

Abstract
As acute infections resolve, most effector CD8+ T cells die, whereas some persist and become memory T cells. Recent work showed that subsets of effector CD8+ T cells, identified by reciprocal expression of killer cell lectin-like receptor G1 (KLRG1) and CD127, have different lifespans. Similar to previous reports, we found that effector CD8+ T cells reported to have a longer lifespan (i.e., KLRG1lowCD127high) have increased levels of Bcl-2 compared with their shorter-lived KLRG1highCD127low counterparts. Surprisingly, we found that these effector KLRG1lowCD127high CD8+ T cells also had increased levels of Bim compared with KLRG1highCD127low cells. Similar effects were observed in memory cells, in which CD8+ central memory T cells expressed higher levels of Bim and Bcl-2 than did CD8+ effector memory T cells. Using both pharmacologic and genetic approaches, we found that survival of both subsets of effector and memory CD8+ T cells required Bcl-2 to combat the proapoptotic activity of Bim. Interestingly, inhibition or absence of Bcl-2 led to significantly decreased expression of Bim in surviving effector and memory T cells. In addition, manipulation of Bcl-2 levels by IL-7 or IL-15 also affected expression of Bim in effector CD8+ T cells. Finally, we found that Bim levels were significantly increased in effector CD8+ T cells lacking Bax and Bak. Together, these data indicate that cells having the highest levels of Bim are selected against during contraction of the response and that Bcl-2 determines the level of Bim that effector and memory T cells can tolerate.
During acute viral infection, naive CD8+ T cells expand vigorously and generate a population of effector T cells. Shortly after viral clearance, most of the effector T cells die, whereas some remain and become memory T cells. Although initial work suggested that Fas/Fas ligand signaling was critical for the death of activated T cells (1–5), more recent work suggests a dominant role for members of the Bcl-2 family (6). More specifically, the proapoptotic molecule Bim and its ability to signal through the downstream proapoptotic executioners Bax and Bak are essential for driving the contraction of T cell responses in vivo (7–10). Failure to eliminate effector T cells in Bim-deficient animals leads to significantly enhanced T cell memory (10) and protective immunity (11). However, mechanism(s) by which some cells succumb to, whereas others resist, Bim-driven death remain unclear.
The effector CD8+ T cell response to acute viral infection is heterogeneous, consisting of two major subsets, identified largely by the reciprocal cell-surface expression of killer cell lectin-like receptor G1 (KLRG1) and IL-7Rα (CD127) (1, 12, 13). Effector CD8+ T cells with increased expression of CD127, but decreased expression of KLRG1, survived more during contraction of the response, and have been referred to as memory precursor effector cells (12). Conversely, effector CD8+ T cells with decreased expression of CD127, but increased expression of KLRG1, survive less during contraction of the response and have been called short-lived effector cells (12). The longer lifespan of KLRG1low CD127high cells has been attributed to their increased expression of the antiapoptotic molecule Bcl-2, although this has yet to be tested experimentally.
Similar to effector T cells, heterogeneity also exists in the memory compartment, as two major subpopulations of memory T cells have been described (14). CD8+ effector memory T cells (TEM) lack CD62L and CCR7 expression, and they reside in peripheral tissues (e.g., liver, lung, etc.) and the spleen. CD8+ central memory T cells (TCM) expressing lymph node-homing receptors CD62L and CCR7 are mostly found in the secondary lymphoid organs. Although IL-7 and IL-15 critically control survival and homeostatic turnover of overall memory T cells (15, 16), the role of Bcl-2 in the survival of memory T cells is somewhat controversial. For example, Y449 in the cytoplasmic domain of IL-7Rα was shown to be critical for survival of memory T cells but not for IL-7–driven Bcl-2 upregulation (17), suggesting that antiapoptotic signals other than Bcl-2 control memory T cell survival. In contrast, using a gene-knockout approach, we showed that Bcl-2 is critical for IL-7– and IL-15–driven survival in vitro and for overall memory T cell survival in vivo (18, 19). However, the requirement of Bcl-2 in the survival of different subsets of memory CD8+ T cells was not determined in either of the previous studies (17, 18). Thus, the role of Bcl-2 in maintaining subsets of memory T cells and the degree to which Bcl-2 combats Bim in such cells remain unclear.
In naive T cells, most Bim is complexed to Bcl-2 (20), and we recently showed that this interaction is critical to promote naïve T cell survival (18). Following activation, levels of Bcl-2 within activated T cells are decreased, which is correlated with enhanced death of effector T cells (21). Recent work, however, has suggested a potentially more complex interplay between Bim and Bcl-2 (22), as it was shown that genetic ablation of Bim leads to decreased levels of Bcl-2 protein, whereas overexpression of Bcl-2 promotes increased expression of Bim mRNA and protein within activated T cells (21, 22). These data suggested that Bim and Bcl-2 could reciprocally affect the other's expression in T cells, although the mechanism(s) underlying such phenomenon remain unclear.
In this study, we examined the role of Bim and Bcl-2 in both effector and memory subsets of CD8+ T cells using both pharmacologic and genetic approaches. We found that both effector and viral-specific memory CD8+ T cells require Bcl-2 to combat the proapoptotic effects of Bim and, by doing so, determine the levels of Bim CD8+ T cells can tolerate and survive.
Materials and Methods
Mice and viral infection
C57BL/6 mice were either purchased from The Jackson Laboratory or Taconic Farms. Bim−/− mice were a gift from P. Bouillet and A. Strasser (Walter and Eliza Hall Institute, Melbourne, VIC, Australia) and have been backcrossed to C57/BL6 mice for at least 14 generations. Breeding pairs of Bcl-2Tm1sjk mice were obtained from The Jackson Laboratory and were mated to Bim−/− mice to generate Bim+/−Bcl-2−/−, Bim+/−Bcl-2+/−, and Bim−/−Bcl-2−/− mice. Lck Cre−Baxf/fBak−/− mice were a gift from Dr. Stanley Korsmeyer. IL-15–deficient mice on a C57BL/6 background were purchased from Taconic Farms. ABT-737 was a generous gift of Abbott Laboratories, dissolved in DMSO, and diluted in 35% polyethylene glycol, 5% Tween-80, and 65% of dextrose in water solution. Mice were injected i.p. once a day with 75 mg/kg in a volume of 0.2 ml. Mice were infected i.p. with 2 × 105 PFU lymphocytic choriomeningitis virus (LCMV). LCMV was grown in BHK-21 cells, and viral titers from spleen and liver homogenates were determined by plaque assay on BHK-21 monolayers as described. For BrdU incorporation, mice were injected i.p. with three doses of 0.7 mg/mouse BrdU (BD Biosciences, San Jose, CA) 2 d before sacrifice (one on day −2 and two on day −1). BrdU incorporation was assessed with the BrdU Flow kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions. Animals were housed under specific pathogen-free conditions in the Division of Veterinary Services at Cincinnati Children's Hospital Research Foundation. Experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Cincinnati Children's Hospital Research Foundation.
MHC tetramer staining and flow cytometry
Spleens from individual mice were harvested and crushed through a 70-μm mesh cup (BD Falcon) to generate single-cell suspensions. A total of 2 × 106 cells were stained with different combinations of the following cell-surface Abs: anti-CD8, CD44, CD62L, KLRG1, CD127 (from either Bio-legend or eBioscience, San Diego, CA) and intracellularly with either anti– Bcl-2 (clone 3F11, produced in-house) or anti-Bim (Cell Signaling Technology, Beverly, MA) as described. Dbgp33 monomers were produced in collaboration with Dr. Aaron Johnson and were coupled to either APC or PE as previously described (10). A minimum of 5 × 105 events was acquired on a BD LSR II flow cytometer and analyzed by either FACSDiva (BD Biosciences) or FlowJo software (Tree Star).
IL-7 and IL-15 manipulation in vivo
For in vivo IL-7 blockade experiments, M25 was grown as ascites, purified by ammonium sulfate precipitation and ion exchange chromatography, and injected i.p. at a dose of 3 mg/mouse every other day. Effectiveness of IL-7 blockade was assessed by measuring the numbers of pre-B cells in the bone marrow of each mouse via flow cytometry using Abs against IgM, B220, and CD24. For IL-7 and IL-15 delivery experiments, human rIL-7/anti–IL-7 immune complexes were mixed in vitro, and the equivalent of 2.5 μg human rIL-7 (PeproTech, Rocky Hill, NJ) was injected i.p. on days 8, 10, 12, and 14 postinfection (p.i.). For IL-15 delivery experiments, IL-15/IL-15Rα (R&D Systems, Minneapolis, MN) was mixed in vitro, and the equivalent of 750 ng IL-15 was injected i.p. on days 8, 10, 12, and 14 p.i.
Retroviral transduction
C57BL/6 mice were injected with 100 μg Staphylococcus enterotoxin B (Toxin Technology, Sarasota, FL), and 1 d later, T cells were isolated by MACS isolation (Miltenyi Biotec, Auburn, CA). Activated T cells were spin-transduced with empty vector (MIT) or MIT–Bcl-2 retroviruses at room temperature for 2 h. Retroviruses were produced as previously described (21). Transduced T cells were stained with Abs against Thy1.1, Vb8, CD4, CD8, and Bim and analyzed by flow cytometry.
In vitro treatments
Splenocytes from either uninfected or day 10 LCMV-infected mice were cultured with 2 μM cycloheximide (CHX) (Sigma-Aldrich, St. Louis, MO) for 6 h at 37°C. To reduce variability in staining, cells were harvested after culture and stained alongside cells that had not been cultured to assess the decline of Bim protein from time zero.
Quantitative real-time PCR
Spleen cells from LCMV-infected mice on day 20 p.i. were harvested and stained with CD8, CD44, CD62L, KLRG1, and CD127 Abs. CD44+ CD62L−KLRG1highCD127low or CD44+CD62L−KLRG1lowCD127high CD8+ T cells from C57BL/6 or Lck-Cre+Baxf/fBak−/− mice were sorted with FACSAria (BD Biosciences), and RNA was isolated from cells using Qiagen's RNeasy Mini Isolation kit (Qiagen) and converted into cDNA using Superscript II Reverse transcriptase (Invitrogen, CA). Real-time PCR was performed with IQ SyBrGreen Supermix (Bio-Rad, Hercules, CA), and primers for Bim were 5′-ACAAACCCCAAGTCCTCCTT-3′ and 5′-GTTTCGTTGAACTCGTCTCC-3′; and for internal control L19: 5′-CCTGAAGGTCAAAGGGAATGTG-3′ and 5′-GCTTTCGTGCTTCCTTGGTCT-3′. Real-time PCR was performed in IQ Cycler PCR Machine (Bio-Rad).
Results
Levels of Bcl-2 and Bim are correlated in subpopulations of effector CD8+ T cells
Although the increased survival of KLRG1lowCD127high effector CD8+ T cells was attributed to their increased expression of Bcl-2, this was not tested (12). In addition, as Bcl-2 is critical to combat Bim to ensure naive T cell survival (18), and Bim is critical for contraction of T cell responses (10, 21), it is possible that decreased levels of Bim could contribute to enhanced survival of KLRG1lowCD127high effector CD8+ T cells. Using a mouse model of LCMV infection, we examined the levels of Bim and Bcl-2 within LCMV-specific (LCMV-sp) effector CD8+ T cell subsets by intracellular flow cytometry using specific Abs (23). First, we found that the levels of Bim and Bcl-2 were largely correlated in both KLRG1highCD127low and KLRG1lowCD127high subsets of effector Dbgp33-specific (gp33-sp) CD8+ T cells at the peak of the response (Fig. 1A). Next, we found that Bcl-2 levels were significantly higher in KLRG1lowCD127high cells compared with KLRG1highCD127low effector Dbgp33-sp CD8+ T cells 10 d p.i. (Fig. 1B, 1C). Surprisingly, we also found that Bim levels were significantly increased in KLRG1lowCD127high effector CD8+ T cells (Fig. 1B, 1C). Thus, both Bim and Bcl-2 are more highly expressed in effector CD8+ T cells with more potential to develop into long-lived memory cells.
Figure 1.
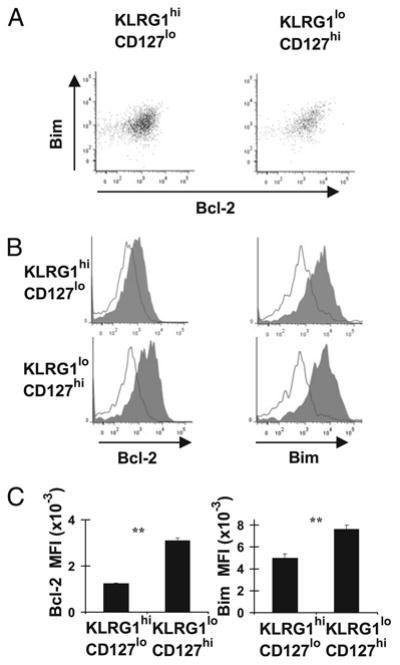
Levels of Bcl-2 and Bim are correlated in subpopulations of effector CD8+ T cells. C57BL/6 mice (n = 4 mice) were infected with 2 × 105 PFU/mouse LCMVArm strain i.p. and sacrificed on either day 8 (A) or 10 (B) p.i. Cells from Bim−/− and Bim+/−Bcl-2−/− mice were included as Ab specificity controls in experiments in B. Single-cell spleen suspensions were stained with Dbgp33 tetramers and Abs against KLRG1, CD127, and CD8 and intracellularly against Bim and Bcl-2 and data acquired on a flow cytometer. A, Representative dot plots show Bim and Bcl-2 levels in subpopulations of CD8+ Dbgp33+ cells gated as KLRG1lowCD127high or KLRG1highCD127low cells. B, Representative histograms of Bim and Bcl-2 in KLRG1lowCD127high or KLRG1highCD127low cells from either C57BL/6 mice (filled histograms) or Bim−/− (open Bim histograms), Bim+/−Bcl-2−/− mice (open Bcl-2 histograms) are shown. C, Bar graphs show the mean fluorescence intensity (MFI) values ± SEM for Bim and Bcl-2 in KLRG1highCD127low or KLRG1lowCD127high cells from C57BL/6 mice on day 10 p.i. Data are pooled from two independent experiments including the one shown in B. p values for statistically significant differences were calculated by Student t test. **p ≥ 0.01.
Bcl-2 is critical to combat Bim for survival of KLRG1low CD127high effector CD8+ T cells
We next used both genetic and pharmacological approaches to determine whether Bcl-2 is critical for the survival of effector CD8+ T cells following LCMV infection. First, we took advantage of Bim+/−Bcl-2−/− mice, as loss of a single allele of Bim has previously been shown to prevent the early lethality and kidney disease of Bcl-2 deficiency (24). We infected groups of C57BL/6 and Bim+/−Bcl-2−/− mice with LCMV and analyzed the effector CD8+ T cell subsets 10 and 23 d later. As expected, ∼75–80% of KLRG1highCD127low and ∼30–40% of KLRG1lowCD127high effector gp33-sp CD8+ T cells were lost between days 10 and 23 p.i. (Fig. 2A). Interestingly, deficiency in Bcl-2 did not exacerbate the loss of KLRG1highCD127low cells, but it significantly enhanced the loss of KLRG1lowCD127high cells (Fig. 2A). This greater loss of effector CD8+ T cells in the absence of Bcl-2 was largely alleviated by the additional loss of the remaining allele of Bim (Fig. 2A). These data show that Bcl-2 is critical to counteract Bim in KLRG1lowCD127high effector CD8+ T cells.
Figure 2.

Bcl-2 is critical to combat Bim for survival of KLRG1low CD127high effector CD8+ T cells. A, Groups of C57BL/6, Bim+/−Bcl-2−/−, and Bim−/−Bcl-2−/− mice (n = 4 mice/group) were infected i.p. with 2 × 105 PFU/mouse LCMV and sacrificed 10 or 23 d p.i. Splenocytes were stained with Dbgp33 tetramers and Abs against KLRG1, CD127, and CD8. CD8+GP33+ KLRG1lowCD127high or CD8+GP33+ KLRG1highCD127low populations were analyzed on days 10 or 23 after LCMV infection by flow cytometry. Bar graphs show the percentage decrease in the numbers of each CD8+ T cell effector subset ± SEM from day 10 to day 23 p.i. B, C57BL/6 mice were infected i.p. with LCMV. Mice were treated either with vehicle or the synthetic Bcl-2 inhibitor ABT-737 (1 mg/mouse/d) (n = 4 mice/group) between days 10 and 23 p.i. Mice were sacrificed 23 d p.i., and splenocytes were stained as in A. Bar graphs show the percentage decrease in the numbers of each CD8+ T cell effector subset ± SEM from days 10–23 p.i. Data are representative of two independent experiments. C, C57BL/6 mice were infected i.p. with LCMV. Mice (n = 9 mice/group) were treated either with vehicle or the synthetic Bcl-2 inhibitor ABT-737 between days 10 and 20 p.i as in B. Mice were sacrificed 110 d p.i., and splenocytes were stained with Dbgp33 tetramers and Abs against CD8, CD62L, and CD44. CD8+ TEM and TCM numbers ± SEM values are shown. Data are pooled from two independent experiments. *p ≤ 0.05.
As the lymphopenic environment and potential developmental abnormalities in T cells from Bim+/−Bcl-2−/− mice could have impacted the above results, we took advantage of a synthetic Bcl-2 antagonist, ABT-737, that inhibits Bcl-2, Bcl-xL, and Bcl-w (25). Our previous data showed that ABT-737 kills naive T cells in C57BL/6 mice in a Bim-dependent manner (18). To determine the role of Bcl-2 in effector T cell survival, we treated C57BL/6 mice with ABT-737 or vehicle between days 10 and 23 after LCMV infection. Pharmacological inhibition of Bcl-2 resulted in a significant reduction in the numbers of gp33-sp KLRG1lowCD127high cells, as 90% of these cells were lost during the contraction phase compared with an ∼45% reduction in vehicle-treated mice (Fig. 2B). In contrast, loss of KLRG1highCD127low cells was slightly exacerbated by Bcl-2 inhibition (Fig. 2B). Thus, similar to our genetic studies, pharmacologic inhibition of Bcl-2 also resulted in a significant loss of effector CD8+ T cells, particularly affecting KLRG1lowCD127high cells.
Bcl-2 is critical for the generation and survival of memory CD8+ T cells
As KLRG1lowCD127high CD8+ effector T cells contribute to the long-term memory cell pool (12, 13), we next determined how inhibition of Bcl-2 during the contraction phase would affect memory development. Again, we treated C57BL/6 mice with ABT-737 between day 10 and 20 after LCMV infection and analyzed gp33-sp CD8+ memory T cells 110 d later. We found that the overall numbers of TCM and TEM were decreased in ABT-737– treated mice compared with those treated with vehicle (Fig. 2C). Thus, these results suggest that generation of normal numbers of memory CD8+ T cells critically depends upon the function of Bcl-2 during the contraction of the response.
Although inhibiting Bcl-2 at the effector stage can have a long-term impact on CD8+ T cell memory, we next determined the roles and expression levels of Bim and Bcl-2 in memory T cells after they were formed. First, we measured Bim and Bcl-2 levels in gp33-sp TEM and TCM CD8+ T cells by flow cytometry. Similar to subsets of effector CD8+ T cells, the levels of Bim and Bcl-2 were directly correlated in both TEM and TCM subsets of CD8+ gp33-sp memory cells (Fig. 3A). Further, levels of both Bim and Bcl-2 were significantly increased in TCM compared with TEM within the CD8+ gp33-sp population (Fig. 3B, 3C). Thus, similar to KLRG1lowCD127high cells, gp33-sp TCM CD8+ T cells exhibited high expression of both Bim and Bcl-2.
Figure 3.
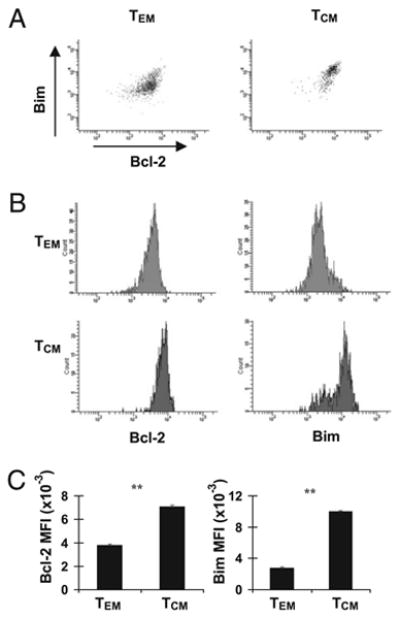
Expression of Bim and Bcl-2 in memory CD8+ T cell subpopulations. C57BL/6 mice were infected i.p. with LCMV. Mice (n = 6 mice/group) were sacrificed 100 d later, and splenocytes were stained with Dbgp33 tetramers and Abs against CD8, CD44, CD62L, and intracellularly against Bim or Bcl-2, and data were acquired on a flow cytometer and analyzed using FACSDiva software after gating on TEM (CD8+CD44high CD62Llow) (TEM) or TCM (CD8+CD44highCD62Lhigh) populations. A, Dot plots for Bim and Bcl-2 in TEM and TCM after gating on CD8+GP33+ cells. Histograms (B) and bar graphs (C) for Bim and Bcl-2 MFI in TEM or TCM CD8+ T cells are shown. Graphs are representative of three independent experiments. **p ≤ 0.01.
Next, we determined the role of Bcl-2 in combating Bim to maintain LCMV-sp memory CD8+ T cells. We infected C57BL/6 and Bim−/− mice with LCMV and, 90 d later, treated them with ABT-737 or vehicle control for 10 d and analyzed gp33-sp memory cell subsets by flow cytometry. In C57BL/6 mice, ABT-737 led to a ∼3-fold loss of gp33-sp CD8+ TCM and a ∼2-fold loss of TEM cells (Fig. 4A). As expected, numbers of TEM and TCM in vehicle-treated Bim-deficient mice were significantly increased compared with vehicle-treated C57BL/6 mice (Fig. 4A). Further, gp33-sp TEM and TCM in Bim-deficient mice were not significantly decreased by ABT-737 (Fig. 4A), suggesting that a Bim/Bcl-2 balance is also critical for the maintenance of LCMV-sp memory cells. As ABT-737 can cause moderate lymphopenia (18) that can in turn cause homeostatic proliferation, we assessed in vivo T cell proliferation using BrdU. gp33-sp CD8+ TCM and, to a lesser extent, TEM in ABT-737–treated C57BL/6 mice underwent significantly greater proliferation in vivo (Fig. 4B). ABT-737 did not increase proliferation of gp33-sp CD8+ T cells in Bim−/− mice (Fig. 4B), as Bim−/− mice are resistant to ABT-737–induced lymphopenia (18). Thus, the actual dependence of viral-specific memory T cells upon Bcl-2 is likely underestimated because of the lymphopenia-induced proliferation following ABT-737 treatment.
Figure 4.

Bcl-2 is critical for survival of memory CD8+ T cells. Groups of C57BL/6 and Bim−/− mice (n = 5 mice/group) were infected i.p. with LCMV, and after 90 d, they were treated with either vehicle or ABT-737 (1 mg/mouse/d) for 10 d and sacrificed. Mice were also injected with BrdU (0.7 mg/mouse i.p.) for 2 d before sacrifice. Spleen cells were stained with Dbgp33 tetramers and Abs against CD8, CD44, and CD62L and analyzed by flow cytometry. A, Bar graphs indicate the total numbers ± SEM values of TEM (CD8+CD44highCD62Llow) or TCM (CD8+ CD44highCD62Lhigh) subsets. B, Splenocytes were stained with the above MHC tetramer and surface stains along with an Ab against BrdU according to the manufacturer's instructions. The percent of TEM and TCM that were BrdU+ ± SEM are shown. C, Splenocytes were also stained with the above MHC tetramer and surface stains along with an Ab against Bim. Bar graphs show Bim MFI ± SEM values in TEM and TCM subsets. Graphs are representative of three independent experiments. D, Groups of C57BL/6, Bim+/−Bcl-2−/−, and Bim+/−Bcl-2+/− mice (n = 4 mice/group) were infected i.p. with LCMV and sacrificed 100 d p.i. Splenocytes were stained with MHC tetramers and surface stains as above and intracellularly for Bim. Bar graphs show Bim MFI ± SEM values in TEM and TCM subsets. *p ≥ 0.05, **p ≤ 0.01.
Bcl-2 determines the level of Bim in effector and memory CD8+ T cells
As Bim and Bcl-2 levels within effector and memory CD8+ T cells are correlated and have been shown to influence expression of each other (21, 22), we next determined the role of Bcl-2 in maintaining Bim expression in memory T cells. To do this, we first treated C57BL/6 mice infected with LCMV 3 mo previously with ABT-737 or vehicle. Interestingly, we found that Bim levels were significantly decreased in both memory CD8+ subsets from ABT-737–treated compared with controls (Fig. 4C). We also assessed the influence of Bcl-2 on Bim levels in gp33-sp CD8+ T cells in groups of LCMV-infected C57BL/6, Bim+/−Bcl-2+/−, and Bim+/− Bcl-2−/− mice. Bim levels in CD8+ T cells from both Bim+/−Bcl-2+/− and Bim+/−Bcl-2−/− mice should be ∼50% of that found in C57BL/6 mice, unless the lack of Bcl-2 has an effect on Bim levels. Similar to the results with ABT-737, we found that the levels of Bim within both gp33-sp TEM and TCM subpopulations of memory cells from Bim+/−Bcl-2−/− mice were significantly decreased compared with cells from either C57BL/6 or Bim+/−Bcl-2+/− mice (Fig. 4D). Thus, using both pharmacologic and genetic approaches, we found that in TCM, and to a lesser extent TEM, Bcl-2 is critical to maintain significant expression of Bim.
As Bim levels were decreased in TEM and TCM in Bim+/−Bcl-2−/− mice and because these mice have a substantial loss of effector T cells, we next inquired whether the low levels of Bim in their memory cells were apparent earlier, during either the peak or after contraction of the response. To do this, we examined the levels of Bim within gp33-sp effector CD8+ T cells on days 10 and 23 after LCMV infection. In C57BL/6 mice, expression of Bim within gp33-sp CD8+ KLRG1highCD127low cells was significantly decreased from days 10–23 p.i. (Fig. 5A). In contrast, Bim levels remained high in the KLRG1lowCD127high subset on days 10 and 23 p.i. (Fig. 5B).
Figure 5.

Bcl-2, IL-7, and IL-15 determine the level of Bim in effector CD8+ T cells. A and B, Groups of C57BL/6, Bim+/−Bcl-2−/−, and Bim−/− Bcl-2−/− mice (n = 4 mice/group) were infected i.p. with LCMV and sacrificed 10 or 23 d p.i. Splenocytes were stained with Dbgp33 tetramers and Abs against KLRG1, CD127, CD8, and intracellularly for Bim. Bar graphs show Bim MFI ± SEM values in KLRG1highCD127low (A) and KLRG1lowCD127high (B) subsets. Significant differences were observed for C57BL/6 mice between days 10 and 23 and between Bim+/−Bcl-2+/− and Bim+/−Bcl-2−/− mice on day 23 (A) and for Bim+/−Bcl-2−/− mice between days 10 and 23 (B). C, Groups of C57BL/6 or IL-15−/− mice (n = 5 mice/group) were infected with LCMV and treated i.p. with either isotype control (MPC11) or anti–IL-7 (M25) Ab (3 mg/mouse) on days 11, 13,15, 17, and 19 and sacrificed on day 20. Splenocytes were stained with Dbgp33 tetramers and Abs against KLRG1, CD8, and Bim. Bar graphs show Bim MFI ± SEM values in KLRG1high and KLRG1low subsets. D, C57BL/6 mice (n = 4 mice/group) were infected with LCMV, treated with PBS, IL-7/anti–IL-7 complexes, or IL-15/IL-15Rα complexes on days 8, 10, 12, and 14 p.i., and sacrificed on day 16 p.i. Splenocytes were stained with Dbgp33 tetramers and Abs against CD127, CD8, and Bim. Bar graphs show Bim MFI ± SEM values in CD127low and CD127high subsets. *p ≥ 0.05, **p ≤ 0.01.
Next, we determined the role of Bcl-2 in influencing the levels of Bim in gp33-sp effector CD8+ T cell subsets. Using LCMV-infected C57BL/6 mice, Bim+/−Bcl-2+/−, and Bim+/−Bcl-2−/− mice, we assessed Bim levels within effector CD8+ subsets. On day 10 p.i., the levels of Bim in both KLRG1highCD127low and KLRG1lowCD127high effector cells in both Bim+/−Bcl-2+/− and Bim+/−Bcl-2−/− mice were roughly half of that observed in C57BL/6 mice (Fig. 5A, 5B). Interestingly, on day 23 p.i., the levels of Bim decreased slightly in KLRG1highCD127low cells from Bim+/−Bcl-2+/− mice, but decreased to a significantly greater extent in cells from Bim+/−Bcl-2−/− mice (Fig. 5A, 5B). Moreover, in KLRG1lowCD127high cells, the levels of Bim were only significantly reduced in mice that completely lacked expression of Bcl-2 (Fig. 5A, 5B). Similar decreases in Bim in effector CD8+ T cells were observed when C57BL/6 mice were treated with ABT-737 during contraction of the response (data not shown). Thus, Bcl-2 is required to maintain normal levels of Bim within effector CD8+ T cells.
IL-7 and IL-15 availability can influence the levels of Bim in effector CD8+ T cells
It is well known that IL-7 and IL-15 (as well as other common γ-chain cytokines) can increase expression of Bcl-2 within effector T cells (15, 26–28). Therefore, we predicted that manipulation of IL-7 and/or IL-15 levels during contraction of the CD8+ T cell response would affect their levels of Bim in a manner that correlates with Bcl-2 expression. First, we examined Bim levels in effector T cells from LCMV-infected C57BL/6 and IL-15–de-ficient mice that were treated with either isotype control Ab or anti–IL-7 neutralizing Ab between days 10 and 20 p.i. We found that, in both KLRG1high and KLRG1low cells, neutralization of IL-7 or loss of IL-15 led to decreased levels of Bim, which was slightly, albeit nonsignificantly, decreased by neutralization of IL-7 in IL-15−/− mice (Fig. 5C). Recently, we showed that IL-7 and IL-15 are critical for maintaining expression of Bcl-2 in effector CD8+ T cells (19). Thus, IL-7 and IL-15 are critical to maintain expression of both Bcl-2 and Bim in effector CD8+ T cells.
Conversely, we reasoned that administration of IL-7 or IL-15, either of which can increase Bcl-2 levels in effector T cells (19, 26, 29), would also increase their expression of Bim. To test this, we infected groups of C57BL/6 mice with LCMV and administered PBS, human IL-7/anti–IL-7, or IL-15/IL-15Rα complexes on days 8, 10, 12, and 14 p.i., sacrificed them on day 16 p.i., and measured Bim levels within effector CD8+ T cells. In contrast to inhibition of IL-7 and IL-15, administration of either IL-7 or IL-15 significantly increased levels of Bim within effector CD8+ T cells (Fig. 5D). Thus, enhancing IL-7 or IL-15 availability increases the levels of both Bim and Bcl-2 in effector CD8+ T cells.
Posttranslational control of Bim by Bcl-2
As Bcl-2 is complexed to Bim in naive T cells (20), it is possible that Bcl-2 influences the levels of Bim through protein–protein interactions that increase the stabilization of Bim (30, 31). We next measured Bim turnover in effector CD8+ T cells by culturing splenocytes from either uninfected or day 10 LCMV-infected C57BL/6 mice with the inhibitor CHX and assessed Bim protein levels over time in culture. We found that 6 h was a reasonable time to assess Bim turnover because: 1) there was a significant loss of Bim at this time point; and 2) there was little cell death occurring during this time (data not shown). After culture with CHX, levels of Bim decreased moderately in naive T cells and substantially in effector T cells (Fig. 6A). Further, the CHX-induced loss of Bim protein was similar between KLRG1high CD127low and KLRG1lowCD127high effector CD8+ T cells (Fig. 6A). We next used Bim+/−Bcl-2−/− mice to test the role of Bcl-2 in stabilization of Bim protein. We found that the lack of Bcl-2 slightly, albeit significantly, exacerbated the CHX-induced loss of Bim in KLRG1lowCD127high effector cells compared with C57BL/6 and Bim+/− controls (Fig. 6B). In contrast, the absence of Bcl-2 did not accelerate the loss of Bim in KLRG1highCD127low effector cells. Thus, Bcl-2 likely contributes somewhat to Bim stabilization, at least in KLRG1lowCD127high effector cells.
Figure 6.

Bcl-2 regulates the amount of Bim effector CD8+ T cells can tolerate. A, C57BL6 mice (n = 3 mice) were infected i.p. with LCMV. Mice were sacrificed 10 d p.i. together with uninfected C57BL/6 mice. Splenocytes were cultured with or without 2 μM CHX for 6 h and then stained with Db gp33 tetramers and Abs against CD8, KLRG1, CD127, and Bim. Bar graphs show the percentage change in Bim MFI levels ± SEM values in CHX-treated cells relative to uncultured cells. B, Groups of C57BL/6, Bim+/−, and Bim+/−Bcl-2−/− mice (n = 3 mice/group) were infected i.p. with LCMV and sacrificed 10 d p.i. Splenocytes were cultured with or without 2 μM CHX for 6 h and then stained as in A. Bar graphs show the percentage change in Bim MFI levels ± SEM values in CHX-treated cells relative to uncultured cells. C–F, Groups of C57BL/6, Lck Cre−Baxf/f Bak−/− and Lck Cre+Baxf/fBak−/− mice (n = 3–6 mice/group) were infected i.p. with LCMV and sacrificed on days 20 and 23 p.i. Data in C, E, and F are pooled from two independent experiments. C, E, and F, Splenocytes were stained with Dbgp33 tetramers and Abs against KLRG1, CD127, and CD8 and intracellularly for Bim (C) and Bcl-2 (E). C, Bar graphs show Bim MFI ± SEM values in KLRG1highCD127low and KLRG1lowCD127high subsets. D, Real-time PCR for Bim mRNA from CD44+CD62L−KLRG1highCD127low or CD44+CD62L−KLRG1lowCD127high cells that were FACS sorted from pooled C57BL/6 mice or three Lck Cre+ Baxf/fBak−/− individual mice on day 23 p.i. Expression levels were normalized to L19 expression. Similar results were obtained in sorted cells on day 20 p.i. E, Bar graphs show Bcl-2 MFI ± SEM values in KLRG1highCD127low and KLRG1lowCD127high subsets. F, Bar graphs show the total numbers of each CD8+ T cell effector subset ± SEM. **p ≥ 0.01.
Lack of cell death allows effector T cells to tolerate higher expression of Bim
Another potential, and not mutually exclusive, explanation is that a threshold level of Bcl-2 is critical to protect effector CD8+ T cells from toxic levels of Bim. A prediction of this model is that inhibition of apoptosis should lead to increased expression of Bim. We tested this prediction using two separate approaches. First, we determined whether overexpression of Bcl-2, which protects activated T cells from death (21), would affect Bim levels in activated T cells. Similar to a recent report (22), retroviral over-expression of Bcl-2 led to significantly increased levels of Bim (Supplemental Fig. 1). However, as Bcl-2 can physically interact and stabilize Bim, we decided to assess Bim levels in mice in which T cells were deficient in Bax and Bak, thereby removing the death signaling apparatus downstream of Bim. Interestingly, we found that the levels of Bim were dramatically higher in both KLRG1highCD127low and KLRG1lowCD127high effector CD8+ T cells in LckCre+Baxf/fBak−/− mice compared with littermate Cre− or C57BL/6 controls (Fig. 6C). Bim mRNA levels were also increased in effector CD8+ T cells from LckCre+ Baxf/fBak−/− mice (Fig. 6D). Importantly, increased levels of Bim were not accompanied by an increase in Bcl-2; in fact, Bcl-2 levels were significantly decreased in effector CD8+ T cells from Cre+ Baxf/f Bak−/− mice compared with C57BL/6 mice (Fig. 6E). Moreover, we found that the total numbers of KLRG1highCD127low and KLRG1lowCD127high effector CD8+ T cells were significantly increased in the absence of Bax and Bak (Fig. 6F). Finally, turnover of Bim was not significantly affected by the loss of Bax and Bak (data not shown). Together, these data strongly suggest that a major role for Bcl-2 is to inhibit cell death and that the level of Bcl-2 within the cell determines the amount of Bim that T cells can tolerate and survive.
Discussion
We and others previously showed that most effector CD8+ T cells have decreased expression of Bcl-2 at the peak of the response (21, 26, 32). Further, our previous data in a superantigen model showed that decreased expression of Bcl-2 correlated with susceptibility to Bim-driven death of effector T cells (21). More recently, it was found that subsets of effector T cells differ in their expression of Bcl-2 (13, 33). In addition, it is well documented that most memory T cells express high levels of Bcl-2 (32). However, in none of these reports was the actual role of Bcl-2 in the survival of effector and memory T cells tested. We recently found that Bcl-2 was critical for the survival of most naive and some memory T cells (18). In this study, we show that Bcl-2 is critical for the survival of particular subsets of effector and memory T cells and, in doing so, allowed these cells to tolerate higher levels of Bim and survive.
A recent report showed that Bim and Bcl-2 can reciprocally affect the expression of each other (22), although the underlying mechanism(s) were unclear. Similar to our results, Marrack's group (22) found that retroviral overexpression of Bcl-2 led to increased levels of Bim mRNA and protein within activated T cells. In this study, we found that Bcl-2 was important to prevent the loss of Bim protein in effector T cells, likely acting at two levels. At one level, Bcl-2 likely stabilizes Bim, a normally unstructured protein (34), and likely protects it from proteasomal degradation. At another level, a consequence of Bcl-2 binding to Bim is the prevention of cell death, which allows cells with higher levels of Bim to survive. Combined, these data suggest a model in which, following T cell activation, Bim levels are transcriptionally increased, but most effector T cells do not maintain sufficient levels of Bcl-2 to protect these T cells from death. The few cells that do maintain Bcl-2 at high levels also appear to have the highest levels of Bim; elimination or inhibition of Bcl-2 in these cells leads to rapid death, and only cells having the lowest expression of Bim survive. Conversely, inhibition of apoptosis downstream of Bim (i.e., in effector T cells that lack Bax and Bak) allowed cells to survive despite having substantially higher Bim mRNA and protein levels. This Bim/Bcl-2 balance in the effector stage is absolutely critical to memory T cell development, as disruptions to this balance affect the generation of normal numbers of memory CD8+ T cells.
Although the loss of Bcl-2 in effector T cells is likely a significant initiating apoptotic event, our data also suggest an activation-induced increase in Bim expression; this has been difficult to observe previously, because the cells die. Indeed, when this death was eliminated by genetic deletion of Bax and Bak, we saw a significant increase in Bim mRNA and protein in both subsets of effector CD8+ T cells. Interestingly, even though death was alleviated, and Bim levels were increased in both effector CD8+ T cell subsets, they were not increased to the same overall level (i.e., Bim levels were still lower in KLRG1highCD127low cells). It is unlikely that different levels of Bcl-2 in the different subsets of CD8+ T cells in Bax/Bak double-deficient mice contribute to this differential expression of Bim, because in both effector subsets, Bcl-2 levels are significantly lower than those observed in C57BL/6 animals. In contrast, the transcriptional profiles of KLRG1highCD127low and KLRG1lowCD127high cells have been reported to be different (12). It is possible that Bim expression is controlled by distinct mechanisms in KLRG1highCD127low and KLRG1lowCD127high cells.
We and others have recently shown that KLRG1highCD127low cells are predominantly maintained by IL-15, whereas IL-7 and IL-15 act redundantly to maintain KLRG1lowCD127high cells (19, 35). We also found that Bcl-2 is a critical antiapoptotic molecule induced/maintained by IL-7 and IL-15 in both of these subsets (19). In this study, we show that Bim levels are also regulated by the availability of IL-7 and IL-15, perhaps because of their ability to maintain Bcl-2. In contrast to previous models in the literature, which suggest that Bim levels are increased in response to cytokine withdrawal (36), we find just the opposite: that Bim levels are positively correlated with cytokine availability. In the cytokine withdrawal models, the forkhead box subclass O transcription factor 3a (Foxo3a) has been shown to be a major transcriptional regulator of Bim (37). The data suggest that Foxo3a is maintained in the cytosol by cytokine signaling through Akt, and upon cytokine withdrawal, Foxo3a translocates to the nucleus and drives expression of Bim (37, 38). However, in primary T cells, the role for Foxo3a in controlling Bim expression is less clear. For example, one report suggested that Bim levels were not affected in primary T cells from Foxo3a-deficient mice (39). Recently, another group reported that levels of phosphorylated (and hence transcriptionally inactive) Foxo were actually higher in KLRG1low CD127high CD8+ T cells relative to KLRG1highCD127low CD8+ T cells when stimulated with IL-15 (40). As KLRG1lowCD127high CD8+ T cells have significantly higher levels of Bim compared with KLRG1highCD127low T cells, these data suggest that Foxos are not a significant regulator of Bim in KLRG1lowCD127high CD8+ T cells. In contrast, another group showed that TCR and cytokine signaling control expression of Bim in human memory CD4+ T cell subsets through an Akt/Foxo3a-dependent mechanism (41). Although this study showed decreased Bim expression in CD4+ TCM, our data clearly show that Bim levels in CD8+ TCM are increased. Whether these differences are due to the differences between humans and mice or between CD4+ and CD8+ T cells remain to be determined. Nonetheless, more work is necessary to further understand the regulation of Bim in effector and memory T cell subsets.
The similarity in Bim and Bcl-2 expression between effector and memory CD8+ T cell subsets is intriguing. At first glance, the simplest explanation for our data would be that CD8+ TEM cells are derived largely from KLRG1highCD127low cells, whereas KLRG1lowCD127high cells give rise to CD8+ TCM. Indeed, the few viral-specific CD62Lhi cells that are present shortly after the contraction of the response are located within the KLRG1lowCD127high cell population (data not shown and Ref. 42). In addition, adoptive transfer studies have suggested that CD8+ TCM are derived from KLRG1lowCD127high cells (12). However, the origin of CD8+ TEM is a little less clear. The ability to track and monitor effector CD8+ T cells based on their level of Bim and/or Bcl-2 we think would be helpful in determining the origins of memory T cell subsets. Unfortunately, as Bim and Bcl-2 are intracellular molecules, their levels cannot be detected while maintaining cell viability without reporter mice.
Our results also suggest that the regulation of effector CD8+ T cell responses is dictated by a dynamic interplay carefully controlled by the levels of pro- and antiapoptotic Bcl-2 family members. Why would this balance be so critical? There are few circumstances in nature whereby a quiescent cell is stimulated to undergo 15–20 rounds of vigorous division and then return to quiescence. During an immune response, expanded naive T cells bear many hallmarks of neoplasia: rapid proliferation, generation of self-renewing cells, and massive epigenetic changes. This balance of a tumor suppressor (Bim) with a proto-oncogene (Bcl-2) is likely part of a highly conserved mechanism to prevent effector T cells from turning into T cell lymphomas. As such, we have found that a highly effective anticancer therapeutic can dramatically curtail effector and memory T cell responses in vivo. Further, we found that the effects of ABT-737 on effector T cell responses are long-lived, impacting the memory compartment long after cessation of the drug. Thus, we envision one potential application of Bcl-2 antagonists could be to target and eliminate autoreactive T cells in autoimmune disease or eliminate alloreactive T cells during organ transplant. In fact, during the course of our studies, it was shown that ABT-737 can protect mice from allograft rejection and prevent diabetes in otherwise susceptible mice (43). Therefore, Bcl-2 family member antagonists offer substantial promise as a way to delete populations of unwanted, disease-causing T cells.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service Grant AI057753 (to D.A.H.).
We thank Drs. Edith Janssen and Claire Chougnet for critical review of the manuscript. We also thank Dr. Aaron Johnson for assistance in generation of Db-gp33 monomers and Dr. Stanley Korsmeyer for the Lck-Cre-Baxfl/fl Bax−/− mice.
Abbreviations used in this article
- CHX
cycloheximide
- Foxo3a
forkhead box subclass O transcription factor 3a
- gp33-sp
gp33-specific
- KLRG1
killer cell lectin-like receptor G1
- LCMV
lymphocytic choriomeningitis virus
- LCMV-sp
lymphocytic choriomeningitis virus-specific
- MFI
mean fluorescence intensity
- p.i
postinfection
- TCM
central memory T cell
- TEM
effector memory CD8+ T cell
Footnotes
Disclosures: The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Gillette-Ferguson I, Sidman CL. A specific intercellular pathway of apoptotic cell death is defective in the mature peripheral T cells of autoimmune lpr and gld mice. Eur J Immunol. 1994;24:1181–1185. doi: 10.1002/eji.1830240526. [DOI] [PubMed] [Google Scholar]
- 2.Mogil RJ, Radvanyi L, Gonzalez-Quintial R, Miller R, Mills G, Theofilopoulos AN, Green DR. Fas (CD95) participates in peripheral T cell deletion and associated apoptosis in vivo. Int Immunol. 1995;7:1451–1458. doi: 10.1093/intimm/7.9.1451. [DOI] [PubMed] [Google Scholar]
- 3.Singer GG, Abbas AK. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 4.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 5.Dhein J, Walczak H, Bäumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 6.Kurtulus S, Tripathi P, Opferman JT, Hildeman DA. Contracting the ‘mus cells’—does down-sizing suit us for diving into the memory pool? Immunol Rev. 2010;236:54–67. doi: 10.1111/j.1600-065X.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci USA. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Dyken SJ, Green RS, Marth JD. Structural and mechanistic features of protein O glycosylation linked to CD8+ T-cell apoptosis. Mol Cell Biol. 2007;27:1096–1111. doi: 10.1128/MCB.01750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol. 2002;3:932–939. doi: 10.1038/ni834. [DOI] [PubMed] [Google Scholar]
- 10.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA. Bim mediates apoptosis of CD127(lo) effector T cells and limits T cell memory. Eur J Immunol. 2006;36:1694–1706. doi: 10.1002/eji.200635897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reckling S, Divanovic S, Karp CL, Wojciechowski S, Belkaid Y, Hildeman D. Proapoptotic Bcl-2 family member Bim promotes persistent infection and limits protective immunity. Infect Immun. 2008;76:1179–1185. doi: 10.1128/IAI.01093-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 15.Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of nïve and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 16.Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, Ahmed R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195:1541–1548. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osborne LC, Dhanji S, Snow JW, Priatel JJ, Ma MC, Miners MJ, Teh HS, Goldsmith MA, Abraham N. Impaired CD8 T cell memory and CD4 T cell primary responses in IL-7R alpha mutant mice. J Exp Med. 2007;204:619–631. doi: 10.1084/jem.20061871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med. 2007;204:1665–1675. doi: 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tripathi P, Kurtulus S, Wojciechowski S, Sholl A, Hoebe K, Morris SC, Finkelman FD, Grimes HL, Hildeman DA. STAT5 is critical to maintain effector CD8+ T cell responses. J Immunol. 2010;185:2116–2124. doi: 10.4049/jimmunol.1000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Y, Swanson BJ, Wang M, Hildeman DA, Schaefer BC, Liu X, Suzuki H, Mihara K, Kappler J, Marrack P. Constitutive association of the proapoptotic protein Bim with Bcl-2-related proteins on mitochondria in T cells. Proc Natl Acad Sci USA. 2004;101:7681–7686. doi: 10.1073/pnas.0402293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 22.Jorgensen TN, McKee A, Wang M, Kushnir E, White J, Refaeli Y, Kappler JW, Marrack P. Bim and Bcl-2 mutually affect the expression of the other in T cells. J Immunol. 2007;179:3417–3424. doi: 10.4049/jimmunol.179.6.3417. [DOI] [PubMed] [Google Scholar]
- 23.Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, Plas DR, Hildeman DA. A major role for Bim in regulatory T cell homeostasis. J Immunol. 2010;186:156–163. doi: 10.4049/jimmunol.1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell. 2001;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 25.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell T, Kappler J, Marrack P. Bystander virus infection prolongs activated T cell survival. J Immunol. 1999;162:4527–4535. [PubMed] [Google Scholar]
- 27.Tripathi P, Mitchell TC, Finkelman F, Hildeman DA. Cutting Edge: Limiting amounts of IL-7 do not control contraction of CD4+ T cell responses. J Immunol. 2007;178:4027–4031. doi: 10.4049/jimmunol.178.7.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yajima T, Yoshihara K, Nakazato K, Kumabe S, Koyasu S, Sad S, Shen H, Kuwano H, Yoshikai Y. IL-15 regulates CD8+ T cell contraction during primary infection. J Immunol. 2006;176:507–515. doi: 10.4049/jimmunol.176.1.507. [DOI] [PubMed] [Google Scholar]
- 29.Nanjappa SG, Walent JH, Morre M, Suresh M. Effects of IL-7 on memory CD8 T cell homeostasis are influenced by the timing of therapy in mice. J Clin Invest. 2008;118:1027–1039. doi: 10.1172/JCI32020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hübner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- 32.Grayson JM, Zajac AJ, Altman JD, Ahmed R. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J Immunol. 2000;164:3950–3954. doi: 10.4049/jimmunol.164.8.3950. [DOI] [PubMed] [Google Scholar]
- 33.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 34.Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, Day CL. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to pro-survival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 35.Rubinstein MP, Lind NA, Purton JF, Filippou P, Best JA, McGhee PA, Surh CD, Goldrath AW. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood. 2008;112:3704–3712. doi: 10.1182/blood-2008-06-160945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 37.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 38.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 39.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, Liu Y, Kaech SM. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riou C, Yassine-Diab B, Van grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 2007;204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obar JJ, Lefrançois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol. 2010;185:263–272. doi: 10.4049/jimmunol.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, Huang DC, Lew AM, Tarlinton DM. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci USA. 2010;107:10967–10971. doi: 10.1073/pnas.1005256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.