

Abstract
Enterobacteria are able to survive under stressful conditions within animals, such as acidic conditions in the stomach, bile salts during transfer to the intestine and anaerobic conditions within the intestine. The glutamate-dependent (GAD) system plays a major role in acid resistance in Escherichia coli, and expression of the GAD system is controlled by the regulatory cascade consisting of EvgAS > YdeO > GadE. To understand the YdeO regulon in vivo, we used ChIP-chip to interrogate the E. coli genome for candidate YdeO binding sites. All of the seven operons identified by ChIP-chip as being potentially regulated by YdeO were confirmed as being under the direct control of YdeO using RT-qPCR, EMSA, DNaseI-footprinting and reporter assays. Within this YdeO regulon, we identified four stress-response transcription factors, DctR, NhaR, GadE, and GadW and enzymes for anaerobic respiration. Both GadE and GadW are involved in regulation of the GAD system and NhaR is an activator for the sodium/proton antiporter gene. In conjunction with co-transcribed Slp, DctR is involved in protection against metabolic endoproducts under acidic conditions. Taken all together, we suggest that YdeO is a key regulator of E. coli survival in both acidic and anaerobic conditions.
Introduction
Enterobacteria such as Escherichia coli, exist in the environment, and in the gut of warm blooded animals. To survive this switch in lifestyles, and upon ingestion by a new host, bacteria are directly exposed to various stresses and hence require sophisticated stress response systems to survive continuous changes in environment such as acidic conditions in the stomach, bile salts, and anaerobic conditions within the intestines [1]. For survival under acidic conditions, E. coli possesses three amino acid-dependent acid resistance systems with glutamate, arginine, and lysine [2], [3], [4]. The resistance mechanism involves the transient consumption of the intracellular proton by glutamate, arginine and lysine decarboxylases, and exchange of the amine products with extracellular amino acids through their respective antiporters [2], [3], [5], [6]. The most effective system of acid resistance is the GAD (glutamic acid-dependent) system which is composed of two glutamate decarboxylase isozymes, GadA and GadB, and the cognate antiporter GadC. Expression of these components is under the control of a complex network of transcription factors, including GadE, GadX, GadW, EvgA, YdeO, and H-NS [1].
YdeO is a transcription factor, belonging to the AraC/XylS family. Knowledge about the regulatory functions of YdeO is limited except that it is known that YdeO activates transcription of the gad system components, gadE, gadA and gadBC [7], [8], [9]. The expression of ydeO is activated by the two-component system EvgSA [9], [10], [11], forming a regulatory cascade, EvgA > YdeO > GadE [9], [12]. In this study, we performed a comprehensive interrogation of YdeO-binding sites in vivo on the E. coli genome using ChIP-chip analysis, and identified a set of YdeO-regulated genes, including four stress-response transcription factors, DctR, NhaR, GadE, and GadW, and several genes involved in respiration. Taking these observations together we propose that YdeO is the regulator which coordinates the response to acid and anaerobic conditions in E. coli.
Materials and Methods
E. coli strains and growth conditions
E. coli strains and plasmids used in this study are shown in Table S1. E. coli cells were grown at 37°C in Luria-Bertani (LB) medium. Cell growth was monitored by measuring the turbidity with a Mini photo 518R spectrophotometer (Taitec). The standard procedure for bacterial cell cultivation in this study was as follows: A single colony was isolated from an overnight culture on a LB agar plate, and inoculated into 5 ml of fresh LB medium. This liquid culture was grown overnight at 37°C, and the overnight culture was diluted 100-fold into fresh LB medium. The culture was incubated at 37°C with reciprocal shaking (160 revolutions min−1) for aerobiosis or without shaking for anaerobiosis.
Introduction of a tagged gene into the E. coli genome
The introduction of a tagged gene into the E. coli genome was carried out using the method of Uzzau et al. [13]. In brief, primers were used to make PCR extensions homologous to the last portion of the targeted gene (forward primer) and to a region downstream of it (reverse primer) as follows; YDEOF-1 (forward) and YDEOR-1 (reverse) for ydeO-3xflag; GADE-F (forward) and GADE-R (reverse) for gadE-3xflag; GADW-F (forward) and GADW-R (reverse) for gadW-3xflag (Table S1). Amplified DNA fragments including the 3′ sequence with flag tag and a kanamycin-resistance gene were amplified by PCR using pSUB11 as a template, a pair of primers, and Ex-Taq DNA polymerase (Takara Bio). PCR products were purified using a QIAquick PCR purification kit (Qiagen), and then used directly for electro-transformation. E. coli carrying a lambda-Red helper plasmid, pKD46, was used to make competent cells, and were grown at 30°C in LB medium supplemented with 100 µg ml−1 ampicillin and 1 mM arabinose to an OD600 of 0.4. Cells were collected by centrifugation, and washed two times with ice-cold sterile deionized water containing 10% glycerol. Aliquots (50 µl) of the bacterial suspensions in 10% glycerol were mixed with more than 1 µg of PCR product in a chilled cuvette (0.2 cm electrode gap) and subjected to a single pulse (2.5 kV) by a Gene pulser Xcell (Bio Rad). After 1 hr recovery at 37°C in 1 mL of SOC medium (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose) containing 1 mM arabinose, half of the volume of electroporated bacteria in SOC media were spread on to LB agar plates supplemented with antibiotics for the selection of kanamycin-resistant recombinants. If none grew on the agar plate after incubation overnight at 37°C, the remainder stored was spread on to LB kan plates. The kanamycin-resistance recombinants were isolated once on LB agar at 37°C, and then examined for ampicillin sensitivity for loss of the helper plasmid.
Construction of YdeO expression plasmids
To construct pYY0401 for YdeO-3xFLAG expression, DNA fragments containing the ydeO coding region were amplified by PCR using E. coli YY5001 genomic DNA, including the 3xflag tag at the end of ydeO as a template, and a pair of primers, YDEOF-2 and YDEOR-3, in which the Bam HI and Eco RI sites were included (see sequences in Table. S1). After digestion of PCR products with Bam HI and Eco RI, the PCR-amplified fragments were cloned into the pTrc99A vector containing an inducible trc promoter between the Bam HI and Eco RI sites. To construct pYdeO for expression of intact YdeO, DNA fragments containing the ydeO coding region were amplified by PCR using E. coli W3110 type A [14] genomic DNA as a template and the primers, YDEOF-2 and YDEOR-2 (see sequences in Table. S1). After digestion of the PCR product with Bam HI and Eco RI, the PCR-amplified fragments were ligated into the pTrc99A vector between appropriate restriction enzyme sites. To construct pYdeO-SUMO for overproduction of SUMO (Small Ubiquitin-related MOdifier) fused YdeO, DNA fragments containing the ydeO coding region were amplified by PCR using E. coli BW25113 genomic DNA as a template and the primers, YDEO-SUMO-F and YDEO-SUMO-R, in which 15-nt homologous to pE-SUMO vector (Life Sensors) digested with Bsa I were included (see sequences in Table. S1). The PCR-amplified fragments were cloned into the pE-SUMO vector using In-Fusion HD cloning kit (Clontech). All of the plasmids were confirmed by DNA sequencing with primers, Trc99A-F and/or Trc99A-R for pTrc99A derivatives and T7 terminator and SUMO forward for pE-SUMO derivatives.
Construction of lacZ and lux reporter plasmids
To construct a lacZ fusion gene, the pRS552 plasmid was used as a vector for the construction of translational fusions [15]. The promoter DNA fragment was amplified by PCR using the genome of E. coli W3110 type-A strain [14] as a template and a pair of primers. The primers used were: APPC-LF and APPC-LR for pAPPC-L; YIIS-LF and YIIS-LR for pYY0503; HYAA-LF and HYAA-LR for pHYAA-L (Table S1). The PCR product was digested with BamH I and/or EcoR I and then ligated into pRS552 at the corresponding sites. A nhaR-lux transcription fusion was also constructed. First, DNA fragments containing the nhaR promoter were amplified by PCR using the primers: NHAR-lux-F and NHA-lux-R, which contained 15-nt homologous to the pLUX vector [16] digested with Xho I and Bam HI were included (see sequences in Table. S1). The PCR-amplified fragments were cloned into the pLUX vector using In-Fusion HD cloning kit (Clontech), resulting in the construction of pLUXnhaR (Table S1). All of the plasmids were confirmed by DNA sequencing using the lacZ-30R primer complementary to lacZ or Lux-R primer complementary to luxC in a vector.
ChIP-chip analysis
The ChIP-chip assay was carried out as described in previous reports [17], [18], [19] with a few modifications. YY0201 (ΔydeO) harbouring pYY0401 (ydeO-3xflag) was grown to an OD600 of 0.4 then re-incubated in LB medium containing formaldehyde (final concentration of 1%) at 37°C for 30 min. The cross-linking reaction was terminated by the addition of glycine, and cells were collected, washed, re-suspended with lysis buffer, and lysed by incubation with Lysozyme. Lysed cells were dissolved in 4 ml of IP buffer containing PMSF. The sample was then sonicated 60 times for 30 sec at 30 sec intervals on ice using a BRANSON Digital Sonifier (Branson). After centrifugation, the supernatant fraction (whole cell extract) was mixed with anti-FLAG antibody (Sigma Aldrich)-coated-protein A Dynal Dynabeads (Invitrogen) and incubated at 4°C overnight. After washing twice with IP buffer and IP salt buffer, the DNA–YdeO-3xFLAG complex bound to the beads was recovered by eluting with elution buffer (50 mM Tris–HCl pH 7.5, 10 mM EDTA, 1% SDS). YdeO-3xFLAG in whole cell extracts and in immunoprecipitated DNA fractions were digested by Pronase (Roche). DNA fragments free of cross-linked DNA–protein were purified using a QIAquick PCR purification kit (Qiagen). Recovered DNA fragments were amplified according to the random DNA amplification method using the primers, PF 43 and PF 44 described by Katou et al. [17]. PCR was performed over 30 cycles, using Phusion high-fidelity DNA polymerase (New England Biolabs). Amplified DNA fragments were terminally labeled and hybridized with the custom-designed Affymetrix oligonucleotide tiling array and raw data (CEL files) were processed using the Array edition of the In Silico Molecular Cloning (IMC) software (In Silico Biology) as previously described [18], [19], [20]. To detect DNA fragments by immunoprecipitation, the signal intensities of ChIP DNA were divided by those of the supernatant (Sup) fraction.
Pufirication of the YdeO protein
In a typical procedure [21], a single colony of transformed E. coli BL21 (DE3) was grown to OD600 = 0.6 at 37°C with shaking in LB medium supplemented with 100 µg ml−1 ampicillin. The culture was then cooled on ice, induced with 4.5 mM IPTG, and incubated at 20°C overnight with shaking. Cells were isolated by centrifugation and resuspended in 400 µL of lysis buffer (100 mM NaCl, 50 mM Tris-HCl pH 8.0) containing 0.2 mM PMSF. Cells were treated with lysozyme and then subjected to sonication. Triton X-100 was added to 1% (v/v) and incubated on ice for 1 hr. The culture was centrifuged, and the supernatant was decanted and stored at 4°C. Supernatant was mixed with 2 ml of 50% Ni-nitrilotriacetic acid (NTA) agarose solution (Qiagen) and loaded onto a column. The column was washed with 10 ml of lysis buffer containing 1%Triton X-100, and then washed with 10 ml of lysis buffer containing 1%Triton X-100 and 25 mM imidazole. Proteins were eluted with 3 ml of each elution buffer (lysis buffer containing 1%Triton x-100 and 0.1 M, 0.2 M, 0.3 M, 0.4 M, or 0.5 M imidazole), and peak fractions of transcription factors were pooled and dialyzed against a storage buffer (50 mM Tris-HCl, pH 7.5 at 4°C, 200 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, and 50% glycerol), and stored at –80°C until use. Protein purity was checked on SDS-PAGE.
Preparation of total RNA from E. coli cells
Total RNA was prepared using the as previously described [22]. A single colony of E. coli was grown in LB medium to OD600 = 0.3 at 37°C with shaking. Cells were harvested and total RNAs were prepared using hot phenol. In brief, total RNA was extracted with H2O-saturated phenol and precipitated with ethanol. After digestion with RNase-free DNase I (Takara Bio), total RNA was extracted with H2O-saturated phenol and precipitated with ethanol, and dissolved in RNase-free water. The concentration of total RNA was determined by measuring the absorbance at 260 nm. The purity of total RNA was checked by agarose gel electrophoresis.
Transcriptome analysis
To prepare fluorescently labeled cDNA, total RNA (5 µg) was used. We used the FairPlay III Microarray Labeling kit (Agilent), CyDye Cy3 mono-reactive Dye, and CyDye Cy5 mono-reactive Dye (GE Healthcare). For all experiments, two sets of RNAs from an independent colony were carried out with a pair of the fluorescence dye. The mixture containing 1 µl of Ramdom hexanucleotide primers, 5 µg of total RNA, and 12 µl of DEPC-treated water was heated at 75°C for 10 min and cooled to room temperature. After addition of 3 µl of Affinity script HC RTase (Agilent), 1X Affinity script RT buffer, 1X dNTP mixture, 75 mM DTT, and 0.5 µl of RNase block to 10 µl of RNA/primer mixture product, cDNA synthesis was carried out at 42°C for 1 hr and stopped by addition of 10 µM NaOH. The mixture was neutralized by addition of 10 µM HCl. The synthesized cDNA was purified by ethanol-precipitation and then labelled by CyDye Cy3 mono-reactive Dye or CyDye Cy5 mono-reactive Dye. The dye-coupled cDNA was purified by attached the micro spin cup.
The E. coli Gene Expression Microarray microarray 8×15 K (Agilent) was used. Each 300 ng of Cy3- and Cy5-labeled cDNA were mixed and added to 1X Blocking Buffer (Agilent) and 1X HI-RPM GE Hybridization Buffer (Agilent). After precipitation of impurities, 40 µl of the labelled-cDNA mixture was applied to the DNA chip, and the hybridization was carried out at 65°C for 17 hr. The DNA chip was washed at room temperature with Agilent Gene Expression Wash Buffer 1 (Agilent) and at 37°C with Agilent Gene Expression Wash Buffer 2 (Agilent). The DNA chip was scanned with an Agilent G2565CA microarray scanner Ver. 8.1, and the intensities of both Cy3 and Cy5 were quantified by Feature Extraction Ver. 8.1. And then, the Cy5/Cy3 ratios were calculated from the normalized values.
RT-qPCR
Total RNAs were transcribed to cDNA with random primers using Primer Script 1st strand cDNA synthesis Kit (Takara Bio). Quantitative PCR (qPCR) was conducted using SYBR Green PCR Master Mix (Applied Biosystems). Pairs of primers used are described in Table S1. The cDNA templates were twofold serially diluted and used in the qPCR assays. The qPCR reaction mixtures, each containing 12.5 µl of 2X Power SYBR Green PCR Master Mix (Applied Biosystems), 0.225 µl of each primer (10 µM stock), 9.55 µl of water, and 2.5 µl of cDNA, were amplified under the following thermal cycle conditions of: 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 15 sec at 95°C and then 60 sec at 60°C. The expression levels of the 16 S rRNA gene were used for normalization of data, and the relative expression levels were quantified using ‘Delta–delta method’ presented by PE Applied Biosystems (Perkin Elmer) as described in previous reports [23], [24]. The results presented are averages of the results from the replicate experiments ± standard errors of the means (SEM).
EMSA
Probes were amplified by PCR using the previously constructed reporter plasmids as templates, with a pair of primers: a specific primer and an FITC-labeled primer. PCR products with FITC at their termini were purified using the QIAquick PCR purification kit (Qiagen). For gel shift assays, mixtures of the FITC-labeled probes and purified SUMO-YdeO were incubated at 37°C for 30 min in gel shift buffer (50 mM Tris-HCl, pH 7.8 at 37°C, 50 mM NaCl, 3 mM Mg acetate, 0.1 mM EDTA, 0.1 mM DTT, and 0.37 µM BSA) containing 0.2 µg ml−1 salmon sperm DNA. After addition of a DNA dye solution, the mixture was directly subjected to 4% or 7% PAGE. Fluorescent-labeled DNA in gels was detected using Typhoon 9410 (Amersham Biosciences).
DNase I footprinting analysis
The probe was amplified by PCR using a pLUXgadWp as a template, primer pairs GADW-F-2 and Lux-R-FITC, and Ex Taq DNA polymerase (Takara). 1.0 pmol of a FITC-labeled probe was incubated at 37°C for 30 min with purified SUMO-YdeO (0.5 to 15 pmol) in 25 µl of gel shift buffer (50 mM Tris-HCl, pH 7.8 at 37°C, 50 mM NaCl, 3 mM Mg acetate, 0.1 mM EDTA, 0.1 mM DTT, and 0.37 µM BSA). After incubation for 30 min, DNA was digested by DNase I (Takara Bio) for 30 s at 25°C, and then the reaction was terminated by addition of phenol. DNA was precipitated by ethanol, dissolved in formamide dye solution, and analyzed by electrophoresis on a DNA analyzer DSQ-2000L (Shimadu).
Measurement of luciferase activity in E. coli
A single colony of a strain freshly transformed with one of the luciferase reporter plasmids (Table S1) was grown in LB medium supplemented with 50 µg ml−1 kanamycin to OD600 = 0.3 at 37°C with shaking. At this point, the culture was transferred to a micro-titer plate (96-well micro-titer) to start monitoring reporter activity measurement in an automated plate reader MTP-880 (Corona). The Lux (luciferase activity) reads were then divided by the equivalent OD reads (Lux/OD) to approximate Lux activity unit per cell mass for each well. The Lux/OD values of the three technical replicate wells of each culture were averaged.
Measurement of β-galactosidase activity in E. coli
E. coli cells were grown in LB medium and subjected to measurement of β-galactosidase activity with o-nitrophenyl-D-galactopyranoside as described in the previous report [11].
Western blotting analysis
E. coli cells grown in LB medium were harvested by centrifugation and re-suspended in 0.4 ml lysis buffer containing 8 M urea and sonicated. After centrifugation, the same volume of supernatant was subjected to 15% SDS-PAGE and blotted on to PVDF membranes using an iBlot semi-dry transfer apparatus (Invitrogen). Membranes were first immuno-detected with anti-FLAG (Sigma), anti-NhaR serum (Lab stock), or anti-α (Neoclone) and HRP-conjugated anti-mouse IgG (Nacalai Tesque) antibodies and then developed with a chemiluminescence kit (Nacalai Tesque). The image was analyzed with a LAS-4000 IR multi colour imager (Fuji Film).
Results
Identification of YdeO associated sites in vivo within the E. coli genome
To identify the genes directly regulated by YdeO, we first determined the genome-wide distribution of YdeO-binding sites by ChIP-chip (Chromatin ImmunoPreciptation-DNA chip) analysis. For this purpose, we inserted a 3xflag tail into the 3′ end of the ydeO gene in the genome and tried to prepare YdeO-DNA complexes for ChIP-chip analysis from the YY5001 strain harbouring ydeO-3xflag grown in LB medium at 37°C with shaking. The level of YdeO-3xFLAG expression was, however, not enough to isolate YdeO-DNA complexes using the anti-FLAG antibody. We then constructed plasmid pYY0401 for the expression of YdeO-3xFLAG and transformed it into the ydeO-deficient mutant. The ydeO-deficient mutant transformed with pYY0401 was grown until it reached log phase and was then treated with formaldehyde for DNA-protein cross-linking. The E. coli cells were disrupted with sonication to prepare a whole cell extract from which YdeO-DNA complexes were isolated, sonicated and subjected to immune-precipitation using anti-FLAG antibody. After the pronase treatment, ChIP DNA fragments were isolated from the YdeO-DNA complexes for mapping on the genome. As an internal reference for the specific binding of YdeO with its targets, we interrogated the association of YdeO with the gadE promoter, the only known target of YdeO. After PCR amplification from the ChIP DNA samples using specific primers, the gadE promoter could be specifically amplified (data not shown).
To identify the genome-wide YdeO-binding sites on the entire E. coli genome, Sup (the whole extract DNA) and ChIP samples were each labelled and subjected to hybridization on a tiling array. Seven chromosomal regions were determined with high-level signal peaks indicating YdeO-binding, which were distinguishable from the background intensities (Fig. 1), including the gadEp2p3 promoters (Fig. 1F), the only known direct target of YdeO [9]. Six additional YdeO-binding sites were identified by ChIP-chip and were located within intergenic chromosomal regions. These included the intergenic spacer between yccA (an inner membrane protein) and hyaA (hydrogenase I) (Fig. 1B); the intergenic spacer upstream of appC (cytochrome bd-II oxidase) (Fig. 1C); the intergenic spacer upstream of the yiiS gene (a conserved protein) (Fig. 1D); the intergenic spacer upstream of the gadW gene (the gad operon regulator) (Fig. 1E); the intergenic spacer upstream of the gadE gene (the gad operon regulator) (Fig. 1F); and the intergenic spacer upstream of the slp gene (an outer membrane lipoprotein) (Fig. 1G). Although one YdeO-binding site was located inside the nhaA ORF another binding site was identified upstream of nhaR (Fig. 1A), in which the nhaR promoter has previously been identified [25]. Thus, all of YdeO binding sites were found in the vicinity of possible promoters (see below).
Figure 1. Genome-wide regulation of the Escherichia coli YdeO protein.

Location of YdeO binding sites. The panel shows detailed YdeO binding data from ChIP-chip experiments at the nhaR (A), hyaA (B), appC (C), yiiS (D), gadW (E), gadE (F), and slp (G) genomic loci. The box indicates the YdeO-binding site.
Identification of YdeO-binding in vitro to the seven targets
In order to confirm the direct interaction of YdeO to the seven target sequences determined by ChIP-chip, we performed the EMSA assay. Firstly we failed to purify the YdeO protein using the pET system, because the over-expressed YdeO proteins formed inclusion bodies in E. coli cells. Next YdeO was over-expressed as a His-SUMO fusion, and the His-SUMO-tagged YdeO protein could be purified in soluble forms by affinity chromatography with Ni-NTA agarose (data not shown). After treatment with SUMO protease to remove the His-SUMO tag, the intact YdeO protein, however, became insoluble. Then we used this His-SUMO-tagged YdeO as the test protein. The purified His-SUMO-YdeO protein bound to the gadEp2p3 promoters, the only known target of YdeO (Fig. 2A-f), in good agreement with the previous report [9]. Besides the gadE promoter, His-SUMO-YdeO formed complexes with the nhaR (Fig. 2A-a), hyaA (Fig. 2A-b), yiiS (Fig. 2A-d), gadW (Fig. 2A-e), and slp (Fig. 2A-g) promoters, which were observed as a smeared band, in the presence of 10-fold molar excess of YdeO over the DNA probes. A detectable level of the YdeO-probe complex was not formed with the appC promoter even in the presence of 35-fold molar excess of YdeO (Fig. 2A-c). These results indicate that YdeO directly binds to at least these six sites. YdeO-DNA was detected as a smeared band in several cases, implying the cooperative binding of YdeO at the higher concentration. Since the association of YdeO with the appC promoter was observed only in vivo (see Fig. 1), this association might require another factor(s) for effective binding.
Figure 2. The binding of YdeO on target promoters.

[A] The binding of YdeO to the target DNA, nhaR (a), hyaA (b), appC (c), yiiS (d), gadW (e), gadE (f), and slp (g). Probes were amplified by PCR using constructed reporter plasmids as templates and a pair of primers as the following; pLUXnhaR and a pair of NHAR-lux-F and Lux-R-FITC for nhaR probe; pHYAA-L and a pair of HYAA-LF and lacZ-30R-FITC for hyaA probe; pAPPC-L and a pair of APPC-LF and lacZ-30R-FITC for appC probe; pYY0503 and a pair of YIIS-LF and lacZ-30R-FITC for yiiS probe; pLUXgadWp and a pair of GADW-F-2 and Lux-R-FITC for gadW probe; pLUXgadEp and a pair of GADE-SCL-F-2 and Lux-R-FITC for gadE probe; and pLUXslpp and a pair of SLP-F-2 and Lux-R-FITC for slp probe; (Table S1). Each FITC-labeled probe (1 pmol) was incubated with YdeO protein (1, 5, 10, 25, or 35 pmol) and then DNA-YdeO complex was analyzed by native PAGE. Solid and dot lines indicate the migration of free DNA probe and DNA-YdeO complex, respectively. [B] The YdeO-binding site on gadW promoter. FITC-labeled probe (1 pmol) was incubated with 0 (lane 1), 0.5 (lane 2), 1.0 (lane 3), 5.0 (lane 4), or 15 (lane 5) pmol YdeO protein and then digested by DNase I. Sanger ladders are synthesized using Lux-R-FITC primer and pLUXgadWp plasmid as a template. A bar indicates the major region protected from DNase I digestion. The numbers represent the position from the transcription start site of gadWp1 promoter. [C] The sequence of YdeO-binding on gadW promoter. Black and gray bars indicate the major and minor YdeO-binding regions, respectively, as shown in [B]. The initiation codon of gadW coding is represented as a bold triplet. The numbers represent the position from the transcription start site of gadWp1 promoter.
Regulation in vivo of the predicted targets by YdeO: Transcriptome and RT-qPCR assays
We analyzed the alteration in the E. coli K-12 transcriptome caused by the over-expression of YdeO from a plasmid. E. coli KP7600 harboring pYY0401 (ydeO-3xflag) or the empty expression vector, pTrc99A, were grown until log phase under the same conditions used for ChIP-chip analysis, and total RNAs from these cultures were subjected to transcriptome analysis. Amongst genes downstream of a YdeO-binding site, 19 genes, (nhaA, nhaR, hyaA, hyaB, hyaC, hyaD, hyaE, hyaF, appC, appB, appA, yiiS, yiiT/uspD, slp, dctR, gadE, mdtE, mdtF, and gadW) were induced more than 3-fold by the over-expression of YdeO; while three genes, yccA, yiiR, and yhiS, were not affected in both duplicate experiments. (Table S2 and see also Table 1). These 19 genes induced by YdeO constitute a total of 7 transcriptional units, nhaAR, hyaABCDEF, appCBA, yiiS-yiiT/uspD, slp-dctR, gadE-mdtEF, and gadW. All 7 of these operons carry promoters containing YdeO-binding sites (see Fig. 2), and thus should be under the direct control of YdeO. We also examined the induction of these transcriptional units by the expression of YdeO by RT-qPCR after expression of YdeO. Transcripts of some representative genes from each operon were measured using specific pairs of the respective primers (Table S1). Transcripts were found to increase for all seven operons, nhaAR, hyaABCDEF, appCBA, yiiS-uspD(yiiT), gadW, gadE-mdtEF, and slp-dctR, in the ydeO-expressing cells (Table 2).
Table 1. Genes up-regulated by YdeO expression.
| Gene name | Transcriptoin unitsa | Wild type (Log10 ratiob) | ΔgadE (Log10 ratiob) | |||
| 1stc | 2ndc | 1stc | 2ndc | |||
| In both wild type and ΔgadE | adiC | adiC | 1.68 | 1.68 | 1.65 | 1.42 |
| appC | appCBA | 1.66 | 1.85 | 1.73 | 1.64 | |
| appB | appCBA | 1.71 | 1.69 | 1.71 | 1.60 | |
| appA | appCBA | 1.34 | 1.33 | 1.04 | 1.12 | |
| dnaK | dnaK-tpke11-dnaJ | 0.74 | 0.57 | –0.01 | 0.46 | |
| dnaJ | dnaK-tpke12-dnaJ | 0.66 | 0.57 | 0.04 | 0.75 | |
| hyaA | hyaABCDEF | 1.46 | 1.54 | 1.87 | 1.17 | |
| hyaB | hyaABCDEF | 1.61 | 1.50 | 2.16 | 1.11 | |
| hyaC | hyaABCDEF | 1.46 | 1.53 | 1.84 | 1.20 | |
| hyaD | hyaABCDEF | 1.22 | 1.29 | 1.57 | 1.03 | |
| hyaE | hyaABCDEF | 1.19 | 1.02 | 1.45 | 0.92 | |
| hyaF | hyaABCDEF | 1.15 | 1.09 | 1.4\5 | 1.09 | |
| ibpA | ibpAB | 0.65 | 0.50 | 0.09 | 0.64 | |
| katE | katE | 0.78 | 0.72 | 0.08 | 0.46 | |
| metK | metK | 0.64 | 0.63 | 0.03 | 0.60 | |
| nhaA | nhaAR | 0.53 | 0.50 | 0.54 | 0.40 | |
| yehX | osmF-yehYXW | 0.67 | 0.63 | 0.22 | 0.46 | |
| slp | slp-dctR | 1.99 | 2.15 | 2.41 | 1.79 | |
| dctR | slp-dctR | 1.85 | 1.16 | 2.08 | 1.82 | |
| thrA | thrLABC | 0.50 | 0.59 | 0.04 | 0.49 | |
| ybaS | ybaST | 1.55 | 1.52 | 1.38 | 0.87 | |
| ybaT | ybaST | 1.31 | 1.22 | 1.29 | 0.73 | |
| ynaI | ynaI | 0.77 | 0.74 | 0.70 | 0.63 | |
| In wild type but not ΔgadE | aidB | aidB | 1.11 | 1.13 | 0.03 | –0.31 |
| blc | blc | 0.61 | 0.64 | 0.35 | –0.27 | |
| cbpA | cbpAM | 0.57 | 0.53 | –0.15 | –0.50 | |
| cbpM | cbpAM | 0.62 | 0.53 | –0.04 | –0.47 | |
| dps | dps | 0.56 | 0.55 | 0.41 | –0.18 | |
| elaB | elaB | 0.60 | 0.53 | 0.10 | 0.08 | |
| gabT | gabDTP | 0.52 | 0.52 | 0.18 | 0.11 | |
| gadA | gadAX | 2.03 | 2.23 | –0.09 | 0.04 | |
| gadX | gadAX | 1.03 | 0.87 | 0.27 | –0.05 | |
| gadC | gadCB | 2.18 | 2.13 | 0.05 | –0.10 | |
| gadB | gadCB | 2.23 | 2.22 | 0.09 | 0.05 | |
| gadW | gadW | 0.51 | 0.50 | 0.15 | 0.02 | |
| hdeA | hdeAB-yhiD | 1.89 | 2.06 | 0.81 | 0.75 | |
| hdeB | hdeAB-yhiD | 1.85 | 2.10 | 1.04 | 0.75 | |
| hdeD | hdeD | 1.72 | 1.84 | 0.60 | 0.27 | |
| mdtE | mdtEF | 1.89 | 1.94 | –0.40 | –0.49 | |
| mdtF | mdtEF | 1.93 | 1.93 | 0.50 | 0.18 | |
| osmF | osmF-yehYXW | 0.61 | 0.60 | 0.11 | 0.17 | |
| yehY | osmF-yehYXW | 0.59 | 0.65 | 0.25 | 0.29 | |
| pagP | pagP | 0.92 | 0.95 | –0.38 | 0.21 | |
| sufA | sufABCDSE | 0.93 | 0.77 | –0.18 | –0.20 | |
| sufB | sufABCDSE | 0.56 | 0.50 | –0.25 | –0.31 | |
| wrbA | wrbA-yccJ | 0.76 | 0.70 | 0.40 | 0.13 | |
| yccJ | wrbA-yccJ | 0.73 | 0.64 | 0.55 | 0.14 | |
| ycaC | ycaC | 0.59 | 0.58 | 0.28 | 0.12 | |
| yfcG | yfcG | 0.68 | 0.61 | 0.18 | 0.05 | |
| ygaM | ygaM | 0.63 | 0.62 | 0.12 | –0.03 | |
| yhiM | yhiM | 1.98 | 1.75 | 0.33 | 0.23 | |
| yjjU | yjjUV | 0.58 | 0.63 | –0.39 | –0.41 | |
| yjjV | yjjUV | 0.58 | 0.62 | –0.11 | –0.28 | |
Transcriptional unit is represented according to the Regulon DB (http://regulondb.ccg.unam.mx/).
The processed intensity was calculated by Agilent Future Extraction. More than 0.5 of log ratio in WT.
Experiment was independently performed twice (each ratio is shown as 1st and 2nd).
Table 2. Induction of gene expression by YdeO.
| Gene name | Log10 ratio |
| nhaA | 0.62±0.02 |
| nhaR | 0.31±0.05 |
| yccA | –0.04±0.04 |
| hyaA | 0.72±0.20 |
| hyaF | 1.66±0.04 |
| appC | 2.64±0.04 |
| appA | 1.72±0.03 |
| yiiT/uspD | 0.70±0.03 |
| yiiS | 0.72±0.02 |
| gadW | 0.39±0.04 |
| gadE | 2.91±0.06 |
| mdtF | 2.36±0.07 |
| slp | 2.86±0.04 |
| dctR | 1.47±0.25 |
Ratio (ydeO +/vector) ± SEM is determined by RT-qPCR as described in Materials and methods.
We also measured the level of mRNAs in the ydeO-deficeint mutant, but detectable differences were not found for the mRNA from YdeO-target genes between the wild-type and the ydeO mutant. Transcript of yccA, an opposite direction gene from hyaA, was also not affected in the presence and absence of the YdeO-expressing plasmid (Table 2). Although the yiiS and uspD genes, encoding conserved proteins with unidentified function, were expressed even without the over-expression of YdeO, their expressions were further increased after YdeO expression. These results altogether indicate that YdeO plays a role as a positive regulator for expression of all seven operons, nhaAR, hyaABCDEF, appCBA, yiiS-yiiT/uspD, gadW, gadE-mdtEF, and slp-dctR.
Regulation in vivo of the predicted targets by YdeO: Reporter assay
To confirm the positive role of YdeO on expression of the newly identified target promoters, we performed the reporter assay using the lacZ reporter [15] and lux reporter [16] systems. The translation fusions, hyaA-lacZ, appC-lacZ, and yiiS-lacZ, on the pRS552 derivative plasmids were introduced at the attachment (att) site of the E. coli YY0201 chromosome using the λRS45 phage, resulting in isolation of HYAA-JL (hyaA-lacZ), APPC-JL (appC-lacZ), and YY1101 (yiiS-lacZ). Three E. coli lysogens containing hyaA, appC, and yiiS translational lac fusions in their chromosomes were transformed with either the YdeO-expression plasmid or the vector plasmid. The β-galactosidase activities in these transformants were measured in log-phase (Fig. 3A). YdeO-expression was found to induce the expression of all these test promoters, hyaA-lacZ, yiiS-lacZ, and appC-lacZ (Fig. 3A and B). In the cases of hyaA-lacZ and yiiS-lacZ, the promoter activity increased approximately 1.5 fold upon expression of YdeO. The detectable level of expression was not observed for appC-lacZ in the absence of YdeO expression but a high-level of appC-lacZ activity was detected upon expression of YdeO (Fig. 3B). The result indicates YdeO has a positive role in activation of the appC, hyaA, and yiiS promoters, in agreement with the observation by transcriptome and RT-qPCR (see above).
Figure 3. Reporter assays for transcriptional regulation by YdeO.
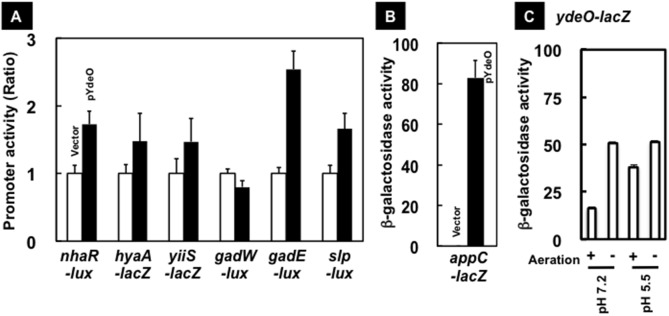
[A] YdeO-expression induces the expression of target promoters. YY0201/pLUXnhaR (nhaR-lux), HYAA-JL (hyaA-lacZ), YY1101 (yiiS-lacZ), YY0201/pLUXgadWp (gadW-lux), YY0201/pLUXgadE (gadE-lux), and YY0201/pLUXslpp (slp-lux), and were transformed with pTrc99A (vector, white bar) and pYdeO (ydeO, black bar). Transformants grew until logarithmic phase and β-galactosidase and luciferase activities of cultures were measured as described in Materials and methods. The data show the average of independent eight experiments with standard deviation as the ratio of a vector-transformant. [B] APPC-JL (appC-lacZ) was transformed with pTrc99A (vector) or pYdeO (ydeO). Transformants grew until logarithmic phase and β-galactosidase was measured as decribed in [A]. The data show the average of independent eight experiments with standard deviation as the Miller unit. [C] The ydeO expression induced under anaerobic conditions. The activity of ydeO promoter was measured in YY0101 growing in LB medium with pH 7.2 and 5.5 under aerobic (+) and anaerobic (−) conditions at logarithmic phase.
The nhaR, slp, gadE and gadW promoters were too weak for quantitation by the LacZ reporter system, so we then employed the more sensitive Lux reporter system. The lux reporter plasmids of four transcription fusions, slp-lux, gadE-lux and gadW-lux (kindly provide by Peter Lund [16]) and the nhaR-lux plasmid [constructed in this study], were introduced into YY0201 E. coli carrying either the vector plasmid or the YdeO-expression plasmid. The expression of nhaR-lux, slp-lux, and gadE-lux was found to be activated in the presence of the YdeO-expressing plasmid (Fig. 3A), indicating that YdeO is also a positive regulator for these promoters. Recently RNA-seq analysis indicated the presence of a novel nhaR promoter inside the coding region of nhaA [25]. The binding site of YdeO is located upstream of this putative promoter (see above). Accordingly the constructed nhaR-lux reporter plasmid containing this novel nhaR promoter was also activated in the presence of YdeO expression (Fig. 3A).
The expression level of gadW-lux stayed unaltered with and without the YdeO-expression plasmid. It is inconsistent with the RT-qPCR result that the mRNA level of gadW increased in the presence of YdeO expression as detected by RT-qPCR (Table 2). This apparent disagreement might be due to translational inhibition of gadW-lux by the anti-sense RNA of gadW, named gadY, encoded in the gadW-lux plasmid.
Recognition sequence of YdeO transcription factor
To identify the YdeO-binding sequence, we performed DNase I footprinting of the gadW promoter with increasing concentrations of YdeO. At low protein levels, YdeO protected the region from –53 to +8 of the gadW promoter (Fig. 2B, lanes 2–4). In the presence of 15-fold molar excess of YdeO, the protected region by YdeO expanded from –53 to +84 of the gadW promoter possibly due to protein-protein interaction (Fig. 2B, lane 5) in agreement with the smeared band formation observed by EMSA (see above). Within the core YdeO-binding region, the inverted repeat of hexa-nucleotides, 5′-ATTTCA-3′, was identified (see Figs. 2C and 4A).
Figure 4. The characterization and location of YdeO-box on target promoters.

We examined the conservation of the inverted repeat across seven YdeO-binding regions detected in vivo by ChIP-chip analysis, 131-bp on nhaR promoter, 216-bp on hyaA promoter, 139-bp on appC promoter, 217-bp on yiiS promoter, 181-bp on gadW promoter, 241-bp on gadE promoter, and 145-bp on slp promoter. [A] The panel shows the DNA sequence, containing the identified hexa-mer repeat (YdeO-box). The YdeO-box identified in all of promoters located on seven binding sites of YdeO. The number indicates the distance from each transcription start site (RegulonDB [http://regulondb.ccg.unam.mx]). [B] Organization of the promoters controlled by YdeO is shown. The locations of a hexamer of YdeO-binding sites (triangle) at relative positions from the transcription initiation site (solid arrow) are shown for the promoters. The filled bars represent open reading frames of the target genes.
Using this YdeO-box sequence, we searched for this inverted repeat within the seven YdeO-binding regions detected by in vivo by ChIP-chip analysis, and identified this inverted repeat sequence of all the YdeO-binding regions at various positions between –131 to –1 with respect to the transcription start site (Fig. 4). The length of spacer between the 5′-ATTTCA-3′ hexa-nucleotide sequence ranges from 9 to 21 nucleotides (Fig. 4A). Recent studies show that YpdB and YehT bind to the direct repeat of their specific sequence separated by a 9- and 13-bp spacer, respectively, in E. coli [26], [27]. Previous work shows that the spacer length of the specific DNA binding region is diverse for the E. coli transcription factor CpxR [28]. Therefore, we have denoted the inverted repeat as the YdeO-box (Fig. 4).
Induction of NhaR, GadE, and GadW by YdeO
Four transcription factors, the LysR-type NhaR, the LuxR-type GadE, and the AraC-type GadW, were found to be under the direct control of YdeO (see Figs. 1–4). NhaR is an activator of a sodium/proton antiporter gene [29] and both GadE and GadW are involved in regulation of the genes for glutamate-dependent acid resistance system [8], [9]. In addition to these three transcription factors, the gene encoding the CadC-like transcription factor DctR is located downstream of the slp gene which codes for a starvation lipoprotein, and is considered to be co-transcribed with the slp gene. DctR is involved in protection against metabolic endproducts under acidic conditions [30]. To examine the involvement of YdeO in control of expression of the three transcription factors, the cellular level of these proteins in E. coli, with or without the YdeO-expressing plasmid, were measured by Western blotting assay.
To perform the Western blotting assay of GadE and GadW using the anti-FLAG antibody, we constructed E. coli strains YY5002 and YY5003 including 3xflag tag at the 3′-terminal end of gadE and gadW, respectively, on the E. coli chromosome. The YdeO-expression plasmid, pYdeO (ydeO), and the empty expression vector pTrc99A were transformed into these E. coli strains and the transformants were grown in LB medium until log phase. The whole-cell lysates were prepared, and subjected to Western blotting assay by using anti-NhaR, anti-FLAG, and anti-RpoA for detection of NhaR, GadE-3xFLAG and GadW-3xFLAG, and RNA polymerase α subunit, respectively. All transformants with or without the YdeO-expressing plasmid retain approximately a constant amount of the α subunit of RNA polymerase (data not shown). The level of GadE increased in the YY5002 harboring the YdeO-expression plasmid, supporting the prediction that the gadE gene is under the direct and positive control of YdeO. However, we failed to detect NhaR and GadW even in the presence of YdeO expression (Fig. 5).
Figure 5. Induction of NhaR, GadE, and GadW by YdeO in E. coli.
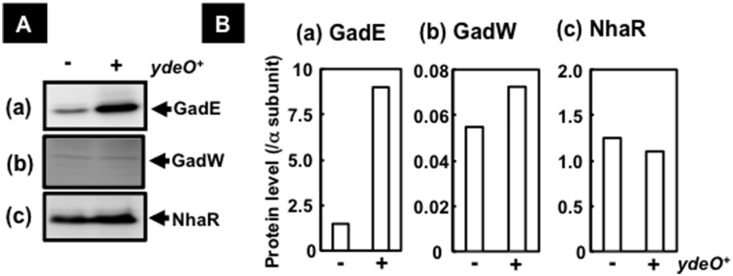
YY5002 (gadE-3xflag) (a), YY5003 (gadW-3xflag) (b), and BW25113, (c) harboring pTrc99A (−) or pYdeO (+), were grown in LB medium until logarithmic phase. After centrifugation the lysate solution was prepared in lysis buffer containing 8 M urea by sonication. The lysates were subjected to western blotting as described in Materials and Methods. Anti-FLAG (SIGMA) and anti-NhaR (Lab preparation) were used for detection of GadE-3xFLAG/GadW-3xFLAG, and NhaR, respectively [A]. The amounts of GadE-3xFLAG, GadW-3xFLAg, and NhaR were represented as the ratio of level of RNA polymerase-α subunit, detected by anti-α (Neoclone) [B].
Search for the whole set of genes regulated by YdeO > GadE
To obtain the gene expression profile of the YdeO > GadE cascade, we performed a transcriptome assay. E. coli wild-type KP7600 and gadE-deficient JD25278 harbouring pTrc99A and pYY0401 (ydeO-3xflag) were incubated in LB medium at 37°C with shaking until log phase and total RNA from these cultures was subjected to transcriptome analysis under standard experimental conditions as described in Materials and methods. The results revealed that a total of 106 genes were markedly affected by YdeO expression in the wild-type and included 53 up- and the same number of down-regulated genes (Tables S2 and S3). Among the 53 genes up-regulated by YdeO expression, clustering analysis showed 23 genes were induced in both the parent strain and the gadE-deficient mutant and 30 genes induced in the wild-type but not the gadE-deficient mutant (Fig. 6). The observed alteration of the transcriptome profile caused by deletion of the gadE gene was similar to that reported by Masuda and Church [31]. Genes induced in both strains are organized into a total of 12 transcriptional units (Table 1), including five transcription units, hyaABCDEF, appCBA, slp-dctR, and nhaAR, that are under the direct control of YdeO (see above). On the other hand, the rest of the 30 up-regulated genes forming 21 transcription units were induced in the wild-type but not in the gadE-deficient mutant (Table 1), indicating that these 21 transcription units are under the direct control of GadE but the indirect control of YdeO. This set of 21 transcription units includes the hitherto identified GadE targets, gadA, gadB, and gadC [9]. On the other hand, detectable change was not observed in the transcription pattern between the parent strain and the gadW-deficient mutant, consistent with the lack of YdeO-dependent GadW expression under the conditions herein employed (Figs. 3 and 5). The yehX gene was induced by the YdeO-expression plasmid in both the parent strain and the gadE mutant but the osmF and yehY genes, and parts of osmF-yehYXW transcription unit, were not induced in the gadE mutant (Fig. 6 and Table 1), implying that GadE activates the known promoters located at the upstream of yehX which is possibly activated by YdeO.
Figure 6. YdeO > GadE cascade regulation in E. coli.

This shows the clustering pattern of expression of genes induced by the ydeO-expression in the parent (KP7600) and gadE-deficient mutant (JD25278) by Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm).
Physiological roles of YdeO in response to environmental stresses
The level of translational control of the YdeO regulator itself was analyzed using a reporter assay with the ydeO-lacZ fusion. In E. coli YY0101 (ydeO-lacZ) grown under aerobic conditions, β-galactosidase activity from ydeO-lacZ increased two-fold under the acidic condition of pH 5.5 compared with pH 7.0 (Fig. 3C). Interestingly the high level of ydeO-lacZ was detected in both pH 5.5 and 7.0 when E. coli were grown under anaerobic conditions (Fig. 3C), implying that YdeO plays a role in E. coli respiration under anaerobic conditions, such as in the animal intestine. Previously, we identified that the transcription of ydeO is induced by exposure to ultraviolet light via the two-component system EvgSA two-component system [11]. In agreement with this finding, ydeO expression was not induced in the evgA-defective mutant under both acidic and anaerobic conditions (data not shown).
Discussion
The YdeO regulon
Here we have identified a total of seven YdeO-binding sites on the E. coli genome using ChIP-chip and transcription analyses in vivo. The EMSA experiments showed that purified YdeO also binds in vitro to these six sites (see Fig. 2). The reporter and RT-qPCR assays indicated that all of the promoters located downstream of these YdeO-binding sites are activated by YdeO (see Table 1 and Fig. 3). The hexa-nucleotide repeat 5′-ATTTCA-3′, which we have named the YdeO box, is conserved in all of YdeO-binding sites we identified experimentally (see Fig. 4). Even though this YdeO-box like sequence exists within the appC promoter, which is located immediately downstream of a YdeO-binding site (see Fig. 1), the binding in vitro of YdeO to the appC promoter probe was not high (see Fig. 2), implying that an as yet unidentified additional transcription factor or DNA secondary structure is needed for efficient binding of YdeO to the target promoter. Since the appC promoter is transcribed in vivo by RNA polymerase containing the RpoS sigma factor and is induced by AppY [32], [33], one possibility is that AppY and/or RpoS sigma are required for the efficient binding of YdeO to the appC promoter. Thus, we conclude that YdeO is a positive regulator for transcription of operons controlled by seven promoters, the nhaR promoter, hyaA promoter, appC promoter, yiiS promoter, slp promoter, gadE promoter, and gadW promoter (Fig. 7).
Figure 7. EvgAS > YdeO > DctR/NhaR/GadE/GadW regulatory network in E. coli.

Transcription cascade: EvgSA > YdeO > NhaR, GadE, GadW
E. coli responds to temporary low pH using the glutamate-dependent acid resistant system, which involves two complex regulatory systems: EvgAS > YdeO > GadE; and Crp> RpoS > GadX > GadW [9], [12]. In this study, we showed that YdeO directly regulates the expression of three transcription factor genes, nhaR, gadE, and gadW (see Fig. 7), proving the novel transcription cascade: EvgAS > YdeO > NhaR/GadE/GadW.
YdeO not only plays a regulatory role in positive feedback loop of EvgAS > YdeO > GadE pathway, but also a positive role in the GadXW pathway, thereby linking the GadE- and GadXW-pathways for acid resistance. The GadXW circuit is believed to function during stationary phase. YdeO-overexpression induced GadE-dependent transcription of the gadW gene but GadW protein was not detected in growing E. coli cell (Fig. 6), suggesting that stationary phase specific factors are required for GadW.
Transcriptome analysis identified the set of genes directly regulated by YdeO or indirectly through the YdeO > GadE cascade (Table 2; see Fig. 7 for the summary model). GadE induced by YdeO stimulated the transcription of hdeAB-yhiD, hdeD, gadAX, gadCB, mdtEF, gadW, and yhiM as well as those previously reported promoters [9], [34], [35]. The GAD cluster including hdeAB-yhiD, hdeD, gadAX, gadCB, gadE, mdtEF, and gadW, is necessary for glutamine-dependent acid resistance [9], [34]. Recently the yhiM gene was reported to be essential for growth at pH 2.5 and is necessary for glutamine- and lysine-dependent acid resistance, but is not required for arginine-dependent acid resistance [36]. In addition of these operons, the YdeO > GadE cascade induced a total of 19 operons including aidB, blc, cbpAM, elaB, gabDTP, pagP, sufABCDSE, ycaC, yfcG, ygaM, and yjjUV (see Table 1), of which the yfcG gene encodes a disulfide reductase [37] and the sufABCDSE operon encodes the complex biosynthetic machinery for iron-sulfur clusters in several enzymes which have critical cysteine residues [38], suggesting a relationship between the function of YdeO and cysteine metabolism.
The physiological role of the YdeO regulon
In addition of the hitherto-identified target gadE, we have identified a total of seven operons belonging to the YdeO regulon. The expression of ydeO is induced under acidic conditions (see Fig. 3C). In good agreement, the gadE operon encodes the master activator for expression of gadA and gadBC, which are involved in the glutamate-dependent acid resistance system which works for consumption of intracellular protons by glutamate decarboxylation. In addition to acid conditions, the expression of ydeO is also induced under anaerobic growth in both neutral and acid conditions. Two YdeO-regulated targets, hyaABCDEF and appCBA, encode a hydrogenase and a quinone oxidase, respectively, both being involved in bacterial respiration. The HyaABC complex oxidizes dihydrogen to two protons, following release of them to the outside of the membrane, and donation of the electrons to the quinone pool. The AppBC complex donates electrons taken by a quinone to intracellular oxygen, consuming an intracellular proton per electron (Fig. 7), resulting in H2O production via oxygen [39]. Thus, the hyaABCDEF operon contributes to the consumption of the intracellular proton while the appCBA operon contributes to the utilization of reduced quinone. Taken together, these physiological systems activated by YdeO stimulate stress response and respiration. These findings also suggest that YdeO activated genes play an important role in primary adaptation, which enables the cell to colonize animal intestines by contributing to adaptation to acidic conditions in the stomach and to anaerobic conditions in the intestine.
Supporting Information
Bacterial strains, phage, plasmids, and oligonucleotides used in this study. E. coli K-12 derivatives used in this study were indicated with characterizations. The used bacteriophage and plasmids were also shown. Oligonucleotides were represented with DNA sequences.
(DOCX)
The genes affected by expression of ydeO gene in E. coli KP7600. Transcriptome analysis was performed using total RNAs from KP7600 harboring pTrc99A (vector) and pYY0401 (ydeO-3xflag) as described in Materials and methods. The E. coli Gene Expression Microarray microarray 8×15 K (Agilent) hybridized by the fluorescent cDNAs was scanned with an Agilent G2565CA microarray scanner Ver. 8.1, the intensities of both Cy3 and Cy5 were quantified by Feature Extraction Ver. 8.1, and then, the Cy5/Cy3 ratios were calculated from the normalized values.
(XLSX)
The genes affected by expression of ydeO gene in the gadE -deficient E. coli mutant. Transcriptome analysis was performed using total RNAs from JD25278 (gadE::mini-Tn10) harboring pTrc99A (vector) and pYY0401 (ydeO-3xflag) as described in Table S2. The intensities quantified by Feature Extraction Ver. 8.1 and the Cy5/Cy3 ratios were represented.
(XLSX)
Acknowledgments
We gratefully acknowledge Jon Hobman (Nottingham University) for valuable comments and proofreading of the manuscript.
This work is supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities and Special Coordination Funds for Promoting Science and Technology and JSPS-DST International Collaborations. We are grateful to Peter Lund for providing lux plasmids. We also thank the National BioResource Project (NBRP) of Japan for providing E. coli strains.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work is supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities and Special Coordination Funds for Promoting Science and Technology and JSPS-DST International Collaborations. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Foster JW (2004) Escherichia coli acid resistance: tales of an amateur acidophile. Nature Rev Microbiol 2: 898–907. [DOI] [PubMed] [Google Scholar]
- 2. Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW (1999) Control of acid resistance in Escherichia coli . J Bacteriol 181: 3525–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iyer R, Williams C, Miller C (2003) Arginine-agmatine antiporter in extreme acid resistance in Escherichia coli . J Bacteriol 185: 6556–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin J, Lee IS, Frey J, Slonczewski JL, Foster JW (1995) Comparative analysis of extreme acid survival in Salmonella typhimurium, Shigellaflexneri, and Escherichia coli . J Bacteriol 177: 4097–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Biase D, Tramonti A, Bossa F, Visca P (1999) The response to stationary-phase stress conditions in Escherichia coli: role and regulation of the glutamic acid decarboxylase system. Mol Microbiol 32: 1198–1211. [DOI] [PubMed] [Google Scholar]
- 6. HershA BM, Farooq FT, Barstad DN, Blankenshorn DL, Slonczewski JL (1996) A glutamate-dependent acid resistance gene in Escherichia coli . J Bacteriol 178: 3978–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hommais F, Krin E, Coppee JY, Lacroix C, Yeramian E, et al. (2004) GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli . Microbiology 150: 61–72. [DOI] [PubMed] [Google Scholar]
- 8. Ma Z, Gong S, Richard H, Tucker DL, Conway T, et al. (2003) GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol Microbiol 49: 1309–1320. [DOI] [PubMed] [Google Scholar]
- 9. Ma Z, Masuda N, Foster JW (2004) Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli . J Bacteriol 186: 7378–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Itou J, Eguchi Y, Utsumi R (2009) Molecular mechanism of transcriptional cascade initiated by the EvgS/EvgA system in Escherichia coli K-12. Biosci Biotechnol Biochem 73: 870–878. [DOI] [PubMed] [Google Scholar]
- 11. Yamanaka Y, Ishihama A, Yamamoto K (2012) Induction of YdeO, a regulator for acid resistance genes, by ultraviolet irradiation in Escherichia coli . Biosci Biotechnol Biochem 76: 1236–1238. [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto K (2014) The hierarchic network of metal-response transcription factors in Escherichia coli . Biosci Biotechnol Biochem 78: 737–747. [DOI] [PubMed] [Google Scholar]
- 13. Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L (2001) Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci USA 94: 13997–14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jishage M, Ishihama A (1997) Variation in RNA polymerase sigma subunit composition within different stocks of Escheichia coli strain W3110. J Bacteriol 179: 959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simon RW, Hausman F, Kleckner N (1987) Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53: 85–96. [DOI] [PubMed] [Google Scholar]
- 16. Burton NA, Johnson MD, Antczak P, Robinson A, Lund PA (2010) Novel aspects of the acid response network of E. coli K-12 are revealed by a study of transcriptional dynamics. J Mol Biol 401: 726–742. [DOI] [PubMed] [Google Scholar]
- 17. Katou Y, Kaneshiro K, Aburatani H, Shirahige K (2006) Genomic approach for the understanding of dynamic aspect of chromosome behavior, Methods Enzymol. 409: 389–410. [DOI] [PubMed] [Google Scholar]
- 18. Uyar E, Kurokawa K, Yoshimura M, Ishikawa S, Ogasawara N, et al. (2009) Differential binding profiles of StpA in wild-type and h-ns mutant cells: a comparative analysis of cooperative partners by chromatin immunoprecipitation-microarray analysis. J Bacteriol 191: 2388–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chumsakul O, Takahashi H, Oshima T, Hishimoto T, Kanaya S, et al. (2011) Genome-wide binding profiles of the Bacillus subtilis transition state regulator AbrB and its homolog Abh reveals their interactive role in transcriptional regulation, Nucleic Acids Res. 39: 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ueda T, Takahashi H, Uyar E, Ishikawa S, Ogasawara N, et al. (2013) Functions of the Hha and YdgT proteins in transcriptional silencing by the nucleoid proteins, H-NS and StpA, in Escherichia coli . DNA Res 20: 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, et al. (2005) Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli . J Biol Chem 280: 1448–1456. [DOI] [PubMed] [Google Scholar]
- 22. Yamamoto K, Ishihama A (2005) Transcriptional response of Escherichia coli to external copper. Mol Microbiol 56: 215–227. [DOI] [PubMed] [Google Scholar]
- 23. Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kailasan Vanaja S, Bergholz TM, Whittam TS (2009) Characterization of the Escherichia coli O157:H7 Sakai GadE regulon. J Bacteriol 191: 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salgado H, Peralta-Gil M, Gama-Castro S, Santos-Zavaleta A, Muñiz-Rascado L, et al. (2013) RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res 41: D203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fried L, Behr S, Jung K (2013) Identification of a target gene and activating stimulus for the YpdA/YpdB histidine kinase/response regulator system in Escherichia coli. J Bacteriol 195: 807–815. J Bacteriol 194: 4272–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraxenberger T, Fried L, Behr S, Jung K (2012) First insights into the unexplored two-component system YehU/YehT in Escherichia coli. [DOI] [PMC free article] [PubMed]
- 28. Yamamoto K, Ishihama A (2006) Characterization of copper-inducible promoters regulated by CpxA/CpxR in Escherichia coli . Biosci Biotechnol Biochem 70: 1688–1695. [DOI] [PubMed] [Google Scholar]
- 29. Rahav-Manor O, Carmel O, Karpel R, Taglicht D, Glaser G, et al. (1992) NhaR, a protein homologous to a family of bacterial regulatory proteins (LysR), regulates nhaA, the sodium proton antiporter gene in Escherichia coli. . J Biol Chem 267: 10433–10438. [PubMed] [Google Scholar]
- 30. Mates AK, Sayad AK, Foster JW (2007) Products of the Escherichia coli acid fitness island attenuate metabolic stress at extreme low pH and mediate a cell density-dependent acid resistance. J Bacteriol 189: 2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Masuda N, Church GM (2003) Regulatory network of acid resistance genes in Escherichia coli . Mol Microbiol 48: 699–712. [DOI] [PubMed] [Google Scholar]
- 32. Atlung T, Brøndsted L (1994) Role of the transcriptional activator AppY in regulation of the cyx-appA operon of Escherichia coli by anaerobiosis, phosphate starvation, and growth phase. J Bacteriol 176: 5414–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brøndsted L, Atlung T (1996) Effect of growth conditions on expression of the acid phosphatase (cyx-appA) operon and the appY gene, which encodes a transcriptional activator of Escherichia coli . J Bacteriol 178: 1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Masuda N, Church GM (2002) Escherichia coli gene expression responsive to levels of the response regulator EvgA. J Bacteriol 184: 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krin E, Danchin A, Soutourina O (2010) RcsB plays a central role in H-NS-dependent regulation of motility and acid stress resistance in Escherichia coli . Res Microbiol 161: 363–371. [DOI] [PubMed] [Google Scholar]
- 36. Nguyen TM, Sparks-Thissen RL (2012) The inner membrane protein, YhiM, is necessary for Escherichia coli (E. coli) survival in acidic conditions. Arch Microbiol 194: 637–641. [DOI] [PubMed] [Google Scholar]
- 37. Wadington MC, Ladner JE, Stourman NV, Harp JM, Armstrong RN (2009) Analysis of the structure and function of YfcG from Escherichia coli reveals an efficient and unique disulfide bond reductase. Biochemistry 48: 6559–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vinella D, Brochier-Armanet C, Loiseau L, Talla E, Barras F (2009) Iron-sulfur (Fe/S) protein biogenesis: phylogenomic and genetic studies of A-type carriers. PLoS Genet 5: e1000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borisov VB, Gennis RB, Hemp J, Verkhovsky MI (2011) The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta 1807: 1398–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial strains, phage, plasmids, and oligonucleotides used in this study. E. coli K-12 derivatives used in this study were indicated with characterizations. The used bacteriophage and plasmids were also shown. Oligonucleotides were represented with DNA sequences.
(DOCX)
The genes affected by expression of ydeO gene in E. coli KP7600. Transcriptome analysis was performed using total RNAs from KP7600 harboring pTrc99A (vector) and pYY0401 (ydeO-3xflag) as described in Materials and methods. The E. coli Gene Expression Microarray microarray 8×15 K (Agilent) hybridized by the fluorescent cDNAs was scanned with an Agilent G2565CA microarray scanner Ver. 8.1, the intensities of both Cy3 and Cy5 were quantified by Feature Extraction Ver. 8.1, and then, the Cy5/Cy3 ratios were calculated from the normalized values.
(XLSX)
The genes affected by expression of ydeO gene in the gadE -deficient E. coli mutant. Transcriptome analysis was performed using total RNAs from JD25278 (gadE::mini-Tn10) harboring pTrc99A (vector) and pYY0401 (ydeO-3xflag) as described in Table S2. The intensities quantified by Feature Extraction Ver. 8.1 and the Cy5/Cy3 ratios were represented.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.