

Abstract
Background
Hypodontus macropi is a common intestinal nematode of a range of kangaroos and wallabies (macropodid marsupials). Based on previous multilocus enzyme electrophoresis (MEE) and nuclear ribosomal DNA sequence data sets, H. macropi has been proposed to be complex of species. To test this proposal using independent molecular data, we sequenced the whole mitochondrial (mt) genomes of individuals of H. macropi from three different species of hosts (Macropus robustus robustus, Thylogale billardierii and Macropus [Wallabia] bicolor) as well as that of Macropicola ocydromi (a related nematode), and undertook a comparative analysis of the amino acid sequence datasets derived from these genomes.
Results
The mt genomes sequenced by next-generation (454) technology from H. macropi from the three host species varied from 13,634 bp to 13,699 bp in size. Pairwise comparisons of the amino acid sequences predicted from these three mt genomes revealed differences of 5.8% to 18%. Phylogenetic analysis of the amino acid sequence data sets using Bayesian Inference (BI) showed that H. macropi from the three different host species formed distinct, well-supported clades. In addition, sliding window analysis of the mt genomes defined variable regions for future population genetic studies of H. macropi in different macropodid hosts and geographical regions around Australia.
Conclusions
The present analyses of inferred mt protein sequence datasets clearly supported the hypothesis that H. macropi from M. robustus robustus, M. bicolor and T. billardierii represent distinct species.
Keywords: Hypodontus macropi, Mitochondrial genome, Kangaroo, Wallaby, Sliding window analysis, Next-generation sequencing, Genetics, Systematics
Background
Hypodontus macropi sensu lato (Strongyloidea) is a nematode that occurs in the terminal ileum, caecum or colon of a range of macropodid marsupials (kangaroos and wallabies) and was first described as a hookworm-like nematode [1,2]. Various studies of H. macropi from hosts across vast geographical distances in Australia [3-7] did not reveal any significant morphological variation in the nematode among different host species, but particular predilection sites in the gut as well as host affiliations suggested that H. macropi represented a complex of morphologically indistinguishable, but genetically distinct species or groups which have a relatively high level of host specificity. To test this proposal, Chilton et al. [8] conducted a multilocus enzyme electrophoretic (MEE) analysis and subsequently [9] determined genetic variation in the second internal transcribed spacer (ITS-2) of nuclear ribosomal DNA (rDNA), and provided evidence of genetic differences in H. macropi.
Using single-strand conformation polymorphism (SSCP) and selective sequence analysis of the ITS-2, genetic variation in the nematode was found [10], with nucleotide sequence variability mainly in loops or bulges of the predicted secondary structure. Moreover, in the most extensive investigation of H. macropi to date [11], a molecular-phylogenetic analysis of 547 specimens representing all ten known macropidid host species of this nematode from across the Australian continent revealed clear genetic clades in each of Macropus agilis, M. dorsalis, M. rufogriseus, M. bicolor, Petrogale persephone, Thylogale billardierii and T. stigmatica. Another clade was represented by all specimens from M. robustus robustus and M. rufus, together with two examples of host switching by the nematode into M. fuliginosus.
These findings showed that H. macropi, as currently defined morphologically, might represent as many as ten cryptic species. However, to date, DNA sequencing studies have been conducted exclusively using the second internal transcribed spacer (ITS-2) of nuclear ribosomal DNA, which represents species prospecting (as defined in [12]), rather than clear evidence for the existence of separate species. Although ITS-2 has consistently provided specific identification of strongyloid nematodes [13-15], there is a need to provide independent evidence of genetic identity and differentiation. Various studies have shown that mt genomes provide barcodes for organisms [16-21]. Although nucleotide variation within species of nematodes is relatively high for the mt genes studied to date [18] and, thus, is not useful for specific identification, this is not the case for mt protein sequences. Indeed, amino acid sequence variation within species of nematodes examined to date is usually very low (0–1.3%) [17-19]. Therefore, amino acid sequences derived from the mt genome provide barcodes for species and for studying the systematics (taxonomy and phylogeny) of nematodes [20]. Importantly, phylogenetic analysis of mt amino acid datasets usually provides strong statistical support for the relationships of nematodes, which is not achieved using data from short sequence tracts.
In the present study, we sequenced the complete mt genomes of individuals of H. macropi, proposed to be distinct cryptic species based on MEE and ITS-2 data sets [8-11] from three different species of macropodid hosts. We undertook a comparative analysis of the amino acid sequence datasets derived from these genomes and conducted a phylogenetic analysis of these datasets using homologous datasets for other strongyloid nematodes for comparison.
Results
Characteristics and comparisons of mt genomes
The three mt genome sequences of three operational taxonomic units (OTUs) OTU-C, OTU-G and OTU-J of H. macropi from M. r. robustus, M. bicolor and T. billardierii and of Macropicola (Ma.) ocydromi (outgroup), were assembled from 73,189, 29,997, 20,3341 and 127,146 individual sequence reads, respectively. For individual OTUs, the number of contigs resulting from each assembly ranged from 69 to 508, with the longest and shortest contigs being 11,170 and 41 bp in length.
The three circular mt genome sequences of OTU-C, OTU-G and OTU-J of H. macropi were 13699 bp, 13655 bp and 13634 bp in length, respectively (Figure 1). The mt genome of a closely related nematode, (Ma. ocydromi, was 13517 bp in length (Figure 1). Consistent with all species of related nematodes sequenced to date [18,20,21], the mt genomes of all three H. macropi OTUs and Ma. ocydromi contained 12 protein coding genes [adenosine triphosphatase subunit 6 (atp6), cytochrome b subunit (cytb), cytochrome c subunits 1–3 (cox1-3), and the nicotinamide dehydrogenase subunits 1–6 and 4 l (nad1-6 and 4 l) genes], two ribosomal subunits [large (rrnL) and small (rrnS) subunit] and 22 transfer RNA (tRNA) genes (including two leucine and two serine tRNA genes). The overall mt genome structure for all OTUs of H. macropi and Ma. ocydromi was consistent with the gene arrangement GA2 (arrangement of mt genes) representing all other nematodes of the order Strongylida investigated to date [18,20]. The positions, lengths and start/stop codons of individual genes as well as amino acid sequence lengths of predicted proteins of the three OTUs are shown in Table 1. As expected for other strongyloid nematodes [18], no atp8 subunit is present, and all genes are transcribed from the forward strand; all protein coding genes had open reading frames (ORFs). For all three OTUs, the lengths of the protein-coding genes were in the following order: nad5 > cox1 > nad4 > cytb > nad1 > nad2 > cox3 > cox2 > atp6 > nad6 > nad3 > nad4L (Table 1). The longest gene was nad5, and the amino acid lengths of cox1, nad4L and cox3 were the same for all OTUs (Table 1). The predicted nucleotide and amino acid sequences of each of the 12 protein-coding genes of H. macropi and Ma. ocydromi were compared. The mt genes of these OTUs had ATT, ATA and ATG as initiation codons; except for ATC as its initiation codon for atp6. All genes had complete termination codons, i.e., TAA and TAG, in contrast to some nematodes, such as Ascaris suum, Necator americanus and Trichinella spiralis, which have abbreviated stop codons (e.g., TA or T) for some genes [22-24].
Figure 1.
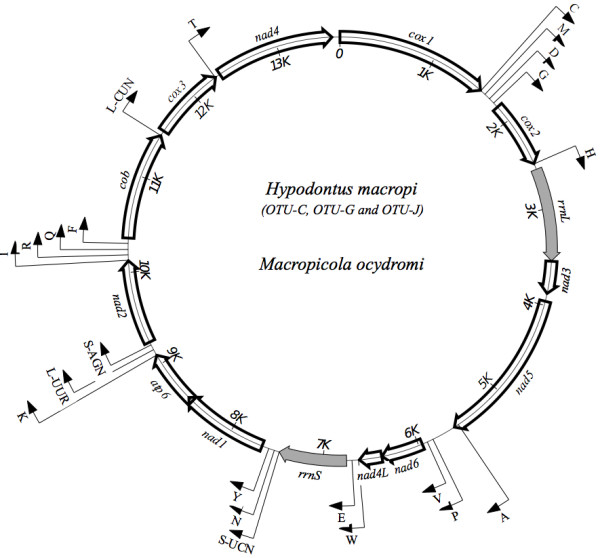
Schematic representation of the circular mitochondrial genome of Hypodontus macropi and Macropicola ocydromi. Three operational taxonomic units (OTUs) of H. macropi were from three different hosts (i.e., OTU-C from Macropus robustus robustus; OTU-G from Macropus bicolor; OTU-J from Thylogale billardierii) and M. ocydromi was from Macropus fuliginosus. Transfer RNA genes are designated using one-letter amino acid codes. Ribosomal genes are shaded.
Table 1.
Summary of mitochondrial genomes of Hypodontus macropi from three different hosts and Macropicola ocydromi
|
Gene |
Positions and nt sequence lengths (bp) |
Initiation/Termination codons and amino acid sequence lengths |
||||||
|---|---|---|---|---|---|---|---|---|
| OTU-C 1 | OTU-G 2 | OTU-J 3 | M. ocydromi | OTU-C | OTU-G | OTU-J | M. ocydromi | |
|
cox1 |
7-1576 |
7-1576 |
7-1576 |
7-1576 |
ATA/TAA (523) |
ATA/TAA (523) |
ATA/TAA (523) |
ATG/TAA (523) |
|
trnC |
1581-1634 |
1583-1639 |
1580-1635 |
1578-1633 |
|
|
|
|
|
trnM |
1636-1695 |
1639-1698 |
1636-1693 |
1633-1692 |
|
|
|
|
|
trnD |
1698-1757 |
1704-1763 |
1694-1752 |
1695-1752 |
|
|
|
|
|
trnG |
1762-1819 |
1765-1822 |
1754-1810 |
1753-1808 |
|
|
|
|
|
cox2 |
1819-2512 |
1822-2515 |
1802-2504 |
1808-2501 |
ATT/TAA (231) |
ATA/TAA (231) |
ATA/TAA (234) |
ATT/TAG (231) |
|
trnH |
2514-2571 |
2518-2573 |
2507-2562 |
2506-2561 |
|
|
|
|
|
rrnL |
2573-3532 |
2578-3537 |
2565-3524 |
2568-3527 |
|
|
|
|
|
nad3 |
3538-3871 |
3542-3875 |
3521-3854 |
3528-3864 |
ATA/TAG (111) |
ATT/TAG (111) |
ATG/TAA (111) |
TTG/TAA (112) |
|
nad5 |
3974-5534 |
3979-5539 |
3961-5521 |
3972-5532 |
ATG/T (536) |
ATG/T (536) |
ATG/T (534) |
ATG/T (536) |
|
trnA |
5535-5591 |
5540-5596 |
5522-5578 |
5533-5589 |
|
|
|
|
|
trnP |
5837-5893 |
5841-5899 |
5828-5882 |
5818-5874 |
|
|
|
|
|
trnV |
5894-5950 |
5905-5961 |
5884-5939 |
5875-5930 |
|
|
|
|
|
nad6 |
5950-6382 |
5961-6393 |
5940-6372 |
5936-6362 |
ATT/TAG (144) |
ATT/TAG (144) |
ATT/TAA (144) |
ATA/TAA (142) |
|
nad4L |
6386-6617 |
6397-6628 |
6378-6609 |
6364-6595 |
ATT/TAG (77) |
ATT/TAA (77) |
ATT/TAG (77) |
ATT/TAG (77) |
|
trnW |
6627-6684 |
6638-6696 |
6613-6669 |
6603-6660 |
|
|
|
|
|
trnE |
6689-6745 |
6705-6761 |
6671-6727 |
6660-6716 |
|
|
|
|
|
rrnS |
6741-7441 |
6760-7460 |
6730-7430 |
6709-7409 |
|
|
|
|
|
trnS (UCN) |
7442-7498 |
7460-7514 |
7418-7482 |
7395-7464 |
|
|
|
|
|
trnN |
7497-7554 |
7513-7570 |
7482-7538 |
7462-7518 |
|
|
|
|
|
trnY |
7563-7617 |
7573-7629 |
7544-7599 |
7522-7576 |
|
|
|
|
|
nad1 |
7690-8527 |
7646-8498 |
7619-8471 |
7593-8445 |
ATG/TAG (279) |
ATA/TAA (284) |
ATA/TAG (284) |
ATG/TAG (284) |
|
atp6 |
8536-9133 |
8501-9098 |
8486-9080 |
8473-9070 |
ATT/TAG (200) |
ATT/TAA (200) |
ATC/TAG (198) |
ATT/TAA (200) |
|
trnK |
9135-9196 |
9101-9163 |
9084-9145 |
9076-9138 |
|
|
|
|
|
trnL (UUR) |
9200-9254 |
9165-9220 |
9148-9202 |
9168-9222 |
|
|
|
|
|
trnS (AGN) |
9255-9308 |
9220-9273 |
9184-9260 |
9223-9275 |
|
|
|
|
|
nad2 |
9320-10151 |
9252-10119 |
9269-10100 |
9288-10119 |
ATA/TAG (277) |
ATA/TAA (289) |
ATA/TAA (277) |
ATA/TAG (277) |
|
trnI |
10156-10214 |
10125-10180 |
10105-10161 |
10124-10180 |
|
|
|
|
|
trnR |
10217-10272 |
10181-10236 |
10162-10216 |
10187-10242 |
|
|
|
|
|
trnQ |
10279-10334 |
10236-10292 |
10219-10273 |
10249-10303 |
|
|
|
|
|
trnF |
10345-10402 |
10298-10356 |
10279-10335 |
10312-10367 |
|
|
|
|
|
cob |
10402-11512 |
10353-11466 |
10333-11446 |
10368-11478 |
ATT/TAA (370) |
ATG/TAG (371) |
ATA/TAA (371) |
ATT/TAA (370) |
|
trnL (CUN) |
11514-11570 |
11468-11523 |
11448-11502 |
11480-11534 |
|
|
|
|
|
cox3 |
11570-12338 |
11523-12291 |
11503-12271 |
11535-12303 |
ATT/TAA (256) |
ATT/TAA (256) |
ATT/TAA (256) |
ATT/TAA (256) |
|
trnT |
12336-12392 |
12289-12346 |
12269-12324 |
12301-12355 |
|
|
|
|
| nad4 | 12420-13620 | 12376-13573 | 12286-13552 | 12380-13583 | ATA/TAA (400) | ATG/TAA (399) | ATA/TAA (422) | ATA/TAA (400) |
1OTU-C = H. macropi from Macropus robustus robustus; 2OTU-G = H. macropi from Macropus bicolor, 3OTU-J = H. macropi from Thylogale billardierii.
Twenty-two tRNA genes were predicted from each mt genome of the three OTUs of H. macropi, and these genes varied from 53–64 nucleotides in length. Their positions in the genome were the same for all OTUs and for Ma. ocydromi (Table 1). The two ribosomal RNA genes (rrnS and rrnL) were separated by nad3, nad5, nad6, nad4L and 5 trn genes (Figure 1). The lengths of these rrn genes were 700 bp and 959 bp, respectively, which is similar to those reported previously for various nematodes, including Ancylostoma duodenale, Necator americanus, Onchocerca volvulus, Strongyloides stercoralis and Xiphinema americanum[23,25-28].
The AT-content of the mt genomes of H. macropi was 70-78%. Within the Strongylida, AT-richness in the mt genome usually ranges from 76.6-77.2% [25,29]. The nucleotide composition of the entire mt genome is biased toward T, and C is least favoured (Table 2), which is similar to other nematodes including Caenorhabditis elegans, Dirofilaria immitis and Onchocerca volvulus[30-32]. The AT content of the rrn genes for H. macropi and Ma. ocydromi ranged from 75–77% (Table 2). Given the AT bias (70-78%) in the mt genome of H. macropi, there was considerable bias in codon usage. Consequently, ATR (methionine), ATY (isoleucine), TTR (leucine), and TTY (phenylalanine) were the most commonly used codons (6.6-7.2%, 6.4-7.6%, 7.2-18% and 12.7-13%, respectively) (Table 3).
Table 2.
Nucleotide composition (%) for the entire or regions of the mitochondrial genomes determined herein
| Species | Length (bp) | A | C | T | G | A + T | |
|---|---|---|---|---|---|---|---|
|
H. macropi (from Macropus robustus robustus) |
Entire sequence |
13928 |
28.0 |
7.2 |
45.6 |
17.6 |
73.6 |
| |
Protein genes |
10520 |
25.5 |
7.4 |
46.9 |
18.5 |
72.4 |
| |
RNA genes |
1689 |
36.1 |
6.6 |
41.2 |
14.5 |
77.3 |
|
H. macropi (from Macropus bicolor) |
Entire sequence |
13883 |
28.8 |
6.7 |
46.1 |
16.8 |
74.9 |
| |
Protein genes |
10553 |
26.4 |
6.9 |
47.4 |
17.7 |
73.7 |
| |
RNA genes |
1689 |
35.8 |
6.2 |
42.0 |
14.3 |
77.8 |
|
H. macropi (from Thylogale billardierii) |
Entire sequence |
13868 |
25.8 |
7.2 |
46.2 |
19.2 |
72.0 |
| |
Protein genes |
10522 |
23.3 |
7.4 |
47.4 |
20.3 |
70.7 |
| |
RNA genes |
1689 |
34.2 |
6.8 |
41.4 |
15.6 |
75.6 |
|
Macropicola ocydromi |
Entire sequence |
13743 |
28.9 |
6.6 |
45.7 |
17.2 |
74.6 |
| |
Protein genes |
10480 |
26.5 |
6.8 |
47.0 |
18.0 |
73.5 |
| RNA genes | 1689 | 36.0 | 6.2 | 41.3 | 14.9 | 77.3 |
Table 3.
Codon usages (%) in mitochondrial protein genes of Hypodontus macropi from three different hosts
|
Amino acid |
Codon |
Number of codons and percentage of codon usage (%) |
|||
|---|---|---|---|---|---|
| OTU-C 1 | OTU-G 2 | OTU-J 3 | Ma. ocydromi | ||
|
Non-polar | |||||
| Alanine |
GCN |
101 (2.9) |
103 (3.0) |
99 (2.9) |
96 (2.8) |
| Isoleucine |
ATY |
260 (7.5) |
247 (7.1) |
221 (6.4) |
252 (7.3) |
| Leucine |
CTN |
361 (10.5) |
14 (0.4) |
30 (0.9) |
12 (0.3) |
| Leucine |
TTR |
621 (18.0) |
261 (7.1) |
500 (14.5) |
264 (7.7) |
| Methionine |
ATR |
233 (6.8) |
245 (7.1) |
226 (6.6) |
248 (7.2) |
| Phenylalanine |
TTY |
447 (13.0) |
438 (12.7) |
450 (13.1) |
438 (12.8) |
| Proline |
CCN |
80 (2.3) |
81 (2.3) |
81 (2.3) |
81 (2.4) |
| Tryptophan |
TGR |
70 (2.0) |
69 (2.0) |
71 (2.1) |
68 (2.0) |
| Valine |
GTN |
274 (8.0) |
285 (8.2) |
310 (9.0) |
274 (8.0) |
|
Polar | |||||
| Aspargine |
AAY |
141 (4.1) |
140 (4.0) |
130 (3.8) |
140 (4.1) |
| Cysteine |
TGY |
45 (1.3) |
45 (1.3) |
51 (1.5) |
47 (1.4) |
| Glutamine |
CAR |
43 (1.2) |
42 (1.2) |
40 (1.2) |
42 (1.2) |
| Glycine |
GGN |
202 (5.9) |
198 (5.7) |
208 (6.0) |
194 (5.7) |
| Serine |
AGN |
225 (6.5) |
221 (6.4) |
226 (6.6) |
226 (6.6) |
| Serine |
TCN |
515 (15.0) |
145 (4.2) |
142 (4.1) |
141 (4.1) |
| Threonine |
ACN |
119 (3.5) |
113 (3.3) |
116 (3.4) |
117 (3.4) |
| Tyrosine |
TAY |
189 (5.5) |
197 (5.7) |
190 (5.5) |
187 (5.4) |
|
Acidic | |||||
| Aspartate |
GAY |
62 (1.8) |
63 (1.8) |
64 (1.9) |
60 (1.7) |
| Glutamate |
GAR |
77 (2.2) |
78 (2.3) |
77 (2.2) |
78 (2.3) |
|
Basic | |||||
| Arginine |
CGN |
31 (0.9) |
31 (0.9) |
31 (0.9) |
31 (0.9) |
| Histidine |
CAY |
57 (1.7) |
57 (1.6) |
56 (1.6) |
56 (1.6) |
| Lysine | AAR | 112 (3.2) | 115 (3.3) | 111 (3.2) | 114 (3.3) |
1OTU-C = H. macropi from Macropus robustus robustus; 2OTU-G = H. macropi from Macropus bicolor; 3OTU-J = H. macropi from Thylogale billardierii.
Genetic differences and phylogenetic relationships
Pairwise comparisons of the amino acid (aa) sequences predicted for each of the 12 protein coding mt genes from OTU-C, OTU-G and OTU-J of H. macropi from the hosts M. robustus, M. bicolor and T. billardierii, respectively, are shown in Table 4. Most variation was detected in NAD6 between OTU-C and OTU-J (29%) and between OTU-G and OTU-J (26.9%), and in NAD4L between OTU-C and OTU-G (15.4%); moderate variation was detected in NAD1, NAD2, NAD3, NAD4 and NAD5 (mean: 9.8%) among the OTUs; least variation was detected in COX1, COX2 and COX3 (1.7-4.3%). Pairwise comparisons of concatenated amino acid sequences revealed sequence differences of 5.8% (OTU-C versus OTU-G), 18% (OTU-C versus OTU-J) and 10.0% (OTU-G versus OTU-J) (Table 4). These percentages are substantially higher than those detected within species of strongyloid nematodes [17,18,23]. Pairwise comparisons of the concatenated amino acid sequence of Ma. ocydromi with those of OTU-C, OTU-G and OTU-J revealed sequence differences of 30.4%, 15.9% and 31.0%, respectively (Table 5). The phylogenetic analysis of the aa sequence data showed that all three OTUs of H. macropi and Ma. ocydromi formed a distinct group, with maximum statistical support (posterior probability [pp] = 1.00), to the exclusion of the other strongyloid nematodes included here (i.e., Oesophagostomum dentatum, Chabertia ovina and Strongylus vulgaris) (Figure 2). In this analysis, OTU-C from M. r. robustus grouped together with Ma. ocydromi (pp = 1.00) to the exclusion of OTU-G (M. bicolor) and OTU-J (T. billardierii). While the three OTUs of H. macropi did form a group with C. ovina, statistical support for this grouping was moderate (pp = 0.74) (Figure 2).
Table 4.
Pairwise comparison of the amino acid sequences of the 12 protein coding mitochondrial genes
| Protein | OTU-G vs. OTU-C | OTU-J vs. OTU-C | OTU-G vs. OTU-J |
|---|---|---|---|
| ATP6 |
4.5 |
13.6 |
15.7 |
| COB |
5.4 |
6.2 |
9.7 |
| COX1 |
2.0 |
4.0 |
3.2 |
| COX2 |
1.7 |
4.3 |
3.9 |
| COX3 |
3.1 |
4.3 |
3.9 |
| NAD1 |
3.0 |
6.4 |
6.7 |
| NAD2 |
13.2 |
16.7 |
16.6 |
| NAD3 |
5.4 |
6.3 |
7.15 |
| NAD4 |
7.4 |
11.8 |
11.7 |
| NAD4L |
15.4 |
14.1 |
9.0 |
| NAD5 |
8.1 |
13.5 |
13.5 |
| NAD6 |
5.6 |
29.0 |
26.9 |
| All genes | 5.8 | 18.0 | 10.0 |
These percentage differences are based on comparisons between three operational taxonomic units (OTUs) of Hypodontus macropi from three different hosts (i.e., OTU-C from Macropus robustus robustus; OTU-G from Macropus bicolor, OTU-J from Thylogale billardierii).
Table 5.
Pairwise comparison of the amino acid sequences of the 12 protein coding mitochondrial genes
| Protein | OTU-C vs. Mo | OTU-G vs. Mo | OTU-J vs. Mo |
|---|---|---|---|
| ATP6 |
5.5 |
5.0 |
14.1 |
| COB |
5.1 |
7.8 |
9.7 |
| COX1 |
2.9 |
2.1 |
3.6 |
| COX2 |
1.7 |
1.7 |
3.9 |
| COX3 |
2.7 |
3.1 |
5.9 |
| NAD1 |
1.9 |
2.6 |
5.6 |
| NAD2 |
10.3 |
13.5 |
16.4 |
| NAD3 |
4.5 |
5.4 |
7.2 |
| NAD4 |
8.9 |
13.9 |
14.6 |
| NAD4L |
9.0 |
11.5 |
10.3 |
| NAD5 |
11.2 |
12.3 |
16.1 |
| NAD6 | 30.4 | 15.9 | 31.0 |
These percentage differences are based on comparisons between three operational taxonomic units of Hypodontus macropi from three different hosts (i.e., OTU-C from Macropus robustus robustus; OTU-G from Macropus bicolor, OTU-J from Thylogale billardierii) and Macropicola ocydromi (Mo) from Macropus fuliginosus.
Figure 2.
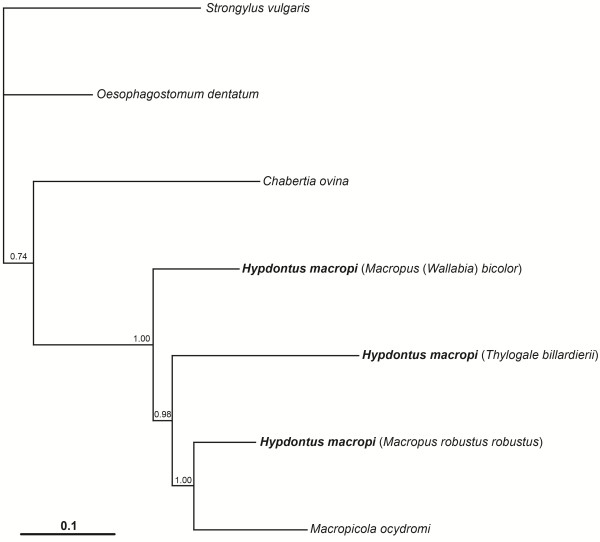
Phylogenetic analysis of the concatenated amino acid sequences for protein coding genes ofHypodontus macropi. Concatenated amino acid sequence data for all protein coding mitochondrial genes for three operational taxonomic units of H. macropi, from Macropus robustus robustus, Macropus bicolor, Thylogale billardierii, and from Macropicola ocydromi sequenced here as well as other concatenated sequence data representing complete mitochondrial genomes of Chabertia ovina, Oesophagostomum dentatum and Strongylus vulgaris (Strongyloidea) were analyzed using Bayesian Inference. The numbers above each tree branch represent the statistical support for each node (based on posterior probability score).
Sliding window analyses
Sliding window analyses were carried out to identify conserved and variable regions in mt genomes among all three OTUs. Results from these analyses are shown in Figure 3 for comparison across all the three OTUs and also for nucleotide diversities calculated from pairwise comparisons across the mt genomes of OTU-C, OTU-G and OTU-J. A nucleotide diversity pattern was broadly similar across the sliding window analyses and diversity within and between genes was relatively consistent across the genera. Greatest nucleotide diversity was detected within AT-rich (AT) region, followed by peaks of variation within NAD3, NAD4, NAD2 and NAD5. Gene-by-gene nucleotide diversity was highly variable, but, by far, the least variation was recorded within COX2. Overall, the full sliding window indicates a wealth of new genes capable of providing high levels of nucleotide variation for H. macropi population genetic studies.
Figure 3.
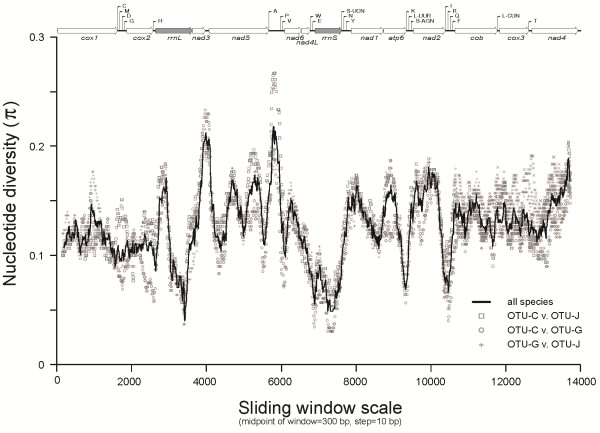
Sliding window analysis of complete mt genome sequences ofHypodontus macropifrom different hosts. The three operational taxonomic units (OTUs) of H. macropi were from Macropus robustus robustus (OTU-C), Macropus bicolor (OTU-G) and Thylogale billardierii (OTU-J). The black line indicates nucleotide diversity comparing all the three OTUs; whereas three other symbols (a square, a circle and a plus sign) indicate pairwise comparisons across the mt genomes of OTU-C, OTU-G and OTU-J. Nucleotide diversity, measured iteratively every 10 bp over 300 bp windows of aligned mtDNA sequence data indicate peaks and troughs of sequence variability. Linearized maps of mtDNA are provided for each aligned data set, with the highest peaks of variability falling within the non-coding AT-rich regions.
Discussion
Molecular tools offer an unprecedented opportunity to include new components in our studies on the discovery and description of parasite species. For example, molecular markers in the identification of parasite species can be used to (i) link morphologically indistinguishable life stages, such as larvae, to the adult stages of known species, (ii) elucidate life cycles by establishing the species that may serve as intermediate or paratenic hosts for larval stages of a parasite, and (iii) search for cryptic species [12]. However, Nadler and Pérez-Ponce de León [33] suggested that, in terms of explanatory power, single locus DNA barcodes and the barcoding-gap are insufficient approaches to delimit species, and concordance of independent information, including other genes, is required. In the present study, we sequenced the whole mt genomes of individuals of H. macropi, proposed to be distinct cryptic species based on MEE analysis and the ITS-2 sequences of specimens [8-11] from three different species of macropodid marsupial hosts. Based on the analysis of the complete mt genomes of three cryptic species, within the morphospecies H. macropi, the present study provides independent support that specimens of H. macropi from three species of macropodid hosts represent three genetically distinct species. The present mt genomic results support previous findings [8-11] from MEE and nuclear DNA-based analyses and indicate that H. macropi is represented by a complex of at least ten sibling species, one in each of M. agilis, M. bicolor, M. dorsalis, M. rufogriseus, P. persephone, T. billardierii and T. stigmatica, with the remaining three species occurring in M. r. robustus and M. rufus.
Although the ITS-2 rDNA has been shown to be a reliable genetic marker to distinguish among a variety of strongyloid nematodes [13-15] and this marker has been useful for discovering sibling species within various cloacinine nematodes in macropodid marsupials [34-38], hypotheses regarding such species need to be independently tested, preferably with sequence data from additional loci [33]. For this purpose, we chose mtDNA genes because of their relatively rapid rate of substitution, coupled to the smaller effective population size of mtDNA and the provision of evidence of lineage exclusivity in a shorter period of time (on average) following speciation [39,40]. Using mt datasets, based upon pairwise comparisons of concatenated amino acid sequences inferred herein, we found a wide range of sequence differences of among different OTUs (5.8 – 18%) (see Table 4), and this magnitude is consistent with the previous findings of Chilton et al. [11] using the ITS-2 rDNA. These authors also found a wide variation in the ITS-2 sequences among three OTUs i.e., genotype (G) 8 (OTU-G, M. bicolor), G13 (OTU-C, M. r. robustus), and G36 (OTU-J, T. billardierii) of H. macropi. Based on the ITS-2 sequences, the highest sequence variation detected was between OTU-C [GenBank: HE866725] and OTU-G [GenBank: HE866720] (33.3%), followed by OTU-C [GenBank: HE866725] and OTU-J [GenBank: HE866748] (27.8%) and OTU-G [GenBank: HE866720] and OTU-J [GenBank: HE866748] (25.8%).
In nematodes, mt DNA is usually more variable in sequence within a species than the ITS-2 and other rDNA regions [14], indicating that mt gene regions are well suited for studying the population genetics of parasitic nematodes [14,17,41]. The sliding window analysis conducted herein displayed distinct patterns of nucleotide diversity among the three mt genomes representing OTUs-C, -G and -J of H. macropi (see Figure 3). In the present study, COX2 was revealed to be the gene with the lowest nucleotide diversity among these three OTUs, whereas NAD2, NAD3, NAD5 and NAD6 showed higher nucleotide diversity, which can be targeted to design primers for studying the population genetics of H. macropi. Given that cryptic species have been detected within the morphospecies H. macropi[8-11], low sequence variability is useful for the design of oligonucleotide primers that flank mt regions with high variability. Utilizing such primer sets, PCR-based SSCP analysis [42] could be applied to screen large numbers of individual specimens representing different host species and populations for cryptic species. Previous studies [10,11] have shown the merit of SSCP for exploring the genetic variation in various populations of H. macropi, and this approach could be applied to large-scale studies of H. macropi specimens representing distinct OTUs. It is hoped that major advances in the next-generation sequencing technology (e.g., Illumina) [43] will allow selective whole genome sequencing of cryptic species of nematodes, thereby helping to re-classify them by employing detailed phylogenetic and comparative genetic analyses.
Conclusions
The present analyses of mt proteomic sequence datasets clearly supported the hypothesis that H. macropi from M. robustus robustus, M. bicolor and T. billardierii represent distinct species. This study emphasizes the importance and utility of the mt genomic datasets for parasites from macropodid marsupials as a basis for systematic, ecological and biological studies, and the specific diagnosis of infections. In particular, studying cryptic species within the morphospecies H. macropi provides a stimulus to re-explore the relationships of sibling species using complete mt genomic and proteomic sequence data sets. Although the present study focused on H. macropi, the approach employed here has major implications for the study of a wide range of invertebrates and vertebrates.
Methods
Collection of nematodes and isolation of genomic DNA
Adult H. macropi individuals were collected opportunistically from the intestines of M. r. robustus, M. bicolor and T. billardierii. The nematodes from these three host species were designated as operational taxonomic units (OTUs), namely OTU-C, OTU-G and OTU-J, respectively, according to Chilton et al. [8]. A specimen of a related species, Macropicola ocydromi, was also collected from Macropus fuliginosus and included for genetic comparison with OTUs of H. macropi. Individual worms were stored (in individual tubes) at −80°C until required. Upon thawing, individual nematodes were washed extensively in physiological saline, pH 7.4. The anterior and posterior ends of each nematode were excised, cleared in lactophenol for identification and then stored in 70% ethanol and retained as voucher specimens, which have been deposited in the South Australian Museum (SAM 46024, 46038 and 46053; see Chilton et al. [11]), Adelaide, Australia. The remainder of each worm was used for the extraction of genomic DNA using a small-scale sodium dodecyl-sulphate (SDS)/proteinase K digestion and column-purification (Wizard DNA Clean-Up Kit, Promega, USA) [42]. The identity of each specimen was verified by PCR-based amplification of the ITS-2 using an established method, followed by mini-column purification (Wizard PCR-Preps, Promega) of the amplicon and subsequent automated (bidirectional) sequencing (BigDye chemistry v.3.1) [42].
Sequencing and assembly of mt genomes
The complete mt genome of each worm was amplified by long-PCR (Advantage 2 kit; BD Biosciences) as two overlapping amplicons (“large” and “small”), using two (relatively) conserved primer sets, 5 F-40R and 39 F-42R, respectively [26,44] and with appropriate positive and negative (i.e., no template) controls. The cycling conditions used were 95°C for 1 min (initial denaturation); then 95°C for 15 s (denaturation), 50°C (for primer set 39 F-42R) or 55°C (for primer set 5 F-40R) for 15 s (annealing), and 60°C for 3 min (for primer set 39 F-42R) or 68°C for 6 min (for primer set 5 F-40R) (extension) for 35 cycles, and a final extension at 60°C for 3 min (for primer set 39 F-42R) or 68°C for 6 min (for primer set 5 F-40R). Products were consistently produced from the positive control samples (Ascaris suum); in no case was a product detected for any of the negative controls. Products were detected in 1% (w/v) agarose gels after ethidium-bromide staining and ultraviolet transillumination, and were then purified over a mini-column (Wizard, Promega) and quantified spectrophotometrically (ND-1000 UV–VIS spectrophotometer v.3.2.1; NanoDrop Technologies). Following electrophoretic verification of quality, the two amplicons (~5 kb and ~10 kb; 2.5 μg of each), spanning the mt genome of each worm specimen, were pooled and subsequently sequenced using the 454 Genome Sequencer FLX (Roche) [45]. The consensus mt genome sequences [GenBank: KF361317-KF361320] were each assembled automatically, using the CAP3 program [46], from individual reads (of ~300 bp).
Bioinformatic pipeline for mt genomic annotation and analysis of sequence data
Following assembly, the genes and features of each mt genome from each worm were annotated using a semi-automated pipeline [21]. In brief, each protein coding mt gene was identified by local alignment comparison (performed in all six reading frames) using amino acid sequences predicted (employing invertebrate mitochondrial code) from the mt genome of a reference species (i.e., Oesophagostomum dentatum [GenBank: NC_013817]. The large and small ribosomal RNA subunit genes (rrnS and rrnL) were identified by local alignment (i.e., using nucleotide sequence data) using the same approach. All transfer RNA (tRNA) genes were detected and identified in a three-step process. Initially, all possible tRNA genes present in each consensus sequence were predicted (from both strands) based on a folding structure, using scalable models based on the standard nematode mt tRNAs [18]. All predicted tRNA genes were clustered into groups based on their anti-codon sequence and provisionally identified based on the amino acid encoded by this anti-codon. Two separate tRNA gene groups were predicted each for leucine (one each for the anticodons CUN and UUR, respectively) and for serine (one each for the anticodons AGN and UCN, respectively), as these tRNA genes have been shown to be duplicated in most invertebrate mt genomes, including those of nematodes [18]. All predicted tRNAs within each amino acid group were ranked based on structural “strength” (as inferred by the number of mismatched nt pairs in each stem), and the 100 best-scoring structures for each group were compared by BLASTn alignment against a database comprising all tRNA gene sequences for each amino acid of all published nematode mt genome sequences (available viahttp://drake.physics.mcmaster.ca/ogre/; [47]). All tRNA genes of each mt genome were then identified and annotated based on having the highest sequence identity to known nematode tRNA genes. Annotated sequence data were imported into the program SEQUIN (available viahttp://www.ncbi.nlm.nih.gov/Sequin/) for final verification of the mt genomic structure and subsequent, direct submission to the GenBank database.
Alignment and phylogenetic analysis of concatenated nucleotide or amino acid sequence data
The amino acid sequences were conceptually translated from the mt genomes of the three individuals of H. macropi and the specimen of Ma. ocydromi, and then concatenated separately. The amino acid sequences derived from published mt genomes from nematodes [Strongylida: Chabertia ovina [GenBank: NC_013831], Oesophagostomum dentatum [GenBank: NC_013817] and Strongylus vulgaris [GenBank: NC_013818] [see [21]] were also used for comparative purposes. These amino acid sequences (considering all homologous characters) were aligned using MUSCLE [48]. The amino acid sequences of H. macropi and Ma. ocydromi were then aligned using BioEdit [49] and subjected to phylogenetic analysis using Bayesian inference (BI), employing the Markov chain Monte Carlo (MCMC) method in MrBayes 3.1.2 [50,51]. The likelihood parameters for BI were based on the Akaike Information Criteria (AIC) test in Prottest v2.4 [52]. The “best” model for the amino acid sequences was a fixed-rate Poisson model [53]. Estimates of the base frequencies, the substitution rate model matrix and the proportion of invariable sites were fixed. Posterior probabilities (pp) were calculated using 2,000,000 generations, employing four simultaneous tree-building chains, with every 100th tree being saved.
Sliding window analysis
This analysis was performed on the aligned, complete mt genome sequences of the three OTUs of H. macropi using DnaSP v.5 [54]. The alignment of these sequences was achieved using MUSCLE v.3.8 [47], as implemented in SeaView v.4 [55]. Keeping the nucleotides in frame, there were no ambiguously aligned regions. A sliding window of 300 bp (steps of 10 bp) was used to estimate nucleotide diversity (π) between the three OTUs of H. macropi; indels were excluded using DnaSP. Nucleotide diversity for the entire alignments was plotted against midpoint positions of each window, and gene boundaries were defined.
Abbreviations
atp6: Adenosine triphosphatase subunit 6; cox: Cytochrome c subunit; cytb: Cytochrome b subunit; ITS-2: Second internal transcribed spacer; Mt: Mitochondrial; MEE: Multilocus enzyme electrophoresis; nad: Nicotinamide dehydrogenase subunit; OTU: Operational taxonomic unit; ORF: Open reading frame; rDNA: Nuclear ribosomal DNA; rrnL: Ribosomal large subunit; rrnS: Ribosomal small subunit; SSCP: Single-strand conformation polymorphism; tRNA: Transfer RNA.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RBG, ARJ & TL conceived the project and attracted the funding; AJ carried out molecular laboratory work; NM assisted with the bioinformatics analysis; TL performed the sliding window analysis; AJ, IB, NBC, ARJ, TL, & RBG carried out data analysis and interpretation; RBG, AJ & IB wrote the draft manuscript. All authors read and approved the final version of the manuscript.
Contributor Information
Abdul Jabbar, Email: jabbara@unimelb.edu.au.
Ian Beveridge, Email: ibeve@unimelb.edu.au.
Namitha Mohandas, Email: namigrt@gmail.com.
Neil B Chilton, Email: neil.chilton@usask.ca.
D Timothy J Littlewood, Email: t.littlewood@nhm.ac.uk.
Aaron R Jex, Email: ajex@unimelb.edu.au.
Robin B Gasser, Email: robinbg@unimelb.edu.au.
Acknowledgements
This project was funded by the Australian Research Council. This project was also supported by a Victorian Life Sciences Computation Initiative (VLSCI) grant number VR0007 on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government. Other support from the Alexander von Humboldt Foundation and Melbourne Water Corporation is gratefully acknowledged (R.B.G). The Early Career Researcher and Collaborative Research grants from The University of Melbourne are also gratefully acknowledged (A.J.).
References
- Mönnig HO. Hypodontus macropi, n. gen., n. sp., a hookworm of the kangaroo. Pretoria: Union of South Africa; 1929. pp. 303–306. (15th Annual Report of the Director of Veterinary Services). [Google Scholar]
- Beveridge I. Hypodontus macropi Mönnig, 1929, a hook-worm like parasite of macropodid marsupials. J Helminthol. 1979;53:229–244. doi: 10.1017/S0022149X00006015. [DOI] [PubMed] [Google Scholar]
- Setasuban P, Arundel JH. Hypodontus macropi Mönnig, 1929 from a Bennett’s wallaby Macropus rufogrisea. . Southeast Asian J Trop Med Public Health. 1996;7:102–105. [PubMed] [Google Scholar]
- Beveridge I, Arundel JH. Helminth parasites of grey kangaroos, Macropus giganteus Shaw and M. fuliginosus (Desmarest), in eastern Australia. Aust Wildlife Res. 1979;6:69–77. doi: 10.1071/WR9790069. [DOI] [Google Scholar]
- Beveridge I, Spratt DM, Close RL, Barker SC, Sharman GB. Helminth parasites of rock wallabies, Petrogale spp. (Marsupialia) from Queensland. Aust Wildlife Res. 1989;39:691–702. [Google Scholar]
- Begg M, Beveridge I, Chilton NB, Johnson PM, O’Callaghan MG. Parasites of the Proserpine Rock Wallaby, Petrogale persephone (Marsupialia: Macropodidae) Aust Mammal. 1996;18:45–53. [Google Scholar]
- Bradley C, Beveridge I, Chilton NB, Johnson PM. Helminth parasites of the purple-necked rock wallaby,Petrogale lateralis purpureicollis, from Queensland. Trans Roy Soc South Aust. 2000;124:37–40. [Google Scholar]
- Chilton NB, Beveridge I, Andrews RH. Detection by allozyme electrophoresis of cryptic species of Hypodontus macropi (Nematoda: Strongyloidea) from macropodid marsupials. Int J Parasitol. 1992;22:271–279. doi: 10.1016/S0020-7519(05)80004-9. [DOI] [PubMed] [Google Scholar]
- Chilton NB, Gasser RB, Beveridge I. Differences in a ribosomal DNA sequence of morphologically indistinguishable species within the Hypodontus macropi complex (Nematoda: Strongyloidea) Int J Parasitol. 1995;25:647–651. doi: 10.1016/0020-7519(94)00171-J. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Zhu X, Beveridge I, Chilton N. Mutation scanning analysis of sequence heterogeneity in the second internal transcribed spacer (rDNA) within some members of the Hypodontus macropi (Nematoda: Strongyloidea) complex. Electrophoresis. 2001;22:1076–1085. doi: 10.1002/1522-2683()22:6<1076::AID-ELPS1076>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Chilton NB, Jabbar A, Huby-Chilton F, Jex A, Gasser RB, Beveridge I. Genetic variation within the Hypodontus macropi (Nematoda: Strongyloidea) complex from macropodid marsupial hosts in Australia. Electrophoresis. 2012;33:3544–3554. doi: 10.1002/elps.201200364. [DOI] [PubMed] [Google Scholar]
- Criscione CD, Poulin R, Blouin MS. Molecular ecology of parasites: elucidating ecological and microevolutionary processes. Mol Ecol. 2005;14:2247–2257. doi: 10.1111/j.1365-294X.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- Chilton NB. The use of nuclear ribosomal DNA markers for the identification of bursate nematodes (order Strongylida) and for the diagnosis of infections. Anim Health Res Rev. 2004;5:171–87. doi: 10.1079/ahr200497. [DOI] [PubMed] [Google Scholar]
- Gasser RB. Molecular tools–advances, opportunities and prospects. Vet Parasitol. 2006;136:69–89. doi: 10.1016/j.vetpar.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Bott NJ, Chilton NB, Hunt P, Beveridge I. Towards practical, DNA-based diagnostic methods for parasitic nematodes of livestock - bionomic and biotechnological implications. Biotechnol Adv. 2008;26:325–34. doi: 10.1016/j.biotechadv.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Le TH, Blair D, McManus DP. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002;18:206–213. doi: 10.1016/S1471-4922(02)02252-3. [DOI] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genomics of parasitic nematodes of socio-economic importance: recent progress, and implications for population genetics and systematics. Adv Parasitol. 2004;56:133–212. doi: 10.1016/s0065-308x(03)56003-1. [DOI] [PubMed] [Google Scholar]
- Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes—progress and perspectives. Trends Parasitol. 2006;22:78–84. doi: 10.1016/j.pt.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Zarowiecki MZ, Huyse T, Littlewood DT. Making the most of mitochondrial genomes – markers for phylogeny, molecular ecology and barcodes in Schistosoma (Platyhelminths: Digenea) Int J Parasitol. 2007;37:1401–1418. doi: 10.1016/j.ijpara.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Jex AR, Littlewood DT, Gasser RB. Toward next-generation sequencing of mitochondrial genomes–focus on parasitic worms of animals and biotechnological implications. Biotechnol Adv. 2010;28:151–159. doi: 10.1016/j.biotechadv.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Jex AR, Hall RS, Littlewood DT, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522–533. doi: 10.1093/nar/gkp883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto R, Macfarlane L, Clary D, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. . Genetics. 1992;130:471–498. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Abs El-Osta YG, Gasser RB. Comparative analysis of mitochondrial genome data for Necator americanus from two endemic regions reveals substantial genetic variation. Int J Parasitol. 2003;33:955–963. doi: 10.1016/S0020-7519(03)00129-2. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM. Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative atp8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–637. doi: 10.1093/genetics/157.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea) Int J Parasitol. 2002;32:145–158. doi: 10.1016/S0020-7519(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Keddie EM, Higazi T, Boakye D, Merriweather A, Wooten MC, Unnasch TR. Onchocerca volvulus: limited heterogeneity in the nuclear and mitochondrial genomes. Exp Parasitol. 1999;93:198–206. doi: 10.1006/expr.1999.4450. [DOI] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genome of Strongyloides stercoralis (Nematoda) – idiosyncratic gene order and evolutionary implications. Int J Parasitol. 2003;33:1393–1408. doi: 10.1016/S0020-7519(03)00130-9. [DOI] [PubMed] [Google Scholar]
- He Y, Jones J, Armstrong M, Lamberti F, Moens M. The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. J Mol Evol. 2005;61:819–833. doi: 10.1007/s00239-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Van der Veer M, de Vries E. A single nucleotide polymorphism map of the mitochondrial genome of the parasitic nematode Cooperia oncophora. Parasitology. 2004;128:421–431. doi: 10.1017/S0031182003004633. [DOI] [PubMed] [Google Scholar]
- Okimoto R, Macfarlane JL, Wolstenholme DR. Evidence for the frequent use of TTG as the translation initiation codon of mitochondrial protein genes in the nematodes, Ascaris suum and Caenorhabditis elegans. Nucleic Acids Res. 1999;18:6113–6118. doi: 10.1093/nar/18.20.6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keddie EM, Higazi T, Unnasch TR. The mitochondrial genome of Onchocerca volvulus : sequence, structure and phylogenetic analysis. Mol Biochem Parasitol. 1998;95:111–127. doi: 10.1016/S0166-6851(98)00102-9. [DOI] [PubMed] [Google Scholar]
- Hu M, Gasser RB, Abs El-Osta YG, Chilton NB. Structure and organization of the mitochondrial genome of the canine heartworm, Dirofilaria immitis. Parasitology. 2003;127:37–51. doi: 10.1017/S0031182003003275. [DOI] [PubMed] [Google Scholar]
- Nadler SA, Pérez-Ponce de León G. Integrating molecular and morphological approaches for characterizing parasite cryptic species: implications for parasitology. Parasitology. 2011;138:1688–1709. doi: 10.1017/S003118201000168X. [DOI] [PubMed] [Google Scholar]
- Beveridge I, Chilton NB, Andrews RH. Sibling species within Macropostrongyloides baylisi (Nematoda: Strongyloidea) from macropodid marsupials. Int J Parasitol. 1993;23:21–33. doi: 10.1016/0020-7519(93)90094-F. [DOI] [PubMed] [Google Scholar]
- Beveridge I, Spratt DM. The helminth fauna of Australasian marsupials: origins and evolutionary biology. Adv Parasitol. 1996;37:136–254. [Google Scholar]
- Chilton NB, Andrews RH, Beveridge I. Genetic evidence for a complex of species within Rugopharynx australis (Mönnig, 1926) (Nematoda: Strongyloidea) from macropodid marsupials. Syst Parasitol. 1996;34:125–133. doi: 10.1007/BF00009686. [DOI] [Google Scholar]
- Beveridge I, Chilton NB. Revision of the Rugopharynx australis (Moennig, 1926) complex (Nematoda: Strongyloidea) from macropodid marsupials. Invert Taxon. 1999;13:805–843. doi: 10.1071/IT98017. [DOI] [Google Scholar]
- Chilton NB, Huby-Chilton F, Johnson PM, Beveridge I, Gasser RB. Genetic variation within species of the nematode genus Cloacina (Strongyloidea: Cloacininae) parasitic in the stomachs of rock wallabies, Petrogale spp. (Marsupialia: Macropodidae) in Queensland. Aust J Zool. 2009;57:1–10. doi: 10.1071/ZO08068. [DOI] [Google Scholar]
- Avise JC. Molecular markers, natural history and evolution. New York: Chapman and Hall; 1994. [Google Scholar]
- Nadler SA. Species delimitation and nematode biodiversity: phylogenies rule. Nematol. 2002;4:615–625. doi: 10.1163/15685410260438908. [DOI] [Google Scholar]
- Anderson TJC, Blouin MS, Beech RN. Populations biology of parasitic nematodes: applications of genetic markers. Adv Parasitol. 1998;41:219–283. doi: 10.1016/s0065-308x(08)60425-x. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AR, Otranto D, Cafarchia C, Beveridge I, Zhu XQ. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1:3121–3128. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- Mardis E. Next-generation DNA sequencing methods. Ann Rev Genomics Human Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2:2339–2344. doi: 10.1038/nprot.2007.358. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage K, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson D, Gibson AP, Hudelot C, Higgs PG. OGRe: a relational database for comparative analysis of mitochondrial genomes. Nucleic Acids Res. 2003;31:202–206. doi: 10.1093/nar/gkg077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [45] [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Posada D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Bishop MJ, Friday AE. Evolutionary trees from nucleic acid and protein sequences. Proc R Soc Lond B Biol Sci. 1985;226:271–302. doi: 10.1098/rspb.1985.0096. [DOI] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]