

Abstract
Genetic mutations in voltage-gated and ligand-gated ion channel genes have been identified in a small number of Mendelian families with genetic generalised epilepsies (GGEs). They are commonly associated with febrile seizures (FS), childhood absence epilepsy (CAE) and particularly with generalised or genetic epilepsy with febrile seizures plus (GEFS+). In clinical practice, despite efforts to categorise epilepsy and epilepsy families into syndromic diagnoses, many generalised epilepsies remain unclassified with a presumed genetic basis. During the systematic collection of epilepsy families, we assembled a cohort of families with evidence of GEFS+ and screened for variations in the γ2 subunit of the γ-aminobutyric acid (GABA) type A receptor gene (GABRG2). We detected a novel GABRG2(p.R136*) premature translation termination codon in one index-case from a two-generation nuclear family, presenting with an unclassified GGE, a borderline GEFS+ phenotype with learning difficulties and autism spectrum disorder (ASD). The GABRG2(p.R136*) mutation segregates with the febrile seizure component of this family's GGE and is absent in 190 healthy control samples. In vitro expression assays demonstrated that γ2(p.R136*) subunits were produced, but had reduced cell-surface and total expression. When γ2(p.R136*) subunits were co-expressed with α1 and β2 subunits in HEK 293T cells, GABA–evoked currents were reduced. Furthermore, γ2(p.R136*) subunits were highly-expressed in intracellular aggregations surrounding the nucleus and endoplasmic reticulum (ER), suggesting compromised receptor trafficking. A novel GABRG2(p.R136*) mutation extends the spectrum of GABRG2 mutations identified in GEFS+ and GGE phenotypes, causes GABAA receptor dysfunction, and represents a putative epilepsy mechanism.
Keywords: GABAA receptors, epilepsy, protein truncating mutations, autism spectrum disorder
Introduction
Genetic generalised epilepsies (GGE) account for approximately 30% of all epilepsies and are considered to have a complex genetic basis. This is reflected in the recent nomenclature revision where idiopathic is deemed to be synonymous with ‘genetic’ (Berg et al., 2010). GGEs are characterised by generalised spike and wave discharges on electroencephalogram (EEG) and comprise a number of distinct and overlapping clinical syndromes that vary in severity. These include specific electro-clinical syndromes such as: childhood absence epilepsy (CAE); juvenile myoclonic epilepsy (JME), epilepsy with generalised tonic-clonic seizures on awakening, and generalised epilepsy with febrile seizures (FS) plus (GEFS+), where FS occur in association with GGE. Over the last decade genetic mutations in voltage-gated ion channels (sodium, calcium and potassium channel subunits) and in ligand-gated ion channels (nicotinic cholinergic and GABAA receptor subunits) have been reported in a small number of GGEs (Heron et al., 2007; Macdonald and Kang, 2009).
GEFS+, also more recently termed genetic epilepsy with febrile seizures plus, is a well-described familial epilepsy syndrome, characterised by heterogeneous epilepsy phenotypes. Although initially described in large families with autosomal dominant inheritance and incomplete penetrance, most GEFS+ families are now considered to follow a complex inheritance pattern and significant overlap with GGE is observed (Scheffer and Berkovic 1997; Singh et al., 1999). GEFS+ is considered genetically-heterogeneous segregating in a small number of GEFS+ families with mutations in genes encoding neuronal voltage-gated sodium channel subunits (SCN1A, SCN2A and SCN1B) and ligand-gated GABAA receptor subunits (GABRG2 and GABRD) (Wallace et al., 1998; Escayg et al., 2000; Baulac et al 2001; Wallace et al 2001). The most common genetic abnormalities identified to date are missense SCN1A mutations, thought to account for approximately 10% of GEFS+ families (Marini et al., 2007).
GABA is the major inhibitory neurotransmitter in the mammalian brain and mutations in genes encoding α1, β3, γ2 and δ GABAA receptor subunits have been associated with different GGE spectrum epilepsies such as simple FS, CAE and GEFS+ (Harkin et al., 2002; Kananura et al., 2002; Dibbens et al., 2004; Adenaert et al., 2006; Sun et al., 2008; Tanaka et al., 2008; Tian et al., 2013). GABAA receptor dysfunction was first demonstrated by the identification of the GABRG2(p.R82Q) mutation in a family with CAE and FS, and the GABRG2(p.K328M) mutation in a GEFS+ family. This was followed by further mutations described in six unrelated index-cases, underscoring the critical role of γ2 subunits for receptor trafficking, targeting, clustering and synaptic maintenance and response to benzodiazepine modulators (Essrich et al., 1998; Moss et al 2001; Schweizer etal., 2003; Keller et al., 2004; Rathenberg and Moss 2004).
Functional studies have suggested that these mutations alter receptor biogenesis or function via impaired receptor subunit mRNA stability, protein folding and stability, or assembly and receptor trafficking (Macdonald and Kang., 2009). Recently, two cellular biological control mechanisms were proposed, nonsense-mediated decay (NMD) and ER-associated degradation (ERAD), that regulate the effects of premature translation-termination codons (PTCs) in GABAA receptor subunit genes associated with epilepsy (Kang et al., 2009).
In this study, we screened SCN1A and GABRG2 in affected index-cases from 24 families with a history of GEFS+ from our growing collection of familial epilepsy in the UK (Johnston et al., 2009; Thomas et al., 2012). A novel nonsense mutation GABRG2(pR136*) was discovered in a borderline GEFS+/unclassified GGE family that segregates with the FS component of the phenotype, and we provide in vitro evidence for pathogenicity of γ2(p.R136*) subunits in relation to the seizure phenotype.
Methods
Clinical Evaluation and Sample Collection
Families for the genetic study were sourced through clinician tertiary referrals, from an established epilepsy clinical database and through self-referral. Written informed consent was obtained from all participants to access the relevant results from investigations such as, electrocardiogram, EEG, or brain imaging (approved by MREC for Wales, 05/MRE09/78). Clinical characteristics and genealogical information was ascertained via a semi-structured interview and a clinical examination was performed. The phenotypes were recorded according to the commission on Classification and Terminology of the International League Against Epilepsy, 1981; and of Epilepsy Syndromes, 1989 with reference to recent revisions (Commission on Classification and Terminology of the International League Against Epilepsy., 1981; Engel., 2001; Engel., 2006). We used the original definition for a GEFS+ families - two or more individuals with a GEFS+ phenotype (Scheffer and Berkovic., 1997). Sets of clinical characteristics were used to subdivide the epilepsy phenotypes into ‘endophenotypes’.
Mutation Analysis
From eighty ascertained epilepsy families who had consented to genetic studies, fourteen were classified as probable GEFS+ and ten as having a borderline GEFS+ family phenotype (Thomas et al., 2012). We tested eighteen participants sourced from nine of these GEFS+ or borderline GEFS+ families for variations in SCN1A (OMIM: 182389) and GABRG2 (OMIM: 137164) genes. Polymerase chain reaction was used to amplify GABRG2 and SCN1A coding regions and splice sites and amplimers were sequenced with ABI capillary technology (Foster City, CA). Population studies on identified gene-mutants were performed using LightScanner™ high-resolution melting (Idaho Technology, USA) and scrutinising bioinformatics databases.
Bioinformatics and structural modeling
Posttranslational modifications and trafficking of wild-type and GABRG2(R136X) sequences were predicted using PSORTII and PROSCAN (Horton and Nakai., 1997; Combet et al., 2000). Structural modelling of wild-type γ2 and mutant γ2(p.R136*) subunits was carried out using a homology modelling pipeline built with the Biskit structural bioinformatics platform, which scans the entire Protein DataBank (PDB) for candidate homologies (Grunberg et al., 2007). The best homology attained for γ2 subunits was based on 21% identity with the crystal structure (PDB: 2BG9) of the nicotinic acetylcholine receptor from Torpedo marmorata (Unwin., 2005). The pipeline workflow incorporates the NCBI tools platform, including the BLAST program for similarity searching of sequence databases (Wheeler et al., 2007; Altschul et al., 1990). T-COFFEE was used for alignment of the test sequence with the template and homology models were generated over 10 iterations of the MODELLER program (Notredame et al., 2000; Eswar et al., 2003). All models were visualized using the molecular graphics program Chimera (Pettersen et al., 2004).
GABAA receptor expression constructs
Complementary DNA (cDNA) was generated from human brain using the Superscript III First-Strand Synthesis System Supermix III (Clontech Cat no. 18080-400). Gene-specific primers (supplementary information) were used to amplify each GABAA receptor subunit. The cDNAs encoding human GABRA1, GABRB2 and GABRG2 were cloned into the expression vector pcDNA3.1/V5-His TOPO TA vector (Invitrogen Cat no. 45-0005). The GABAA receptor tagged or untagged cDNAs and minigenes used for flow cytometry, biochemistry, electrophysiology and confocal microscopy have been described before (Kang et al., 2009). In the transfection of α1, β2 and the mutant γ2 subunits with different ratios, the total amount of cDNA was normalized by the empty vector pcDNA. The GABRG2(p.R136*) mutation was generated using the QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing. In this paper all mutations were identified in the immature peptide, which has been the convention in the literature for α1, β3 and δ subunit mutations and variants but not for γ2 subunit mutations. In most reports, γ2 subunit mutations have been identified in the mature peptide. For example, the γ2 subunit mutation p.K328M has been reported as the p.K289M mutation (the signal peptide is 39 aa long).
Electrophysiology and the surface protein biotinylation
Lifted whole-cell recordings were obtained from transfected HEK 293T cells as previously described (Kang and Macdonald, 2004). Cells were voltage-clamped at -50 mV and ECl was 0 mV. Zinc (10 μm) was co-applied with GABA (1 mM) after pre-application for 6 sec. The cells were washed with control solution for 45 sec after zinc and GABA co-application, and only currents that returned to baseline were included in the analysis. The GABAA receptor subunit surface biotinylation was performed as previously described (Kang and Macdonald, 2004).
Cell culture, magnetofection and immunocytochemistry
PC12 cells were grown in DMEM with sodium pyruvate, pyridoxine and without L-glutamate (Gibco #21969-035) containing 10% foetal bovine serum (Gibco #10106-110), 5% horse serum (Sigma #H1270), 1x penicillin, streptomycin, and L-glutamate (Sigma #G1146) and 1% 1M HEPES, pH 7.4 (Fisher #H/2230/48). The cells were plated onto collagen-coated glass coverslips and 24 hours later were transfected using a magnetofection protocol (Buerli et al., 2007). Sixty hours after magnetofection, the medium was replaced with differentiation medium (DMEM with 1% horse serum, 1× penicillin, streptomycin, and L-glutamate and 1% 1M HEPES, pH 7.4) containing 100 ng/ml nerve growth factor (NGF, Calbiochem #480354) inducing neuronal differentiation (Shafer et al., 1991a, 1991b).Seventy-two hours later, PC12 cells were fixed with ice-cold 100% methanol for 3 minutes, followed by washing with PBS and permeabilization for 10 minutes at room temperature using 0.5% Triton-X 100 in PBS. Fixed cells were rinsed in PBS and incubated for 10 minutes at room temperature in blocking solution (0.1% Triton-X 100 in PBS and 2% BSA). Cells were incubated for 90-120 minutes at room temperature with the following primary antibodies diluted in blocking solution: rabbit polyclonal anti-γ2 subunit (Sigma #G0545, diluted 1:50), and mouse monoclonal anti-β2,3 subunit (bd17, Chemicon #MAB341, diluted 1:75). Cells were subsequently rinsed and incubated with secondary antibody coupled to AlexaFluor 488 or 568 (Molecular Probes, diluted 1:400) for 30-60 minutes at room temperature in blocking solution. Cells were dried, mounted in DAPI (Vector laboratories) and stored at 4°C. The Cos-7 cells or rat cortical neurons were cultured and transfected with γ2YFP or γ2(p.R136*)YFP using the Ca2+-phosphate precipitation method. Live Cos-7 cells were visualized at 2 days after transfection. Live neurons were transfected at day 7 in culture and were directly visualized at 8 days after transfection (15 days in culture) (Kang et al., 2010). All samples and controls were analysed and imaged by confocal laser-scanning microscopy using a Zeiss LSM510 META confocal system.
Results
Molecular Genetics
A novel GABRG2 mutation was identified in a borderline GEFS+/GGE family (Fig. 1A) and no SCN1A variation was identified beyond known SNPs. A novel heterozygous GABRG2 mutation was generated by a C>T substitution at position 406 in the nucleotide sequence, (c.406C>T), and introduced a premature stop codon (TGA) at a highly-conserved arginine residue at position 136 in the immature GABRG2 polypeptide sequence (p.R136*). This results in a truncated GABRG2 protein with loss of all four transmembrane domains and C-terminus and stunted retention of the N-terminus elements (Fig. 1B,C). This novel GABRG2(p.R136*) mutation was subsequently found in three other nuclear family members segregating strongly with the FS component of the clinical presentation (Fig. 1D). The GABRG2 c.406C>T genotype joins a small compendium of other GABRG2 mutants (Fig. 1E) in the literature and was not present in 190 healthy unrelated control samples or database resources.
Figure 1. A novel GABRG2 gene-mutation.
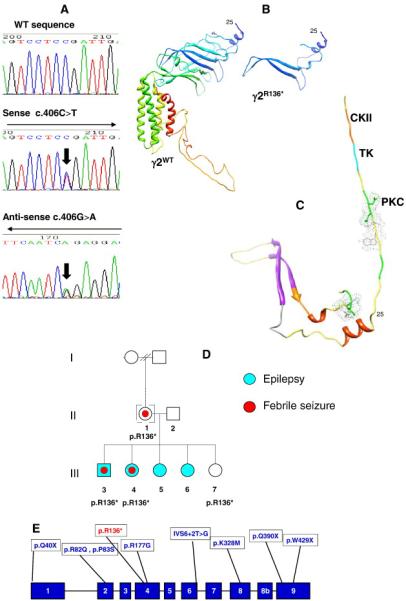
A) Sequencing of exon 4 of GABRG2 in a control (a) and affected index -case DNA (b+c) reveals a new GABRG2 gene-mutant (c.406C>T) resulting in a nonsense mutation outcome - GABRG2(p.R136*). This is illustrated by the double nucleotide peak and marked with an arrow; B) Structural modelling of γ2 WT illustrating the position of R136 and posttranslational modification sites of the truncated GABRG2(p.R136*) subunit. Residues 1-25 could not be reliably modelled due to a lack of coverage for this section by homologues in the Protein Data Bank. The truncated GABRG2(p.R136*) subunit section composes several sections of beta turn, two short regions of alpha helix, retention of functional domains, and two beta strands. C) Structural modelling of γ2 WT illustrating the posttranslational modification sites of the truncated GABRG2(p.R136*). CKII = casein kinase II phosphorylation (orange); TK = tyrosine kinase phosphorylation (cyan); PKC = protein kinase C phosphorylation (green); and the side chains for N-glycosylation and N-myristoylation are shown. D) Pedigree structure and outcomes of GABRG2 sequencing. A pedigree of the nuclear GEFS+ family harbouring the novel GABRG2(p.R136*) mutation and the affection status for epilepsy (cyan) or febrile seizures (red dot) indicated. E) Spectrum of known mutations in GABRG2 relative to the novel GABRG2(p.R136*) mutation. An exonic representation of the GABRG2 gene illustrates the relative positions of the GABRG2(p.R136*) mutation to previously-identified GABRG2 mutations.11-21
Clinical Segregation Analysis
From the family pedigree and clinical characteristics (Fig. 2, Table 1), the GABRG2(p.R136*) mutation does not segregate purely with those clinically-affected with epilepsy. Further analysis through seizure-type endophenotyping was performed considering six main seizure presentations including FS, afebrile seizures, generalised tonic-clonic seizures, absence seizures, myoclonic jerks and eyelid myoclonia. Endophenotyping of GABRG2(p.R136*) suggested that the mutation segregates most closely with presentation of FS (Fig. 2).
Figure 2. Endophenotyping of the GEFS+ Family and the segregation with the GABRG2 p.R136* mutation.
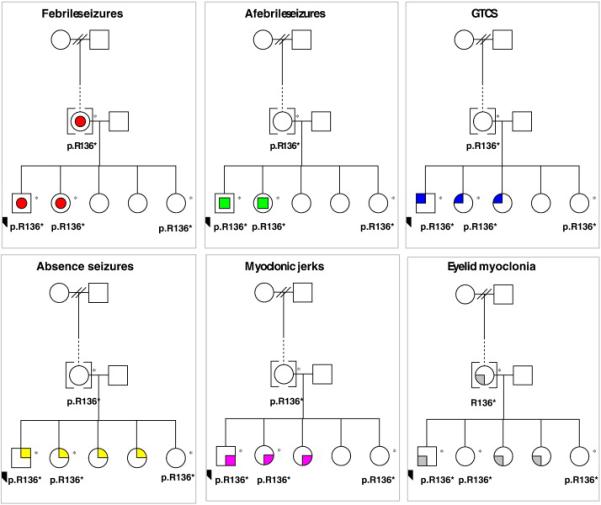
Each presenting phenotype within the multiplex family is represented in a panel, including febrile seizures (red circles), afebrile seizures (green squares), generalised tonic-clonic seizures (GTCS – blue quarter), absence seizures (yellow quarter), myclonic jerks (magenta quarters), and eyelid myoclonia (grey quarters).
Table 1.
Clinical characteristics of the nuclear family members.
| Pedigree Reference |
Age† | Neurological Examination |
Epilepsy Diagnosis |
Seizure Onset |
Seizure type | EEG | RI | Treatment | Other diagnoses |
GABRG2 (p.R136*) |
|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | 33 | N | No | NK | FS, unw EyeM | Not done | ND | - | Migraine | Positive |
| II-2 | 41 | N | No | - | - | Not done | ND | - | - | Negative |
| III-3 | 8 | LD | Yes | 9 months | A, FS+, GTCS, MJ, EyeM | Atypical SW, GSW | AD | VAL LEV | Autistic Spectrum | Positive |
| III-4 | 7 | LD | Yes | 18 months | A, FS, FS+, GTCS, MJ, Uncl sz | Background SW+spikes | ND | VAL LEV | Autistic spectrum | Positive |
| III-5 | 6 | LD | Yes | 24 months | A, GTCS, EyeM, MJ, Uncl sz | Background SW+spikes | ND | CBZ LEV | Educational needs | Negative |
| III-6 | 2 | N | Yes | 12 months | A, CPS, EyeM, Uncl sz | ND | AD | CBZ | - | Negative |
| III-7 | 19 months | N | No | - | - | ND | ND | - | - | Positive |
FS = febrile seizures; FS+ = febrile seizures plus; A = absences; CPS = complex partial seizures; DA = drop attacks; EyeM = eyelid myoclonia; GTCS = generalised tonic -clonic seizures; MJ = myoclonic jerks; LD = learning difficulties; GSW = generalised spike and wave; SW = slow waves; CBZ = carbamazepine LEV = levetiracetam; VAL = sodium valproate; N = normal; NAD = no abnormality detected; NK = not known; ND = Not Done; Uncl = unclassified; unw = unwitnessed
in years at study onset.
Clinical Summaries of the Affected Family Members
Individual II-1 has a history of childhood seizures suggestive of FS and atypical eyelid myoclonia. Suffers from Migraine.
Individual II-2 is not clinically affected.
Individual III-3 is the index case harbouring the GABRG2 p.R136* mutation and suffers from generalised epilepsy, not typical of classical GGE. He has a history of FS and afebrile GTCS seizures. He also has intellectual disability and autism spectrum disorder (ASD). Educationally he requires a high level of one-to-one support. He is treated with sodium valproate and levetiracetam.
Individual III-4 has an unclassified generalised epilepsy with moderate to severe intellectual disability and ASD. She has an educational statement and is in special needs schooling. She is treated with sodium valproate and levetiracetam.
Individual III-5 has an unclassified generalised epilepsy and mild intellectual disability. She also has an educational statement at school and requires full time learning support; her main difficulties are in literacy and numeracy skills. She is treated with carbamazepine and levetiracetam.
Individual III-6 has an unclassified generalised epilepsy. She has been assessed by a community paediatrician and had a Connors scale assessment, however, does not meet with the criteria for ASD or attention deficit hyperactivity disorder (ADHD). She is treated with carbamazepine.
Individual III-7 has joint hypermobility and mild delay in motor skills but does not have epilepsy at present.
All of the children with epilepsy continue to have multiple seizures and are so far refractory to medication despite trying different AEDs, alone or in combination.
Molecular Modeling and Bioinformatics
The full-length 467aa wild-type GABRG2 subunit and the novel mutant GABRG2(p.R136*) subunit were modelled by 21% homology with the crystal structure of the nicotinic acetylcholine receptor (Fig. 1C).31 N-terminal regions are characteristically highly flexible, and the modelled structure of residues 1-97 appear highly unravelled. Residues 1-25 could not be reliably modelled due to a lack of coverage for this section by homologues in the PDB. The truncated mutant would be expected to run to the middle of the main beta sheet of the N-terminal region. Bioinformatic analysis of the mutant γ2(p.R136*) subunit demonstrated an ER retention signal KKXX-like motif (IKVL) in its C-terminal section in contrast to wild-type subunits. The truncated protein following the signal cleavage site was also rich in sites for at least 9 potential posttranslational modifications including 2 N-glycosylation sites (13-16 and 90-93), 3 protein kinase C phosphorylation sites (12-14, 19-21, 92-94), 2 casein kinase II phosphorylation sites (3-6 and 81-84), one tyrosine kinase phosphorylation site (2-10) and one N-myristoylation site (47-52).
In vitro Functional Analysis of the γ2(R136X) Subunit
The cellular fate of mutant γ2(p.R136*) subunits was studied using both recombinant subunit cDNAs and nonsense-mediated mRNA decay (NMD) competent minigene strategies. Since nonsense mutations in early exons may activate NMD and the R136X mutation is located in one of the early exons, it could activate NMD to degrade the mutant γ2 subunit mRNA. We thus included a minigene strategy to study the γ2(p.R136*) subunit mutation as previously used for γ2 subunit truncation mutations.22 We have demonstrated in our previous study that the minigene strategy is feasible to study GABAA receptor truncation mutations because it contains an intron that is required for mRNA processing if nonsense-mediated mRNA decay is involved.
Mutant γ2(R136X) subunits were produced but were not trafficked to the cell surface
HEK 293T cells were co-transfected with α1, β2 and wild-type γ2SHA or mutant γ2S(p.R136*)HA subunit cDNAs. Surface expression of wild-type γ2SHA or mutant γ2S(p.R136*)HA subunits was measured with the quantitative method of flow cytometry using Alexa 647 fluorophores (Fig. 3A). Mutant γ2S(p.R136*)HA subunits only had minimal surface-expression relative to wild-type γ2SHA subunits (0.05 ± 0.04% of wild-type fluorescence) (Fig. 3B). The lack of surface expression of mutant γ2S(p.R136*)HA subunits could be due to compromised subunit trafficking or due to activation of NMD by the p.R136* PTC since it was located in an early exon. We then studied the total protein expression of γ2(p.R136*) subunits with both intronless cDNAs and intron-containing minigenes. We transfected α1 and β2 with γ2SHA or γ2S(p.R136*)HA subunits in cDNAs (Fig. 3C, left) and with γ2 or γ2(p.R136*) subunit minigenes (Fig. 3C, right). As expected, wild-type γ2HA subunits migrated at about 50 KDa. Interestingly, mutant γ2S(p.R136*HA subunits migrated in the 10-30 KDa area as multiple bands. The molecular mass of the mutant truncated γ2(p.R136*) subunit was predicted to be 11.8 KDa after signal peptide cleavage. The nature of these multiple bands is unclear but is likely to be due to different glycosylation forms of γ2(p.R136*) subunits as monomers or dimers. Further study is required to determine whether mutant γ2(p.R136*) subunits dimerize or form oligomers as observed in other γ2 truncated subunits (Kang and Macdonald, 2009). Nevertheless, compared to wild-type subunits, levels of both mutant γ2S(p.R136*)HA subunit cDNA (1 vs 0.67±0.05, n = 4) and γ2(p.R136*) subunit minigene (1 vs 0.42 ± 0.02, n = 4) were reduced (Fig. 3D).
Figure 3. The mutant γ2S(p.R136*) subunit protein was produced and presented as multiple bands but had minimal surface expression.
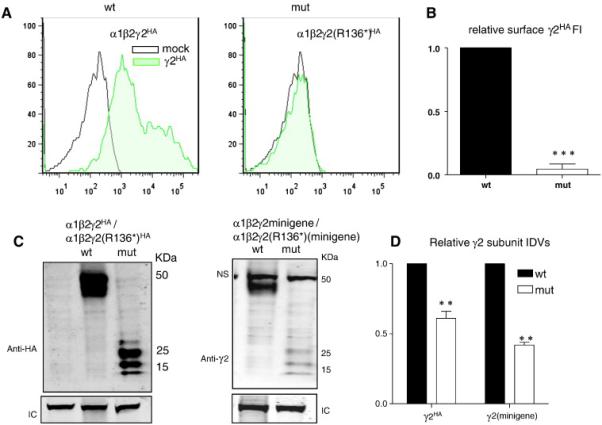
HEK 293T cells were co-transfected with α1 and β2 subunits and γ2SHA(wt) or γ2S(p.R136*)HA (mut) subunits or co-transfected with α1 and β2 subunits and γ2 (wt) or γ2(p.R136*) (mut) minigenes. A) The flow cytometry histograms depict surface expression levels of wild-type γ2SHA or mutant γ2S(p.R136*)HA subunits from HEK 293T cells expressing either α1, β2 and γ2SHA or α1, β2 and γ2S(p.R136*)HA subunits. The surface wild-type and mutant γ2S subunits were fluorescently conjugated with anti-human HA antibody (HA–Alexa Fluor-647). B) The relative fluorescence intensity of HA signals of from mutant γ2S(p.R136*)HA subunits were normalized to those obtained with wild-type γ2SHA subunits. C, D) The total lysates of HEK 293T cells expressing α1, β2 and γ2SHA subunits from expression of cDNAs (C, left panel) or minigenes (C, right panel) were analyzed by SDS-PAGE. LC stands for loading control. NS stands for nonspecific band. D) The total IDVs of γ2SHA subunit cDNA or minigene transfections were quantified, and the data were presented after normalized to the total wild-type γ2HA or γ2 subunits obtained with expression of minigenes (In B and D, **p < 0.001 ***p < 0.001 vs wt).
Mutant γ2(p.R136*) subunits did not assemble into functional receptors, and the receptors formed were likely α1β2 receptors
γ2 subunits are not required for GABAA receptor assembly (Angelotti et al., 1993a). When γ2 subunits are absent or have impaired trafficking, α1β2 receptors will form that have smaller channel conductance and current amplitude and different pharmacological properties (Angelotti and Macdonald, 1993b). We compared the current amplitudes from HEK 293T cells transfected with α1, β2 and γ2 subunit or α1, β2 and γ2(p.R136*) subunit cDNAs in a 1:1:1 cDNA ratio (Fig. 4A). Peak amplitudes of α1β2γ2S(p.R136*) receptor currents (907 ± 238, n = 14) were smaller than those of wild-type currents (4563 ± 467, n = 9) (Fig. 4B). As demonstrated in Figure 3, mutant γ2S(p.R136*) subunits had minimal surface-expression, suggesting that currents recorded with co-expression of α1, β2 and γ2S(p.R136*) subunits were likely from α1β2 receptors, which are very sensitive to zinc inhibition. With 6 sec co-applications of GABA (1 mM) and zinc (10 μM), peak amplitudes of currents recorded from receptors formed with wild-type α1, β2 and γ2 subunits had minimal reduction compared with control currents (Fig. 4C) (7.2% ± 3.4%, n = 4). However, currents recorded from receptors formed with α1, β2 and γ2(p.R136*) subunits were almost abolished with co-application of GABA (1 mM) and zinc (10 μM) (93.4% ± 17%, n = 4, suggesting that α1β2 receptors are the major receptor present with co-expression of α1, β2 and γ2(p.R136*) subunits. The formation of α1β2 receptors was further supported by the evidence that the α1 subunit surface expression was not altered in the mutant α1β2γ2(p.R136*) receptors compared with the wildtype α1β2γ2 receptors (Figure 4D, E). This also suggests that there is no dominant negative suppression from the mutant γ2(p.R136*) subunits on the wildtype partnering subunits like α1. With the increased mutant γ2(p.R136*) subunits, the total expression of α1 and β2 subunits was reduced at the ratio of 1:1:5 (α1:β2:γ2(p.R136*)) (Figure 4F to I), suggesting the dominant negative effect of the mutant γ2(p.R136*) subunits on the wildtype partnering subunits only at a high amount. This dominant negative suppression of γ2(p.R136X) subunit at high amounts was smaller compared with γ2(Q390X), another mutant subunit associated with a more severe epilepsy phenotype Dravet syndrome (Kang et al., 2009; Harkin et al., 2002) (Figure 4F to I).
Figure 4. Current amplitudes recorded with co-expression of α1 and β2 subunits with mutant γ2S(p.R136*) subunits had reduced peak current amplitudes and were more sensitive to zinc inhibition while the muant γ2S(p.R136*) subunits reduced the α1 and β2 subunit expression at high amounts.
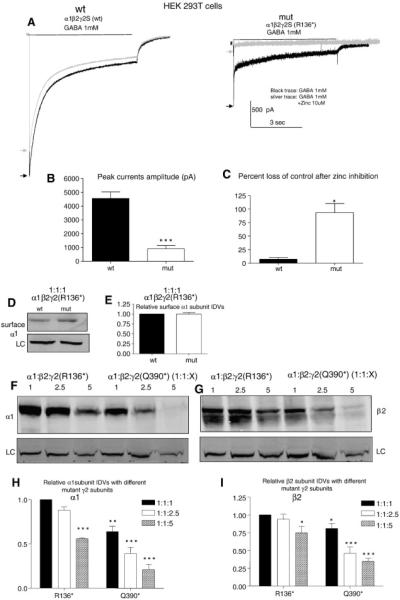
A,B) GABAA receptor currents were obtained from HEK 293T cells expressing wild-type α1 and β2 subunits with γ2S (1:1:1 cDNA ratio; wt) or γ2S(p.R136*) (1:1:1 cDNA ratio, mut) subunits with application of 1 mM GABA applied for 6 sec. The representative traces of GABAA receptor currents were evoked with application of 1 mM GABA for 6 sec (black trace, black arrow) and co-application of 1mM GABA with 10 μM zinc after 6 sec pre-application of 10 μM zinc (silver traces, silver arrow). (B) The amplitudes of GABAA receptor currents from (A) were plotted. Values were mean ± SEM (n = 9-14 patches from 4 different transfections). C) The percent loss of control current or reduction of current amplitude after coapplication of zinc and GABA was presented by the currents evoked by GABA subtracting the currents evoked by GABA and zinc (*p<0.05***p < 0.001 vs wt, n= 4). D,E). The surface α1 subunit was extracted from HEK 293T cells expressing wild-type α1 and β2 subunits with γ2S (1:1:1 cDNA ratio; wt) or γ2S(p.R136*) (1:1:1 cDNA ratio, mut) subunits by surface biotinylation and analyzed by SDS-PAGE. The membranes were immunoblotted with a mouse anti- α1 antibody. (E). The relative amount of the α1 subunit expression in the mutant α1β2γ2S(p.R136*) receptors was normalized to the wildtype, which is arbitrarily taken as 1. F, G, H, I). The total lysates from HEK 293T cells expressing wild-type α1 and β2 subunits with γ2S(p.136*) or γ2S(p.Q390X) at 1:1:1, 1:1:2.5 and 1:1:5 cDNA ratio were harvested and analysed by SDS-PAGE. The membranes were then immunoblotted with either mouse anti-α1 or rabbit anti β2 antibody. The relative amount of the α1 or β2 subunit expression in other conditions were normalized to the α1β2γ2S(p.R136*) receptors at 1:1:1 which was arbitrarily taken as 1 ( in H and I,*p<0.05; **p < 0.01 ;***p < 0.001 vs α1β2γ2S(p.R136*) at 1:1:1, n= 4).
After co-expression of α1, β2, and wild-type γ2 or mutant γ2(p.R136*) subunits in PC12 cells, we determined the surface and intracellular distribution of γ2(p.R136*) and γ2 subunits by co-labelling with anti-γ2 (red) and anti β2,3 antibodies (green) and using confocal microscopy. Wild-type, γ2 subunit immuno-fluorescence had a smooth uniform distribution with clusters that could be detected mainly on the surface (cell membrane), with less prominent intracellular staining (Fig. 5 A, B) and co-localizing with β2 subunit immunoreactivity (Fig. 5C, D). Expression of γ2(p.R136*) subunits resulted in less cell-surface immunoreactivity and more diffuse labelling (Fig. 5E). We also observed the presence of intracellular aggregates, consistent with staining within ER and peri-nuclear regions (Fig. 5F-H).
Figure 5. Expression analysis of γ2(p.R136*) in vitro using differentiated PC12 cells.
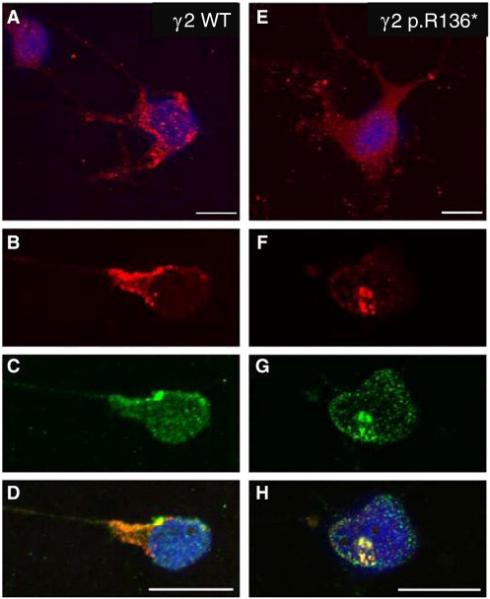
GABAA receptor subunits α1, β2 and γ2-WT (A-D) or γ2 p.R136* mutant (E-H) subunits were transiently co-transfected in PC12 cells, and immunocytochemically stained with anti-γ2 and anti-β2,3-antibodies as analysed by confocal microscopy. α1β2γ2 receptor immunoreactivity (IR) revealed an even distribution with punctuate staining on cell-surface, less prominent intra-cellular staining (A, B) and co-localized with GABAAR β2-IR (C, D). Expression of α1β2γ2(p.R136*) receptors resulted in more diffuse labelling with little cell-surface IR (E) and was predominantly localised within intracellular domains (F), whilst retaining co-labelling patterns for GABAAR β2-IR (G, H). Scale bars = 20 μm.
Consistent to what has been observed in PC12 cells, the coexpression of α1, β2 and wild-type or the mutant γ2(p.R136*) subunits tagged with the yellow fluorescent protein (YFP) in COS-7 had similar expression pattern as seen in PC12 cells. The subcellular localization of α1β2γ2YFP receptors in COS-7 cells were determined with confocal microscopy 2 days later after transfection. Similar to the the observation in PC 12 cells, wild-type γ2YFP subunits were present both intracellularly and on the cell-surface while mutant γ2(p.R136*)YFP subunits were localized around the nuclear region in aggregated fluorescence dots without cell surface-expression (Fig. 6A). Total fluorescence intensity was lower for mutant γ2(p.R136*)YFP subunits than for wild-type γ2YFP subunits (0.61 ± 0.077 vs 1). We then transfected the wild-type γ2YFP or mutant γ2(p.R136*)YFP subunits in cultured cortical neurons and visualized the neurons 8 days after transfection (Fig. 6B). Wild-type γ2YFP subunits had widespread distribution in both somata and neuronal processes including dendrites and axons while mutant γ2(p.R136*)YFP subunits were distributed only in neuronal somata. Compared with wild-type γ2YFP subunits, the fluorescence intensity ratio (process divided by soma fluorescence intensities) was much lower in neurons expressing γ2(p.R136*)YFP subunits (1.4 ± 0.063 vs 0.09 ± 0.05). However, both the wild-type and the mutant γ2 subunits appeared as puncta in neurons.
Figure 6. γ2S(p.R136*)YFP subunit protein was reduced and confined to intracellular region or soma in live COS-7 cells and cortical neurons.
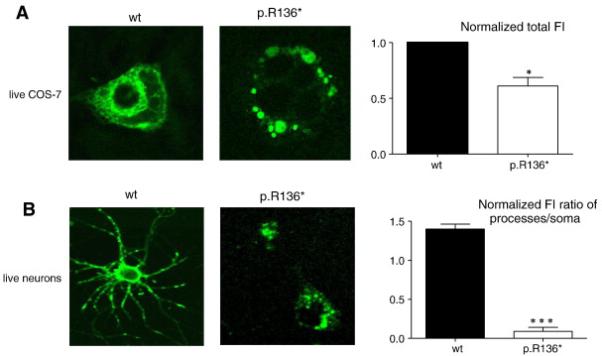
COS-7 cells or cultured cortical neurons were cotransfected with wild-type γ2SYFP (wt) or mutant γ2S(p.R136*)YFP (mut) subunits with (in COS-7 cells) or without (in neurons) α1 and β2 subunits using the calcium phosphate precipitation method with different amounts of subunit cDNAs. Images were acquired 2 days (COS-7) or 8 days (neurons) after transfection. The total fluorescence intensities of cells were measured using the MetaMorph imaging software. In the upper panel, the total fluorescence from COS-7 cells expressing wild-type γ2SYFP subunit-containing receptors (wt) was arbitrarily taken as 1, and the total fluorescence from cells expressing mutant γ2S(p.R136*)YFP subunit-containing receptors (mut) was normalized to wt levels. In lower panel, the total fluorescence of both neuronal somata and processes (including both axon and dendrites) were measured, and the fluorescence intensity ratios of the areas of processes over the somata were quantified. (n = 8 for COS-7 and for neurons from three different transfections; *p < 0.05; ***p < 0.001 vs wt).
Discussion
Families with evidence of GEFS+ or FS have been collected, from which a panel of index-cases were assembled. By mutation screening the coding regions of SCN1A and GABRG2, we discovered a novel p.R136* PTC mutation in the γ2 subunit of the GABAA receptor segregating with the familial FS endophenotype. The in vitro functional characterisation of the novel GABRG2(p.R136*) mutation provides cellular evidence in support of GABAA receptor functional impairment. The co-expression of mutant γ2(p.R136*) subunits with wild-type α1 and β2 subunits, demonstrated that receptors formed had reduced current amplitudes and reduced cell surface expression levels with greater intracellular retention compared to wild-type γ2 subunits. This suggests that the GABRG2(p.R136*) mutation impairs receptor channel function and alters receptor trafficking. The increased zinc sensitivity of currents recorded with co-expression of the mutant γ2(p.R136*) subunit with wild-type α1 and β2 subunits suggests that α1β2 receptors were trafficked to the surface rather than α1β2γ2(p.R136*) receptors. A similar observation of α1β2 receptor formation has been made in other γ2 subunit truncation mutations (Kang, et al., 2013) This is also consistent with the previous finding that the formation of αβ receptors in the GABRG2 heterozygous knockout condition (Crestani et al., 1999). Furthermore, γ2(p.R136*) subunit immunoreactivity was localised to the nuclear membrane and the ER providing evidence to support the bioinformatic structural model that predicted a novel ER retention motif in the C-terminus of the γ2(p.R136*) subunit.
The functional assays suggested that co-expression of the mutant γ2(p.R136*) subunit rather than the wild-type γ2 subunit resulted in reduced channel function due to impaired incorporation of γ2(p.R136*) subunits into receptors trafficked to the cell-surface. What is the basis of the reduced mutant subunit surface-expression? The structural model suggests that the signal peptide, possessed by both wild-type and mutant subunits, would traffic both proteins to the ER where following enzymatic cleavage of the 39 amino-acid long signal peptide, the 97 amino acid polypeptide would remain. Wild-type γ2 subunits would thereafter be oligomerized, assembled into receptors and trafficked to the cell-surface membrane to form functional GABAA receptors. However, the putative KKXX-like retention motif would retain mutant γ2(p.R136*) subunits within the ER, leading to accumulation and eventual degradation of the truncated subunits. Short linear sequences similar to this motif, which occupy the extreme COOH-terminal position of transmembrane ER proteins, have been implicated as retention signals (Nilsson et al., 1989). This suggests that despite potential instability, the γ2(p.R136*) subunit polypeptide may accumulate appreciably in the ER. Bioinformatic analysis also demonstrated that the truncated γ2(p.R136*) subunit following the signal cleavage site would be rich in posttranslational modification sites and given the predicted unravelled structure of the γ2(p.R136*) subunit, it is likely that all of these posttranslational modification sites would be accessible (Fig. 1D).
The in vitro experiments also suggest that γ2(p.R136*) subunits have the capacity to assemble with β2 subunits. The co-localisation of γ2 and β2,3 subunit immunoreactivity, which was especially prominent in the intracellular retentions, suggests that γ2(p.R136*) subunits may oligomerize with β2 subunits, which are potentially retained. It is known that N-terminal sequences are critical for the proper assembly of GABAA receptors and arginine in particular has been reported to be involved in the regulation of ER export (Kang and Macdonald, 2004). The γ2 subunit is also considered to be critical for receptor trafficking, clustering and synaptic maintenance (Crestani et al., 1999). Therefore, it is likely that any disruption of the γ2 subunit would have consequences potentially on the surface-expression of other GABAA receptor subunits and subsequently impair GABAergic inhibition and lower seizure threshold. The impaired GABAA receptor function is due to the loss of the mutant γ2(p.R136*) subunits on the cell surface and synapse and potentially due to the reduced functional wild-type subunits and altered receptor composition.
Most GABAA subunit missense mutations impair surface-expression of GABAA receptors or impair channel kinetics. Similar in vitro experimental outcomes of impaired or altered receptor trafficking are observed with other GABRG2 mutations, including K328M and Q390X found in GEFS+ and Dravet syndrome families and the GABRG2 mutation R82Q found in a family with CAE and FS (Harkin et al., 2002; Kang et al., 2006). In both COS cells and hippocampal neurons, the GABRG2(p.R82Q) mutation reduced cell-surface subunit expression and exhibited impaired temperature-dependent trafficking (Frugier et al., 2007). Cell-surface subunit expression was also reduced with the nonsense GABRG2(p.Q390X) mutation, with ER retention of the mutant protein, no GABA-evoked currents recorded from injected oocytes and reduced GABA-evoked current in transfected HEK 293T cells (Kang et al., 2009). The GABRG2(p.Q40X) and GABRG2(p.W429X) nonsense mutations cause PTCs and may trigger NMD and cause epilepsy by haploinsufficency. The GABRG2(IVSG+2T-G) splice donor site mutation also has the potential to trigger NMD and cause epilepsy by haploinsufficency (Kananura et al., 2002).
The heterozygosity of the GABRG2(p.R136*) mutation would be expected to reduce the number and density of functional GABAA receptor complexes at inhibitory synapses because the mutant γ2(p.R136*) subunits were retained inside somata regions and could not be trafficked to dendrites and synapses as shown in Figure 5B. This potentially would cause a decrease in inhibitory responses to GABA, giving increased neuronal excitability and susceptibility to seizures in individuals harbouring the mutation. However, we cannot rule out the possibility that in vivo expression of both wild-type and mutant γ2 subunits may differ from the in vitro effects observed. Functional analysis of the previously-identified GABRG2(p.Q390X) mutation showed a dominant-negative suppression of the biogenesis of wild-type γ2 subunits (Kang et al., 2006; 2009). We suggest that similar cellular control mechanisms may affect γ2(p.R136*) subunit expression.
The discovery of this novel GABRG2(p.R136*) mutation extends the spectrum of GABGR2 mutations to a wider GGE phenotype. Within the studied family there was a broad-range of seizure types including FS, afebrile seizures, generalised tonic-clonic seizures, absence seizures, myoclonic jerks and eyelid myoclonia, with marked interfamilial phenotypic variability – which is typical of GEFS+. Given the small family size and the adoption of individual II-1, it is difficult to draw conclusive genotype-phenotype correlations; however, consistent with previous findings, the GABRG2(p.R136*) mutation segregated most closely with FS. The epilepsy trait in this family cannot simply be explained by autosomal dominant inheritance. The presence of two epilepsy phenocopies in the nuclear family (Fig. 2 individuals III5 and III6); individuals diagnosed with epilepsy yet who tested negative for the GABRG2(p.R136*) mutation, serves to illustrate that the mutation does not exert a pure dominant effect. Phenocopies, although not typical of GABGR2 gene-positive families, have previously been described (Wallace et al., 1998; Baulac et al., 2001; Harkin et al., 2002). The asymptomatic individual in this studied family harbouring the novel GABRG2(p.R136*) mutation (Fig. 2; individual III7) illustrates that the mutation is either partially-penetrant or potentially that the individual has not been exposed to the necessary environmental triggers to cause seizures, such as an appropriate temperature change to potentially impair receptor subunit trafficking. Since this study was a focussed candidate gene approach, it is possible that GABRG2 is only one of several genes in interplay in this family and that genetic loading across several loci is producing a varied phenotype and skewed segregation analysis. Nevertheless, we add here the in-depth analysis of a rare GABRG2 PTC mutation that has at best a major effect on the affected cases in the family or at least is contributory to the overall phenotypic spectrum.
Acknowledgements
We wish to thanks and acknowledge the families from across Wales and from further afield, who willingly participated in this gene discovery project. This study was funded from the National Institute of Social Care and Health Research (NISCHR) to MIR and PEMS, the Waterloo Foundation to MIR, Epilepsy Research UK (ERUK) Fellowship to SKC, research grants from CURE and National Institute of Health (NS082635) to KJQ and a National Institutes of Health grant (NS33300) to RLM.
Abbreviations
- CAE
childhood absence epilepsy
- EEG
electroencephalogram
- ER
endoplasmic reticulum
- FS
febrile seizures
- GEFS+
generalised epilepsy with febrile seizures plus
- GGE
genetic generalised epilepsy
- JME
juvenile myoclonic epilepsy
- PTC
premature translation termination codon
- GABRG2
γ2 subunit gene of the γ-aminobutyric acid type A receptor
Footnotes
Competing Interests
All authors have confirmed that there are no competing interests in connection to this work.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Angelotti TP, Uhler MD, Macdonald RL. Assembly of GABAA receptor subunits: Analysis of transient single cell expression utilizing a fluorescent substrate/marker gene combination. J. Neurosci. 1993a;13:1418–1428. doi: 10.1523/JNEUROSCI.13-04-01418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits: α1β1 and α1β1γ2S subunits produce unique ion channels with dissimilar single-channel properties. J Neurosci. 1993b;13:1429–1440. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 2001;1:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross HJ, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Buerli TPC, Baer K, Lardi-Studler B, Chudotvorova I, Fritschy JM, Medina I, et al. Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat. Protoc. 2007;2:3090–3101. doi: 10.1038/nprot.2007.445. [DOI] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: Network Protein Sequence Analysis. TIBS. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Lüscher B, Mohler H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci. 1999;2:833–9. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum. Mol. Genet. 2004;13:1315–1329. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- Engel E. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;49:796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- Engel J. Report of the ILAE Classification Core Group. Epilepsia. 2006;47:1558–1568. doi: 10.1111/j.1528-1167.2006.00215.x. [DOI] [PubMed] [Google Scholar]
- Escayg AMB, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Essrich CLM, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, et al. Tools for comparative protein structure modeling and analysis. Nucleic. Acids. Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier G, Coussen F, Giraud MF, Odessa MF, Emerit MB, Boué-Grabot E, et al. A gamma2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell-surface. J. Biol. Chem. 2007;282:3819–3828. doi: 10.1074/jbc.M608910200. [DOI] [PubMed] [Google Scholar]
- Grunberg R, Nilges M, Leckner J. Biskit -a software platform for structural bioinformatics. Bioinformatics. 2007;23:769–770. doi: 10.1093/bioinformatics/btl655. [DOI] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, et al. Truncation of the GABA(A)-receptor gamma-2 subunit in a family with generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 2002;2:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics. 2007;2:295–304. doi: 10.1016/j.nurt.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Horton P, Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbours classifier. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997;5:147–152. [PubMed] [Google Scholar]
- Johnston JA, Thomas RH, Hammond CL, Morris HR, Smith PEM, Rees MI. A novel GABRG2 mutation in an epilepsy family”. Conference proceedings American Epilepsy Society Boston. 2009 Abst. [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, et al. A Splice-Site Mutation in GABRG2 Associated With Childhood Absence Epilepsy and Febrile Convulsions. Arch. Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABA A receptor gamma2 subunit mutations associated with idiopathic generalised epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci. 2006;9:2590–2597. doi: 10.1523/JNEUROSCI.4243-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. Making sense of nonsense GABA(A) receptor mutations associated with genetic epilepsies. Trends. Mol. Med. 2009;19:430–438. doi: 10.1016/j.molmed.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J. Neurosci. 2004;24:8672–8687. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J. Neurosci. 2009;9:2845–2856. doi: 10.1523/JNEUROSCI.4772-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann. Neurol. 2013 doi: 10.1002/ana.23947. doi: 10.1002/ana.23947 (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M, et al. The γ2 subunit of GABA A receptors is a substrate for palmitoylation by GODZ. J. Neurosci. 2004;24:5881–5891. doi: 10.1523/JNEUROSCI.1037-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ. Molecular pathology of genetic epilepsies associated with GABA(A) receptor subunit mutations. Epilepsy Curr. 2009;1:18–23. doi: 10.1111/j.1535-7511.2008.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini C, Mei D, Temudo T, Ferrari AR, Buti D, Dravet C, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007;9:1678–1685. doi: 10.1111/j.1528-1167.2007.01122.x. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat. Rev. Neurosci. 2001;4:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Nilsson T, Jackson M, Peterson PA. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell. 1989;58:707–718. doi: 10.1016/0092-8674(89)90105-0. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J, Mol, Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera -A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rathenberg JKJ, Moss SJ. Palmitoylation regulates the clustering and cell-surface stability of GABAA receptors. Mol. Cell. Neurosci. 2004;26:251–257. doi: 10.1016/j.mcn.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, et al. The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 2003;24:442–450. doi: 10.1016/s1044-7431(03)00202-1. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. BRAIN. 1997;120:479–90. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- Shafer TJ, Atchison WD. Methylmercury blocks N-and L-type Ca++ channels in nerve growth factor-differentiated pheochromocytoma (PC12) cells. J. Pharmacol. Exp. Ther. 1991a;258:149–157. [PubMed] [Google Scholar]
- Shafer TJ, Atchison WD. Transmitter, ion channel and receptor properties of pheochromocytoma (PC12) cell: a model for neurotoxicology studies. Neurotoxicology. 1991b;12:473–492. [PubMed] [Google Scholar]
- Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann. Neurol. 1999;45:75–81. doi: 10.1002/1531-8249(199901)45:1<75::aid-art13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhang Y, Liang J, Liu X, Ma X, Wu H, et al. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalised epilepsy with febrile seizures plus. J. Hum. Genet. 2008;53:769–74. doi: 10.1007/s10038-008-0306-y. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am. J. Hum. Genet. 2008;6:1249–61. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Mei D, Freri E, Hernandez CC, Granata T, Shen W, Macdonald RL, Renzo G. Impaired surface αβγ GABAA receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol. Dis. 2013;50:135–141. doi: 10.1016/j.nbd.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RH, Johnston JA, Hammond CL, Bagguley S, White C, Smith PE, Rees MI. Genetic epilepsy with febrile seizures plus: definite and borderline phenotypes. J. Neurol. Neurosurg. Psych. 2012;83:336–348. doi: 10.1136/jnnp-2011-300405. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005;346:967–89. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 1998;19:366–70. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Wheeler DLT, Barrett DA, Benson SH, Bryant K, Canese V, Chetvernin DM, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]