

Abstract
Pharmacologically, vasoactive agents targeting endothelial and/or smooth muscle cells (SMC) are known to cause acute drug-induced vascular injury (DIVI) and the resulting pathology is due to endothelial cell (EC) perturbation, activation, and/or injury. Alteration in EC structure and/or function may be a critical event in vascular injury and, therefore, evaluation of the circulatory kinetic profile and secretory pattern of EC-specific proteins such as VWF and VWFpp could serve as acute vascular injury biomarkers. In rat and dog models of DIVI, this profile was determined using pharmacologically diverse agents associated with functional stimulation/perturbation (DDAVP), pathological activation (lipopolysaccharide [LPS]/endotoxin), and structural damage (fenoldopam [FD], dopamine [DA], and potassium channel opener (PCO) ZD6169). In rats, FD caused moderate DIVI and time-related increase in plasma VWF levels ∼33% while in control rats VWF increased ∼5%. In dogs, VWF levels transiently increased ∼30% when there was morphologic evidence of DIVI by DA or ZD6169. However, in dogs, VWFpp increased >60-fold (LPS) and >6-fold (DDAVP), respectively. This was in comparison to smaller dynamic 1.38-fold (LPS) and 0.54-fold (DDAVP) increases seen in plasma VWF. Furthermore, DA was associated with a dose-dependent increase in plasma VWFpp. In summary, VWF and VWFpp can discriminate between physiological and pathological perturbation, activation, and injury to ECs.
Keywords: endothelial cell, biomarker, DIVI, rat, dog, perturbation, activation, VWF, VWFpp, LPS, DDAVP
Introduction
Vascular damage is relatively common in rats, dogs, and humans as a spontaneous natural disease or as a result of toxic injury (rats and dogs) induced by a wide range of chemically and pharmacologically diverse vasoactive agents. Evidence from morphologic pathology-based studies combined with molecular investigations suggested that endothelial cell (EC) perturbation with subsequent damage may be an early and obligatory step in development of vascular injury. In humans and animals, the absence of a specific and reliable biomarker of EC perturbation, activation, and/or damage represents an unmet need preclinically as well as in the clinical setting. Because vascular injury and/or EC activation and perturbation is an occult pathology rarely associated with adverse clinical signs in animals, there is a strong interest in evaluating blood (cells, plasma, and/or serum) to identify and characterize potential circulating biomarkers derived from ECs, which when activated, release circulating proteins that reflect structural alteration and/or functional disturbances. Such a molecule would bridge this biomarker deficit/gap, particularly if it is EC-specific and released during periods of EC activation, perturbation, and/or injury.
Of the many products produced by ECs, von Willebrand factor (VWF) and VWF propeptide (VWFpp) have the requisite attributes of sensitivity, specificity, and analytical stability to be potentially useful biomarkers of EC perturbation, activation, and vascular injury; however, simultaneous evaluation of both of these proteins has not been rigorously explored in models of EC activation or drug-induced vascular injury (DIVI). In humans, data from several studies suggest that measurement and determination of the secretory pattern of plasma VWF and VWFpp can be used to assess and discriminate acute versus chronic EC activation, perturbation, and/or injury (van Mourik et al. 1999; van Mourik and Romani deWit 2001). However, in studies using animal models, simultaneous measurement and comparative analysis of VWF and VWFpp during periods of EC perturbation, activation, and DIVI are not well documented. The significance of VWF as a biomarker of EC activation, perturbation, and injury has been challenged, because the adhesive properties of VWF cause binding to platelets and the subendothelial connective tissue; therefore, systemic VWF concentrations are not an accurate measurement of EC secretory activity during activation, perturbation, or injury. Furthermore, species differences in the cellular source of VWF make it difficult to attribute increases in plasma levels as definitive evidence of EC activation, perturbation, or injury (McCarroll et al. 1988; Sanders et al. 1995). It has been suggested that rat and human platelets contain abundant VWF while conversely only a tiny amount is found in dog platelets (McCarroll et al. 1988; Sanders et al. 1995). In animal models of cardiovascular injury and disease, relatively scant attention has been given to the potential utility of VWF as a biomarker of EC activation, perturbation, or vascular damage. An exception is a study in a rat model of acute mesenteric arterial injury, using the dopaminergic vasodilator fenoldopam (FD). FD was associated with marginal and transient increases in plasma VWF during periods of arterial damage but a definitive conclusion could not be drawn because repeated venipuncture was also associated with a time-dependent increase in plasma VWF concentrations in saline-treated rats (Newsholme et al. 2000). Because of this procedural-related increase in VWF associated with venipuncture in rats, interpretation was confounded and the increase in plasma VWF was attributed in part, to an acute phase inflammatory response. In order to generate data that would provide a clearer picture, experiments were conducted to assess the potential relevance of VWF in animal models during periods of EC perturbation, activation, and DIVI.
There is growing interest in assessment of VWF and VWFpp when EC perturbation, activation, and/or vascular injury are suspected because both molecules are processed and released from the endothelium in an equimolar basis, basally, constitutively, or during periods of EC activation and injury that causes release through the regulated pathway (Blann 2006; Meigs et al. 2006; Ware et al. 2004). In response to secretagogue, stimulation of the regulated pathway results in VWF and VWFpp release from Weibel–Palade Bodies (van Buul-Wortelboer et al. 1989; van Mourik et al. 1999). VWFpp offers several distinct advantages over the mature protein because unlike VWF, VWFpp does not bind or get trapped by the subendothelial connective tissues or platelets after secretion, it is found in low levels in normal plasma because it has rapid turnover and a short half-life, and as a result, systemic propeptide level more accurately reflects endothelial secretion and release. Therefore, in response to the type of stimuli and/or levels of vascular perturbation and injury, the differential kinetics in secretion and release pattern of VWF and VWFpp could be used to assess the extent and time course of EC activation under physiological or pathological conditions. These characteristics formed the basis for evaluating the circulatory kinetic profile and temporal secretory release pattern of VWF and VWFpp under (1) physiological EC stimulation/activation in response to a single bolus dose of DDAVP; (2) pathological EC perturbation induced by a low dose of lipopolysaccharide (LPS)/endotoxin not associated with structural/morphologic damage; and (3) VWF/VWFpp profile when there is structural/morphological damage following administration of chemically and structurally diverse pharmacological agents known to cause drug-induced vascular SMC and/or EC injury in dogs.
Additional experiments were conducted in rats to exclude the procedural-related increases in VWF from activated platelets and repeated venipuncture during periods of DIVI caused by FD.
Materials and Methods
All studies were approved by the appropriate institutional animal care and use committee and were designed to minimize severe clinical signs of toxicity. Animal care, husbandry, and cage specifications conformed to the current guidelines of ILAR/AALAC and the “Guide for the Care and Use of Laboratory Animals.”
Animals
Rats
Male Han Wistar rats, 8 to 10 weeks of age (Charles River, Wilmington, MA), were used for all studies. The rats were housed individually in standard polycarbonate cages or in Culex® cages. For intravenous infusion studies (IV), catheters were implanted in the jugular and femoral vein (double catheterization), and the jugular vein catheter was connected to a tethered system and a Harvard pump (KD Scientific, Holliston, MA), while the femoral vein catheter was connected to the Culex® blood collection system (Bioanalytical Systems Inc., West Lafayette, IN). All rats were housed in a temperature-controlled room with 12-hr light/dark cycle and free access to water and food (Purina rodent chow).
Dogs
Female beagle dogs (AstraZeneca Pharmaceutical, Alderley Park, Manchester, UK; Marshall Farms, Inc., North Rose, NY; or Covance Research Products, Inc. Kalamazoo, MI, or Cumberland, VA) were housed in pairs in stainless steel cages in a controlled environment with access to food and water.
Test Article Preparation and Administration
(R)-6-Chloro-2,3,4,5-tetrahydro-1-(4-hydroxyphenyl)-1H-3-benzazepine-7,8-diol hydrobromide (FD; Sigma, St. Louis, MO) was dissolved in the vehicle that consisted of sterile 20% (v/v) propylene glycol:80% (v/v) 0.9% (w/v) sodium chloride (saline). (LPS; Endotoxin) from Escherichia coli (E. coli) and [deamino-Cys, D-Arg8]-vasopressin (DDAVP; Sigma, St. Louis, MO) were dissolved in 0.9% (w/v) sodium chloride (saline) at 2 mg/ ml and 10 μg/ml, respectively. The PCO (ZD6169) was formulated as a suspension in 0.5% (w/v) hydroxypropyl methylcellulose (HPMC) solution containing 0.1% (w/v) aqueous polysorbate 80. Dopamine (DA) was dissolved in 0.9% (w/v) sodium chloride (saline).
Study Design, Blood, and Tissue Collection
Rat studies
Three studies were conducted in rats to evaluate VWF. VWFpp was not evaluated in the rat due to lack of a rat-specific or cross-reactive assay.
First study: In the first experiment, blood samples were collected via the tail vein to determine whether multiple venipunctures in the same rats affected circulating plasma VWF levels. Eight näive rats were restrained in domed heating units (Narco Bio-Systems, Houston, TX) and blood samples were collected from the tail vein using 21-gauge winged infusion needles (MINISET, Travenol Labs, Deerfield, IL) into 3.2% sodium citrate tubes at 0, 2, 4, 6, and 24 hr. The samples were processed to measure plasma VWF and stored at −80°C until analyzed.
Second study: The objective of the second experiment was to investigate the findings of the first study by comparing the amount of VWF in normal plasma versus platelets and determine the platelet VWF:plasma VWF ratio. Briefly, eleven näive rats were euthanatized, and blood samples were collected from the caudal vena cava. Blood samples were collected into 3.2% sodium citrate tubes, processed to measure plasma and platelet VWF levels, and stored at −80°C until analyzed.
Third study: The objective of this experiment was to exclude the contribution of platelet VWF that occurs during repeated venipuncture (tail vein) and determine the plasma VWF levels associated with EC injury/damage following administration of vasotoxic doses of FD. Briefly, rats (5/group) were administered vehicle or 100 μg/kg/min FD by IV infusion. These rats were double cannulated, as previously described, and the catheterized jugular vein was used for dosing and the catheterized femoral vein used for blood collection to measure plasma VWF levels at predose (0 hr), 0.75, 2, 4, 6, and 24 hr after initiation of dosing. Patency of the catheters were maintained 1% heparin after predose, 0.75-, 2-, and 4-hr blood collection and with 100% heparin after the 6-hr blood collection. Rats were necropsied 24-hr postdose and the mesenteric arteries collected and processed for microscopic evaluation to determine the incidence and extent of vascular injury.
Studies in Beagle Dogs
Six studies were conducted in beagle dogs to evaluate VWF and VWFpp.
First study: The objective of the first study was to determine the effects of repeated venipuncture (cephalic vein) on plasma VWF levels in dogs. Briefly, 5 näive dogs were bled from the cephalic vein at 0, 1, 2, 3, 5, 24, 48, 72, and 96 hr. The blood samples were collected into 3.2% sodium citrate tubes, processed to measure plasma VWF, and stored at −80°C until analyzed.
Second study: The second study was designed to (1) compare the amount of VWF in normal plasma versus platelets and (2) determine the platelet VWF:plasma VWF ratio in dogs. Briefly, 3 näive dogs were bled from the cephalic vein and blood samples were collected into 3.2% sodium citrate tubes, processed for measuring plasma and platelet VWF levels, and stored at −80°C until analyzed.
Studies (3, 4, 5, and 6): Four additional studies in dogs were conducted to better understand the VWF/ VWFpp circulatory kinetic profile and temporal secretory release pattern under 3 conditions: (1) physiological EC stimulation/activation using DDAVP; (2) pathological EC perturbation not associated with structural/morphologic damage following administration of a low dose (2 mg/kg) of LPS; and (3) structural/morphological damage following administration of chemically, structurally, and pharmacologically diverse agents (ZD6169 and DA) known to cause DIVI in dogs.
Briefly, in studies 3 and 4, groups of dogs (3/group) were treated with a single IV bolus dose of DDAVP (5 μg/kg), endotoxin (2 mg/kg), or saline. In study 5, groups of dogs (2/group) were treated orally with 15 (low dose), 60 (mid dose), or 240 (high dose) mg/kg of ZD6169. In study 6, groups of dogs (5/group) were treated with DA via a 6-hr IV infusion at doses of 3.6, 10.8, and 27 mg/kg. The control group received vehicle, and blood samples were collected at predose (0 hr) and 0.25-, 1-, 3-, 6-, 12-, and 24-hr postdose. Additionally, in study 5, hemodynamic parameters, such as heart rate (HR) and mean arterial blood pressure (MAP), were manually recorded 1-hr postdose at the approximate time of maximum plasma concentration of ZD6169. All dogs were necropsied 24 hr after dosing.
Necropsy, tissue collection, and histopathology
All rats were euthanatized with carbon monoxide or isoflurane and subjected to a full necropsy. The mesenteric arteries were collected, fixed in 10% buffered formalin, paraffin embedded, sectioned (5 μm), and stained with hematoxylin and eosin (H&E) for microscopic examination.
All dogs were anesthetized with pentobarbitone sodium and euthanatized by exsanguination. A full necropsy was performed on all dogs and the heart and coronary arteries examined at necropsy. Tissues were collected, fixed in 10% buffered formalin, paraffin embedded, sectioned (5 μm), and stained with H&E for microscopic examination.
Immunohistochemistry
Sections were incubated for 10 min with 0.4% trypsin containing 1% (w/v) calcium chloride, blocked for 2 min with 3% hydrogen peroxide, followed by a 10-min protein block (Dako, Carpinteria, CA). Sections were then incubated for 1 hr with rabbit anti-VWF (1:600 diluted; Dako, Carpinteria, CA) or isotype control antibody followed by 30-min incubation with goat anti-rabbit-horseradish peroxidase (HRP; 1:50 diluted; Jackson ImmunoResearch, West Grove, PA). The color was developed using 3,3′ diaminobenzidine tetrahydrochloride (DAB).
Measurement of plasma VWF levels
Sodium citrate anticoagulated blood was centrifuged at 2,150 × g at 4°C for 15 min, and the resulting plasma also referred to as platelet poor plasma (PPP) was stored at −80°C until analyzed.
Measurement of platelet VWF levels
Lysed platelets were prepared by centrifuging blood at 250 × g at 4°C for 10 min, the resulting platelet-rich plasma (PRP) was removed and recentrifuged at 1,250 × g at 4°C for 15 min to obtain a platelet pellet that was then washed twice in phosphate-buffered saline (PBS) and resuspended in PBS. The presence of intact platelets was verified microscopically and the number of platelets determined. Because of the marked differences in platelet counts between rats and dogs the PRP was adjusted by diluting with PBS so that the platelet counts for both rat and dog samples were equivalent. Equal portions of this platelet suspension and 0.1% Triton X-100 were mixed and incubated at 37°C for 20 mins and briefly sonicated. Lysis of platelets was verified microscopically and the samples were frozen at −80°C.
VWF (Antigen) analysis. Plasma or lysed platelet samples were evaluated on the STA Compact® (Diagnostica Stago, Parrsippany, NJ) using the STA-Liatest VWF kit. The STA-Liatest kit is an immunoturbidimetric method used for the semiquantitative determination of VWF antigen (VWF: Ag) in relation to normal species-specific plasma.
VWFpp analysis. A 96-well plate was coated overnight at 4°C with 50 μl of 5 ng/ml antibody 323.2 (from Dr. R Montgomery, Blood Center, Milwaukee WI), the plate was rinsed with washing buffer (0.02% donor serum in PBS) and then blocked for 2 hr at room temperature with 0.05% Tween in PBS. The plates were washed twice with buffer, incubated 2 hr with 50 ul sample or pooled plasma/well, washed 4 times, incubated 90 mins at room temperature with 50 μl/well (5 μg/ml) detection antibody conjugated with biotin (323.1 from Dr. R. Montgomery). After incubation, the plate was washed 4 times, incubated 30 mins with streptavidin-alkaline phosphatase, developed, and read at 405nm. VWFpp level was based on extrapolation from a titrated normal canine-pooled plasma and reported as percentage of normal pooled plasma.
Data analysis
Mean and standard errors were calculated for studies with ≥ 2 animals per group. One-way analysis of variance (ANOVA) was used when comparing more than 2 groups with a Spearman Rank Correlation Coefficient applied to test the dependency between two variables.
Results
The Effect of Multiple Venipuncture on Plasma VWF Levels in Näive Rats
In a published rat study, evaluation of plasma VWF provided mixed and inconclusive data because in saline-treated rats, repeated venipuncture (tail vein) was associated with increases in plasma VWF (Newsholme et al. 2000). This was most likely a response to the minimal injury from venipuncture that activated platelets and caused this proceduralrelated increase in VWF. This was examined when, over a 24-hr time period, VWF was measured in 5 serial blood samples collected via repeated venipuncture from a group of näive rats (Figure 1).
Figure 1.
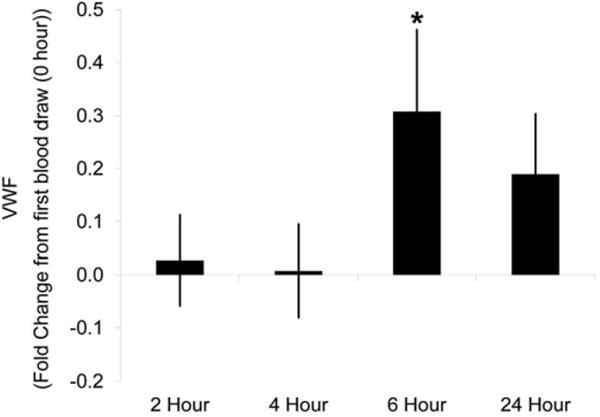
Plasma von Willebrand Factor (VWF) levels with repeat tail vein blood collection in näive rats. Blood was collected from the tail vein at 0, 2, 4, 6, and 24 hr into sodium citrate anticoagulated vacutainers. Samples were centrifuged to obtain platelet poor plasma. Plasma VWF levels were determined using the STA Compact®. Plotted is the average fold difference from 0 hr with standard error. N = 8 rats. * p < 0.05
In general, VWF plasma levels increased in a statistically significant manner (p < .05) with repeated venipuncture over time. When compared to the initial sampling time point (time point 1), VWF levels increased 30 to 50% at the fourth time point (6 hr after the first sample) and were still elevated (20–30%) at the last sampling time point (24 hr after the first sample). After the initial sampling time point, marginal increases (<10%) in plasma VWF were seen at the second and third time points 2 and 4 hr, respectively. In contrast, repeated venipuncture was not associated with increases in plasma VWF in dogs (Figure 2).
Figure 2.
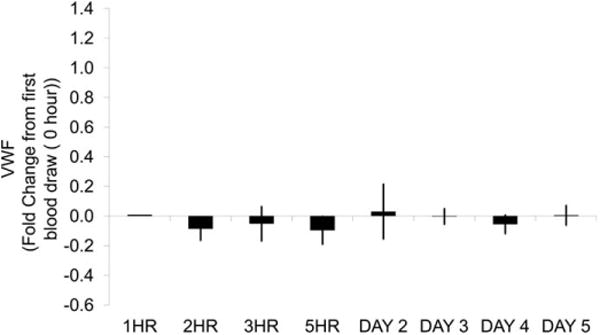
Canine plasma VWF levels from multiple venipunctures within a day and over multiple days. Blood was collected from the jugular vein at 0, 1, 2, 3, 5, 24, 48, 72, and 96 hr into 3.8% sodium citrate anticoagulated vacutainers. Samples were centrifuged to obtain platelet poor plasma. Plasma VWF levels were determined using the STA Compact®. Plotted is the average fold difference from 0 hr with standard error. N = 5 dogs. No statistical differences at any time
Comparative Analysis of VWF Levels in Platelets from Rats versus Dogs
Because repeated venipuncture affects plasma VWF levels in the rat and not the dog, the ratio of platelet VWF to plasma VWF was determined in both species (Figure 3). Rat platelets have ∼ 78% (0.78 ± 0.28, n = 11) equivalent of the amount of VWF found in normal plasma in comparison to dog platelets that have ≤ 10% on or in platelets.
Figure 3.
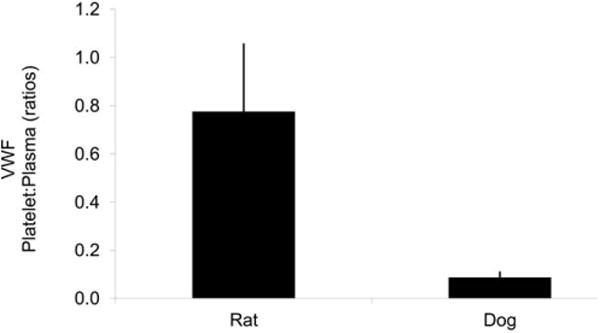
Platelet to plasma VWF ratio in rat and dog. Platelet-free plasma (plasma) and lysed platelets in saline (platelets) were evaluated for VWF on a STA Compact®. N = 11 and 3 for rat and dog, respectively. Plotted are the average and standard error. Note: Rat platelets contain much more VWF than dog platelets, compared to the respective plasma levels.
Evaluation of Plasma VWF in FD-induced Acute Vascular Injury in Rats
Increases in plasma VWF occur following repeated venipuncture in näive rats. Therefore, repeated venipuncture is not a suitable route for blood collection when evaluating VWF during periods of DIVI in rats. Using a double-venous cannulated method for blood collection, plasma VWF was evaluated in rats infused for 24 hr with vehicle or 100 μg/kg/min FD (Figure 4). In FD-treated rats, plasma VWF levels increased in a statistically significant, time-dependent manner; between 2 and 6 hr and at 6 hr (peak increases)VWF levels increased ∼ 35–45%, returning to baseline by 24 hr. In vehicle-treated (saline) rats, VWF levels were stable over the various time points and remained virtually unchanged during the 24-hr infusion period (Figure 4).
Figure 4.
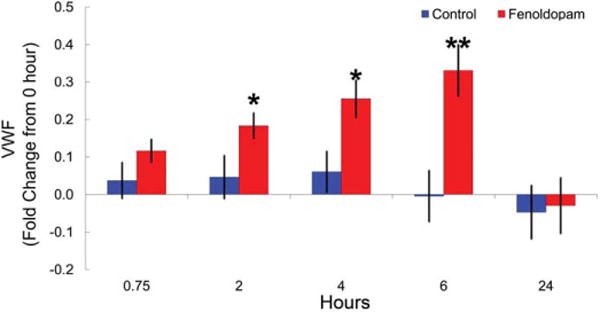
Plasma VWF levels in control or fenoldopam (FD)-treated double venous cannulated rats. Double cannulated (jugular vein for dosing and femoral vein for blood collection) rats were treated with vehicle or 100 mg/kg/min FD for 24 hr with blood collected 0 (pre-dose), 0.75, 2, 4, 6, and 24-hr postdose initiation. Sodium citrate antic-oagulated blood was centrifuged to obtain platelet poor plasma. Plasma von Willebrand Factor (VWF) levels were determined using the STA Compact®. Plotted is the average fold difference from 0 hr with standard error. N = 5 per group. * p < 0.05, ** p < 0.01
Evaluation of Plasma VWF in Dogs Given ZD6169
Blood samples were collected from dogs by repeated venipuncture because, in dogs, this method does not cause a procedural-related increase in plasma VWF levels. Plasma VWF levels were measured in dogs following administration of single oral doses (low, mid, and high) of the PCO ZD6169 that is known to induce vascular injury. In dogs, the high dose (240 mg/kg) of ZD6169 was associated with transient increases (30–35%) in VWF levels up to 6-hr postdose (Figure 5A). Interestingly, plasma VWF levels declined at all doses in all dogs 24-hr postdose when histopathology confirmed morphologic evidence of medial necrosis and hemorrhage (vascular damage).
Figure 5.
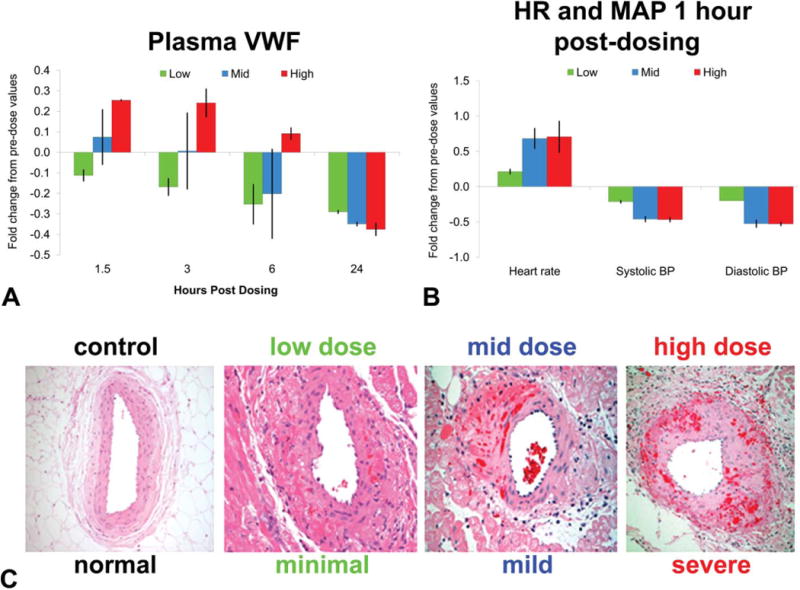
Evaluation of plasma von Willebrand Factor (VWF) as a drug-induced vascular injury (DIVI) biomarker. Dogs were treated with 15, 60, or 240 mg/kg ZD6169, a PCO, with blood collected 0- (predose), 1.5-, 3-, 6-, and 24-hr postdose. Sodium citrate anticoagulated blood was centrifuged to obtain platelet poor plasma with VWF analyzed using the STA Compact1. Plotted is the average fold difference from 0 hr with standard error. Heart rate (HR) and mean arterial blood pressure (MAP) were recorded 1-hr postdose. Dogs were euthanatized 24-hr postdose, and hearts were processed, fixed, sectioned, and hematoxylin and eosin (H&E) stained. N = 2 dogs per group.
Measurement of Hemodynamic Parameters (HR and Mean Arterial Pressure) in Dogs
PCOs mediate pharmacologic effects on vascular tone through direct relaxation of vascular SMC, resulting in vasodilatation. As a result, measurement of MAP and HR have been long accepted as the surrogate toxicity biomarkers for DIVI, so coupling physiological and biochemical endpoints provides a convenient tool to study the correlative relationship between plasma VWF, morphologic pathology, and hemodynamics following administration of ZD6169 in dogs.
ZD6169 induced a slight, dose-related increase in HR (25–80%) which was apparent 1 hr after dosing (Figure 5A). The HR changes were associated with the expected concomitant decrease in MAP (20–50%). The hemodynamic changes (HR and MAP), though clearly treatment related, were considered within normal physiological range for these parameters in the dog (Figure 5B).
Drug-induced Arterial Histopathology
FD (a dopaminergic agonist) and ZD6169 (a PCO) are potent vasodilatators that readily and consistently induced vascular lesion and these chemicals provide an excellent tool to study the correlative relationship between plasma VWF levels and vascular damage.
Rat mesenteric arterial injury induced by FD
Histologic assessment of mesenteric arteries from rats was conducted only at the 24-hr time point because at shorter postdosing intervals, previous study data (Newsholme et al. 2000) indicate that mesenteric arterial pathology induced by FD was an infrequent occurrence. Histologic lesions induced by FD in rats have been previously described (Kerns et al. 1989a; Kerns, Arena, and Morgan 1989b; Yuhas et al. 1985). Briefly, medial necrosis and intramural hemorrhage with perivascular edema was the predominant feature. The endothelium appeared intact and there was no evidence of thrombosis (data not shown).
Dog coronary arterial lesions induced by ZD6169 (PCO)
As expected, ZD6169 induced vascular lesions with a dose-dependent increase in severity (Figure 5C). Histologic changes in the right extramural coronary arteries and along the coronary groove (circumflex artery) were comparable to the lesions reported with other vasodilatators in dogs (Greaves 1998; Louden et al. 2000; Louden and Morgan 2001; Mesfin et al. 1989). Briefly, lesions were characterized histologically by multifocal medial hemorrhage, SMC necrosis, and acute perivascular inflammation. The endothelium remained relatively intact (Figure 5C).
In situ detection of VWF following acute DIVI
In rats and dogs, plasma VWF transiently increased during periods of DIVI. However, in both species, when there is confirmed evidence of microscopic vascular injury, plasma VWF levels declined. Because breakdown in the structural integrity of the vessel wall could lead to passive extravasation of soluble proteins immunohistochemistry was performed to determine the in situ presence and signal strength of extravascular VWF in vascular injury induced by FD in rats or ZD6169 in dogs.
As was previously reported (Brott et al. 2005), immunoreactivity to VWF was present in all vessels with microscopic evidence of vascular injury (Figure 6). In injured vessels, the signal strength of extravascular VWF immunoreactivity correlated with the histopathology grade of severity.
Figure 6.
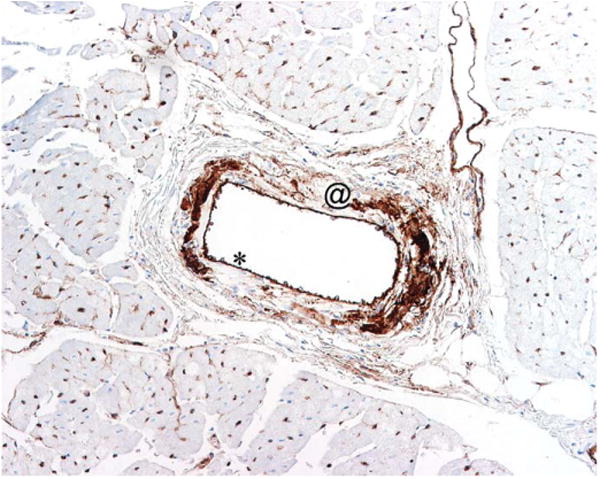
von Willebrand Factor (VWF) immunohistochemistry staining of canine coronary artery 24 hr after treatment with the PCO ZD6169. Dogs were euthanatized 24 hr after bolus treatment with 60 mg/kg ZD6169. Heart was processed, fixed, sectioned, and immunohistochemically stained for VWF. Note: VWF within the smooth muscle layer (@) of the vascular wall. * is showing endothelial cells expressing VWF.
Determination of Plasma VWF and VWFpp Kinetic Profile during Periods of EC Stimulation and/or Activation and DIVI in Dogs
During the process of injury or stimulation, VWF and VWFpp are released from EC and both proteins can be measured in plasma. In a population study of dogs (n = 80), the basal levels of VWF and VWFpp were low with minimal variability (data not shown) and therefore these proteins were measured in plasma from dogs following administration of DDAVP, LPS/endotoxin, and DA a chemical well-known to cause vascular wall medial hemorrhage and necrosis of vascular SMC in dogs. In dogs, VWF levels in plasma increased in a statistically significant manner, ∼ 1.5-fold at 1, 3, and 6 hr following treatment with DDAVP or endotoxin (Figure 7). When compared to DDAVP, endotoxin caused a greater magnitude of increase in plasma VWF (1.5- to 3.5-fold) and VWF values did not return to baseline within 24 hr.
Figure 7.
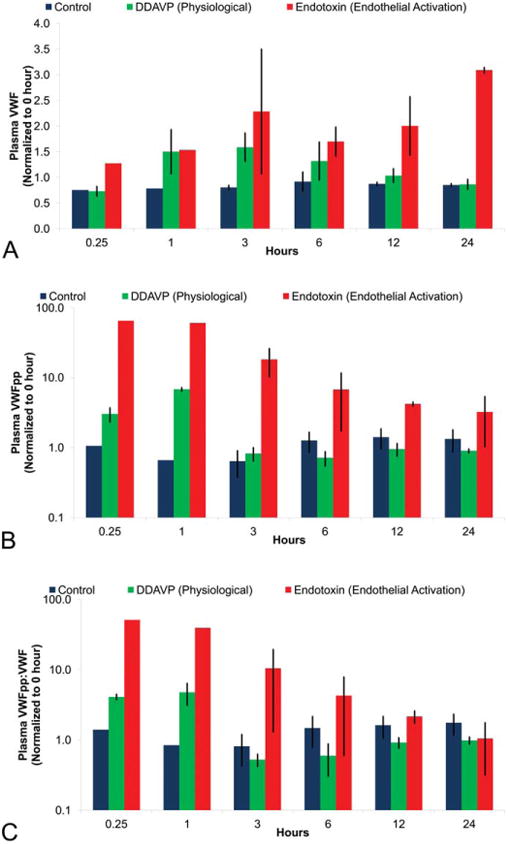
Plasma VWF, VWFpp, and VWFpp:VWF ratio levels as biomarkers of canine endothelial perturbation. Blood was collected from the jugular vein at predose (0 hr) and 0.25, 1, 3, 6, 12, and 24 hr after dosing with vehicle, 5 μg/kg DDAVP (physiological), and 2 mg/kg endotoxin (pathological). Sodium citrate anticoagulated blood was centrifuged to obtain platelet poor plasma with VWF analyzed using the STA Compact®. Plotted is the average fold difference from 0 hr with standard error. N = 3 dogs per group. Statistics was not done because of blood unavailability from all dogs at all time points.
Both DDAVP and endotoxin caused a statistically significant increase in plasma VWFpp levels early, but the increase associated with DDAVP (5- to 8-fold) was transient, returning to baseline values 3-hr postdosing, while endotoxin caused a more prolonged and sustained increase in VWFpp levels greater than 10-fold up to 24-hr postdosing (Figure 7). Furthermore, the VWFpp:VWF ratio shows a discriminatory and distinct pattern between physiological (DDAVP) versus pathological (endotoxin) EC damage (Figure 7).
The hallmark of DIVI is medial hemorrhage and necrosis of vascular SMC visible with light microscopy. Because DA can induce these lesions in dogs the VWF and VWFpp plasma profile was determined during periods of vascular injury as a consequence of intravenous exposure to DA. Plasma VWF levels increased transiently in a slightly dose-dependent (statistically significant at the high dose) fashion 1-hr postdosing; however, values for VWF were comparable to controls at 6-hr postdosing (Figure 8). In contrast, VWFpp plasma levels increased in a dose-dependent fashion and values were statistically significant at the mid- and high-dose 1-hr postdosing, and the increase was sustained at 6 hr. However, values decreased in a statistically significant manner 12- and/or 24-hr postdosing (Figure 8). A clear dose–response in the VWFpp:VWF ratio was evident at 1-hr postDA treatment, and this ratio was marked at 6-hr postdosing (Figure 8).
Figure 8.
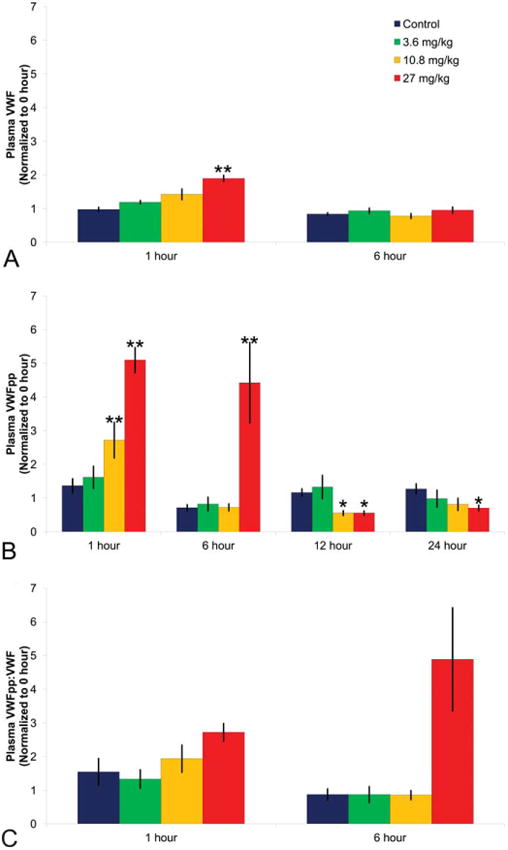
Canine plasma VWF, VWFpp, and VWFpp:VWF ratio as DIVI biomarkers. (A) VWF; (B) VWFpp, and (C) VWFpp:VWF. Dogs were treated with 0, 3.6, 10.8, or 27 mg/kg dopamine with blood collected 0- (predose), 1-, 6-, 12-, and 24-hr postdose. Sodium citrate anticoagulated blood was centrifuged to obtain platelet poor plasma and VWF (0, 1, and 6 hr only) analyzed using the STA Compact®. VWFpp was analyzed by the enzyme-linked immunosorbent assay (ELISA). Plotted are the mean and standard error values normalized to predose. N = 5 dogs per group. *p < .05. **p < .01.
Because medial hemorrhage and necrosis of vascular SMC is the light microscopic hallmark of DIVI, there is strong interest in determining the relationship between the magnitude of increase in plasma VWF, VWFpp, the VWFpp:VWF ratio and the incidence and severity of vascular lesions that developed in dogs during exposure to DA. Vascular lesions were observed only in dogs treated with DA and lesions were present in all dogs given the high dose and in one mid-dose treated dog (Table 1). Furthermore, the dogs with the most severe vascular lesions had the highest plasma VWFpp and VWFpp:VWF ratio (Table 1).
Table 1. Dose-response of plasma VWF, VWFpp, VWFpp:VWF ratio, and incidence of vascular lesions in dogs administered dopamine (DA).
| Groupa | Dopamine (mg/kg) | VWFb | VWFppb | VWFpp:VWFb | Lesion (%)c |
|---|---|---|---|---|---|
| 1d | 0 | 96–157 | 55–91 | 0.36–0.95 | 0 |
| 2 | 3.6 | 127–173 | 76–136 | 0.58–1.03 | 0 |
| 3 | 10.8 | 103–215 | 165–250 | 0.86–2.13 | 20 |
| 4 | 27 | 149–228 | 248–467 | 1.43–2.43 | 100 |
Five dogs per group.
Plasma samples collected at 1-hr postdose initiation. VWF and VWFpp are reported as percentage of normal pooled dog plasma.
Necropsy at 24-hr postdose initiation.
One dog was dropped from group analysis due to extremely low VWF value (25).
Discussion
The current investigation had 3 major aims: (1) reliably and accurately measure plasma VWF concentrations in a rat model of DIVI that excludes the procedural-related artifactual effects of repeated venipuncture; (2) determine the plasma profile and potential diagnostic value of measuring the concentrations of both VWF and VWFpp in dog models of EC perturbation physiologically or pathologically with or without light microscopic evidence of morphologic damage; and (3) evaluate the diagnostic significance of the VWFpp:VWF ratio as a potential means to discriminate types and severity of DIVI. Data from these experiments provide clear evidence that physiological and/or pathological perturbation with or without injury to vascular EC can be associated with increases in plasma VWF and/or VWFpp. Furthermore, physiological stimulation or pathological perturbation and/or injury to vascular EC are associated with a unique and discriminatory plasma profile for VWF, VWFpp, and the VWFpp:VWF ratio.
VWF is a large multifunctional, multimeric glycoprotein with strong adhesive properties that mediate adhesion of platelets to the subendothelial collagen at the site of vascular damage. It is one of the more important circulating proteins produced by EC and because of its cellular source, in vascular disorders, it is frequently used as a biomarker of EC dysfunction, injury, or damage. Because of these biological characteristics and clinical relevance as a biomarker of spontaneous vascular disease in humans, VWF holds promise as a preclinical to clinical translational biomarker if data are provided that demonstrates its performance when measured in models of acute vascular injury. In a published study, this hypothesis was tested in a rat model of acute DIVI using the DA agonist FD (Newsholme et al. 2000). In this study, FD was associated with marginal and transient increases in plasma VWF during periods of arterial damage, but a definitive conclusion could not be drawn because repeated venipuncture was also associated with a time-dependent increase in plasma VWF concentrations in saline-treated rats. This procedural-related artifactual increase in VWF, confounded interpretation, and the increases in plasma VWF were attributed in part to the acute phase inflammatory response (Newsholme et al. 2000). In the current study, we provide strong data showing that plasma VWF is increased significantly over time during periods of DIVI when blood samples are not collected via repeated venipuncture but instead with the use of a double cannulated system. In addition, we provide confirmatory data that support the findings from a previous study that in näive rats (Newsholme et al. 2000), the procedure of repeated venipuncture causes a statistically significant elevation in plasma VWF over time. The most likely explanation for this finding is that VWF is released from damaged EC and activated platelets as a result of venipuncture. Interestingly, repeated venipuncture in dogs is not associated with increases in plasma VWF and so the species-specific response is most likely due to the differences of platelet VWF concentration of rats versus dogs. Data from the current studies support this hypothesis because rat platelet VWF concentrations are much higher when compared to dog platelets, resulting in a marked difference between rat and dog VWF platelet:plasma ratio. Therefore, repeated venipuncture is unlikely to increase plasma VWF concentrations in dogs because the VWF level from dog platelets is very low. Our data as well as reports by others (Newsholme et al. 2000) provide conclusive evidence that when measuring plasma VWF in rats, venipuncture is not the preferred method of blood collection.
As previously reported and observed in these studies, in both dogs and rats, during the later phase of DIVI when there is morphologic evidence of vascular damage, plasma VWF levels decrease significantly. This finding raises concern about the suitability and significance of VWF as a biomarker of EC damage, because of the lack of concordance between the plasma levels of VWF and positive histopathology findings of vascular damage. Data from other studies as well as ours strongly suggest that in vascular injury systemic VWF concentration does not accurately reflect the release and/or secretory capacity of damaged EC. There are several plausible explanations for this observation (1) subendothelial connective tissue extraction of VWF from circulating plasma, (2) endocytic uptake of factor VIII–VWF complex by macrophages, (3) enhanced binding of factor VIII to VWF to increase factor VIII–VWF complexes, (4) binding of VWF to glycoprotein receptor 1b (GP1b) on the surface of circulating platelets, and (5) potential reuptake of VWF by platelets. EC secretion of VWF occurs in a bidirectional fashion and at sites of vascular injury substantial amounts of released VWF may get trapped by the extracellular matrix (Sporn, Marder, and Wagner 1987; van Mourik and Romani deWit 2001; Wagner et al. 1987). VWF is required for binding of platelets to the subendothelial collagen and this can also contribute to the reduction in plasma VWF following injury. Vasculotoxic agents cause changes in shear stress and recently published data show that shear stress is required for macrophages to internalize the factor VIII–VWF complex as well as both (FVIII and VWF) constituents (Castro-Nŭnez et al. 2012), and this could also contribute to the reduction in plasma VWF as a result of injury. Furthermore, because of the close association between factor VIII and VWF to form the active “molecular complex,” increases in plasma VWF will also cause an increase in the level of this circulating complex. In response to injury, VWF will bind GP1b on platelets and occupation of this receptor initiates activation and subsequent aggregation that requires binding of GPIIb-IIIa as well as VWF (Ruggeri 1991). Data from studies with platelets suggest that uptake of VWF by platelets may contribute to the regulatory control of circulating plasma VWF (Montgomery and Gill 2000). It must also be noted that EC has limited VWF storage capacity and it is possible that repeated activation, perturbation, and/or injury to the EC cause exhaustion of the cytoplasmic pool of VWF from Weibel-Palade Bodies (WPBs) and so a transient increase followed by a decrease is plausible (Olsen et al. 2003; van Mourik et al. 1999; van Mourik and Romani deWit 2001; Wagner et al. 1987).
In preclinical toxicology studies, DIVI has a predisposition for the susceptible vascular beds and size of arteries affected; extramural coronary arteries in dogs and the mesenteric vascular bed in rats (Kerns et al. 1989a; Louden et al. 2000; Mesfin et al. 1989; Newsholme et al. 2000). The vascular bed “site selectivity predisposition” limits the number of arteries that can be affected, which then determines the concentration of “susceptible pool of VWF” that could be released from ECs during periods of vascular damage. In our studies, it is likely that DIVI in the first 6 hr exhausted this finite “susceptible pool of VWF” while in contrast endotoxin affects all vascular beds and consequently a larger pool of ECs. Therefore, endotoxin not only caused a greater increase but also produced a sustained elevation of plasma VWF for a longer period of time. In humans, a similar VWF profile was observed following administration of low doses of endotoxin, so that our current data are not without precedence (Borchiellini et al. 1996; van Mourik and Romani deWit 2001; van Mourik et al. 1999). As mentioned previously, the bidirectional secretion of EC VWF allows VWF to bind platelets to the subendothelial collagen at the site of vascular injury and as a result VWF can get trapped in the connective tissue extracellular matrix. That is, because, in response to vascular injury, VWF will bind to platelets and subendothelial collagen as it forms the hemostatic plug to seal the site of vascular damage and prevent further blood loss (Van Buul-Wortelboer et al. 1989). Our data as well as that from other reports (Brott et al. 2005; Brott, Richards, and Louden 2012; Jones, Bjökman, and Schofield 2013) support this hypothesis because in the extravascular space, significant extracellular matrix VWF was detected by immunohistochemistry in damaged vascular wall. It has also been suggested that during the process of DIVI damaged ECs are released from the site of injury into systemic circulation (McFarland et al. 2004; Scicchitano et al. 2003; Zhang et al. 2010) and this may further deplete the limited cellular source of available VWF proteins. There is debate regarding the significance and contribution of released EC and/or EC microparticles into circulation following DIVI because this would most likely trigger release of tissue factor that would initiate the coagulation cascade, increase fibrinogen concentration in plasma, and possibly cause disseminated intravascular coagulopathy. In our experience, hematology changes indicative of activation of the coagulation cascade were only seen with endotoxin not DA or FD. Therefore, based on the available data, measurement of plasma VWF as a biomarker of very early DIVI has merits, but its utility is limited because of the narrow diagnostic window.
As an alternative to measuring only VWF, there is emerging evidence that suggests measurement of VWFpp in conjunction with VWF and determining the VWFpp:VWF ratio can be used diagnostically to potentially distinguish acute from chronic EC perturbation and possibly DIVI (Borchiellini et al. 1996; Louden et al. 2006; van Mourik and Romani deWit 2001; van Mourik et.al. 1999 Vischer et al. 1997). Biologically, release of VWF from ECs into the systemic circulation occurs via one of 3 pathways: (1) regulated, secretagogue-stimulated release from WPBs, (2) basal secretagogue-independent release of VWF from WPBs, and (3) constitutive, secretagogue-independent release of VWF from nonWPB anterograde carriers (Nightingale and Cutler 2013). In the EC, de novo synthesized VWF undergoes endoproteolytic cleavage of its propeptide, with the mature VWF and its propeptide (VWFpp) stored in WPBs (van Mourik and Romani deWit 2001; Wagner et al. 1987; Weibel and Palade 1964; Weibel 2012). Under normal conditions, that is, in the absence of secretagogue stimulation, most of the VWF released from WPBs is via basal secretion (Giblin, Hewlett, and Hannah 2008). VWFpp and VWF are essentially released in equimolar concentrations, both in the resting and in the stimulated EC, but VWFpp differs from VWF in that VWFpp has a short half-life with rapid turnover and following vascular EC stimulation, the concentration of VWFpp increases in plasma, but returns to baseline values much faster than does the mature protein (Borchiellini et al. 1996; van Mourik et al. 1999; van Mourik and Romani deWit 2001). On the basis of these observations, the circulatory kinetic and release profile of both VWF and VWFpp were measured in dogs and VWFpp:VWF ratio determined to provide a means of assessing the extent and time course of endothelial activation through physiological and/or pathological stimulation. Our data provide strong evidence to support endothelial activation/perturbation was provoked by administration of DDAVP, endotoxin, and chemicals known to cause arterial damage in selected vascular beds (DIVI). In dogs, VWF and VWFpp levels in plasma increased following treatment with DDAVP or endotoxin, but endotoxin caused a greater magnitude of increase in plasma VWF (1.5- to 3.5-fold) and VWFpp (>10-fold) with VWF and VWFpp values not returning to baseline within 24 hr. This is most likely due to the mode of action of the secretory and release pattern induced by DDAVP versus endotoxin. The exact mode of action by which DDAVP and endotoxin causes release of VWF is not well known, but it is possible that DDAVP may act primarily through the stimulated-secretagogue regulated pathway. However, the mode of action by which endotoxin causes persistent and prolonged activation of the endothelium with subsequent increases of VWF and VWFpp is more complex and probably reflects enhanced secretory activity through a combination of the basal, constitutive, and regulated pathways. This interpretation, however, would suggest that endotoxin stimulated EC-specific upregulation and increased transcriptional activity to increase de novo synthesis of VWF. This hypothesis is plausible because data from published reports suggest that endotoxin mediates VWF release through both the constitutive and the regulated pathway, the latter requiring second messenger cell signaling (Borchiellini et al. 1996; Suffredini et al. 1989; Van Deventer et al. 1990). Furthermore, ECs can be stimulated to specifically increase transcriptional activity and enhance VWF synthesis (Jahroudi and Lynch 1994; Jahroudi, Ardekani, and Greenberger 1996). Under DDAVP or endotoxin stimulation, the profile and secretory release patterns of VWF and VWFpp in humans and dogs are comparable (Borchiellini et al. 1996; van Mourik et al. 1999), and therefore this provides evidence for the hypothesis that elevation of both VWF and VWFpp are biomarkers of acute transient EC activation.
Both DDAVP and endotoxin caused a statistically significant increase in plasma VWFpp levels early, but the increase associated with DDAVP was transient returning to baseline values 3-hr postdosing, while endotoxin caused a more prolonged and sustained increase in VWFpp levels greater than 10-fold up to 24-hr postdosing. VWFpp has a short half-life, and because DDAVP causes release from stored vesicles, the rapid decline in plasma VWFpp associated with DDAVP is expected (Borchiellini et al. 1996; van Mourik et al. 1999; Wagner et al. 1987). Furthermore, the pharmacodynamics action of DDAVP is short-lived with duration of 1 to 2 hr (Lundin and Vilhardt 1986). As previously mentioned, endotoxin, through second messenger signaling, stimulates both synthesis and release of VWF and VWFpp. The prolonged stimulation and activation of ECs is expected to increase VWF and VWFpp levels in plasma over time. The differences in circulatory plasma profile in VWF, VWFpp, and the VWFpp:VWF ratio show a discriminatory and distinct pattern between physiological (DDAVP) versus pathological (endotoxin) EC stimulation.
One of the aims of our study was to determine the relationship between increases in plasma VWF, VWFpp, the VWFpp:VWF ratio and the incidence and severity of vascular lesions that developed in dogs during exposure to DA. In dogs administered DA, plasma VWFpp levels increased sharply in a dose-dependent fashion, and dogs with the most severe vascular lesions had the highest plasma VWFpp (>6-fold) and VWFpp:VWF ratio and vascular lesions were observed only in dogs treated with DA. These data provide strong evidence that the composite profile of plasma VWF and VWFpp and determining the VWFpp:VWF ratio can be used diagnostically in dog preclinical toxicology studies as a potential biomarker for DIVI. Plasma levels of VWFpp declined over time and this was most likely influenced by several factors including its short half-life, size of the susceptible vascular bed (coronary arterial bed) subject to DIVI, exhaustion of EC stores of VWF and VWFpp from the limited number of damaged EC, and possibly some extraction from plasma. In vitro data from published studies suggest that extraction of VWFpp from plasma is not likely to be a cause of reduced plasma levels because it is unlikely that VWFpp would bind platelets, the endothelium, or the mature VWF protein (Wagner et al. 1987). Data from recent studies provide evidence that VWFpp does bind to extracellular matrix proteins such as collagens and lamin or to mature VWF itself (Fujisawa et al. 1991; Usui, Takagi, and Saito 1993; Vischer and Wagner 1994). Therefore, a combination of several factors is responsible for the decline in plasma VWFpp in models of DIVI. The short half-life of VWFpp allows the discrimination between acute and chronic EC activation and injury. Therefore, persistent elevation of VWFpp would suggest continuous injury.
An additional finding in our studies is that values for plasma VWFpp were higher in dogs administered the mid dose 10.8 mg/ kg DA when compared to the VWFpp plasma values seen in vehicle controls or dogs administered low dose 3.6 mg/kg. Furthermore, a minimal vascular lesion was seen in one dog administered 10.8 mg/kg, and this suggests that indeed VWFpp is an early indicator of vascular damage. However, morphologic evidence of vascular injury was not seen in the other 4 dogs that had comparable levels of VWFpp. This lack of concordance is most likely due to a combination of the minimal nature of the lesion with only few vessels affected and the limited number of sections evaluated histologically. Therefore, lack of concordance is potentially a sensitivity issue, in that concordance could be increased with evaluation of more sections.
In summary, we have clearly demonstrated that local or systemic perturbation of the vascular endothelium causes sharp, transient elevations in VWF, VWFpp, and increases in the VWFpp:VWF ratio. DDAVP does not induce vascular pathology, but in response to administration, ECs become activated (Kanwar et al. 1995). DDAVP is also used therapeutically to treat some forms of von Willebrand disease and diabetes insipidus so that the effects of increased VWF are considered nonadverse and physiological. Endotoxin is a well-known agonist of EC activation that can also cause platelet aggregation in dogs and humans (Meyers, Boehme, and Inbar 1982; van Mourik et al. 1999). This could cause release of stored VWF and VWFpp into plasma; therefore, the increased plasma levels may not be only of EC origin. This is not the case in dogs, because as our data show dog platelets have only a small quantity of VWF and while human platelets are a rich source of VWF and the concentration of VWFpp in platelets is too small to significantly increase plasma levels (Nichols et al. 1995; van Mourik et al. 1999). Taken together, these data suggest that activation of EC will cause release of VWF proteins, and the circulatory kinetic profile in plasma can be used to discriminate between physiological and pathological perturbations.
Acknowledgments
Robert R. Montgomery received financial support from AstraZeneca Pharmaceuticals, for the development and production of the VWFpp reagents and kit.
Abbreviations
- ANOVA
analysis of variance
- DA
Dopamine
- DDAVP
[deamino-Cys1, D-Arg8]-vasopressin
- DIVI
drug-induced vascular injury
- EC
endothelial cell
- FD
fenoldopam
- H&E
hematoxylin and eosin
- HPMC
hydroxypropyl methylcellulose
- HR
heart rate
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- PBS
phosphate-buffered saline
- PCO
potassium channel opener
- PPP
platelet poor plasma
- PRP
platelet rich plasma
- SMC
smooth muscle cells
- VWF
von Willebrand Factor
- VWFpp
VWF propeptide
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Blann AD. Plasma von Willebrand factor, thrombosis, and the endothelium: The first 30 years. Thromb Haemost. 2006;95:49–55. [PubMed] [Google Scholar]
- Borchiellini A, Fijnvandraat K, ten Cat JW, Pajkrt D, van Deventer SJ, Pasterkamp G, Meijer-Huizinga F, Zwart-Huinink L, Voorberg J, van Mourik JA. Quantitative analysis of von Willebrand factor propeptide release in vivo: Effect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88:2951–8. [PubMed] [Google Scholar]
- Brott D, Gould S, Jones H, Schofield J, Prior H, Valentin JP, Bjurstrom S, Kenne K, Schuppe-Koistinen I, Katein A, Foster-Brown L, Betton G, Richardson R, Evans G, Louden C. Bio-markers of drug-induced vascular injury. Toxicol Appl Pharmacol. 2005;207:S441–S5. doi: 10.1016/j.taap.2005.04.028. [DOI] [PubMed] [Google Scholar]
- Brott DA, Richardson RJ, Louden CS. Evidence for the nitric oxide pathway as a potential mode of action in fenoldopam-induced vascular injury. Toxicol Pathol. 2012;40:874–86. doi: 10.1177/0192623312444027. [DOI] [PubMed] [Google Scholar]
- Castro-Nŭnez L, Dienava-Verdoold I, Herczenik E, Mertens K, Meijer AB. Shear stress is required for the endocytic uptake of the factor VIII-von Willebrand factor complex by macrophages. J Thromb Haemost. 2012;10:1929–37. doi: 10.1111/j.1538-7836.2012.04860.x. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, Takagi J, Sekiya F, Goto A, Miake F, Saito Y. Monoclonal antibodies that inhibit binding of propolypeptide of von Willebrand factor to collagen, localization of epitopos. Eur J Biochem. 1991;196:673–7. doi: 10.1111/j.1432-1033.1991.tb15864.x. [DOI] [PubMed] [Google Scholar]
- Giblin JP, Hewlett LJ, Hannah MJ. Basal secretion of von Willebrand factor from human endothelial cells. Blood. 2008;112:957–64. doi: 10.1182/blood-2007-12-130740. [DOI] [PubMed] [Google Scholar]
- Greaves P. Patterns of drug-induced cardiovascular pathology in the beagle dog: Relevance for humans. Exp Toxicol Pathol. 1998;50:283–93. doi: 10.1016/S0940-2993(98)80008-0. [DOI] [PubMed] [Google Scholar]
- Jahroudi N, Ardekani AM, Greenberger JS. Ionizing irradiation increases transcription of the von Willebrand factor gene in endothelial cells. Blood. 1996;88:3801–14. [PubMed] [Google Scholar]
- Jahroudi N, Lynch DC. Endothelial-cell-specific regulation of von Willebrand factor gene expression. Mol Cell Biol. 1994;14:999–1008. doi: 10.1128/mcb.14.2.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HB, Bjökman JA, Schofield J. Coronary and systemic arterial physiology and immunohistochemical markers related to early coronary arterial lesions in beagle dogs given the PCO, ZD6169, or endothelin receptor antagonist, ZD1611. Toxicol Pathol. 2013;41:722–35. doi: 10.1177/0192623312464123. [DOI] [PubMed] [Google Scholar]
- Kanwar S, Woodman RC, Poon MC, Murohara T, Lefer AM, Davenpeck KL, Kubes P. Desmopressin induces endothelial p-selectin expression and leukocyte rolling in postcappillary venules. Blood. 1995;86:2760–6. [PubMed] [Google Scholar]
- Kerns WD, Arena E, Macia RA, Bugelski PJ, Matthews WD, Morgan DG. Pathogenesis of arterial lesions induced by dopaminergic compounds in the rat. Toxicol Pathol. 1989a;17:203–13. doi: 10.1177/019262338901700116. [DOI] [PubMed] [Google Scholar]
- Kerns WD, Arena E, Morgan DG. Role of dopaminergic and adrenergic receptors in the pathogenesis of arterial lesions induced by fenoldopam mesylate and dopamine in the rat. Am J Pathol. 1989b;135:339–49. [PMC free article] [PubMed] [Google Scholar]
- Louden C, Brott D, Katein A, Kelly T, Gould S, Jones H, Betton G, Valentin JP, Richardson R. Biomarkers and mechanisms of drug-induced vascular injury in non-rodents. Toxicol Pathol. 2006;34:19–26. doi: 10.1080/01926230500512076. [DOI] [PubMed] [Google Scholar]
- Louden C, Morgan DG. Pathology and pathophysiology of drug-induced arterial injury in laboratory animals and its implications on the evaluation of novel chemical entities for human clinical trials. Pharmacol Toxicol. 2001;89:158–70. doi: 10.1111/j.0901-9928.2001.890404.x. [DOI] [PubMed] [Google Scholar]
- Louden CS, Nambi P, Pullen MA, Thomas RA, Tierney LA, Solleveld HA, Schwartz LW. Endothelin receptor subtype distribution predisposes coronary arteries to damage. Am J Pathol. 2000;157:123–34. doi: 10.1016/S0002-9440(10)64524-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin S, Vilhardt H. Absorption of intragastrically administered DDAVP in conscious dogs. Life Sci. 1986;38:703–9. doi: 10.1016/0024-3205(86)90584-9. [DOI] [PubMed] [Google Scholar]
- McCarrol DR, Waters DC, Steidley KR, Clift R, McDonald TP. Canine platelet von Willebrand factor: Quantification and multimeric analysis. Exp Hematol. 1988;16:929–37. [PubMed] [Google Scholar]
- McFarland DC, Scicchitano MS, Thomas RA, Narayanan PK, Schwartz LW, Thomas HC. 6-color flow-sorting of rat circulating endothelial cells and taqman® real-time PCR analysis. Cytometry. 2004;59A:65. [Google Scholar]
- Meigs JB, O'Donnell CJ, Tofler GH, Benjamin EJ, Fox CS, Lipinska I, Nathan DM, Sullivan LM, D'Agostino RB, Wilson PW. Hemostatic markers of endothelial dysfunction and risk of incident type 2 diabetes: The Framingham offspring study. Diabetes. 2006;55:530–7. doi: 10.2337/diabetes.55.02.06.db05-1041. [DOI] [PubMed] [Google Scholar]
- Mesfin GM, Piper RC, DuCharme DW, Carlson RG, Humphrey SJ, Zins GR. Pathogenesis of cardiovascular alterations in dogs treated with minoxidil. Toxicol Pathol. 1989;17:164–81. doi: 10.1177/019262338901700113. [DOI] [PubMed] [Google Scholar]
- Meyers KM, Boehme M, Inbar O. Binding of 124I-labeled endotoxin to bovine, canine, and equine platelets and endotoxin-induced agglutination of canine platelets. Am J Vet Res. 1982;43:1721–8. [PubMed] [Google Scholar]
- Montgomery RR, Gill JC. Interactions between von Willebrand factor and Factor VIII: Where did they first meet? J Pediatr Hematol Oncol. 2000;22:269–75. doi: 10.1097/00043426-200005000-00017. [DOI] [PubMed] [Google Scholar]
- Newsholme SJ, Thudium DT, Gossett KA, Watson ES, Schwartz LW. Evaluation of plasma von Willebrand factor as a biomarker for acute arterial damage in rats. Toxicol Pathol. 2000;28:688–93. doi: 10.1177/019262330002800508. [DOI] [PubMed] [Google Scholar]
- Nichols TC, Samama CM, Bellinger DA, Roussi J, Reddick RL, Bonneau M, Read MS, Bailliart O, Kockh GG, Vaiman M. Function of von Willebrand factor after crossed bone marrow transplantation between normal and von willibrand disease pigs: Effect on arterial thrombosis in chimeras. Proc Natl Acad Sci USA. 1995;92:2455–9. doi: 10.1073/pnas.92.7.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells: An increasingly complicated story. J Thromb Hae-most. 2013;11:192–201. doi: 10.1111/jth.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen EH, McCain AS, Merricks EP, Fischer TH, Dillon IM, Raymer RA, Bellinger DA, Fahs SA, Montgomery RR, Keith JC, Jr, Schaub RG, Nichols TC. Comparative response of plasma VWF in dogs to upregulation of VWF mRNA by interleukin-11 versus Weibel–Palade body release by Desmopressin (DDAVP) Blood. 2003;102:436–41. doi: 10.1182/blood-2003-01-0290. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Structure and function of von Willebrand factor: Relationship to von Willebrand's disease. Mayo Clin Proc. 1991;66:847–61. doi: 10.1016/s0025-6196(12)61204-x. [DOI] [PubMed] [Google Scholar]
- Sanders WE, Jr, Reddick RL, Nichols TC, Brinkhous KM, Read MS. Thrombotic thrombocytopenia induced in dogs and pigs. The role of plasma and platelet vwf in animal models of thrombotic thrombocytopenia purpura. Arterioscler Thomb Vasc Biol. 1995;15:793–800. doi: 10.1161/01.atv.15.6.793. [DOI] [PubMed] [Google Scholar]
- Scicchitano M, Thomas R, McFarland D, Narayanan P, Thomas H, Tierney L, Schwartz L. Transcriptional phenotyping of circulating endothelial cells form sorted rat whole blood. Vet Pathol. 2003;40:626a. [Google Scholar]
- Sporn LA, Marder VJ, Wagner DD. Differing polarity of the constitutive and regulated secretory pathways for von Willebrand factor in endothelial cells. J Cell Biol. 1987;108:1283–9. doi: 10.1083/jcb.108.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suffredini AF, Fromm RE, Parker MM, Brenner M, Kovacs JA, Wesley RA, Parrillo JE. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med. 1989;321:280–7. doi: 10.1056/NEJM198908033210503. [DOI] [PubMed] [Google Scholar]
- Usui T, Takagi J, Saito Y. Propolypeptide of von Willebrand factor serves as a substrate for factor XIIIa and is cross-linked to laminin. J Biol Chem. 1993;268:12311–6. [PubMed] [Google Scholar]
- Van Buul-Wortelboer MF, Brinkman HJ, Reinders JH, van Aken WG, van Mourik JA. Polar secretion of von Willebrand factor by endothelial cells. Biochim Biophys Acta. 1989;1011:129–33. doi: 10.1016/0167-4889(89)90199-7. [DOI] [PubMed] [Google Scholar]
- Van Deventer SJ, Büller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in human: Analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76:2520–6. [PubMed] [Google Scholar]
- van Mourik JA, Boertjes R, Huisveld IA, Fijnvandraat K, Pajkrt D, van Genderen PJ, Fijnheer R. von Willebrand factor in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999;94:179–85. [PubMed] [Google Scholar]
- van Mourik JA, Romani de Wit T. von Willebrand factor propeptide in vascular disorders. Thromb Haemost. 2001;86:164–71. [PubMed] [Google Scholar]
- Vischer UM, Ingerslev J, Wollheim CB, Mestries JC, Tsakiris DA, Haefeli WE, Kruithof EK. Acute von Willebrand factor secretion from the endothelium in vivo: Assessment through plasma propeptide (VWF:AgII) levels. Thromb Haemost. 1997;77:387–93. [PubMed] [Google Scholar]
- Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83:3536–44. [PubMed] [Google Scholar]
- Wagner DD, Fay PJ, Sporn LA, Sinha S, Lawrence SO, Marder VJ. Divergent fates of von Willebrand factor and its proplypeptide (von Willebrand antigen II) after secretion from endothelial cells. Proc Natl Acad Sci USA. 1987;84:1955–9. doi: 10.1073/pnas.84.7.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware LB, Eisner MD, Thompson BT, Parsons PE, Matthay MA. Significance of von Willebrand factor in septic and nonseptic patients with acute lung injury. Am J Respir Crit Care Med. 2004;170:766–72. doi: 10.1164/rccm.200310-1434OC. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Palade GE. New cytoplasmic components in arterial endothelia. J Cell Biol. 1964;23:101–12. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER. Fifty years of Weibel-Palade bodies: The discovery and early history of an enigmatic organelle of endothelial cells. J Thromb Haemost. 2012;10:979–84. doi: 10.1111/j.1538-7836.2012.04718.x. [DOI] [PubMed] [Google Scholar]
- Yuhas EM, Morgan DG, Arena E, Kupp RP, Saunders LZ, Lewis HB. Arterial medial necrosis and hemorrhage induced in rats by intravenous infusion of fenoldopam mesylate, a dopaminergic vasodilator. Am J Pathol. 1985;119:83–91. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Defelice AF, Hanig JP, Colatsky T. Biomarkers of endothelial cell (EC) activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol Pathol. 2010;38:856–71. doi: 10.1177/0192623310378866. [DOI] [PubMed] [Google Scholar]