: implications in the pathogenesis of psoriasis. J Immunol. 2013;190:4324–4336. doi: 10.4049/jimmunol.1202045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manthei DM, et al. Protection from asthma in a high-risk birth cohort by attenuated P2X7 function. J Allergy Clin Immunol. 2012;130:496–502. doi: 10.1016/j.jaci.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Atarashi K, et al. ATP drives lamina propria TH17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 86.Gulbransen BD, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nature Med. 2012;18:600–604. doi: 10.1038/nm.2679. Provides evidence that activation of neuronal P2X7R through pannexin 1 underlies neuron death and the subsequent development of abnormal gut motility in IBD, suggesting that this pathway could be targeted to ameliorate the progression of gut dysmotility during intestinal inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurashima Y, et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nature Commun. 2012;3:1034. doi: 10.1038/ncomms2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lucattelli M, et al. P2X7 receptor signalling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol. 2010;44:423–429. doi: 10.1165/rcmb.2010-0038OC. [DOI] [PubMed] [Google Scholar]
- 89.Zimmermann H, Zebisch M, Strater N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012;8:437–502. doi: 10.1007/s11302-012-9309-4. Summarizes what is known about the cellular and molecular functions of ATP-hydrolysing ectonucleotidases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kohler D, et al. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–1794. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 91.Pinsky DJ, et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Flogel U, et al. Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci Transl Med. 2012;4:146ra108. doi: 10.1126/scitranslmed.3003717. Here the authors developed a selective ADORA2A agonist that requires the presence of CD73 to become activated. This compound suppresses joint inflammation in experimental rheumatoid arthritis, while avoiding ADORA2A-mediated vasodilation. [DOI] [PubMed] [Google Scholar]
- 93.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. First study to provide genetic evidence for a non-redundant role for the P1 adenosine receptor ADORA2A in the physiological negative feedback mechanism for limitation and termination of both tissue-specific and systemic inflammatory responses. [DOI] [PubMed] [Google Scholar]
- 94.Sitkovsky MV, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 95.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–175. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Friedman DJ, et al. CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. 2009;106:16788–16793. doi: 10.1073/pnas.0902869106. Provides evidence that CD39 deficiency exacerbates murine colitis and suggests that CD39 polymorphisms are associated with IBD in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eckle T, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 98.Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood. 2013;121:29–37. doi: 10.1182/blood-2012-07-409755. [DOI] [PubMed] [Google Scholar]
- 99.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. Identifies CD39 and CD73 as surface markers of Tregsand implicates extracellular adenosine generation in an autocrine signalling loop critical for the suppressor functions of Tregs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ehrentraut H, et al. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J. 2013;27:2207–2219. doi: 10.1096/fj.12-225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nikolova M, et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog. 2011;7:e1002110. doi: 10.1371/journal.ppat.1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beldi G, et al. Natural killer T cell dysfunction in CD39-null mice protects against concanavalin A-induced hepatitis. Hepatology. 2008;48:841–852. doi: 10.1002/hep.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sansom FM, et al. A bacterial ecto-triphosphate diphosphohydrolase similar to human CD39 is essential for intracellular multiplication of Legionella pneumophila. Cell Microbiol. 2007;9:1922–1935. doi: 10.1111/j.1462-5822.2007.00924.x. [DOI] [PubMed] [Google Scholar]
- 104.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nature Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eltzschig HK, et al. Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood. 2006;108:1602–1610. doi: 10.1182/blood-2006-02-001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 107.Schingnitz U, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010;184:5271–5279. doi: 10.4049/jimmunol.0903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eltzschig HK, Bonney SK, Eckle T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med. 2013;19:345–354. doi: 10.1016/j.molmed.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eckle T, et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nature Med. 2012;18:774–782. doi: 10.1038/nm.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eckle T, et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 2013;27:3078–3089. doi: 10.1096/fj.13-228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 112.Frick JS, et al. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182:4957–4964. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Csoka B, et al. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol. 2010;185:542–550. doi: 10.4049/jimmunol.0901295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Belikoff BG, et al. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J Immunol. 2011;186:2444–2453. doi: 10.4049/jimmunol.1001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang K, et al. Structure of the human P2Y12receptor in complex with an antithrombotic drug. Nature. 2014 Mar 23; doi: 10.1038/nature13083. http://dx.doi.org/10.1038/nature13083. [DOI] [PMC free article] [PubMed]
- 116.Zhang J, et al. Agonist-bound structure of the human P2Y12receptor. Nature. doi: 10.1038/nature13288. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.