

Highlights
-
•
Fasting induces liver TG accumulation accompanied by a decrease of L-FABP and SREBP-1.
-
•
Physostigmine (an AchE inhibitor) dose-dependently aggravates fasting-induced liver TG accumulation.
-
•
Physostigmine increases fasting-decreased protein levels of L-FABP and SREBP-1.
-
•
These physostigmine effects are blocked by muscarinic antagonist atropine.
Abbreviations: ACC, acetyl coenzyme-A carboxylase; ACh, acetylcholine; AChE, acetylcholinesterase; CPT-1, carnitine palmitoyltransferase 1; FA, fatty acid(s); FAS, fatty acid synthase; IRS-2, insulin receptor substrate; L-FABP, liver fatty acid-binding protein; PEPCK, phosphoenolpyruvate carboxykinase; PGC-1α, peroxisome proliferator activated receptor gamma coactivator 1-alpha; PPAR-α, peroxisome proliferator activated receptor alpha; SREBP, sterol regulatory element binding proteins; TG, triglyceride(s); VLDL, very low-density lipoprotein(s)
Keywords: Fatty liver, Parasympathetic nerve, Metabolic syndrome, Triglyceride, Lipogenesis, Lipolysis
Abstract
Although fasting induces hepatic triglyceride (TG) accumulation in both rodents and humans, little is known about the underlying mechanism. Because parasympathetic nervous system activity tends to attenuate the secretion of very-low-density-lipoprotein-triglyceride (VLDL-TG) and increase TG stores in the liver, and serum cholinesterase activity is elevated in fatty liver disease, the inhibition of the parasympathetic neurotransmitter acetylcholinesterase (AChE) may have some influence on hepatic lipid metabolism. To assess the influence of AChE inhibition on lipid metabolism, the effect of physostigmine, an AChE inhibitor, on fasting-induced increase in liver TG was investigated in mice. In comparison with ad libitum-fed mice, 30 h fasting increased liver TG accumulation accompanied by a downregulation of sterol regulatory element-binding protein 1 (SREBP-1) and liver-fatty acid binding-protein (L-FABP). Physostigmine promoted the 30 h fasting-induced increase in liver TG levels in a dose-dependent manner, accompanied by a significant fall in plasma insulin levels, without a fall in plasma TG. Furthermore, physostigmine significantly attenuated the fasting-induced decrease of both mRNA and protein levels of SREBP-1 and L-FABP, and increased IRS-2 protein levels in the liver. The muscarinic receptor antagonist atropine blocked these effects of physostigmine on liver TG, serum insulin, and hepatic protein levels of SREBP-1 and L-FABP. These results demonstrate that AChE inhibition facilitated fasting-induced TG accumulation with up regulation of the hepatic L-FABP and SREBP-1 in mice, at least in part via the activation of muscarinic acetylcholine receptors. Our studies highlight the crucial role of parasympathetic regulation in fasting-induced TG accumulation, and may be an important source of information on the mechanism of hepatic disorders of lipid metabolism.
1. Introduction
Mammals that often face starvation have evolved ingenious metabolic systems. A key aspect of the adaptive response to food shortage is the shift from carbohydrates to ketones as a primary fuel source. In the fasting state, fatty acids are mobilized from adipose tissue to the liver and oxidized to acetyl coenzyme-A, which is a source of ketogenesis. Although the mechanisms are not fully understood, 12–36 h of fasting can increase liver triglyceride (TG) levels in both mice [1,2] and humans [3]. Hepatic steatosis, which is the excessive accumulation of TG in the liver, is one of the risk factors leading to the onset and progress of metabolic syndrome and arteriosclerosis [4,5].
Hepatic steatosis is determined by balancing fatty acid (FA) supply to the liver and intrahepatic lipogenesis with the rate of β-oxidation and lipid export by the liver [6]. Liver fatty acid-binding protein (L-FABP; also known as FABP1), which represents a large family of ∼15 kDa proteins capable of binding hydrophobic lipid ligands, is an abundant protein expressed in hepatocytes and enterocytes [7–9]. L-FABP may play a critical role in FA trafficking and metabolism, as L-FABP-deficient mice exhibit resistance to diet-induced steatosis and L-FABP deletion in hepatocytes disrupts net FA uptake and utilization [9,10]. Sterol regulatory element binding proteins (SREBP), which comprise three isoforms, SREBP-1a, SERBP-1c, and SREBP-2, are synthesized as ∼125 kDa precursors bound to endoplasmic reticulum membrane, and are cleaved to generate soluble NH2-terminal fragments (the mature form of ∼68 kDa) that translocate to the nucleus as transcriptional regulators [4,11,12]. Although SREBP-1a and SREBP-1c are produced from the same gene (SREBP-1) by alternative splicing, SREBP-1c is expressed more in tissues of high lipogenic capacity, such as liver and adipose tissue, than SREBP-1a. SREBP-1c may be a pivotal transcriptional regulator of genes for FA and TG synthesis, including fatty acid synthase (FAS) and acetyl coenzyme-A carboxylase (ACC) [4,11,13]. Furthermore, it is reported that SREBPs suppress insulin receptor substrate 2 (IRS-2)-mediated hepatic insulin signaling [14], and may have an effect on fatty acid oxidation via activating peroxisome proliferator activated receptor alpha (PPAR-α), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and Carnitine palmitoyltransferase I (CPT1) [15,16]. In addition, phosphoenolpyruvate carboxykinase (PEPCK), which is known to be a key enzyme in gluconeogenesis, is necessary for the integration of hepatic energy metabolism [17].
Many factors, including insulin resistance, altered lipogenic factors, lipotoxic free fatty acids, and biological stress, have been implicated in the development of hepatic steatosis. Autonomic nervous regulation is also a factor. It has long been known that the sympathetic and parasympathetic nervous systems may oppose or complement each other, but tend to balance each other overall. In hepatic glucose metabolism, the activation of hepatic sympathetic nerves increases blood glucose levels by stimulating glycolysis, whereas the activation of hepatic parasympathetic nerves represses hepatic glucose output by promoting glycogen synthesis [18]. In hepatic lipid metabolism, the importance of sympathetic drive has been attracting attention recently. Several studies consistently demonstrated that sympathetic activation stimulates very-low-density-lipoprotein-triglyceride (VLDL-TG) production and secretion from the liver into the blood circulation, and increases blood TG levels [19–21]. A clinical study reported that the predominance of sympathetic over parasympathetic activity is strongly associated with metabolic syndrome and might be partly involved in unfavorable consequences such as the incidence of cardiovascular disease [22]. Fasting beyond a certain period also leads to a state of sympathetic nerve stimulation [21]. For example, the ratio between low and high frequency components of heart rate variability, that represents an index of sympathetic nerve activity, was elevated under fasting conditions [23]. However, few studies have examined the role of the parasympathetic nervous system in hepatic lipid metabolism during fasting. Postprandial plasma TG levels were increased by parasympathetic denervation [20]. This has led to the assumption that the parasympathetic nervous system tends to attenuate the secretion of VLDL-TG from the liver and thus increases liver TG stores.
Inhibitors of acetylcholinesterase (AChE) are chemicals that prevent AChE from breaking down the parasympathetic neurotransmitter acetylcholine (ACh), thereby promoting ACh activity. Physostigmine (eserine), which is the oldest known AChE inhibitor, is used clinically for the treatment of glaucoma and myasthenia gravis, and as an antidote for atropine intoxication. It has attracted some attention for its potential role in the amelioration of Alzheimer’s disease [24,25]. Serum cholinesterase activity is elevated in fatty liver diseases and has been accepted as a useful indicator in the diagnosis of hepatobiliary diseases [26,27]. Although AChE inhibitors may also have some influence on lipid metabolism, to our knowledge, few studies have attempted to clarify the effects of AChE inhibitors on lipid metabolism under fasting conditions where a sympathetic activity may dominate. In the present study, we investigated the effect of the AChE inhibitor physostigmine on the fasting-induced increase in liver TG in mice.
2. Results
2.1. Time course of fasting-induced liver TG accumulation in ICR mice
We first validated the effects of fasting periods on the accumulation levels of hepatic TG in ICR mice, because the TG accumulation in the liver may vary with strain, sex, and age. Non-fasted mice showed low TG levels (7.0 ± 5.46 mg/g liver; Fig. 1). Mice fasted for 24 h and 30 h showed considerably high liver TG levels (41.7 ± 6.84 and 33.6 ± 2.29 mg/g liver, respectively). In accordance with the results, 30 h fasting, which stably induced hepatic TG accumulation, was used for the following examinations.
Fig. 1.
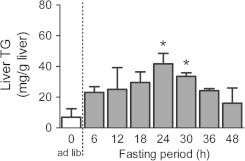
Time course of fasting-induced liver TG accumulation in ICR mice. First we validated the effects of fasting periods on the accumulation levels of hepatic TG in 8-week-old female ICR mice, because the TG accumulations to liver may be different at a strain, sex, and week old of mice. At the 6-h, 12-h, 18-h 24-h, 30-h, 36-h, or 48-h after the fasting respectively, the mice were sacrificed and collected a piece of liver tissues. The extract from each animal liver tissue was measured by an enzymatic colorimetric method. Values shown are the means ± S.E.M. of 4–11 mice. ∗Significant difference from ad libitum-fed mice (one-way ANOVA, Dunnett’s post hoc test, P < 0.05).
2.2. Time course of plasma AChE activity and ACh concentration in fasted and fed mice after the injection of physostigmine
We next validated the effects of physostigmine on plasma AChE activity and ACh concentration in fasted and fed mice. The plasma AChE activity decreased to 60% of the pre-injection (0 min) level at 30 min after the physostigmine injection, to 70% of the pre-injection level at 60 min, and to 90% of the pre-injection level at 60 min (Fig. 2A). Normal plasma AChE activity was restored at 240 min. There was no difference between fed and fasted mice. The plasma ACh concentration of 53.9 ± 8.92 μM in ad libitum-fed mice decreased to 31.9 ± 6.20 μM at 30 min after the physostigmine injection, and restored at 120 min (Fig. 2B). On the other hand, the plasma ACh concentration of 40.6 ± 3.81 μM in 24 h-fasted mice decreased to 31.9 ± 6.20 μM at 30 min after physostigmine injection, and restored at 120 min. In neither evaluation point until 120 min after physostigmine injection, the significant difference was detected between fed and fasted mice.
Fig. 2.
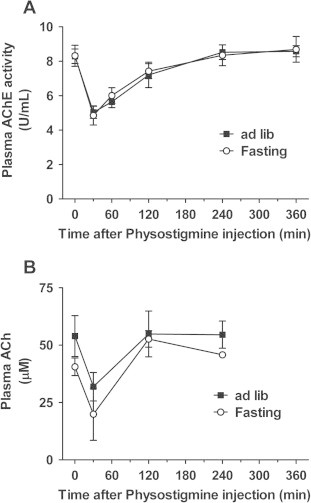
Time course of plasma AChE activity and ACh concentration in fasted and fed mice after the injection of physostigmine (0.3 mg/kg, s.c.). We validated the effects of physostigmine on plasma AChE activity and ACh concentration in fasted and fed mice. At the 24-h after the fasting (or ad libitum feeding), the mice were given a subcutaneous (s.c.) injection of 0.3 mg/kg physostigmine. At each point, mice were sacrificed and collected whole blood. The plasma specimen measured by the fluorometric enzyme assay for AChE or ACh. Values shown are the means ± S.E.M. of 4–12 mice. In neither evaluation point, the significant difference was detected between fed and fasted mice.
2.3. Effect of the AChE inhibitor physostigmine on the 30 h fasting-induced increase in liver TG levels
As shown in Fig. 3A, 30 h fasting increased the liver TG in mice compared with ad libitum-fed mice. Administration of the AChE inhibitor physostigmine increased the 30 h fasting-induced liver TG levels in a dose-dependent manner, and the increase was significantly different at a dose of 0.3 mg/kg once daily (semel in die, SID; 47.6 ± 4.76 mg/g liver) or 0.3 mg/kg twice daily (bis in die, BID; 54.2 ± 4.17 mg/g liver), whereas there was no difference in the plasma TG levels between either group (Fig. 3B). The plasma glucose level of 158.1 ± 4.32 mg/dL in ad libitum-fed mice decreased to 45.4 ± 4.87 mg/dL in 30 h-fasted mice (Fig. 4A), while the insulin concentration of 127.2 ± 30.43 pmol/L in ad libitum-fed mice simultaneously decreased to 42.6 ± 6.16 pmol/L in 30 h-fasted mice (Fig. 4B). Physostigmine additionally reduced the 30 h fasting-induced decrease in the plasma insulin concentration, but raised the 30 h fasting-induced decrease in the plasma glucose level. Significant effects on the plasma glucose level and insulin concentration were observed when physostigmine was administered at 0.3 mg/kg, bis in die (BID) (86.4 ± 10.21 mg/dL and 19.1 ± 3.24 pmol/L, respectively). The muscarinic receptor antagonist atropine (1 mg/kg, BID) blocked the potentiating effect of physostigmine (0.3 mg/kg, BID) on the 30 h fasting-induced increase in liver TG level, with a significant increase in the plasma insulin concentration (liver TG, 34.0 ± 3.01 mg/g liver; plasma insulin, 31.2 ± 6.37 pmol/L). However, the plasma glucose level was not restored by injection of atropine (1 mg/kg, BID). Treatment with atropine alone during fasting failed to affect liver TG levels and plasma insulin concentrations, but increased plasma glucose levels.
Fig. 3.
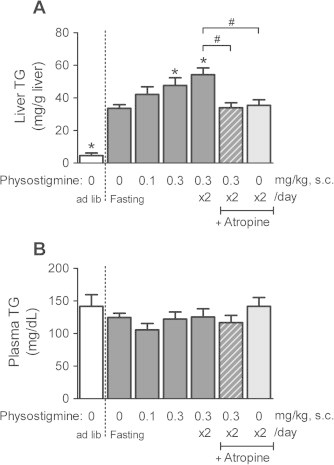
Effect of AChE inhibitor physostigmine on liver TG (A) and plasma TG (B) levels in the 30-h fasted mice. Mice underwent a 30-h fast from ZT6 (14:00) to ZT36 (the next day 20:00), and given a subcutaneous (s.c.) injection of physostigmine (0.1 or 0.3 mg/kg) or saline at ZT24 and ZT30. Muscarinic receptor antagonist atropine (1 mg/kg, s.c.) was administered 15 min before the physostigmine injections. At the 30-h after the fasting, the mice were sacrificed and collected whole blood and a piece of liver tissues. The plasma specimen and the extract from each animal liver tissue were measured by an enzymatic colorimetric method, respectively. Values shown are the means ± S.E.M. of 7–11 mice. ∗Significant difference from 30 h-fasted mice with saline (described 0 mg/kg physostigmine in figure) (one-way ANOVA, Dunnett’s post hoc test, P < 0.05). #Significant difference between two groups (Student’s t-test, P < 0.05).
Fig. 4.
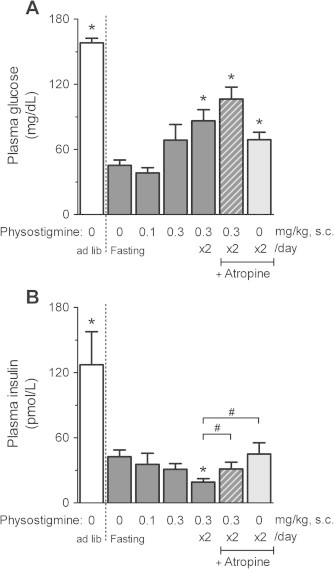
Effect of AChE inhibitor physostigmine on plasma glucose (A) and plasma insulin (B) in the 30-h fasted mice. Mice underwent a 30-h fast from ZT6 to ZT36, and given a subcutaneous (s.c.) injection of physostigmine (0.1 or 0.3 mg/kg) or saline at ZT24 and ZT30. The muscarinic receptor antagonist atropine (1 mg/kg, s.c.) was administered 15 min before the physostigmine injections. At the 30-h after the fasting, the mice were sacrificed and collected whole blood (as mentioned in Section 2 and the legend of Fig. 3). Plasma glucose level and insulin concentration in the specimen, which used for measurement of plasma TG levels in Fig. 3B, were measured by a glucose dehydrogenase method and enzyme immunoassay, respectively. Values shown are the means ± S.E.M. of 7–11 mice. ∗Significant difference from 30 h-fasted mice with saline (described 0 mg/kg physostigmine in figure) (one-way ANOVA, Dunnett’s post hoc test, P < 0.05). #Significant difference between two groups (Student’s t-test, P < 0.05).
2.4. Effect of the AChE inhibitor physostigmine on the expression of lipid-related genes in the liver
To characterize the effect of physostigmine on hepatic lipid metabolism, we examined the expression of genes related to TG synthesis (Srebp1c, Acc), β-oxidation of fatty acids (Pparα, Pgc1α, Cpt1), gluconeogenesis (Pepck), FA trafficking and metabolism (Fabp1), and hepatic insulin signaling (Irs2). As reported previously [8,26], gene expression of Srebp1c, Acc, and Fabp1 in the liver was significantly decreased in the 30 h-fasted mice compared with ad libitum-fed mice (Fig. 5A–C), whereas Pparα, Pgc1α, Cpt1, and Irs2 were significantly increased in the 30 h-fasted mice (Fig. 5D, F and H). The liver mRNA levels of Fabp1 and Acc were higher in mice administered physostigmine (0.3 mg/kg BID) than in fasting-only mice, but did not exceed the mRNA levels in the liver of ad libitum-fed mice. The effect of physostigmine on Fabp1 and Acc expression in the liver was not changed by pre-injection of atropine (1 mg/kg, BID). The liver mRNA levels of Pgc1α and Pepck were not affected by the 30 h of fasting (Fig. 5E and G). The liver mRNA level of Pepck in mice pre-injected with atropine before physostigmine increased by more than twice the mRNA level in mice injected with physostigmine only.
Fig. 5.
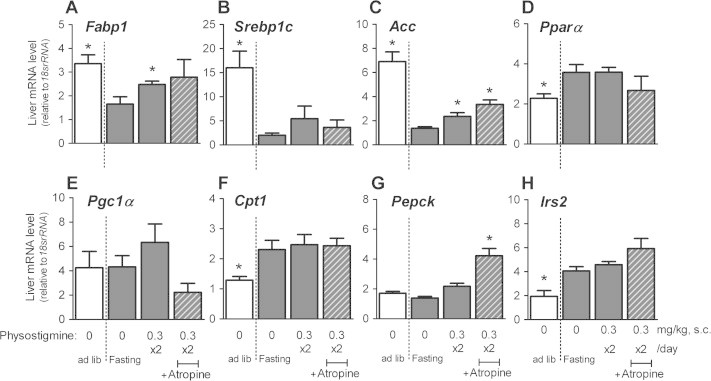
Effect of AChE inhibitor physostigmine on the gene expression of L-Fabp (A), Srebp1c (B), Acc (C), Pparα (D), Pgc1α (E), Cpt1 (F) Pepck (G), and Irs2 (H) in liver extracts from the 30-h fasted mice. Mice underwent a 30-h fast from ZT6 to ZT36, and given twice a day subcutaneous (s.c.) injection of 0.3 mg/kg physostigmine or saline at ZT24 and ZT30. Muscarinic receptor antagonist atropine (1 mg/kg, s.c.) was administered 15 min before the physostigmine injections. At the 30-h after the fasting, the mice were sacrificed and collected a piece of liver tissues. Total RNA was extracted from a piece of the liver tissues and performed PCR as described in Section 2. The relative levels of the target gene PCR product were normalized to those of 18srRNA. Values shown are the means ± S.E.M. of 4–6 mice. ∗Significant difference from 30 h-fasted mice with saline (described 0 mg/kg physostigmine in figure) (t-test or Wilcoxon–Mann–Whitney test, based on the F value, P < 0.05).
2.5. Effect of the AChE inhibitor physostigmine on the protein levels of L-FABP and SREBP-1 in the liver
As mentioned above, physostigmine may have an influence on TG synthesis-related genes and Fabp1. Therefore, we next analyzed the protein expression levels of SREBP-1 and L-FABP in the liver. In addition, we analyzed the 68 kDa protein equivalent to the mature form of SREBP-1, similar to previous reports [11,12]. Fig. 6A shows typical membrane immunoreactivity to anti-L-FABP, anti-SREBP-1, and β-actin (as an internal control). The protein levels of L-FABP and mature SREBP-1 in the liver were significantly decreased in the 30 h-fasted mice compared with ad libitum-fed mice (Fig. 6B and C). Physostigmine (0.3 mg/kg, BID) increased the 30 h fasting-induced relative protein levels of L-FABP and mature SREBP-1, and the physostigmine-induced increase in both protein levels was blocked by pre-injection of atropine (1 mg/kg, BID).
Fig. 6.
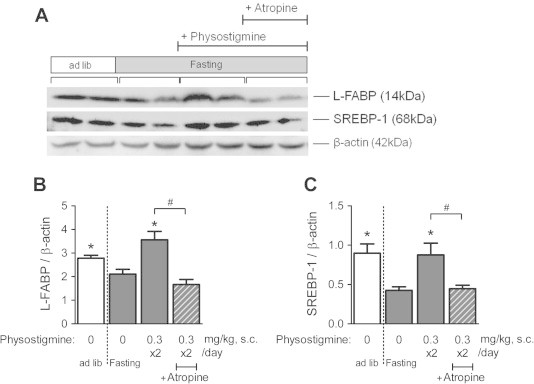
Effect of AChE inhibitor physostigmine on the protein levels of L-FABP and SREBP-1 in liver extracts from the 30-h fasted mice. Mice underwent a 30-h fast from ZT6 to ZT36, and given twice a day subcutaneous (s.c.) injection of 0.3 mg/kg physostigmine or saline at ZT24 and ZT30. Muscarinic receptor antagonist atropine (1 mg/kg, s.c.) was administered 15 min before the physostigmine injections. At the 30-h after the fasting, the mice were sacrificed and collected a piece of liver tissues. The preparation of liver extracts and Western blot analyses was carried out as described in Section 2. The relative expression levels of the L-FABP and SREBP-1 proteins were normalized to those of β-actin using same gels and membranes. (A) A representative immunoblot. (B) Quantification of immunoreactivity of L-FABP. (C) Quantification of immunoreactivity of SREBP-1. Values shown are the means ± S.E.M. of 4–6 mice. ∗Significant difference from 30 h-fasted mice with saline (described 0 mg/kg physostigmine in figure) (t-test or Wilcoxon–Mann–Whitney test, based on the F value, P < 0.05). #Significant difference between two groups (Student’s t-test, P < 0.05).
2.6. Effect of physostigmine or atropine on the liver protein levels of L-FABP and SREBP-1, liver TG, plasma TG, plasma glucose, and plasma insulin in ad libitum-fed mice
As mentioned above, physostigmine may have facilitated fasting-induced liver TG accumulation with up-regulation of hepatic L-FABP and SREBP-1. On the other hand, treatment with atropine alone during fasting significantly increased plasma glucose levels (Fig. 4). We have evaluated the effect of physostigmine or atropine alone during ad libitum state on liver protein levels of L-FABP and SREBP-1, liver TG, plasma TG, plasma glucose, and plasma insulin. Fig. 7A shows typical membrane immunoreactivity to anti-L-FABP, anti-SREBP-1, and β-actin. The protein levels of L-FABP and mature SREBP-1 in the liver of ad libitum-fed mice were not influenced neither by physostigmine nor by atropine (Fig. 7B and C). Neither physostigmine nor atropine alone during fasting affected liver TG level plasma TG, plasma glucose, and plasma insulin (Fig. 7D–G).
Fig. 7.
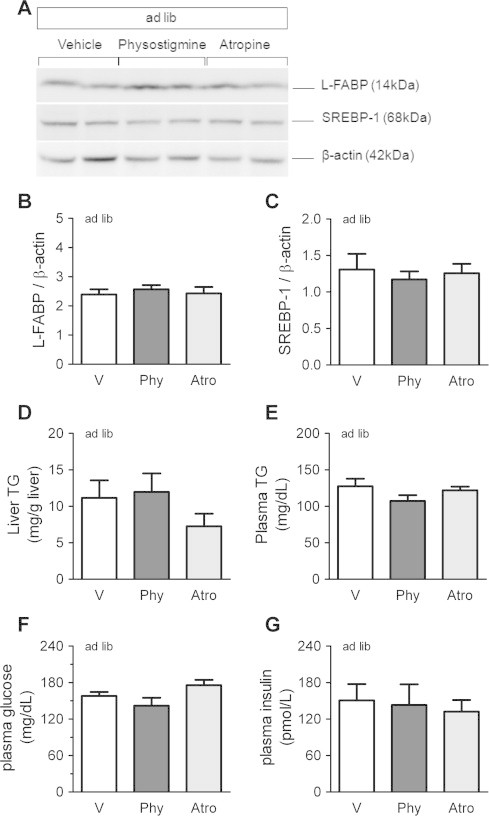
Effect of physostigmine or atropine on the liver protein levels of L-FABP and SREBP-1 (A–C), liver TG (D), plasma TG (E), plasma glucose (F), and plasma insulin (G) in ad libitum-fed mice. Mice were given twice a day subcutaneous (s.c.) injection of AChE inhibitor physostigmine (0.3 mg/kg; Phy), muscarinic receptor antagonist atropine (1 mg/kg; Atro) or saline (5 mL/kg; V) at ZT0 and ZT6. Mice were sacrificed and collected a piece of liver tissues and whole blood at ZT12. All of the analyses were carried out as mentioned above. Values shown are the means ± S.E.M. of 4–8 mice. The significant difference was not detected.
2.7. Effect of AChE inhibitor physostigmine on plasma glucagon, plasma LDL/VLDL, and plasma FFA in fasted and fed mice
We also investigated the effect of physostigmine on plasma glucagon, plasma LDL/VLDL, and plasma FFA in fasted and fed mice. There were no significant differences between fasted and fed mice in plasma glucagon and plasma LDL/VLDL (Fig. 8A and B). Injection of physostigmine also failed to affect in these parameters. On the other hand, the plasma FFA level in 24 h-fasted mice was significantly higher than that in ad libitum fed mice (1.8 ± 0.11 mEq/L and 1.2 ± 0.09 mEq/L, respectively) (Fig. 8C). At 30 h after fasting, the plasma FFA in fasted mice decreased to 1.3 ± 0.10 mEq/L, but that in mice with physostigmine maintained significantly high level (1.6 ± 0.07 mEq/L) in comparison to non-injected mice.
Fig. 8.
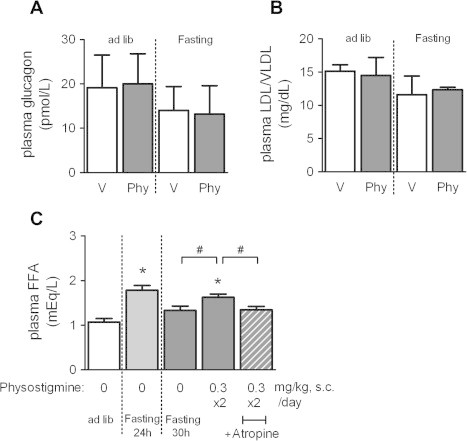
Effect of AChE inhibitor physostigmine on plasma glucagon (A), plasma LDL/VLDL (B), and plasma FFA (C) in fasted and fed mice. Mice underwent a 30-h fast from ZT6 to ZT36, and given twice a day subcutaneous (s.c.) injection of 0.3 mg/kg physostigmine or saline at ZT24 and ZT30. Muscarinic receptor antagonist atropine (1 mg/kg, s.c.) were administered 15 min before the physostigmine injections. To examine the effect of fasting duration, we also measured the plasma FFA concentration in 24 h-fasted mice. At the 30-h or 24-h after the fasting, the mice were sacrificed and collected whole blood. The plasma specimen measured by the fluorometric or colorimetric enzyme assay kit for glucagon, LDL/VLDL, or FFA, respectively. Values shown are the means ± S.E.M. of 6–16 mice. ∗Significant difference from ad lib-fed mice (t-test, P < 0.05). #Significant difference between two groups (t-test, P < 0.05).
2.8. Effect of the AChE inhibitor physostigmine on the protein levels of IRS-1 and IRS-2 in the liver
According to the experiments described above (Figs. 4 and 5), physostigmine (0.3 mg/kg, BID) reduced plasma levels of insulin, which is also a strong initiator of hepatic lipogenesis. Nevertheless, physostigmine increased fasting-induced liver TG accumulation and the expression of the Acc gene and mature SREBP-1 protein in the liver. It has been reported that SREBPs directly bind to the IRS-2 promoter and suppress IRS-2, leading to hepatic insulin resistance [14,27]. Therefore, physostigmine might have an influence on insulin sensitivity in the liver. In a previous report, we have shown by Western blot analysis that dietary protein deprivation increases the amount of insulin receptor β-subunit, IRS-1, and IRS-2 [28]. As shown in Fig. 9, 30 h fasting increased the protein levels of IRS-2. The 30 h fasting-induced increase in IRS-2 protein levels was not influenced by the administration of physostigmine alone, but was influenced by the combination of physostigmine and atropine. In contrast, IRS-1 protein levels in the liver were not influenced by fasting or drug administration.
Fig. 9.
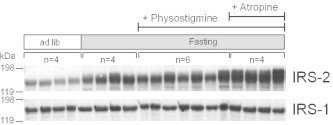
Effect of AChE inhibitor physostigmine on the protein levels of IRS-1 and IRS-2 in liver extracts from the 30-h fasted mice. Mice underwent a 30-h fast from ZT6 to ZT36, and given twice a day subcutaneous (s.c.) injection of 0.3 mg/kg physostigmine or saline at ZT24 and ZT30. Muscarinic receptor antagonist atropine (1 mg/kg, s.c.) and recombinant human insulin (1 U/kg, i.p.) were administered 15 min before the physostigmine injections. At the 30-h after the fasting, the mice were sacrificed and collected a piece of liver tissues. The preparation of liver extracts and Western blot analyses was carried out as described in Section 2. A representative immunoblot was shown.
3. Discussion
Because parasympathetic activity tends to attenuate the secretion of VLDL-TG and increase the stores of TG in the liver [18,29], and serum cholinesterase activity is elevated in human fatty liver disease [31,32], AChE inhibitors may have some influence on hepatic lipid metabolism. In the present study, administration of the AChE inhibitor physostigmine did not induce the accumulation of TG in normal liver from mice fed ad libitum, but did exacerbate the accumulation of TG in the fatty liver caused by 30 h of fasting. To our knowledge, this study is the first report revealing that the AChE inhibitor physostigmine aggravates fasting-induced hepatic TG accumulation.
During fasting, it is thought that sympathetic activity is increased in most peripheral tissues [21], and that parasympathetic activity is reduced concomitantly. At least in our experimental conditions, the plasma ACh level in 30 h-fasted mice was not significantly different from that in ad libitum-fed mice, but slightly lower than that in ad libitum-fed mice. Sympathetic nerve signals are transmitted to each organ involved in TG metabolism. This increases FFA secretion from white adipose tissue and promotes VLDL-TG production and secretion from liver leading to elevated blood TG levels. Injecting an AChE inhibitor might restore the activity of parasympathetic nerves weakened during fasting. There is also the possibility that, while fasting stimulates the sympathetic nervous system and increases FFA secretion from white adipose tissue to the blood circulation, physostigmine retains hepatic parasympathetic activity and keeps TG in the liver. In the present experiment, physostigmine significantly increased hepatic expression level of FA trafficking protein L-FABP in 30 h-fasted mouse. The effect of the physostigmine contributing to fasting-induced liver TG accumulation might be relatively high, because plasma FFA as an important source was significantly higher in physostigmine-injected mice than non-injected mice. Since FFA release from adipose tissue is high and LDL/VLDL secretion from liver is kept constant, the up-regulation of FFA incorporation to the liver during fasting might be likely to facilitate TG accumulation.
Physostigmine-induced increase in liver TG accumulation during fasting was clearly blocked by atropine, an antagonist of muscarinic ACh receptors. In addition, we found a clue to the molecular mechanisms of hepatic TG accumulation; physostigmine up regulated the expression of L-FABP and mature SREBP-1 accompanied by TG accumulation in the liver. This result suggests that the activation of parasympathetic nerves stimulates TG synthesis in fatty liver by the maturation of SREBP-1, a transcriptional regulator. Parasympathetic nerve activation also facilitates the incorporation of FA, which is mobilized from adipose tissues during fasting, into the liver by increasing L-FABP expression. The protein levels of L-FABP and mature SREBP-1 was blocked by atropine, a muscarinic receptor antagonist. Moreover, atropine attenuated both the physostigmine-induced increase in liver TG accumulation and decrease in plasma insulin concentration. These results suggest that physostigmine directly stimulates L-FABP expression and SREBP-1 activation in the fatty liver, at least in part through the muscarinic ACh receptors. However, current experiments do not allow conclusions about the degree of lipogenesis effectively occurring within the tissue. The use of radioactive precursors (water or acetate) would allow for a direct assessment of hepatic de novo fatty acid- or triglyceride synthesis. Therefor future experiment would help determine whether the degree of fatty acid uptake is affected.
In the present study, the administration of atropine (1 mg/kg, BID) alone to mice fed ad libitum did not alter liver or plasma TG levels. Furthermore, atropine alone neither aggravated nor ameliorated the accumulation of TG in the liver after 30 h of fasting. Atropine inhibited the decrease in plasma glucose caused by fasting, but did not inhibit the effect of physostigmine on the fasting-induced decrease in plasma glucose levels. There is a possibility that atropine exerts a positive action on systemic lipid metabolism as well as on the antagonistic action on physostigmine. Although the target tissue of atropine remains unclear, it is suggested that tissues involved in glucose homeostasis, i.e. pancreas, fat cells, and/or intestine, but not liver, are critical candidates. Atropine acts on muscarinic receptors only, while physostigmine acts on both muscarinic and nicotinic receptors. In general, 0.3 mg/kg dose of physostigmine is extremely high dose, which cannot assume clinical application for human. However in mouse and rat, 0.3 mg/kg dose is often used in experiment, which clearly exerts drug efficacy include with central action and/or behavioral changes [24]. Thus, the effects of physostigmine, which were not blocked by atropine, may be exerted via activation of nicotinic receptors.
Our previous reports showed that dietary glucose deprivation promoted the effect of insulin on glucose uptake in the muscle and inhibited gluconeogenesis by the enhancement of insulin signals through the IR–IRS–PI3K pathway in the liver [28,30]. This suggests that the decrease in glucose content and availability promotes tissue-specific insulin sensitivity as an adaptive response to a critical situation. In the present study, the amount of IRS-2 in the liver increased in 30 h-fasted mice, indicating the possibility that fasting itself increases the amount of the insulin signaling molecule in the mouse liver, which could enhance insulin action. In the present study, physostigmine did not affect IRS-2 mRNA expression and IRS-2 protein levels in fatty liver. This suggests that the effect of physostigmine is not directly concerned with IRS-2 related signaling pathway in the liver. On the other hand, the expression of IRS-2 in the liver seems to be increased by an additional atropine injection. The increase in protein levels of L-FABP and SREBP-1 in the liver was clearly blocked by atropine, whereas atropine injection was ineffective on the physostigmine-induced increase in mRNA expression, such as Fabp1, Acc, and Irs2. The reason is unclear why there is no correlation between mRNA expression and protein levels. There is a possibility that physostigmine effects on not only the liver muscarinic ACh receptors but also other plural complicated targets and/or metabolic pathways. Alternately, the expression time course may not be identical between mRNA expression and protein levels. It has been reported that in the isolated rat liver, ACh increases glucose production from the liver and enhances glycogen synthesis [33], whereas in white adipose tissues, ACh attenuates insulin-stimulated glucose uptake and the release of glycerol from adipocytes [29]. The mechanism underlying the effect of parasympathetic agents is unclear. However, it is assumed that the alteration of tissue-specific insulin sensitivity is at least partially involved in the stimulatory effect of physostigmine on the 30 h fasting-induced increase in TG levels in the mouse liver. Further investigation into TG metabolism in adipose tissue is required to clarify the systemic effect of physostigmine on lipolysis in adipose tissue and characterize the role of parasympathetic regulation in hepatic lipid disorders.
We initially hypothesized that parasympathetic activation by physostigmine has effects on the pancreas and augments insulin secretion. This hypothesis was based on evidence from various animal models showing that increased parasympathetic input to the pancreas contributes to hyperinsulinemia, which is independent of blood glucose levels [34]. In the present study, physostigmine promoted the decrease in plasma insulin concentrations caused by 30 h of fasting, but restored the fasting-induced decrease in plasma glucose levels. Our data demonstrated that atropine and physostigmine did not alter plasma insulin levels in ad libitum-fed mice. Furthermore, one of the opposite or contrasting hormones glucagon, which is produced from pancreatic alpha-cells and signals the liver to break down glycogen stores, was also not altered by atropine and physostigmine. Although the mechanisms contributing to this contradictory effect of physostigmine on insulin-producing pancreatic beta-cells remains unknown, it is suggested that the target organ of physostigmine in the present experiment is not the pancreas.
When mice were fed ad libitum, the secretion of incretins, which are gastrointestinal hormones including gastric inhibitory peptide and glucagon-like peptide-1, is promoted by the physical stimulation of food intake [35,36]. Because incretins are also controlled by the parasympathetic nervous system, the restoration of parasympathetic activity by physostigmine might stimulate the secretion of incretins resulting in the insulin-dependent accumulation of lipids. Furthermore, inhibition of AChE may also have effects unrelated to the parasympathetic system, i.e., ghrelin is activated during fasting and degraded by ACh.
In the present study, the levels of Pparα, Pgc1α, and Cpt1, the genes associated with β-oxidation of FA, were not significantly influenced by physostigmine injection to fasted mice, although Pgc1α was not also influenced by 30-h fasting alone. This result suggests that physostigmine has no discernible effect on hepatic β-oxidation. On the other hand, it is reported that SREBP-1 promotes the transcription of TG synthesis genes and stimulates β-oxidation of FA through the direct activation of PPAR-α and PGC-1α [15,16]. Mature SREBP-1 protein, which is increased in the liver by physostigmine administration, may influence hepatic β-oxidation and TG synthesis. Further investigation is required to clarify the effect of physostigmine on hepatic β-oxidation.
In conclusion, this study demonstrated that AChE inhibition aggravated fasting-induced acute TG accumulation with up regulation of hepatic L-FABP and SREBP-1 in mice, at least in part via activation of the muscarinic ACh receptors, although at present it is unclear whether next to the liver other organs are involved in this TG accumulation promoting effect of AChE inhibition. Our studies highlight the critical role of parasympathetic regulation in fasting-induced TG accumulation, and may be an important source of information on the mechanism of hepatic lipid disorders.
4. Materials and methods
4.1. Animals and experimental protocols
Eight-week-old female ICR mice (Tokyo Laboratory Animals, Tokyo, Japan) were housed at 22 ± 2 °C in an atmosphere with a humidity of 60 ± 5%, subjected to a 12 h light/12 h dark cycle, and provided with food and water ad libitum before the experiments. The lights-on time was 8:00 and was assigned Zeitgeber time (ZT) 0. Experimental animal care was conducted with permission from the Committee for Animal Experimentation of the School of Science and Engineering at Waseda University. All drugs were freshly prepared before injection. In the experiments, mice underwent a 30 h fast from ZT6 (14:00) to ZT36 (20:00 the next day), and given a subcutaneous (s.c.) injection of physostigmine (0.1 or 0.3 mg/kg) or saline at ZT24 and ZT30. The muscarinic receptor antagonist atropine (1 mg/kg, s.c.) was administered 15 min before the physostigmine injections. At 30 h after fasting, the mice were anesthetized with ether and sacrificed, and whole blood and liver tissue samples were collected. An assay for liver TG levels was performed as described previously [37,38]. Each blood sample was centrifuged to obtain plasma, and a plasma specimen from each animal was used to calculate ACh concentration using Choline/Acetylcholine Quantification Colorimetric/Fluorometric Kit (BioVision, CA, USA), AChE activity using Acetylcholinesterase Activity Colorimetric Assay Kit (BioVision, CA, USA), TG concentration using Triglyceride E-test Wako (Wako Pure Chemical Industries, Osaka, Japan), glucose concentration using Glucose Pilot (Aventir Biotech, Carlsbad, CA), insulin concentration using Mouse Insulin ELISA (Mercodia AB, Sylveniusgatan, Sweden), glucagon concentration using Mercodia Glucagon ELISA (Mercodia AB, Sylveniusgatan, Sweden), LDL/VLDL concentration using HDL and LDL/VLDL Quantification Colorimetric/Fluorometric Kit (BioVision, CA, USA), and FFA concentration using NEFA C-test Wako (Wako Pure Chemical Industries, Osaka, Japan).
4.2. Real-time quantitative PCR analysis
A liver sample from each animal was dissolved in RNA-Solv Reagent (Omega Bio-Tek, Norcross, GA), and total RNA was extracted. PCR was performed as described previously [37,38]. Specific primer pairs were designed based on published data for the 18srRNA, Srebp-1c, L-Fabp, Irs2, Acc, Fas, Pgc-1α, and Pepck genes [37–39]. The relative levels of the target gene PCR product were normalized to those of 18srRNA. Data were analyzed by the delta-delta Ct method. Melt curve analysis was performed to check for nonspecific products. The results indicated no amplification of nonspecific products.
4.3. Western blot analysis
Anti-SREBP-1 antibody (sc-8984), anti-IRS-1 antibody (sc-559), and anti-IRS-2 antibody (sc-8299) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-L-FABP antibody (#5352) was purchased from Cell Signaling Technology (Danvers, MA). Anti-β-actin antibody (#622102; Poly6221) was purchased from BioLegend (San Diego, CA). The preparation of liver extracts and Western blot analyses were carried out according to a slight modification of methods described previously [11,40]. The blotted signals were visualized using LAS-4000 UVmini systems (Fujifilm, Tokyo, Japan) and signal intensity was analyzed densitometrically (Multi Gauge V3.0; Fujifilm, Tokyo, Japan). The relative expression levels of the L-FABP and SREBP-1 proteins were normalized to those of β-actin using same gels and membranes. The first blot used anti-L-FABP or anti-SREBP-1 antibody was stripped by conventional methods, and then provided for β-actin evaluations.
4.4. Data analysis
All data are expressed as means ± standard error of the mean (S.E.M.). One-way analysis of variance (ANOVA) followed by the Dunnett’s test were used for intragroup comparisons of the physostigmine dose-dependency and fasting period-dependency. For the other comparisons between two different groups, the data were assessed by ANOVA. Depending on the F value, either the Student’s t-test or the Wilcoxon–Mann–Whitney test was used to determine statistical significance. Differences were considered significant when P < 0.05.
Author contributions
Conceived and designed the experiments: SY and SS. Acquired the data: SY KM MA HK. Analyzed and interpreted the data: SY and SS. Contributed reagents/materials/analysis tools and supervised the manuscript: FH and ST. Wrote the paper: SY.
Acknowledgments
This work was supported in part by grants to S.S. in the form of Grants-in-Aid for Scientific Research from JSPS (23300278 and 23659126), the Fuji Foundation for Protein Research (2010 and 2012), and the Program for the Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry. No additional external funding was received for this study.
References
- 1.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Sokolović M., Sokolović A., van Roomen C.P., Gruber A., Ottenhoff R., Scheij S., Hakvoort T.B., Lamers W.H., Groen A.K. Unexpected effects of fasting on murine lipid homeostasis—transcriptomic and lipid profiling. J. Hepatol. 2010;52:737–744. doi: 10.1016/j.jhep.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 3.Moller L., Stodkilde-Jorgensen H., Jensen F.T., Jorgensen J.O. Fasting in healthy subjects is associated with intrahepatic accumulation of lipids as assessed by 1H-magnetic resonance spectroscopy. Clin. Sci. (Lond.) 2008;114:547–552. doi: 10.1042/CS20070217. [DOI] [PubMed] [Google Scholar]
- 4.Hooper A.J., Adams L.A., Burnett J.R. Genetic determinants of hepatic steatosis in man. J. Lipid Res. 2011;52:593–617. doi: 10.1194/jlr.R008896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carallo C., Mancuso G., Mauro G., Laghi F., Madafferi B., Irace C., Gnasso A., Scavelli F., Dell’Aquila F., Bartone M. Hepatic steatosis, carotid atherosclerosis and metabolic syndrome: the STEATO Study. J. Gastroenterol. 2009;44:1156–1161. doi: 10.1007/s00535-009-0125-8. [DOI] [PubMed] [Google Scholar]
- 6.Roden M. Mechanisms of disease: hepatic steatosis in type 2 diabetes pathogenesis and clinical relevance. Nat. Clin. Pract. Endocrinol. Metab. 2006;2:335–348. doi: 10.1038/ncpendmet0190. [DOI] [PubMed] [Google Scholar]
- 7.Furuhashi M., Hotamisligil G.S. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iseki S., Kondo H., Hitomi M., Ono T. Localization of liver fatty acid-binding protein and its mRNA in the liver and jejunum of rats: an immunohistochemical and in situ hybridization study. Mol. Cell. Biochem. 1990;98:27–33. doi: 10.1007/BF00231364. [DOI] [PubMed] [Google Scholar]
- 9.Newberry E.P., Xie Y., Kennedy S., Han X., Buhman K.K., Luo J., Gross R.W., Davidson N.O. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J. Biol. Chem. 2003;278:51664–51672. doi: 10.1074/jbc.M309377200. [DOI] [PubMed] [Google Scholar]
- 10.Newberry E.P., Kennedy S.M., Xie Y., Sternard B.T., Luo J., Davidson N.O. Diet-induced obesity and hepatic steatosis in L-Fabp/mice is abrogated with SF, but not PUFA, feeding and attenuated after cholesterol supplementation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G307–G314. doi: 10.1152/ajpgi.00377.2007. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto E., Ishihara A., Tamai S., Nemoto A., Iwase K., Hiwasa T., Shibata S., Takiguchi M. Time of day and nutrients in feeding govern daily expression rhythms of the gene for sterol regulatory element-binding protein (SREBP)-1 in the mouse liver. J. Biol. Chem. 2010;285:33028–33036. doi: 10.1074/jbc.M109.089391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park H.J., Georgescu S.P., Du C., Madias C., Aronovitz M.J., Welzig C.M., Wang B., Begley U., Zhang Y., Blaustein R.O. Parasympathetic response in chick myocytes and mouse heart is controlled by SREBP. J. Clin. Invest. 2008;118:259–271. doi: 10.1172/JCI32011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toth J.I., Datta S., Athanikar J.N., Freedman L.P., Osborne T.F. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol. Cell. Biol. 2004;24:8288–8300. doi: 10.1128/MCB.24.18.8288-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ide T., Shimano H., Yahagi N., Matsuzaka T., Nakakuki M., Yamamoto T., Nakagawa Y., Takahashi A., Suzuki H., Sone H. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 2004;6:351–357. doi: 10.1038/ncb1111. [DOI] [PubMed] [Google Scholar]
- 15.Finck B.N., Gropler M.C., Chen Z., Leone T.C., Croce M.A., Harris T.E., Lawrence J.C., Jr., Kelly D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Peterson T.R., Sengupta S.S., Harris T.E., Carmack A.E., Kang S.A., Balderas E., Guertin D.A., Madden K.L., Carpenter A.E., Finck B.N. MTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.She P., Shiota M., Shelton K.D., Chalkley R., Postic C., Magnuson M.A. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol. Cell. Biol. 2000;20:6508–6517. doi: 10.1128/mcb.20.17.6508-6517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Püschel G.P. Control of hepatocyte metabolism by sympathetic and parasympathetic hepatic nerves. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004;280:854–867. doi: 10.1002/ar.a.20091. [DOI] [PubMed] [Google Scholar]
- 19.Bruinstroop E., Pei L., Ackermans M.T., Foppen E., Borgers A.J., Kwakkel J., Alkemade A., Fliers E., Kalsbeek A. Hypothalamic neuropeptide Y (NPY) controls hepatic VLDL-triglyceride secretion in rats via the sympathetic nervous system. Diabetes. 2012;61:1043–1050. doi: 10.2337/db11-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruinstroop E., la Fleur S.E., Ackermans M.T., Foppen E., Wortel J., Kooijman S., Berbée J.F., Rensen P.C., Fliers E., Kalsbeek A. The autonomic nervous system regulates postprandial hepatic lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2013;304:E1089–1096. doi: 10.1152/ajpendo.00614.2012. [DOI] [PubMed] [Google Scholar]
- 21.Geerling J.J., Boon M.R., Kooijman S., Parlevliet E.T., Havekes L.M., Romijn J.A., Meurs I.M., Rensen P.C. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. J. Lipid Res. 2014;55:180–189. doi: 10.1194/jlr.R045013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Licht C.M., Vreeburg S.A., van Reedt Dortland A.K., Giltay E.J., Hoogendijk W.J., DeRijk R.H., Vogelzangs N., Zitman F.G., de Geus E.J., Penninx B.W. Increased sympathetic and decreased parasympathetic activity rather than changes in hypothalamic–pituitary–adrenal axis activity is associated with metabolic abnormalities. J. Clin. Endocrinol. Metab. 2010;95:2458–2466. doi: 10.1210/jc.2009-2801. [DOI] [PubMed] [Google Scholar]
- 23.Kawahara K., Yamamoto H., Ebana S., Tsukui K., Sasaki A., Kumano H., Suematsu H. A study on autonomic nervous function under fasting therapy. Jpn. J. Psychosom. Med. 1997;37:407–415. [Google Scholar]
- 24.Triggle D.J., Mitchell J.M., Filler R. The pharmacology of physostigmine. CNS Drug Rev. 1998;4:87–136. [Google Scholar]
- 25.Singh M., Kaur M., Kukreja H., Chugh R., Silakari O., Singh D. Acetylcholinesterase inhibitors as Alzheimer therapy: from nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013;70C:165–188. doi: 10.1016/j.ejmech.2013.09.050. [DOI] [PubMed] [Google Scholar]
- 26.Le Martelot G., Claudel T., Gatfield D., Schaad O., Kornmann B., Sasso G.L., Moschetta A., Schibler U. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009;7:e1000181. doi: 10.1371/journal.pbio.1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakagawa Y., Shimano H., Yoshikawa T., Ide T., Tamura M., Furusawa M., Yamamoto T., Inoue N., Matsuzaka T., Takahashi A. TFE3 transcriptionally activates hepatic IRS-2, participates in insulin signaling and ameliorates diabetes. Nat. Med. 2006;12:107–113. doi: 10.1038/nm1334. [DOI] [PubMed] [Google Scholar]
- 28.Toyoshima Y., Tokita R., Ohne Y., Hakuno F., Noguchi T., Minami S., Kato H., Takahashi S. Dietary protein deprivation upregulates insulin signaling and inhibits gluconeogenesis in rat liver. J. Mol. Endocrinol. 2010;45:329–340. doi: 10.1677/JME-10-0102. [DOI] [PubMed] [Google Scholar]
- 29.Yang T.T., Chang C.K., Tsao C.W., Hsu Y.M., Hsu C.T., Cheng J.T. Activation of muscarinic M-3 receptor may decrease glucose uptake and lipolysis in adipose tissue of rats. Neurosci. Lett. 2009;451:57–59. doi: 10.1016/j.neulet.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 30.Toyoshima Y., Ohne Y., Takahashi S.I., Noguchi T., Kato H. Dietary protein deprivation decreases the serine phosphorylation of insulin receptor substrate-1 in rat skeletal muscle. J. Mol. Endocrinol. 2004;32:519–531. doi: 10.1677/jme.0.0320519. [DOI] [PubMed] [Google Scholar]
- 31.Turecky L., Kupcova V., Mojto V., Smutny M., Uhlikova E., Vozar I. Serum cholinesterase activity and proteosynthetic function of liver in patients with diabetes mellitus. Bratisl. Lek. Listy. 2005;106:266–269. [PubMed] [Google Scholar]
- 32.Ogunkeye O.O., Chuhwak E.K., Otokwula A.A. Serum cholinesterase activity in the diagnosis of nonalcoholic fatty liver disease in type 2 diabetic patients. Pathophysiology. 2010;17:29–32. doi: 10.1016/j.pathophys.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Vatamaniuk M.Z., Horyn O.V., Vatamaniuk O.K., Doliba N.M. Acetylcholine affects rat liver metabolism via type 3 muscarinic receptors in hepatocytes. Life Sci. 2003;72:1871–1882. doi: 10.1016/s0024-3205(02)02506-7. [DOI] [PubMed] [Google Scholar]
- 34.Vozarova de Courten B., Weyer C., Stefan N., Horton M., DelParigi A., Havel P., Bogardus C., Tataranni P.A. Parasympathetic blockade attenuates augmented pancreatic polypeptide but not insulin secretion in Pima Indians. Diabetes. 2004;53:663–671. doi: 10.2337/diabetes.53.3.663. [DOI] [PubMed] [Google Scholar]
- 35.Burcelin R. The incretins: a link between nutrients and well-being. Br. J. Nutr. 2005;93:S147–156. doi: 10.1079/bjn20041340. [DOI] [PubMed] [Google Scholar]
- 36.D’Alessio D.A., Kieffer T.J., Taborsky G.J., Jr, Havel P.J. Activation of the parasympathetic nervous system is necessary for normal meal-induced insulin secretion in rhesus macaques. J. Clin. Endocrinol. Metab. 2001;86:1253–1259. doi: 10.1210/jcem.86.3.7367. [DOI] [PubMed] [Google Scholar]
- 37.Kudo T., Tamagawa T., Kawashima M., Mito N., Shibata S. Attenuating effect of clock mutation on triglyceride contents in the ICR mouse liver under a high-fat diet. J. Biol. Rhythms. 2007;22:312–323. doi: 10.1177/0748730407302625. [DOI] [PubMed] [Google Scholar]
- 38.Fuse Y., Hirao A., Kuroda H., Otsuka M., Tahara Y., Shibata S. Differential roles of breakfast only (one meal per day) and a bigger breakfast with a small dinner (two meals per day) in mice fed a high-fat diet with regard to induced obesity and lipid metabolism. J. Circadian Rhythms. 2012;10:4. doi: 10.1186/1740-3391-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubota N., Kubota T., Itoh S., Kumagai H., Kozono H., Takamoto I., Mineyama T., Ogata H., Tokuyama K., Ohsugi M. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008;8:49–64. doi: 10.1016/j.cmet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Fukushima T., Nakamura Y., Yamanaka D., Shibano T., Chida K., Minami S., Asano T., Hakuno F., Takahashi S. Phosphatidylinositol 3-kinase (PI3K) activity bound to insulin-like growth factor-I (IGF-I) receptor, which is continuously sustained by IGF-I stimulation, is required for IGF-I-induced cell proliferation. J. Biol. Chem. 2012;287:29713–29721. doi: 10.1074/jbc.M112.393074. [DOI] [PMC free article] [PubMed] [Google Scholar]