

Supplemental Digital Content is available in the text.
Keywords: angiogenesis, gastric cancer, metastasis, mammalian target of rapamycin inhibitor, proliferation
Abstract
Although preclinical work with rapalogs suggests potential in the treatment of gastric cancer, they have been less successful clinically. In this study, we report the impact of the investigational drug PP242, a potent and selective small-molecule active-site TORC1/2 kinase inhibitor, on tumor growth and metastasis. The antiproliferative effect of PP242 was assessed using the Cell Counting Kit-8 assay. The migration and invasion potential were analyzed using wound-healing and transwell assays, respectively. The Matrigel capillary tube formation assay was performed to mimic in-vivo angiogenesis. Immunoblotting and immunofluorescence were used to observe protein levels and distribution of actin fibers. Finally, S2448p-mammalian target of rapamycin (mTOR) expression was detected on gastric cancer tissues using immunohistochemistry. First, PP242 potently inhibited cell proliferation in gastric cancer cell lines and in human endothelial cells in vitro at the IC50 ranged from 50 to 500 nmol/l. Then, an inhibitory effect of PP242 on metastasis was observed in gastric cancer cell AGS, along with the cytoskeletal rearrangements and suppression of the phosphorylation of PI3K downstream factors including AKT, mTOR, and P70S6K. Furthermore, PP242 was found to decrease the tube formation and migration of human umbilical vein endothelial cells. Using immunohistochemistry, we found that S2448p-mTOR staining was observed in 41.8% (82/196) of gastric cancer tissues and correlated with depth of mural invasion, lymph node metastasis, tumor node metastasis stage, and vascular invasion. These results show that PP242 suppresses cell proliferation and angiogenesis of gastric cancer through inhibition of the PI3K/AKT/mTOR pathway, which might be an effective novel therapeutic candidate against gastric cancer in the future.
Background
Gastric cancer is the fourth most common cancer worldwide and the second most common cause of cancer death, with over seven million deaths annually worldwide 1. The poor outcomes suggest a need for targeted agents that may confer a better survival benefit. However, there is hardly any effective targeted therapy, except trastuzumab, in the field of gastric cancer. Considering that the positive expression rate of HER-2 is only 4–28%, only a small proportion of patients could benefit from it 1,2. Thus, it is necessary to find novel targets for gastric cancer therapy. The mammalian target of rapamycin (mTOR) is a serine/threonine downstream mediator in the phosphatidylinositol 3-kinase signaling pathway, which is a central controller of eukaryotic cell growth and plays a critical role in regulating important cellular functions, including proliferation, growth, survival, mobility, and angiogenesis 3,4. Aberrant activation of mTOR signaling has been reported in many cancers including gastric cancer, which could be attributed to several mechanisms including mutational activation of PIK3CA and loss of the phosphatase–tensin homolog deleted from chromosome 10 (PTEN) 5,6. It exists in two distinct complexes – mTORC1 and mTORC2. mTORC1 controls cell autonomous growth in response to nutrient availability and growth factors, whereas mTORC2 is considered to mediate cell proliferation and cell survival 7,8. When active, mTORC1 promotes cell growth by directly phosphorylating the translational regulators ribosomal protein S6 kinase (P70S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1). Phosphorylation of 4E-BP1 inhibits its binding to eukaryotic initiation factor 4E (eIF4E), which enables the cap-dependent translation of multiple mRNAs such as Bcl-2 and vascular endothelial growth factor, thereby increasing cell proliferation, survival, and angiogenesis 9,10. This provides a strong rationale for targeting mTORC1 in cancer and this led to clinical trials of several rapamycin analogues 11. However, the first-generation mTOR inhibitor Everolimus as a single agent was thwarted in advanced gastric cancer patients possibly in part by strong mTORC1-dependent negative feedback loops 7,8. It is reported that the shorter time-to-progression was associated with hyperactivation of Akt in response to rapamycin in PTEN-deficient glioblastoma patients, thus decreasing its beneficial effect as an anticancer drug 12.
Although mTORC1 is in the vanguard of mTOR research, mTORC2 is emerging as a pivotal player in many cancers. It was discovered that mTORC2 directly phosphorylates AKT on ser473, which is required for maximal activation of the antiapoptosis kinase, leading to enhanced cell survival, proliferation, and migration 7. This led to efforts to develop selective ATP-competitive small-molecule mTOR inhibitors with the expectation of completely blocking both mTORC1 downstream effectors and mTORC2 substrates, which may outperform rapamycin analogues as anticancer drugs 7. For these reasons, we initiated this study by testing the potential efficacy of an inhibitor, PP242, which specifically inhibits the mTOR kinase domain and significantly suppresses mTORC2 as well as mTORC1 activity.
Materials and methods
Cell lines and reagent
Gastric cancer cell lines AGS, MKN45, MKN28, KATO3, and N87 were obtained from ATCC (American Type Culture Collection, Manassas, Virginia, USA) and the cell line SGC7901 was established in China and obtained from the Cell Research Institute, Shanghai, China. Human microvascular endothelial cells (HMEC) and human umbilical vein endothelial cells (HUVEC) were purchased from Lonza (Walkersville, Maryland, USA). All cell lines were carefully maintained in a humidified tissue culture incubator at 37°C in 5% CO2. The cell lines were grown in culture media containing supplements as suggested by the suppliers.
PP242 was purchased from BioVision (Zurich, Switzerland). For in-vitro experiments, pp242 was dissolved in dimethyl sulfoxide (DMSO).
Measurement of cell proliferation
Cell viability was assessed using the Cell Counting Kit-8 assay (Dojindo Laboratories, Kumamoto, Japan). Briefly, AGS, N87, MKN28, MKN45, KATO3, SGC7901, HUVEC, and HMEC cells were plated at a density of 3–5×103 cells/well in a 96-well plate for 24 h. The medium was removed, and cells were treated with either DMSO as a control or increasing concentrations of PP242 in a total volume of 100 μl. After the cells had been incubated for 24 or 48 h, WST-8 solution (10 μl) was added to each well and the cultures were incubated at 37°C for 120 min. Absorbance at 450 nm was measured using an immunoreader. Three replicate wells were used for each analysis. The median inhibitory concentration (IC50, defined as the drug concentration at which cell growth was inhibited by 50%) was assessed from the dose–response curves.
Western blotting
Cells were washed three times with ice-cold PBS before lysis. Cells were lysed with buffer containing 1% Triton X-100, 1% NonidetP-40 (NP-40), and the following protease and phosphatase inhibitors: phenylmethylsulfonyl fluoride (1.72 mmol/l), aprotinin (10 mg/ml), leupeptin (10 mg/ml), NaF (100 mmol/l), and Na3VO4 (500 mmol/l). Equal amounts of protein were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes. Immunostaining of the blots was performed using the primary antibodies, followed by the secondary antibody conjugated to horseradish peroxidase and detection by enhanced chemiluminescence reagent (Pierce, Rockford, Illinois, USA). Quantification was performed using Image Lab software version 3.0 (Bio-Rad, Hercules, California, USA).
Primary antibodies were mouse monoclonal antibody phosphorylated P70 [p-P70 (T389), #9206] and rabbit antibodies: total p70S6 kinase (t-P70, #2708), total AKT (t-AKT, #9272), phosphorylated AKT [p-AKT (S473), #4508], total mTOR (t-mTOR, #2972), phosphorylated mTOR [p-mTOR (S2481), #2974], and phosphorylated mTOR [p-mTOR (S2448), #2971] (Cell Signaling Technologies, Danvers, Massachusetts, USA). The secondary antibodies were purchased from Santa Cruz (Dallas, Texas, USA).
Tube formation assay
10 mg/ml (200 μl) of Matrigel (BD Biosciences) was polymerized for 30 min at 37°C in a 24-well plate. HUVECs were suspended in 1640 medium at a density of 3×105 cells/ml and 0.2 ml of cell suspension was added to each well coated with Matrigel, together with or without the indicated concentrations of PP242 for 20 h. The morphological changes in the cells and tubes formed were observed under a phase-contrast microscope and photographed at ×100 magnification. Images were then processed using Adobe Photoshop CS2 9.0.2 (Adobe Systems, San Jose, California, USA) with the Image Processing Tool Kit plug-ins (Reindeer Graphics, Asheville, North Carolina, USA) and analyzed using Angiosys 1.0 (TCS Cellworks, Buckingham, UK). Results from triplicate wells were expressed as mean vessel area per field±SEM (relative units) from four random fields per well. Each assay was performed three times.
Wound-healing assay
AGS and HUVEC cells plated on 60 mm-diameter culture dishes at 90% confluence were wounded with a 200-μl pipette tip and marked at the injury line. After wounding, the peeled-off cells were removed with a serum-free medium and further incubated in a medium with DMSO or various concentration of PP242. The width of this wound line was photographed using an inverted microscope (ECLIPSE TS100; Nikon, Tokyo, Japan) at a 24 h interval. The motility speed of cells was determined using the ImageJ program (National Institutes of Health, Bethesda, Maryland, USA) as an average closed area of the wound relative to the initial wound area at 48 h after wounding.
Tissue samples
The records of a total of 196 gastric cancer patients who underwent surgical resection from February 1998 to January 2007 at Peking University Cancer Hospital were reviewed in the context of clinicopathological and follow-up information. The stage of gastric cancer was classified according to the American Joint Committee on Cancer Stage (7th ed.). The OS was calculated starting from the date of the initial surgery to the time of death, counting death from any cause as the end point or the last date of follow-up as the end point, if no event was documented. All patients were followed up until November 2010. None of the patients received preoperative chemotherapy or radiation therapy. Informed consent was obtained from all of the patients. The study was approved and supervised by the Medical Ethics Committee of Beijing Cancer Hospital/Institute.
IHC
Staining of gastric cancer specimen sections for phosphorylated mTOR was performed with an antibody specific for mTOR when it was phosphorylated on S2448 (clone 49F9 used at a 1 : 50 dilution; Cell Signaling Technologies). Paraffin-embedded sections of 5 μm thickness were deparaffinized with xylene and rehydrated through a graded alcohol series. Endogenous peroxidase activity was then blocked by incubation in 3% hydrogen peroxide–methanol for 10 min. Antigen retrieval was achieved by microwaving (0.1 mol/l citrate buffer, pH 6.0). EnVision (Dako, Carpinteria, California, USA) was used as the secondary antibody. Antibody binding was visualized by a standard streptavidin immunoperoxidase reaction, followed by chromogen detection with diaminobenzidine for 30 s and hematoxylin counterstaining. Immunoreactivity in the membrane was evaluated. Each histological section was examined at ×40 magnification to identify areas of maximum tumor positivity. At ×200 or ×400 magnification, cells were analyzed from five areas of maximum tumor positivity in each case and the average percentage of positive cells was recorded. The specificity of phospho-mTOR staining was validated in serial negative control sections.
Immunofluorescence assay
To determine the effect of PP242 on cell morphology and actin stress fibers, AGS and HUVEC cells (8×104 cells per well) were plated in six-well plates and grown for 24 h so that they attached to the surface of the plates completely. PP242 was added to cells grown at 37°C for 24 h. After the exposure period, media were removed and cells were washed with PBS. Cells were then fixed with 3.7% paraformaldehyde in PBS for 15 min and incubated with 0.1% Triton X-100/in PBS for 30 min. Cells were incubated with 1% BSA in PBS for 30 min (blocking) and then with 70 nmol/l rhodamine phalloidin for 20 min to stain the actin filaments. Nuclei were stained with 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole) in PBS for 1 min and the culture plates were examined and photographed by immunofluorescence microscopy.
Cell invasion assay
Cell invasion assay was carried out using the cytoselect 24-well cell invasion assay (CBA-110; Cell Biolabs, San Diego, California, USA). AGS and HUVEC cells were treated with various concentrations of PP242. After 24 h incubation, cells resuspended in serum-free medium, also containing the corresponding concentrations of PP242, were carefully transferred onto the top chamber of each transwell apparatus at a density of 2×105 cells/ml (100 μl/chamber). Cells were allowed to migrate for 24 h at 37°C. After removing the cells that remained in the top chamber, the top surface of each membrane was cleared of cells with a cotton swab. Cells that had penetrated to the bottom side of the membrane were then fixed in methanol, stained using hematoxylin, and counted in nine randomly selected microscope fields (×200) per well.
Statistical analysis
For most experiments, data are expressed as mean±2 SD of three independent experiments. Statistical significance was evaluated by Student’s t-test using PRISM software (GraphPad, San Diego, California, USA). A two-tailed P-value of less than 0.05 was considered statistically significant.
Clinicopathologic variables were extracted from histopathological reports. A standard χ2-test was performed to assess the association between S2448p-mTOR expression and clinicopathological parameters, except for age, which was assessed by Student’s t-test. Survival were estimated by the Kaplan–Meier method and compared with the log-rank test. Multivariate analysis was performed using the Cox regression model to assess whether a factor was an independent predictor of disease-free survival. Hazard ratios with 95% confidence intervals were estimated. Statistical analyses were performed using SPSS version 13.0 software (SPSS Inc., Chicago, Illinois, USA).
Results
Antiproliferative effect of PP242 in gastric cancer cells
To determine whether PP242 could function as a new therapeutic compound, we tested the cell growth inhibition on six gastric cancer cell lines (AGS, MKN45, MKN28, KATO3, SGC7901, and N87 cells) using the Cell Counting Kit-8 assay. Gastric cancer cells were exposed to various concentrations (0, 50, 100, 200, 500, and 1000 nmol/l) of PP242 for 24 or 48 h. PP242 treatment clearly reduced cell viability in the gastric cancer cell lines in a time-dependent and dose-dependent manner (Fig. 1c–h). At 24 h, PP242 only had cytotoxic effects with 50% growth-inhibitory concentrations of 200–500 nmol/l in AGS and MKN45 cells. However, at 48 h, 50 nmol/l of PP242 induced a strong reduction in the growth rate of the four gastric cancer cell lines (AGS, MKN45, KATO3, and N87), which ranged between 40 and 60% (P<0.05). Moreover, this drug suppressed cell growth by ∼80% in AGS and N87 cells lines at higher concentrations and by ∼70% in KATO3 and SGC7901 cells. To further elucidate the antiangiogenic effects of PP242 in vitro, we examined HUVEC and HMEC proliferation and survival. As shown in Fig. 1a and b, PP242 showed ∼50% growth inhibition of HMEC at 50 nmol/l and HUVEC cells at 200 nmol/l in 48 h (P<0.05), and a significant reduction was observed at a higher concentration of PP242. Therefore, noncytotoxic concentrations of PP242 (under 200 nmol/l at 24 h) in the presence of serum were used, and AGS and HUVEC cells were selected for use in the following in-vitro experiments.
Fig. 1.
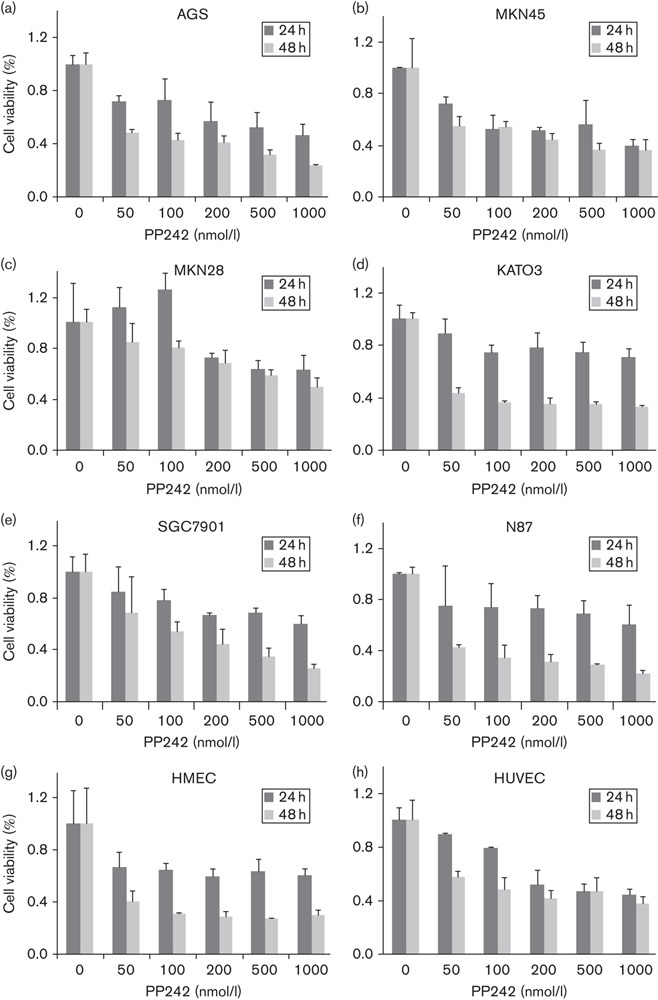
Effects of PP242 on cell proliferation. Gastric cancer cell lines (a–e) and vascular endothelial cells (c, h) were treated with indicated concentrations of PP242 for 24 or 48 h and the number of viable cells was assessed using an MTS assay. Data are presented as percentage of control (no inhibitor treatment) as mean±SD from the triplicate wells. HMEC, Human microvascular endothelial cells; HUVEC, human umbilical vein endothelial cell.
Inhibition of cell motility and invasion by PP242 treatment
We investigated the inhibitory effects of PP242 on the chemotactic motility of AGS and HUVEC cells using a wound-healing assay. Cells were carefully wounded and a continuous cell-free ‘wound’ region was observed. After 48 h incubation with increasing concentrations of PP242, the motility capacity of gastric cancer cells and human endothelial cells to move into the ‘wound’ region was significantly reduced in a dose-dependent manner (Fig. 2) as metastasis is one of most the critical events in tumor progression and migration is essential for endothelial cells to form blood vessels in angiogenesis and is necessary for tumor growth and metastasis. To examine the effect of PP242 on tumor invasion and metastasis (Fig. 3), we performed the invasion assay and the results were consistent with the observations obtained with the wound-healing assay. PP242 significantly inhibited AGS and HUVEC migration and invasion in a dose-dependent manner (Fig. 3, P<0.05).
Fig. 2.
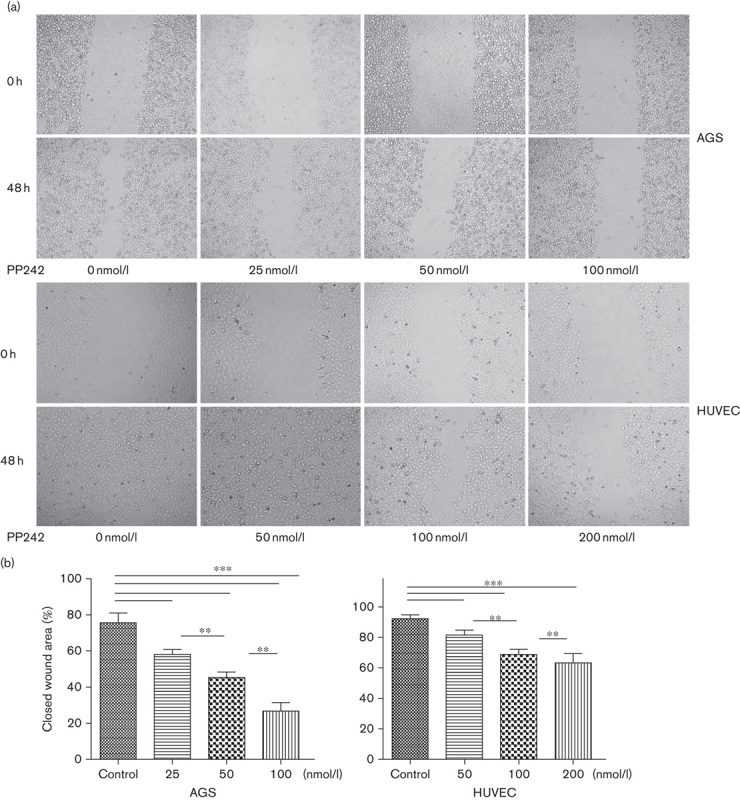
Effect of PP242 on the motility capability of gastric cancer cell AGS and endothelial cell HUVEC. (a) Representative wound-healing images at 0 and 48 h. Wounds were created with a pipette tip in confluent monolayers. (b) Quantification of wound-healing rates. Data are mean±SD. *P<0.05, **P<0.01, ***P<0.001. HUVEC, human umbilical vein endothelial cell.
Fig. 3.
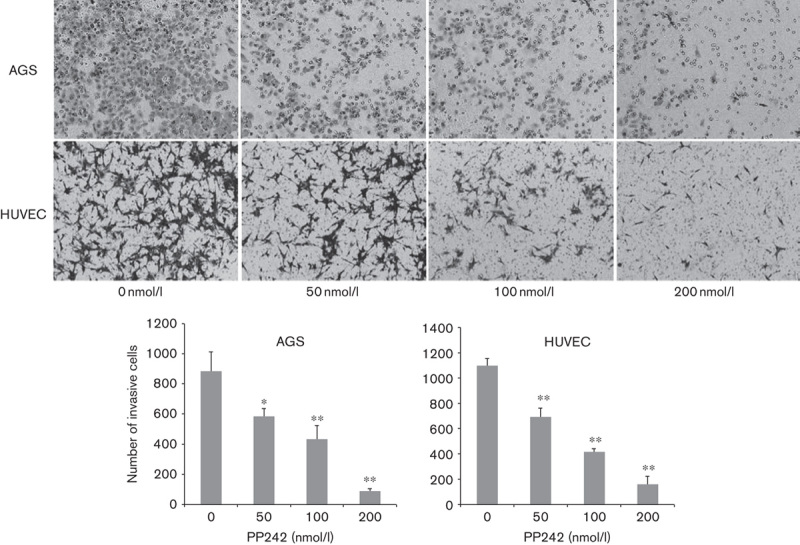
Invasion inhibition of PP242 on gastric cancer cell AGS and endothelial cell HUVEC pretreated with indicated concentrations of PP242. Top panel: representative illustrations for motility and invasion assays (original magnification, ×200). Bottom panel: quantification of migrating and invasive cells. The experiments were repeated at least three times independently. Error bars represent SEs of the mean value (*P<0.05; **P<0.01). HUVEC, human umbilical vein endothelial cell.
PP242 induces rearrangement of cytoskeleton
In the process of cancer cell metastasis including many biological changes, cytoskeletal rearrangement is one of the most important. Polymerization of actin filaments at the leading edge of the malignant cancer cells promotes the formation of membrane protrusions, which are associated with increased cell migration and invasion. As shown in Fig. 4a, actin filaments were mostly assembled at the leading edge and presented as protrusions in the gastric cancer cells AGS without PP242. After incubation with 100 nmol/l PP242 for 24 h, the cytoskeleton was remodeled and abundant actin filament staining was found throughout the cells, some of which even showed a regular filament skeleton. However, PP242 induced cytoskeleton rearrangement in the HUVEC cells, showing assembly and fracture of F-actin in the cytoplasm and membrane, which had the normal cytoskeleton without PP242. This result shows that PP242 can affect the rearrangement of F-actin in AGS and HUVEC. In addition, PP242 reduced the level of phospho-paxillin (Tyr181) in AGS and HUVEC cells, which was reported to be one of the mTORC2 targets in the regulation of actin dynamics (Fig. 4b) 13,14.
Fig. 4.
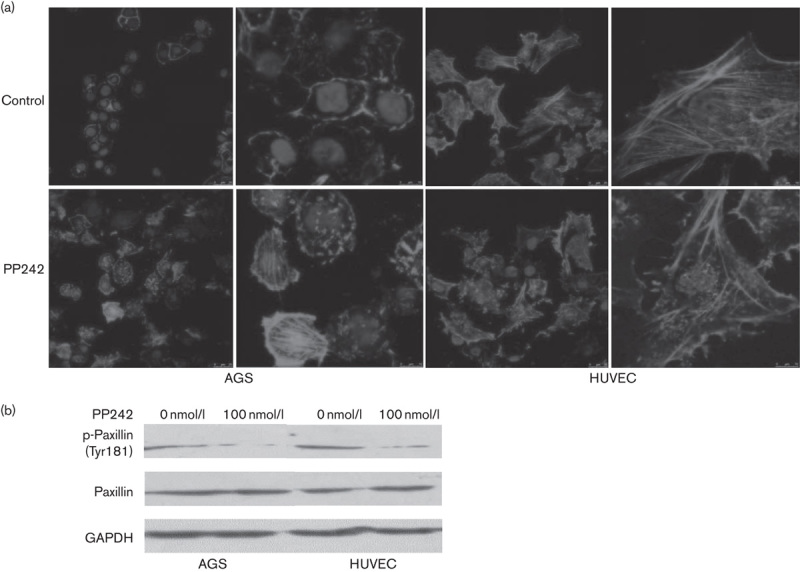
Rearrangement effects of PP242 on the cytoskeleton of gastric cancer cell line AGS and endothelial cells HUVEC. (a) F-actin staining was visualized by immunolocalization using rhodamine phalloidin (red). Cell nuclei were stained with DAPI (blue). Scale bar represents 25 μm. (b) Immunoblotting for phospho-paxillin in AGS and endothelial cells HUVEC that were treated with or without 100 nmol/l PP242. HUVEC, human umbilical vein endothelial cell.
PP242 suppressed tube formation of HUVECs
Tube formation of endothelial cells is also one of the key steps of angiogenesis, although angiogenesis is a very complex procedure. To examine the effect of PP242 on the angiogenesis, a dimensional Matrigel capillary tube formation assay using HUVECs was performed to mimic in-vivo angiogenesis. When HUVECs were seeded on the growth factor-reduced Matrigel, robust tubular-like structures were formed. PP242 inhibited the formation of vessel-like structures, consisting of the elongation and alignment of the cells at the indicated concentrations. Approximately 100 nmol/l PP242 inhibited 50% tube formation of HUVECs on Matrigel and 200 mmol/l PP242 almost completely inhibited tube formation (Fig. 5, P<0.05). These results indicate that PP242 may block angiogenesis in vitro by inhibiting the migration and tubular structure formation of endothelial cells.
Fig. 5.

Effects of PP242 on endothelial tube formation in vitro. HUVEC cells were plated on Matrigel (200 μl/well) and treated with increasing concentrations of PP242. Capillary tube formation was assessed after 24 h. The morphological changes in the cells and tube formation were observed and photographed at ×200. HUVEC, human umbilical vein endothelial cell.
Inhibition of mTORC1 and mTORC2 activation by PP242
PP242 is an ATP-competitive inhibitor that binds the mTOR catalytic site and, thus, theoretically curtails both TORC1 and TORC2 activity. Therefore, we investigated the effects of PP242 on the mTOR pathway in AGS and HUVEC cells. p70S6K is a direct substrate of TORC1 and phosphorylation of P70S6K on threonine 389 serves as a read-out for TORC1 activity. Also, AKT phosphorylation at the S473 residue is specific as the TORC2 substrate. Immunoblot assay for phospho-mTOR yielded comparable results. Phosphorylation of mTOR on serine 2481 is an active form of the TORC2 complex whereas mTOR on serine 2448 is the active form of the TORC1 complex 15–17. Compared with the control, when AGS and HUVEC cells were treated with various concentrations of PP242, the phosphorylation level of mTOR and its downstream factor were effectively suppressed in a dose-dependent manner (Fig. 6).
Fig. 6.
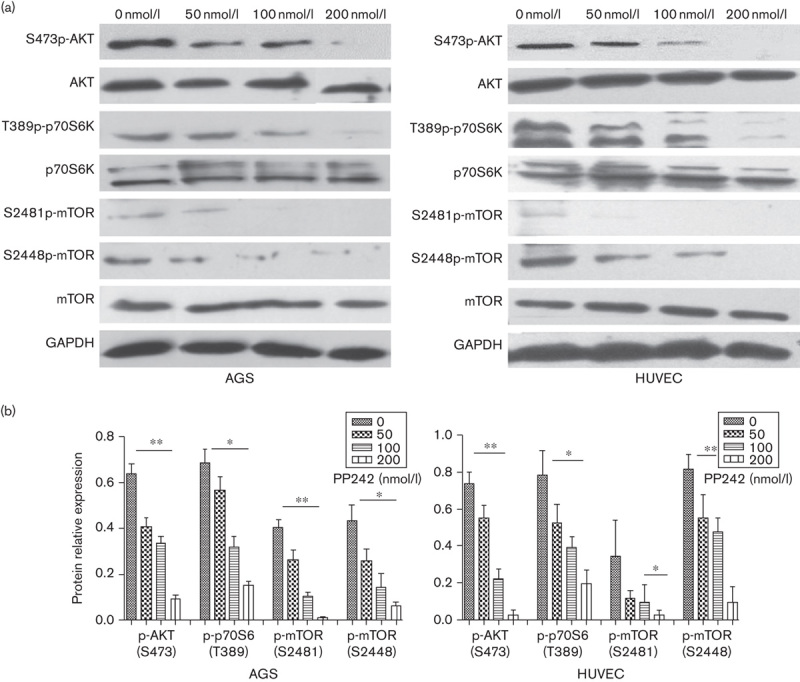
Influence of mTORC1 and mTORC2 activity by PP242. AGS and HUVEC cells were pretreated with increasing concentrations of PP242. After 1 h incubation, protein lysates was immunoblotted for the expression of total AKT (t-AKT), phosphorylated AKT [p-AKT (S473)], total p70S6 kinase (t-p70S6), phosphorylated P70 [p-p70S6 (T389)], total mTOR (t-mTOR), phosphorylated mTOR [p-mTOR (S2481)], and phosphorylated mTOR [p-mTOR (S2448)]. GAPDH was used as controls for equal loading. (a) Representative images of western blotting. (b) Quantification of phosphorylated protein expression relative to the total protein expression. Expressions of p-AKT (S473), p-p70S6 (T389), p-mTOR (S2481), and p-mTOR (S2448) protein were determined relative to t-AKT, t-p70S6, t-mTOR, and t-mTOR, respectively. Data are mean±SD of three independent experiments. *P<0.05, **P<0.01, ***P<0.001. HUVEC, human umbilical vein endothelial cell; mTOR, mammalian target of rapamycin.
Expression of S2448p-mTOR in human gastric cancer and vascular endothelial cells
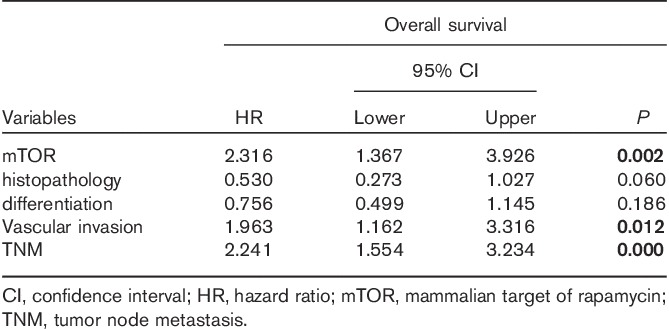
To confirm that mTORC activity is upregulated in gastric cancer, we performed immunohistochemistry on archived gastric cancer samples of 196 cases. Using the primary antibody of S2448p-mTOR, cytomembrane staining was observed in 41.8% (82/196) of gastric cancer tissues (Fig. 7). Statistical analysis showed that S2448p-mTOR expression correlated with the depth of mural invasion, lymph node metastasis, tumor node metastasis (TNM) stage, and vascular invasion (Table 1, P<0.05). The S2448p-mTOR expression rate in advanced diseased (stages III and IV) was significantly increased compared with that in the early stages (I and II) of carcinoma (54.0 vs. 25.3%, P<0.05). Patients with a positive S2448p-mTOR expression showed poorer 5-year overall survival than those with a negative expression (Table 2, Supplementary 1, Supplemental digital content 1, http://links.lww.com/ACD/A78; P=0.000). Also, S2448p-mTOR was found to be expressed in the vascular endothelial cells (Fig. 7d and e). These results indicate that the highly activated mTORC pathway could be a potential target in gastric cancer.
Fig. 7.
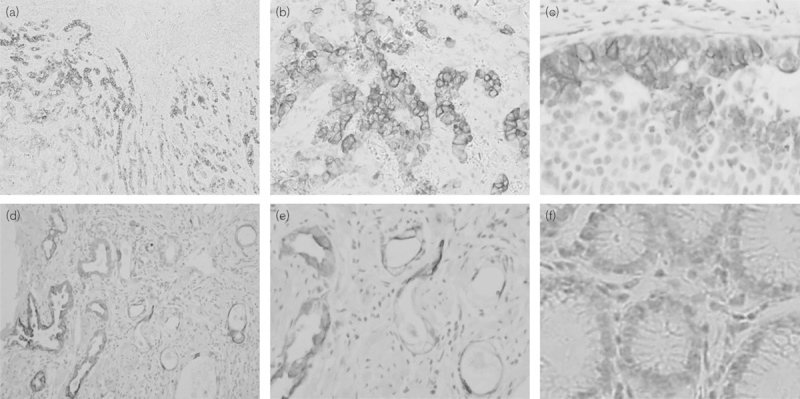
Expression of the in-situ mTORC1 active form in human gastric cancer and vascular endothelial cells. A positive expression of S2448p-mTOR was detected predominantly in the cytoplasm and membrane of gastric cancer (a, ×40; b, ×100; c, ×200). mTORC1 activation was also observed in the vascular endothelial cells (d, ×100; e, ×200). (f) Negative expression in normal gastric mucosa. mTOR, mammalian target of rapamycin.
Table 1.
Association of S2448p-mTOR expression with clinicopathological parameters in gastric cancer
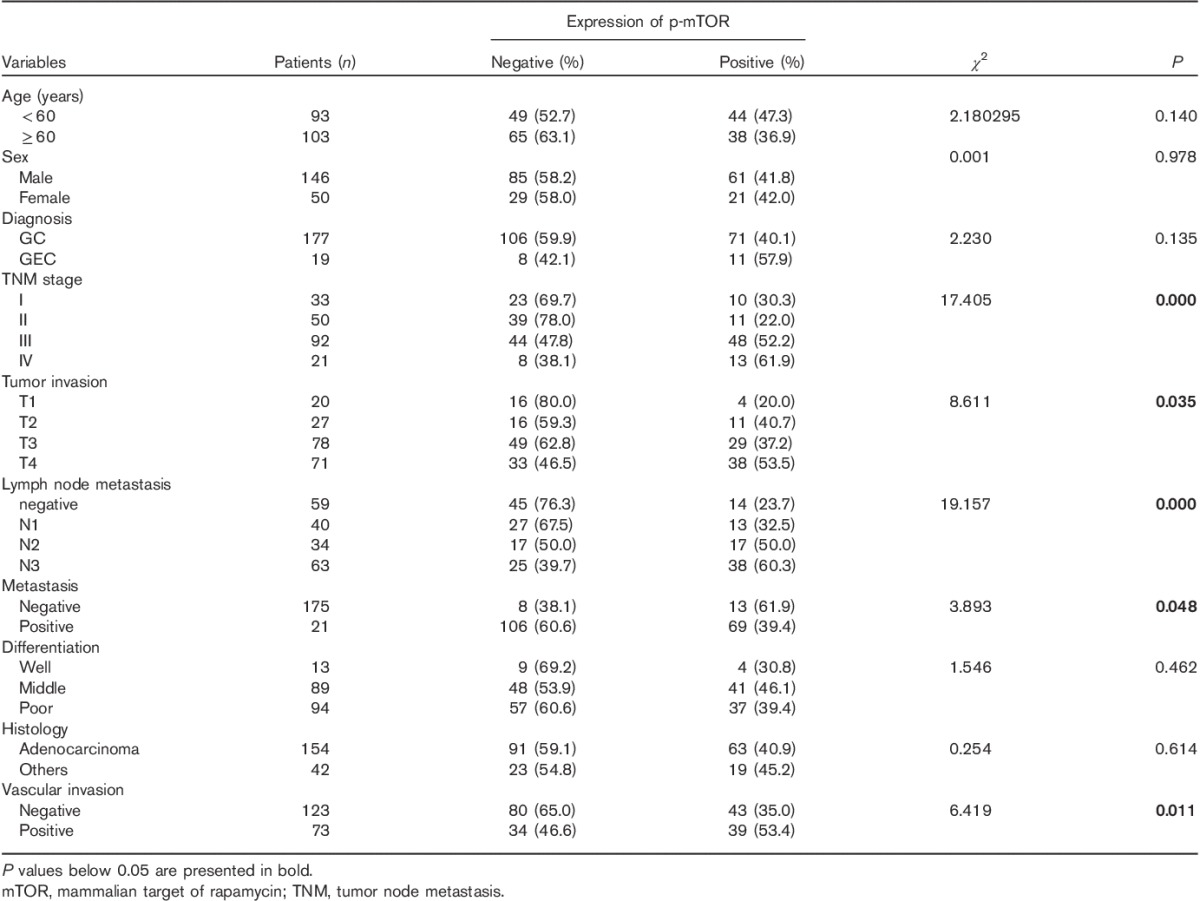
Table 2.
Multivariate analysis of survival in gastric cancer according to clinicopathologic factors and S2448p-mTOR overexpression
Discussion
As a downstream molecule of the PI3K/AKT pathway, mTOR has long been investigated in cancer treatment for its low toxicity and stable efficacy 18–20. Despite its critical role in cancer proliferation and angiogenesis, the first-generation mTOR inhibitors, rapalogs such as sirolimus and everolimus, have had limited efficacy in the clinic 21. This might be attributed to the lack of inhibition of mTORC2 signaling and activation of Akt through an IGF-1R/PI3K-dependent negative feedback 22. Discovering that mTORC2 directly phosphorylates AKT led to speculation that mTORC2-specific inhibitors might also be valuable cancer drugs 7. Further molecular characterization of the mTOR complex (mTORC1/2) resulted in the development of additional mTOR inhibitors, which are ATP-competitive inhibitors and target the kinase domain of mTOR blocking both mTORC1/2 activity. These so-called mTOR kinase domain inhibitors (TORKinibs) might overcome the undesired effects of rapalogs and have a broad impact in cancer therapy 8.
In this study, we show that PP242 is a potent and dual inhibitor of mTORC1/2 in gastric cancer cells. PP242 inhibited gastric cancer cell proliferation in a time-dependent and dose-dependent manner. The greater cytoreduction of PP242 was possibly not only because of a greater inhibitory effect on TORC1 substrates but was also mediated by its targeting of TORC2. In pathway detection, PP242 inhibits both the phosphorylation of P70S6K, the downstream substrates of mTORC1, and selectively inhibits phosphorylation of AKT at Ser473, the downstream substrate of mTORC2. It was observed that PP242 was more effective than rapamycin against multiple myeloma cells and its greater efficacy was shown by a more complete decrease and a significant induction of apoptosis that is primarily mediated by its targeting of TORC2 23,24. Then, we observed that the motility and invasion ability of AGS cells was reduced by PP242 treatment, suggesting that the mTOR pathway also participates in the process of tumor invasion and metastasis. F-actin is also rearranged in AGS cells with PP242, which may partly explain the reduced motility. Recent studies have reported that the downstream nodules of mTOR-dependent metastasis may include vimentin, CD44, and MTA1, that as effectors further promote cancer initiation and metastasis 3. It has also been reported that rapamycin suppressed migration and MMP production in gastric cancer with peritoneal dissemination 25. In addition, the prolonged treatment with rapamycin was found to impair the assembly of mTORC2 and the subsequent phosphorylation of STAT3 at Ser 727, resulting in a decrease in MMP2 and Twist1 in sarcomatoid cholangiocarcinomas that are associated with metastasis 26. In the preclinical models of breast cancer, a similar ATP-competitive mTOR1/2 inhibitor MLN0128 could inhibit both invasion and metastatic colonization. Further studies are still necessary to determine the complex regulatory role of TORC1/TORC2 signaling in the metastatic cascade 27.
In this study, we did not focus on the effector molecules, but investigated the inhibition of angiogenesis upon PP242 treatment. First, an antiproliferative effect of exponentially proliferating PP242 on normal endothelial cells (HUVEC and HMEC) in a dose-dependent manner was observed in our study. Considering that mTOR serves as a central regulator of cell growth, proliferation, and survival, the inhibitory effect may depend on the proliferative status; in which case, the normal endothelial cells may possibly be less affected. Accumulating evidence supports the role of the PI3K/Akt/mTOR pathway in tumor angiogenesis 28,29. In angiogenesis, PI3K/AKT kinases are activated and regulate multiple critical steps by phosphorylating different downstream substrates, such as mTOR. This pathway has also been reported to play a key role in hypoxia, expression of hypoxia-inducible factor-1α (HIF-1α) and HIF-1α-responsive genes including vascular endothelial growth factor, and regulation of tumor vascularization 30. It is known that endothelial cells undergo a cascade of events, including proliferation, survival, migration, and maturation, to achieve new blood vessel formation. In the present study, the antiangiogenic action of PP242 cannot be explained only by the antiproliferative effect as some experimental models do not allow endothelial proliferation but instead endothelial migration and reorganization. These effects may be probably because of the reduced phosphorylation level of mTOR1/2 and its substrates P70S6K and AKT. On staining the F-actin, we found that the cytoskeleton was rearranged accompanied by a decreased level of phosphorylated paxillin, which plays a critical role in actin dynamics. PP242 also reduced in-vitro tube formation in a concentrate-dependent manner, providing evidence of the effect of PP242 on angiogenesis inhibition. Taken together, these results support the concept that PP242 exerts its antitumor effect both directly through proliferation and indirectly through interference with the angiogenic process.
To strengthen the rational for targeting mTOR, we further performed immune-histochemistry of phosphorylated mTOR at ser2448 in a series of gastric cancer tissue samples. mTORC1 is activated when being phosphorylated at Ser2448 31. The results showed a positive expression of S2448P-mTOR in 41.8% (82/196) of gastric cancer tissues. It also correlated with the depth of mucosal invasion, lymph node metastasis, TNM stage, and vascular invasion. The S2448p-mTOR expression rate in advanced disease was significantly increased compared with that in the early stages. Patients with a positive S2448p-mTOR expression showed poorer 5-year overall survival than those with a negative expression. Although we did not detect the activity level of mTORC2, another report found that immunohistochemical staining indicated a high frequency of in-situ TORC2 activation in gastric cancer as shown by immunodetection of S473p-AKT phosphorylation. Also, the S473p-AKT expression level was correlated with vessel infiltration, lymph node metastasis, and prognosis 32,33. These results indicate that the highly activated mTORC pathway could be a potential target in gastric cancer. Furthermore, we also observed that S2448p-mTOR was expressed in the vascular endothelial cells (Fig. 7d and e). Taken together with its correlation with vascular invasion, lymph node metastasis, and TNM stage, inhibition of mTOR1/2 by PP242 may occur in both gastric cancer cells and vascular endothelial cells.
Conclusion
Our study confirmed the hypothesis that PP242 has antitumor efficacy by antiproliferative and antiangiogenesis. Indeed, preclinical data with PP242 indicate that it is superior to rapamycin and may have more promising potential in the future.
Acknowledgements
This work was supported by the WuJieping cancer target therapy research foundation (320.6700.09009), the Specialized Research Fund for the doctoral Program of Higher Education (No. 20110001120067), the National Nature Science Foundation of China (No. 81101881 and No. 81341072), and the National High Technology Research and Development Program of China (863 Program, No. 2014AA020603).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.anti-cancerdrugs.com).
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008, cancer incidence and mortality worldwide 2010Lyon, France: International Agency for Research on Cancer. [Google Scholar]
- 2.Bouche O, Penault-Llorca F. HER2 and gastric cancer: a novel therapeutic target for trastuzumab. Bull Cancer 2010; 97:1429–1440. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012; 485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab 2014; 19:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korets SB, Czok S, Blank SV. Curtin JP.Schneider RJ. Targeting the mTOR/4E-BP pathway in endometrial cancer. Clin Cancer Res 2011; 17:7518–7528. [DOI] [PubMed] [Google Scholar]
- 6.Kremer CL, Klein RR, Mendelson J, Browne W, Samadzedeh LK, Vanpatten K, et al. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate 2006; 66:1203–1212. [DOI] [PubMed] [Google Scholar]
- 7.Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 2010; 29:3733–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shor B, Gibbons JJ, Abraham R, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle 2009; 8:3831–3837. [DOI] [PubMed] [Google Scholar]
- 9.Marinov M, Ziogas A, Pardo OE, Tan LT, Dhillon T, Mauri FA, et al. AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res 2009; 15:1277–1287. [DOI] [PubMed] [Google Scholar]
- 10.Trinh XB, Tjalma WA, Vermeulen PB, Van den Eynden G, Van der Auwera I, Van Laere SJ, et al. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer 2009; 100:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25:6436–6446. [DOI] [PubMed] [Google Scholar]
- 12.Jhanwar-Uniyal M, Albert L, McKenna E, Karsy M, Rajdev P, Braun A, et al. Deciphering the signaling pathways of cancer stem cells of glioblastoma multiforme: role of Akt/mTOR and MAPK pathways. Adv Enzyme Regul 2011; 51:164–170. [DOI] [PubMed] [Google Scholar]
- 13.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6:1122–1128. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Hau AM, Beach JR, Harwalker J, Mantuano E, Gonias SL, et al. Mammalian target of rapamycin complex 2 (mTORC2) is a critical determinant of bladder cancer invasion. PLoS One 2013; 8:e81081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu G, Wang J, Chen Y, Wang X, Pan J, Li G, et al. Overexpression of phosphorylated mammalian target of rapamycin predicts lymph node metastasis and prognosis of Chinese patients with gastric cancer. Clin Cancer Res 2009; 15:1821–1829. [DOI] [PubMed] [Google Scholar]
- 16.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009; 27:2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood 2010; 116:4560–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubio-Viqueira B, Hidalgo M. Targeting mTOR for cancer treatment. Adv Exp Med Biol 2006; 587:309–327. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS. mTOR mediated anti-cancer drug discovery. Drug Discov Today Ther Strateg 2009; 6:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bu Z, Ji J. Therapeutic implications of mTOR inhibitors in the treatment of gastric cancer. Curr Cancer Drug Targets 2013; 13:121–125. [DOI] [PubMed] [Google Scholar]
- 21.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 2009; 8:567–572. [DOI] [PubMed] [Google Scholar]
- 22.Gruppuso PA, Boylan JM, Sanders JA. The physiology and pathophysiology of rapamycin resistance: implications for cancer. Cell Cycle 2009; 10:1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blaser B, Waselle L, Dormond-Meuwly A, Dufour M, Roulin D, Demartines N, et al. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer 2012; 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng Z, Shi YX, Tsao T, Qiu Y, Kornblau SM, Baggerly KA, et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012; 120:2679–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto I, Koizumi K, Tatematsu M, Minami T, Cho S, Takeno N, et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur J Cancer 2008; 44:1022–1029. [DOI] [PubMed] [Google Scholar]
- 26.Hong SM, Park CW, Cha HJ, Kwon JH, Yun YS, Lee NG, et al. Rapamycin inhibits both motility through down-regulation of p-STAT3 (S727) by disrupting the mTORC2 assembly and peritoneal dissemination in sarcomatoid cholangiocarcinoma. Clin Exp Metastasis 2012; 30:177–187. [DOI] [PubMed] [Google Scholar]
- 27.Gokmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat 2012; 136:673–682. [DOI] [PubMed] [Google Scholar]
- 28.Baldo P, Cecco S, Giacomin E, Lazzarini R, Ros B, Marastoni S. mTOR pathway and mTOR inhibitors as agents for cancer therapy. Curr Cancer Drug Targets 2008; 8:647–665. [DOI] [PubMed] [Google Scholar]
- 29.Azim H, Azim HA, Jr, Escudier B. Targeting mTOR in cancer: renal cell is just a beginning. Target Oncol 2010; 5:269–280. [DOI] [PubMed] [Google Scholar]
- 30.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008; 8:851–864. [DOI] [PubMed] [Google Scholar]
- 31.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2010; 12:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murayama T, Inokuchi M, Takagi Y, Yamada H, Kojima K, Kumagai J, et al. Relation between outcomes and localisation of p-mTOR expression in gastric cancer. Br J Cancer 2009; 100:782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers 2008; 23:1–9. [DOI] [PubMed] [Google Scholar]