

Abstract
To locate the biosensor peptide DPc10 bound to ryanodine receptor (RyR) Ca2+ channels, we developed an approach that combines fluorescence resonance energy transfer (FRET), simulated-annealing, cryo-electron microscopy, and crystallographic data. DPc10 is identical to the 2460–2495 segment within the cardiac muscle RyR isoform (RyR2) central domain. DPc10 binding to RyR2 results in a pathologically elevated Ca2+ leak by destabilizing key interactions between the RyR2 N-terminal and central domains (unzipping). To localize the DPc10 binding site within RyR2, we measured FRET between five single-cysteine variants of the FK506-binding protein (FKBP) labeled with a donor probe, and DPc10 labeled with an acceptor probe (A-DPc10). Effective donor positions were calculated from simulated-annealing constrained by both the RyR cryo-EM map and the FKBP atomic structure docked to the RyR. FRET to A-DPc10 was measured in permeabilized cardiomyocytes via confocal microscopy, converted to distances, and used to trilaterate the acceptor locus within RyR. Additional FRET measurements between donor-labeled calmodulin and A-DPc10 were used to constrain the trilaterations. Results locate the DPc10 probe within RyR domain 3, ∼35 Å from the previously docked N-terminal domain crystal structure. This multiscale approach may be useful in mapping other RyR sites of mechanistic interest within FRET range of FKBP.
Introduction
Muscle contraction is triggered by a massive release of Ca2+, via activation of the ryanodine receptor (RyR) Ca2+ channels embedded in the sarcoplasmic reticulum membrane. RyR isoforms expressed in skeletal (RyR1) and cardiac (RyR2) muscle are key components of the excitation-contraction coupling mechanism in these tissues (1,2). Elevated Ca2+ leak through dysfunctional RyRs results in several widely spread pathologies characterized by abnormally high cytosolic [Ca2+] (3).
The 2.3 MDa RyRs are the largest ion channels identified in natural membranes, and they consist of tetrameric assemblies of identical protomers (∼5000 amino acids each) presenting an enormous domain to the cytosol that extends >100 Å from the small transmembrane (TM) domain. The channel function residing within the RyR TM-domain is regulated via long-range conformational changes from the RyR cytosolic domain resulting from binding of cellular modulators, such as Ca2+, Mg2+, and ATP, or small accessory proteins like the ∼12 kDa FK506 binding proteins (FKBP, isoforms 12.0 and 12.6) or calmodulin (CaM) (4,5).
Cryo-electron microscopy (cryo-EM) three-dimensional reconstructions at ∼10 Å resolution have revealed important information about the structural domains of RyR1, including some structure-function correlations (6), although most secondary and tertiary structural features are difficult to discern. Thus far, some of these structural elements have been deduced based on computational secondary structure prediction (7,8) and docking of partial atomic structures into the cryo-EM map (9–11). Significant research efforts are currently devoted to locating channel regulatory sites in the RyR three-dimensional map (12).
FKBP12.6 binds tightly to both the skeletal and cardiac channel isoforms (RyR1 and RyR2, respectively), and behaves essentially as a constitutive RyR subunit (13). A density corresponding to FKBP is clearly resolved in the RyR cryo-EM map, and the atomic structure of FKBP has been docked in this density (14). Using fluorescence resonance energy transfer (FRET), we have previously shown that FKBP12.0 and 12.6 bind at equivalent locations and orientations within the RyR1 and RyR2 complexes (15). These characteristics of the FKBP-RyR interaction have enabled the placing fluorescent probes at precisely determined locations within the RyR three-dimensional structure, for use as FRET donors in studies aiming to correlate RyR structural and functional information (13,16).
A prominent working model postulates that in the resting RyR2, there is a tight interaction (domain zipping) between an N-terminal 150 kDa domain (17,18) (which includes the docked ABC-domain (9)) and a central domain (residues 2000–2500). Although this theory may be an oversimplification, pathophysiological RyR leaky states have been connected to weakened or disrupted physical contacts between these two domains (domain unzipping), caused by RyR disease-linked mutations or abnormal intracellular conditions (19–22). Conversely, inhibitors of the RyR under resting-state conditions also restore tight domain-domain interactions (zipping) (19,23–25). A 36-residue domain-peptide (DPc10), with the same sequence as residues 2460–2495 of the RyR2 central domain, induces a pathologically leaky RyR2 state, and is one of the molecular tools that have been used to develop the zipping-unzipping theory (18). We have shown that a fluorescent DPc10 derivative used as FRET acceptor (A-DPc10) can be an accurate biosensor of the RyR2 functional state (26). Using FRET to A-DPc10 from donor-labeled FKBP (D-FKBP) or CaM (D-CaM), we have found that DPc10 binds at a sterically restricted RyR2 site. We ruled out a previously proposed location of this site in the “clamp”, and deduced that it must lie within, or close to, the “handle” (domain 3), flanked by FKBP and CaM. Whereas we detected no significant cross-talk between DPc10 and FKBP binding to RyR2, we found reciprocal allosteric inhibition between DPc10 and CaM binding to RyR2 (26).
To localize the DPc10 binding site, and possibly the N-terminal domain zipping interface, within the RyR cryo-EM map, we have conducted detailed confocal FRET studies in cardiac myocytes. We hypothesized that, by using multiple distance measurements from different known spatial coordinates, it is possible to resolve the location of DPc10 on the RyR cryo-EM map using a method termed “trilateration” (although “triangulation” has been casually used to indicate the same process). We developed a computationally efficient protocol for simulated-annealing calculations (with constraints from the RyR1 cryo-EM map and FKBP atomic structure) to determine the effective coordinates of donors attached at different sites within FKBP. A set of distances derived from confocal FRET measurements were then used to determine the location of the A-DPc10 probe, and thus infer the position of a modulatory interface within RyR2.
Materials and Methods
Protein expression and labeling
Single-Cys mutants of FKBP12.6 and CaM were expressed and purified as previously described in Cornea et al. (15). Alexa Fluor 488 C5 maleimide (AF488; Invitrogen, Carlsbad, CA) was reacted to single Cys introduced by site-directed mutagenesis at residues 14, 32, 44, 49, or 85 of the C22A/C76I variant of the human isoform of FKBP12.6 (Fig. 1). These AF488-labeled FKBPs were used as FRET donors (denoted D-FKBP, or D14-, D32-, D44-, D49-, and D85-FKBP, respectively, to indicate the donor’s location within FKBP) (27). AF488 was reacted to a CaM mutant containing a single Cys at position 34, and used as FRET donor (denoted D34-CaM). Alexa Fluor 568 C5 maleimide (AF568) was reacted to a CaM mutant containing a single Cys at position 110, and then used as FRET donor (D110-CaM). We have previously shown that labeling at these sites does not significantly alter binding relative to unlabeled controls (15). DPc10 peptides either unlabeled, or labeled with HiLyte Fluor 647 (HF647) were synthesized at AnaSpec (Fremont, CA), and used as FRET acceptors (A-DPc10), as described in Oda et al. (26). The molecular model of DPc10 (Fig. 1), sequence 2460-GFCPDHKAAMVLFLDRVYGIEVQDFLLHLLEVGFLP-2495 was generated by the HHPred structure prediction server (28). Full-length RyR cDNA constructs, each containing a His10 tag inserted at a specific position within the N-terminal ABC-domain of RyR1, were created as previously described in Girgenrath et al. (27).
Figure 1.
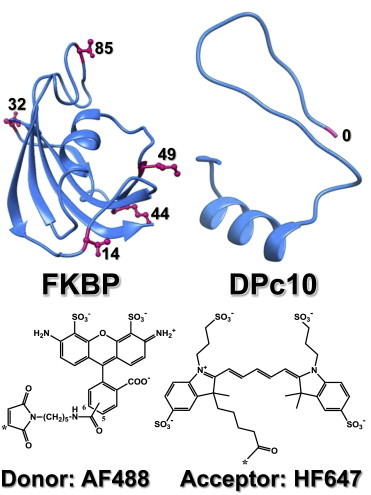
Crystal structure of FKBP and predicted molecular structure of DPc10 showing the labeling sites for the FRET probes (red). Chemical structures of the FRET probes AF488 and HF647 are shown as indicated. To see this figure in color, go online.
FRET imaging
FRET measurements within RyR1 constructs expressed in HEK-293T (HEK) cells were conducted using procedures we recently described in Girgenrath et al. (27). HEK cells were permeabilized and imaged in 125 mM NaCl, 5 mM KCl, 6 mM glucose, 25 mM HEPES (pH 7.6), 0.1% saponin, 3 μM Cy3NTA, and 10 nM D-FKBP. FRET within native RyR2 was measured in isolated rat ventricular myocytes, isolated, and permeabilized as previously described in Oda et al. (26). All procedures were performed according to the Guiding Principles in the Care and Use of Animals, approved by the Council of the American Physiological Society (Bethesda, MD). Confocal imaging of permeabilized myocytes and FRET between D-FKBPs, or D-CaMs, and A-DPc10 was measured in a Olympus FV1000 confocal microscope (Olympus, Center Valley, PA) equipped with Argon and Green HeNe lasers. AF488 donor was excited with the 488 nm laser, and donor fluorescence was recorded through a 500–530 nm bandpass filter. AF568 donor was excited at 543 nm, and donor fluorescence was detected through a 560–620 nm bandpass filter. HF647 acceptor fluorescence was recorded through a 660–760 nm bandpass filter.
Acceptor photobleach was performed using a 652-nm laser. Media contained 1 mM EGTA, 10 mM HEPES, 120 mM potassium aspartate, 5 mM ATP, 1 mM MgCl2, and 10 mM GSH, with CaCl2 added to obtain 30 nM free Ca2+ (calculated using the software MAXCHELATOR, Stanford University, Stanford, CA; http://maxchelator.stanford.edu/), 10 mM phosphocreatine, 5 I.U./mL creatine phosphokinase, and 4% dextran (Mr 40,000), at pH 7.2. Fluorescently labeled FKBP, CaM, and DPc10 were added to the bath at the indicated concentrations (26). FRET was measured using both the donor quenching and the acceptor photobleaching methods, as previously described in Oda et al. (26) and Guo et al. (29). The FRET efficiency was calculated according to
| (1) |
where FD and FDA are the fluorescence intensities of donor-only and donor-acceptor samples in the donor quenching measurement, respectively, and
| (2) |
where FDpre and FDpost are the fluorescence intensities before and after acceptor photobleaching, respectively. From E, we calculated donor-acceptor using
| (3) |
where R0 is, by definition, the distance at which E = 0.5, and is calculated from
| (4) |
where n is the refractive index (for a protein in aqueous solution, n = 1.4), κ2 is the orientation factor (κ2 = 2/3 when random orientation can be assumed), and ϕD is the fluorescence quantum yield of the donor (ϕD(AF488) = 0.92 (30)). J is the normalized spectral-overlap integral of donor emission FD(λ) and acceptor absorbance ε(λ) spectra, and was calculated from
| (5) |
by numerical integration using a Microcal ORIGIN template (OriginLab, Northampton, MA; http://www.originlab.com/). For the AF488-HF647 donor-acceptor pair, we used εHF647(652 nm) = 250,000 (mol−1 cm−1) to calculate R0 = 54 Å.
Docking of FKBP into the RyR1 EM map
The atomic structure of FKBP12 (Protein Data Bank (PDB) PDB:1D6O (31)) was docked to the cryo-EM reconstruction of the closed state of RyR1 (EMDB:1606 (6)). This was achieved by manually placing the structures in the RyR1 map at positions previously published in Samsó et al. (14), then fitting in the density using the “Fit in Map” function of the software UCSF CHIMERA (University of California at San Francisco, San Francisco, CA; https://www.cgl.ucsf.edu/chimera/) (32). The fit was optimized for correlation with a map simulated from the FKBP coordinates using the same resolution as the RyR map, i.e., 10.2 Å. All other settings were as default.
Selection of EM map coordinates around FKBP
The CHIMERA Segment Map tool (33) was used to select the map region corresponding to the FKBP density and to subtract that selected region from the closed RyR EM map. The grid points of the new map without the FKBP density were converted to dummy atoms in a PDB file using the software package VOL2PDB program from the SITUS software package (Old Dominion University, Norfolk, VA; http://situs.biomachina.org/) (34). Dummy atoms within 30 Å from FKBP were used to restrict the conformational space of the fluorescent probe.
Donor parameters for simulation with CNS
A model for AF488 was built using GAUSSVIEW 5 and geometry-optimized using GAUSSIAN 09 (Gaussian, Wallingford, CT; http://www.gaussian.com/) at the HF/6-31+g∗ level of theory. This model was used with XPLO2D (Uppsala Software Factory, Uppsala University, Uppsala, Sweden; http://xray.bmc.uu.se/usf/xplo2d_man.html) (35) to generate topology and parameter files needed for simulations with the software CRYSTALLOGRAPHY & NMR SYSTEM 1.3 (CNS) (Stanford University, Stanford, CA; http//cns-online.org/) (36,37). To allow the probe to move more freely, the XPLO2D energy parameter for the torsional rotation in the linker region was lowered from the default value of 750 kcal/mol to 2 kcal/mol, which is the standard CNS parameter for the methylene linkage. The maleimide linkage to cysteine was adopted from the CNS dyes topology and parameter files (38,39).
Determining the fluorophore average position by simulated annealing
In the atomic structure of FKBP12, the five residues at position 14, 32, 44, 49, and 85 that were used for probe attachment in the fluorescence experiments were mutated to Cys using DISCOVERY STUDIO VISUALIZER (Accelrys, San Diego, CA). AF488 was attached to, respectively, each of the five Cys, generating five starting conformations for simulated annealing using CNS. The models were initially minimized with CNS (36,37) for 100 steps. To obtain good sampling of all possible conformations, we implemented published methods involving a high-temperature multitrial simulated annealing protocol with a slow cooling schedule (40).
Each trial started with a cycle of six torsion-angle dynamics steps at 300,000 K, followed by cooling at a rate of 500 K per cycle. The integration time step was 0.75 fs. The atomic coordinates are saved at the end of each trial. The fluorescent probe, the Cys sulfur of attachment, and (depending on labeling site) the side chains of 2–6 neighboring residues of FKBP, were free to move while everything else in the model remained fixed. For each probe location, 72 simulated-annealing calculations, each for 150 trials, were run in parallel. Different initial velocities (obtained from a random-number generator given a different seed value) and initial starting conformations were used in each calculation to ensure different sampling of the conformational space. The additional starting conformations were taken from the resulting conformations of the first calculation. The simulations produced a total of 10,800 conformations at each location. Data analysis—including calculation of the average probe position, root-mean-square distance (RMSD), and order parameters—was carried out using custom software we developed for this particular application. The volume of the conformational space sampled was calculated using the VDW program from the VOSSVOLVOX software package (https://code.google.com/p/vossvolvox/) (41).
Trilateration
The method of using distances to determine a location in space is termed trilateration. Such methods have been previously described for macromolecular complexes, e.g., the Nano-Positioning System developed by Muschielok et al. (42) and Muschielok and Michaelis (43). Their method was named based on similarity with the Global Positioning System, where known satellite positions are used to determine an antenna location. From the simulated-annealing calculations above, we obtained coordinates of the average probe locations, which we are using as effective donor locations for trilateration. We used effective AF488 locations calculated for the five FKBP attachment sites (14, 32, 44, 49, and 85), and distances calculated from FRET values as constraints, to determine a locus in space corresponding to the acceptor attached to the N-terminus of DPc10.
We developed software that performs trilateration using an arbitrary number of donor sites and distances. The method used was to identify grid points on a three-dimensional grid that falls within a range defined by the minimum and maximum distances (Rmin and Rmax, respectively) calculated using Eq. 3 from the FRET measurement standard deviation (SD) for each donor position. The SD-based distance ranges can be widened symmetrically by a constant entered on the program command line. This feature may be used, alternatively, to set a range centered at the average distance (Ravg) calculated from the average E. The spacing between the grid points was 1 Å. Inasmuch as in some cases there may not be a location in space that satisfies all distance constraints, the software automatically determined an acceptor location in space based on any combination of n, n-1, and n-2 distances. The results were saved in PDB format, and were also exported to a volumetric map after conversion via the program PDB2VOL from the SITUS software package (http://situs.biomachina.org/) (34). The trilateration software, is available from Bengt Svensson at the University of Minnesota, Minneapolis, MN (http://ddt.umn.edu/).
Results
Determining the effective fluorophore locations
Our first goal was to determine, in silico, the effective locations of the FKBP-attached donor probes as accurately as possible. Usually, the effective probe location is set at the Cα or Cβ atom of the labeled cysteine, or displaced from the labeled cysteine by an arbitrary distance (44). That may be grossly inaccurate for the Alexa Fluors, due to relatively long linker chains between the fluorophore and reactive moieties, as long as 21 Å for a fully extended linker. We achieved more realistic predictions of the fluorophore location using a computationally efficient simulated-annealing method that utilizes short, high-temperature molecular dynamics (MD) simulations to sample the conformational space, developed based on methods previously described by Choi et al. (38) and Vrljic et al. (39).
To account for steric hindrance of the probe location due to proximity to FKBP or RyR, we used the RyR1 cryo-EM three-dimensional map to restrict the space that can be sampled by the FKBP-attached AF488 (Fig. 2 A). For this, we placed dummy atoms at the RyR1 map’s grid points in the region surrounding FKBP. This was done by first docking the atomic structure of FKBP12 (31) into the cryo-EM map corresponding to the RyR1 complex in closed conformation (6) (Fig. 2 A). Then the map density corresponding to FKBP was removed, inasmuch as that region would be represented by the FKBP atomic coordinates (Fig. 2 B). We then placed Ar atoms (inert atoms whose radii match the cryo-EM map grid-point spacing) at grid points that are proximal to FKBP and may directly come into contact with FKBP residues or with the attached AF488 probes (Fig. 2 C). The van der Waals radii of these atoms represent the RyR protein volume and thus replace the cryo-EM map to restrict the region sampled by the chromophore in our simulations (Fig. 2 D).
Figure 2.
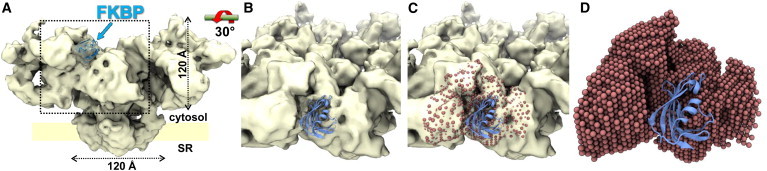
(A) Cryo-EM map of RyR1 in the closed state (6), with the atomic structure of FKBP docked into the previously published location (14). This map was rotated as indicated to generate the views in panels B–D. (B) RyR1 map with docked FKBP (blue ribbon). The density corresponding to FKBP was removed. (C) Grid points of the map without the FKBP density were converted to Ar dummy-atoms using the VOL2PDB program (SITUS software package). Dummy-atoms within 30 Å from FKBP are shown (red). (D) The atomic system used in the simulated annealing calculations consists of the FKBP coordinates and the dummy atoms, which provide steric constraints corresponding to RyR. To see this figure in color, go online.
Five variants of AF488-labeled FKBP were built by substituting Cys at residues 14, 32, 44, 49, or 85 of FKBP, and then attaching the AF488 probe to these sites by molecular modeling. Running simulated-annealing using the CNS software requires force-field parameters for the fluorophore. Initial values of these parameters for AF488 were determined using XPLO2D, then manually optimized before performing simulated-annealing calculations.
In previous reports, a few hundred trials/conformations of the simulated-annealing were deemed sufficient to obtain good conformational sampling (39). We initially ran five simulations, each for 200 trials, with different initial velocities, to generate 1000 conformations. We observed that after ∼200–300 conformations we already had achieved convergence to within ∼1 Å RMSD of the 1000 conformation average. It was considered that an RMSD of <1 Å would provide sufficient precision for our purpose, when compared with the SD typical for FRET.
To test whether sufficient sampling had occurred, additional simulations were conducted with different starting conformations of AF488. Surprisingly, considering that each of the five 200-trial runs with different initial velocities all converged to within <1 Å RMSD, we observed that simulations with different starting conformations did not converge to the same average location. The discrepancy of the average chromophore positions between different runs was in the worst cases >10 Å, depending on the labeling site and starting conformation. The maximum variability ranges seen in the results from different starting conformation simulations were similar. We concluded that different starting configurations sample similar volumes in the conformational space but with different density distributions. One might expect that a simulated-annealing run with the MD portion starting at high temperature (300,000 K) would quickly become degenerate and completely sample the conformational space. However, this is not what we observed.
To correct this problem, and find an average chromophore position representing a complete sampling of the available conformational space, we varied two parameters in the simulation protocol:
-
1.
We extended the number of trials per run and observed that even when using as many as 3400 trials in a single run, i.e., ∼10 times more sampling than previously recommended (39), the average position of the chromophore was similar to a 200-trial run with the same starting conformation.
-
2.
We ran simulations using combinations of the numbers of trials per run and the number of different seeds each generating 4800 conformations. These combinations were: two simulations with different seeds run for 2400 trials, 12 simulations with different seeds run for 400 trials, 16 simulations with different seeds run for 300 trials, 24 simulations with different seeds run for 200 trials, and 48 simulations with different seeds run for 100 trials. The RMSD between the average center positions of the chromophore was ≤ 0.2 Å for all these cases. Based on these results, we concluded that running simulations with the same starting conformation with different seeds still gives a possibly biased result.
To test whether our parameters for AF488 were the cause of this unexpected behavior, we ran simulations where the torsional-energy parameters for the linker had been decreased 10-fold to 0.2 kcal/mol. Decreased torsional barriers should allow the chromophore to sample the conformational space more efficiently and eliminate the apparent bias associated with the starting conformation. However, the resulting average chromophore position was still similar to simulations run with the same starting conformation and the higher torsional energy barriers (RMSD <0.2 Å). We concluded that the torsional-energy parameters did not cause the bias. Furthermore, we ran simulations with the Cy3 and Cy5 fluorescent probes, parameterized previously (38,39), to test whether their starting conformations affected the resulting average chromophore location. We arrived at the same result as with AF488, i.e., different starting conformations yielded different average chromophore locations. Taken together, these results indicate that the simulated-annealing procedure, as implemented in the CNS protocol (39), is not sufficient to uniquely determine an average chromophore position.
A possible way to correct this problem is to run multiple trials with different starting conformations. To determine how many starting conformations are needed before convergence on an average position is achieved, we ran four independent simulations with 12, 16, 24, and 48 starting conformations, each generating 4800 conformations. We found that the average position does converge toward one position in space when increasing the number of starting conformations. To determine how many calculations with different starting conformations are needed to determine the average location of the chromophore, we ran 103 calculations for each 150 trials. By randomly selecting a specific number of calculations from this pool and calculating the average position of that sample, we determined the average and maximum RMSD error versus the average location calculated from all 103 calculations (Fig. 3). As expected, increasing the number of calculations with a different starting conformation led to lowering the RMSD error. Considering the available computing resources and the queue time limitations (<24 h wall time), as well as minimizing the estimated error, we opted for 72 starting conformations each run for 150 trials, generating 10,800 conformations for the simulations on all labeling sites. The average and maximum errors for this combination are 0.3 and 1.4 Å, respectively (Fig. 3).
Figure 3.
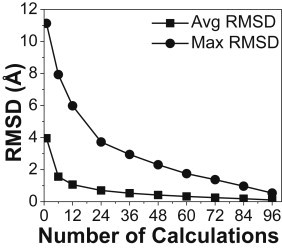
Estimated average (squares) and maximum (dots) error in the prediction of the average chromophore center position from the simulated-annealing calculations, as shown by the RMSD distance to the average position.
Effective positions of the FKBP-attached donors
Our simulated-annealing calculations show that the AF488 probe can adopt a wide range of conformations in the available space around the RyR-associated FKBP. For FRET-based distance analysis and trilateration of acceptors bound to RyR, we need to determine the effective positions of the AF488 probe attached to position 14, 32, 44, 49, and 85 of FKBP. This was done by calculating the average location of the chromophore from the simulated-annealing calculations (Fig. 4). Results show effective probe positions that are located 10–16 Å away from the Cα atom of the labeled Cys (Table 1). The conformational space sampled by the AF488 chromophore and average positions corresponding to each attachment site are shown in Fig. 4.
Figure 4.
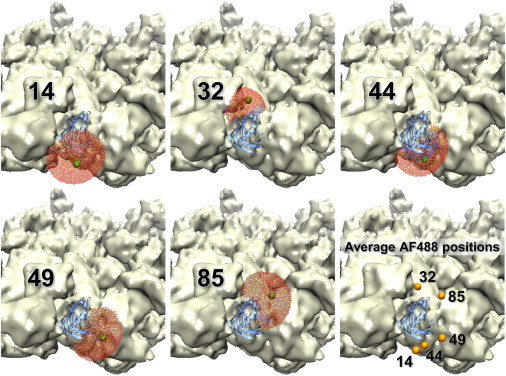
Conformational distribution of AF488 attached at specific FKBP sequence positions, as indicated. The conformational space sampled by the fluorophore (red) was used to calculate the coordinates of its average position (green sphere). Average AF488 positions corresponding to all FKBP labeling sites are shown (orange, bottom right). To see this figure in color, go online.
Table 1.
Parameters from simulated-annealing calculations of AF488 attached at Cys residues substituted within the FKBP sequence
| FKBP labeling site | Distance to Cα (Å) | Sampled volume (Å3) | Sa | θcb |
|---|---|---|---|---|
| 14 | 11.2 | 35,419 | 0.017 | 88.1 |
| 32 | 15.9 | 13,487 | 0.033 | 86.4 |
| 44 | 12.8 | 27,443 | −0.014 | 91.6 |
| 49 | 12.3 | 35,695 | 0.024 | 87.4 |
| 85 | 10.4 | 36,959 | −0.010 | 90.1 |
Defined in Eq. 6.
Defined in Eqs. 7 and 8.
The volume sampled by AF488 depends on the labeling site and is spatially restricted by both FKBP and RyR (Table 1). The volume sampled by AF488 is >35,000 Å3 at sites 14, 49 and 85, where little restriction from the RyR is observed. AF488 at site 44 shows a slight (∼20%) reduction in the sampled volume due to spatial restriction by the RyR map. AF488 at site 32 shows a large reduction in its sampled volume (by >60%) because of steric restriction due to the labeling site being located near an apparent interface between FKBP and RyR. Even though the label attached at position 32 appears to be more spatially confined, we have shown that the binding of AF488-FKBP to RyR is similar to unlabeled FKBP for all tested labeling sites (15).
Order parameters for AF488 attached to FKBP
The large number of possible conformations and large volume sampled by the AF488 probes suggest that the motion of the probe is unrestricted, i.e., isotropic. This was quantitatively analyzed based on the order parameters (S) derived from the simulated-annealing data (Table 1). S can be used to describe how restricted the motion of the chromophore is, and can be experimentally determined from fluorescence anisotropy measurements. However, such measurements are not possible in our system. Therefore, we estimated S from the simulated annealing calculations, by determining the orientation of the dipole vectors and calculating θeA, the angle between the dipole vector of the chromophore in each conformation and the mode vector. The mode vector has the lowest sum of the great-circle distances to all other vectors:
| (6) |
In the wobble-in-a-cone model, S depends on the cone angle, θc, according to
| (7) |
so θc can be expressed as
| (8) |
S is nearly zero for donors attached at all the FKBP sites used in this study (Table 1), thus indicating that all orientations are sampled, i.e., fluorophore dynamics is isotropic. Although we cannot compare these results with experimental fluorescence anisotropy data (which cannot be measured in myocytes or isolated sarcoplasmic reticulum samples, due to light scattering), the near-zero order parameter and wide cone-angles justifies the use of the orientation factor κ2 = 2/3 for determining R0 according to Eq. 4. The value κ2 = 2/3 is based on the assumption that the probes undergo isotropic motion on a timescale that is short compared with the excited-state lifetime (several nanoseconds). Considering that the acceptor fluorophores are attached to their respective labeling sites via linkers similar in length to the donors, they likely also have a low order parameter. Therefore, our distance calculations from E assume random orientation of fluorophores. Even if the acceptor is completely immobilized, which is unlikely in practice, the effect on κ2 will result in a ≤10% error in the distance estimation (45).
Method validation using acceptor-labeled RyR sites within the docked N-terminal ABC-domain
To test the computationally optimized donor coordinates (Fig. 4), we used our previously published FRET-derived distances between D-FKBPs and the FRET acceptor Cy3NTA (46) bound to His10 tags within the N-terminal domain of RyR1 (27). We trilaterated the positions of Cy3NTA targeted to His10 tags inserted at residues 76 (His10-76, domain A), 181 (His10-181, domain A), 500 (His10-500, domain C), and 519 (His10-519, domain C) (Fig. 5). All four loci in the ABC-domain were identified using the procedure described in Materials and Methods, which relies on FRET measurements from all five D-FKBP constructs to each donor. For each FRET measurement, we used Eq. 3, the average E, and corresponding SD to calculate Ravg, Rmin, and Rmax. These values are from Table 1 of Girgenrath et al. (27) for His10-76, -181, and -519, and Table 2 of this article for His10-500.
Figure 5.
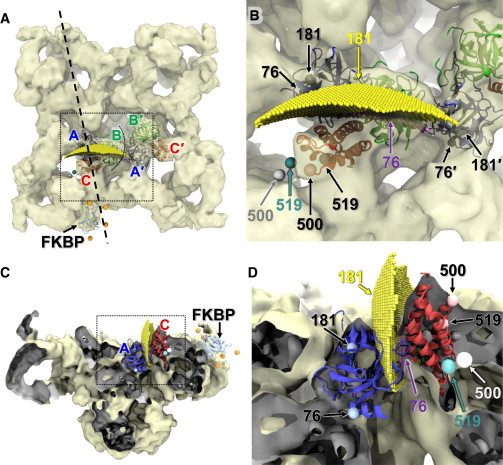
Trilaterated loci of Cy3NTA acceptors attached to His10 tags in the RyR1 N-terminal domain. (A) RyR1 is viewed from cytosolic side. Trilaterated acceptor positions are shown for His10 inserted at positions 76 (purple) and 181 (yellow) in the A-domain (blue), and at positions 500 (gray sphere) and 519 (cyan sphere) in the C-domain (red), relative to the RyR1 cryo-EM map (EMDB:1606), the docked FKBP, and the docked N-terminal ABC-domain, as indicated. Two adjacent ABC domains are represented, as indicated by A, B, C, and A′, B′, C′ in their respective colors. A vertical sectioning plane (dashed line) was used to create the cut-out views in panels C and D. (B) Magnified view of the region indicated in panel A (dashed box). Trilaterated acceptor locations are indicated in their respective color, whereas the His10 insertion site locations are represented (spheres) within the ABC-domain crystal structure (black arrows and labels). Because residue 500 is within a flexible loop that is not visible in the crystal structure, we are indicating residue 499 instead. (C) Cut-out view perpendicular to the plane indicated in panel A. (Dark gray shading) Inside surfaces of the EM density revealed in cut-off. (D) Magnified view of the region (dashed box) in panel C. To see this figure in color, go online.
Table 2.
FRET E values and calculated distances between D-FKBP and His10 inserted at RyR1 residue 500
| FKBP labeling site | Predicted Ea, site 500 | Predicted Ea, site 500′ | Observed Eb | Ravg (Å)c | Rmin (Å)c | Rmax (Å)c |
|---|---|---|---|---|---|---|
| 14 | 0.10 | 0.01 | 0.20 ± 0.05(47) | 75 | 71 | 79 |
| 32 | 0.94 | 0.02 | 0.69 ± 0.05(9) | 52 | 49 | 54 |
| 44 | 0.25 | 0.01 | 0.28 ± 0.06(41) | 70 | 66 | 73 |
| 49 | 0.19 | 0.02 | 0.24 ± 0.06(34) | 72 | 68 | 76 |
| 85 | 0.71 | 0.04 | 0.43 ± 0.06(53) | 62 | 59 | 64 |
Calculated using Eq. 3, based on distances measured from the effective positions of the FKBP-attached donors to the His10 acceptor-insertion positions 500 and 500′ in domains C and C′, respectively, of two adjacent RyR protomers (Fig. 5A).
Observed FRET efficiencies (means ± SD, n indicated in parentheses) were calculated from the increase in donor fluorescence after selective acceptor photobleaching, using Eq. 2.
Distances (R) were calculated using Eq. 3. The average distance (Ravg) was calculated based on the average E. The minimum (Rmin) and maximum (Rmax) distances were calculated based on the average E, to which we added or subtracted the maximum SD of the measurement of E, respectively.
The distance ranges for His10-76 and -181 (Rmax–Rmin) are relatively broad (averaging 15 and 18 Å, respectively), and their resulting loci are composed of many grid units (represented as small spheres in Fig. 5, and see Fig. S1 in the Supporting Material). Comparison of Fig. 5 in this article and Fig. 5 from Girgenrath et al. (27) shows that loci for Cy3NTA bound to His10-76 and His10-181 constructs are in similar locations. Fig. S1 depicts only the His10-76 locus, without the overlapping larger volume of the His10-181 locus that obscures it in Fig. 5. Both these loci are slightly closer to FKBP than the corresponding His10 insertion sites appear in the docked crystal structure of the ABC-domain, and are located between their corresponding insertion points in the A- and A′-domains (Fig. 5). This result may be due to comparable FRET contributions from acceptor bound within both the A- and A′-domain in the neighboring RyR protomers (27), as indicated by arrows in Fig. 5 B. However, using the revised set of donor coordinates, we obtained smaller loci (1300 vs. 5200 Å3 for the His10-76 site, and 16,900 vs. 25,000 Å3 for the His10-181 site), thus confirming our prediction of improved precision.
The locus for Cy3NTA attached at His10-519 was also determined based on previously reported FRET results (27). However, the FRET measurement SD translated into 50% narrower distance ranges than for His10-76 and -181, and yielded a locus only upon a slight broadening of these ranges (by 1.7 Å). This locus consists of a single grid unit (indicated by a single sphere in Fig. 5, B and D) that is found in a void near domain C, 23 Å from the insertion site at residue 519. This location is consistent with the predicted cytosolic exposure of the insertion site. Upon further broadening of the distance ranges, this locus would gradually expand in this void, away from the His10 insertion site. As predicted, for the locus of Cy3NTA attached to site 519 there is no evidence of FRET contribution from multiple acceptors, by comparison with the trilaterations of sites 76 and 181 above. This precision and apparent accuracy seemed quite remarkable.
To further test the consistency of this method, we trilaterated Cy3NTA attached at His10-500, also in domain C. For this site also, we observed good consistency between the profiles of predicted and measured FRET from D-FKBP (Table 2). FRET measurement SDs for the 500 locus were similar to those observed for 519, and also needed to be slightly broadened (by 1.3 Å) to yield a locus, which consisted of a single grid unit. This was 27 Å from the His10 insertion site, in the same void of the RyR1 map as the locus corresponding to the 519 insertion site (Fig. 5, B and D). The offsets between the 500 and 519 acceptor loci and their predicted attachment site locations are consistent with the length of the linker between the Cy3 fluorophore and the NTA moiety.
Therefore, we conclude that the revised donor coordinates provide significantly improved precision and accuracy versus donor positions set at the Cβ coordinates.
Trilateration of acceptor-labeled DPc10 bound to RyR2, based on confocal FRET measurements in permeabilized cardiomyocytes
Using confocal FRET measurements in permeabilized cardiomyocytes, we have previously determined that the acceptor probe covalently linked to the N-terminus of DPc10 may localize along a broad arc intersecting RyR domain 3 between FKBP and CaM (26). In that study, FRET was measured only from D14-FKBP to A-DPc10, and the result was further constrained using FRET between CaM and DPc10 (26). Here, we aimed to resolve a discrete location of the A-DPc10 probe using FRET measurements from each of our five donor-labeled FKBP variants to the same acceptor-labeled DPc10 bound to RyR2, in permeabilized rat cardiomyocytes. FRET efficiencies, E, were measured using two methods—acceptor photobleach and donor quench—with highly consistent results, summarized in Table 3. All FRET measurements were carried out under conditions that saturate the DPc10 binding sites ([F-DPc10] = 5 μM), as shown before in Oda et al. (26). Measured FRET efficiencies ranged between 0.4 and 0.7, which is well within the quasi-linear range of the R-versus-E relationship (Eq. 1).
Table 3.
Confocal FRET measurements from FKBP-attached AF488 donors to a DPc10-attached HF647 acceptor in permeabilized cardiomyocytes
| FKBP labeling site | E(photobleaching)a | E(D quench)b | Ravg (Å)c | Rmin (Å)c | Rmax (Å)c |
|---|---|---|---|---|---|
| 14 | 0.51 ± 0.01 | 0.52 ± 0.03 | 54 | 52 | 55 |
| 32 | 0.41 ± 0.01 | 0.47 ± 0.02 | 57 | 54 | 57 |
| 44 | 0.51 ± 0.01 | 0.52 ± 0.02 | 54 | 53 | 54 |
| 49 | 0.63 ± 0.01 | 0.66 ± 0.01 | 49 | 48 | 50 |
| 85 | 0.57 ± 0.02 | 0.55 ± 0.01 | 52 | 51 | 53 |
E was calculated using Eq. 2.
E was calculated using Eq. 1.
Distances (R) were calculated using Eq. 3, as described in Materials and Methods and Table 2.
To determine the DPc10 binding site location, we used a cryo-EM map of RyR1, which has been solved to a much higher resolution than RyR2 (10.2 vs. ∼30 Å). This approach is based on the assumption that the RyR1 and RyR2 channel isoforms have similar overall structures and features (47,48), as has been revealed by cryo-EM image reconstructions of comparable resolution (∼30 Å) and by crystal structures of isolated soluble RyR fragments (10,49). Using FRET, we have previously shown that FKBP12.0 and 12.6 bind at equivalent sites (16), and in the same orientation within the RyR1 and RyR2 complexes (15).
A-DPc10 loci were calculated based on all five FRET-derived distances between the D-FKBP variants and A-DPc10, and for all five sets of four FRET-derived distances. Solutions were constrained by the RyR1 cryo-EM map, by the CaM N- and C-domain locations previously determined within RyR1 (Fig. 6, A and B) from FRET measurements and the cryo-EM map of the CaM-RyR1 complex (13,15,29), and by the FRET-derived distances between D34-CaM or D110-CaM and A-DPc10 (63 ± 1 and 53 ± 1 Å, respectively) (26).
Figure 6.
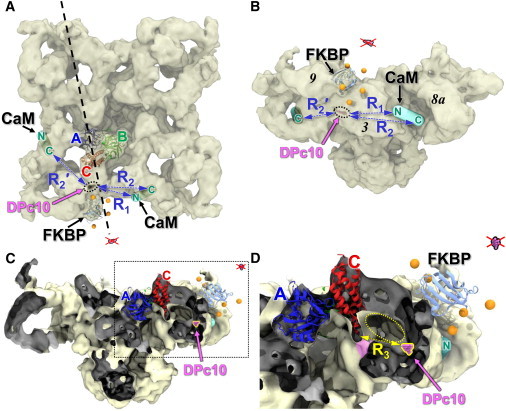
Location of HF647 attached to DPc10 bound to RyR2. (A) RyR1 viewed from the cytosolic side. The trilaterated position of the DPc10-attached acceptor is shown (magenta within black-dotted oval) relative to the cryo-EM structure of the closed RyR1 (EMDB:1606), the docked FKBP, the docked RyR ABC-domain, and the predicted locations of the CaM N- and C-lobes, as indicated (13). Distances between A-DPc10 and the N- and C-lobe of CaM on the same face of RyR are indicated by R1 and R2. R2′ indicates the distance between DPc10 and the C-lobe of CaM bound on adjacent faces of RyR. A vertical sectioning plane (dashed line) was used to create the cut-out views in C and D. (B) RyR1 is shown in side view. The trilaterated position of the DPc10-attached acceptor (dotted oval) was found in RyR domain 3 (the handle). A second solution generated by the same distances is on the opposite side of FKBP, well outside the cryo-EM map (red x). (C) Cut-out view perpendicular to the plane indicated in panel A. (Dark gray shading) Inside surfaces of the EM map revealed in cut-off. (D) Magnified view of the region (dashed box) in panel C. The distance of nearest approach between the C-domain (at residue 455) and the A-DPc10 locus (magenta) is R3 = 35 Å. A gallery that communicates with the cytosol (dotted yellow oval) allows DPc10 access to space around domain C, where it may perturb an interface (magenta shading) formed between the lower tip of the C-domain and a noncontiguous RyR domain. To see this figure in color, go online.
The five-distance trilateration of DPc10 yielded a locus only after widening the Rmax-Rmin range by 14 Å, but this locus is ∼40 Å away from the surface of the RyR cryo-EM map, and therefore this is not a possible solution (see Fig. S2). Multiple loci were found based on different combinations of four distances. The loci found by excluding the measurements from donor attached at FKBP sites 14, 49, or 85 were implausible solutions because they were >25 Å away from the cryo-EM map surface and also incompatible with the CaM-lobe placements (see Fig. S2). The locus found by excluding the measurement from donor at FKBP position 32 was found within domain 9 of the RyR1 cryo-EM map (see Fig. S2). However, that location was incompatible with the FRET measurements from CaM-attached donors to A-DPc10 (26). Only the set of distances from donors at FKBP residues 14, 32, 49, and 85 (44 was excluded) yielded a solution that was acceptable when constrained by the cryo-EM map (Fig. 6).
This A-DPc10 locus was found within RyR domain 3 (Fig. 6, magenta shading), the large handle density that connects the FKBP and CaM binding sites. The distance between this locus and D34-CaM is R1 = 60 Å (Fig. 6, A and B), which matches well with the 63 ± 1 Å calculated from FRET (26). Distances between the A-DPc10 locus and the D110-CaM on the same and adjacent faces of the RyR1 map are R2 = 81 Å and R2′ = 76 Å, respectively, which are significantly different from the apparent distance derived from FRET (53 ± 1 Å). This inconsistency is likely due to a higher uncertainty in the C-lobe placement (versus N-lobe). The relationship among this buried A-DPc10 locus, FRET distance analysis (including from donor bound to CaM’s C-domain), and the RyR2 location of CaM proposed by Huang et al. (50), is further analyzed in the Discussion.
Discussion
Trilateration method development
We have developed a trilateration method that uses spectroscopic distance measurements from an ensemble of probes placed at known locations within a three-dimensional protein structure to a probe bound at an unknown location, aiming to determine its three-dimensional location. We previously used the Cβ coordinates of the labeled Cys residue to place the known-location probes. That method does not take into account the linker between the labeling site and the fluorophore itself. To more accurately estimate the effective probe location, and improve the precision of trilateration, we developed a simulated-annealing protocol designed to determine the space occupied by all probe conformations, and determine the average probe position. We started from an existing computational-annealing protocol (39) and developed additional steps to minimize the potential dependence in the final average position on the starting conformation. We determined that this major source of error in probe placement can be almost completely eliminated by averaging the results from as many simulated annealing calculations as practically afforded by the computational resources. Based on queue time limitations, we chose to average the results from 72 annealing calculations using different starting conformations, which results in acceptable predicted RMSD error (0.3 Å average, 1.4 Å maximum error; Fig. 3).
Calculated donor positions improve trilateration precision
We used this simulated-annealing protocol to determine the average locations of AF488 probes covalently bound to several Cys residues substituted at surface sites of FKBP (Fig. 4), chosen to be away from the RyR-FKBP binding interface, such that their chemical modification would not interfere with binding. Analysis of the simulated-annealing data indicates that there is little steric restriction of the probes by either the RyR or FKBP, so fluorophore MD can be assumed to be almost completely isotropic (Table 1). This justifies using κ2 = 2/3 for distance calculations based on FRET data, thereby facilitating conversion to donor-acceptor distances from the measured FRET efficiencies. We found that the calculated effective location of the AF488 fluorophore moiety can be relatively far (10–16 Å) from the Cβ, which can potentially affect the trilateration result. These calculated fluorophore positions present the advantage that they are more broadly separated in space (than when set at the Cβ atoms), so we predicted that this spatial expansion of the reference-probe set would significantly improve the precision of trilateration. This is what we actually observed for the acceptors attached at known locations in the ABC domain—in all cases, the believed-new trilateration method yielded smaller loci than our previous method. The smallest and most plausibly positioned loci were obtained when the acceptor attachment sites were in the RyR1 C-domain, which is the farthest from the channel’s axis of symmetry, and thus least affected by FRET cross-talk between adjacent RyR protomers (Fig. 5). Nevertheless, the general trilateration locations for the ABC domain acceptor attachment sites were similar between the previously used Cβ (27) and the computed effective coordinates of the donor probes used here.
DPc10 locus relative to RyR structural features revealed by the cryo-EM map
Having validated our improved trilateration method, we then used it to determine the location of an acceptor probe attached to DPc10 bound to RyR2 based on FRET measurements carried out in permeabilized myocytes (Table 3). It has been postulated that DPc10 directly competes with intersubunit contacts (51) between the RyR2 N- and central domains (zipping) by binding to the N-domain sequence (to unzip) (18). Taken literally, this model implies that DPc10 should bind in close proximity to the N-domain zipping surface. Because DPc10 is related to the RyR1-derived peptide DP4 (18), both probes may bind at equivalent three-dimensional locations within RyR1 and RyR2. Our FRET-based trilateration results indicate that A-DPc10 is deep within RyR domain 3 (which forms the handle domains in the cryo-EM map). This buried location of the A-DPc10 probe is consistent with the previous finding that DPc10 access to its RyR2 binding site is sterically restricted and under negative allosteric control by CaM (26). To place the acceptor at that location, DPc10 may bind to domain 3 on its distal face relative to the fourfold axis of symmetry of RyR. In this placement, direct contact between DPc10 and the centrally located ABC domain can be ruled out. This model is plausible but incompatible with the prevalent version of the domain zipping model (52). Alternatively, DPc10 may bind to the proximal face of domain 3 by diffusing through a wide gallery formed between peripheral RyR domains (i.e., 3, 8, 9, and 10) and more central domains (i.e., 2 and 4).
Indeed, the DPc10 locus is nearest (∼35 Å) to residue Pro455 in the RyR1 C domain (R3 in Fig. 6 D). Assuming that RyR2-bound DPc10 adopts the predicted compact structure (Fig. 1, ∼25 Å diameter) and that acceptor might be situated ∼12 Å from its N-terminal attachment site, it is possible that DPc10 can lodge in the cytosol-accessible gallery separating domains C and 3 (Fig. 6 D, yellow oval). Here, it may perturb an apparent interface (Fig. 6 D, magenta shading) between the RyR C domain and the densities (columns) that connect the large RyR cytoplasmic domain to the TM domain, which contains the channel pore. Thus, the DPc10 binding site may be at the convergence of structural pathways transducing regulatory gating signals from the RyR cytosolic headpiece to the channel pore in the TM domain. Mutations or posttranslational modifications (oxidation, phosphorylation) associated with pathologies may structurally alter one or more of these pathways, thus leading to a more accessible DPc10 binding site (domain unzipping), as observed for arrhythmia-linked leaky RyR2 mutants (20), or WT-RyR2 from animal models of heart failure (22,52).
Relationship between the A-DPc10 locus and the N- and C-lobes of CaM
Two FRET measurements from CaM-attached donors to A-DPc10 are available (26)—from AF488 attached at CaM position 34 (N-lobe), and from AF568 attached at CaM position 110 (C-lobe). Distances derived from these measurements are 63 ± 1 Å and 53 ± 1 Å, respectively. The distance R1 = 61 Å (Fig. 6, A and B) between the A-DPc10 locus and the previously determined location of CaM’s N-lobe (13) is consistent with the FRET measurement. However, the distance R2 = 81 Å (Fig. 6, A and B) between the A-DPc10 locus and the C-lobe location (13) on the same face of RyR, is inconsistent with the corresponding FRET measurement. This discrepancy may be due to DPc10 altering the structure of RyR2-bound CaM (and there may be a reciprocal effect).
Alternatively, this apparent discrepancy may be due to CaM binding to RyR2 in an extended conformation, with the C-lobe projecting deep into the groove formed between domains 3 and 8 (Fig. 6, A and B). This would place the donor attached at CaM residue 110 nearer the A-DPc10 locus in the adjacent RyR protomer (R2′ in Fig. 6, A and B). This is consistent with the mapping of Ca2+-CaM (inhibitory in both RyR1 and RyR2) within RyR1 (53). This placement of the C-lobe-attached donor does not contradict the results reported by Huang et al. (50), who noted in their detailed cryo-EM analysis of CaM binding to RyR2 versus RyR1 that the mass corresponding to Ca2+-CaM bound to RyR2 was weaker than expected, suggesting that a significant portion of CaM was not visible in their cryo-EM map, possibly due to structural disorder. More detailed determinations of the RyR-bound CaM structure will be the focus of future studies.
Perspective
The multiscale mapping process demonstrated in this article enables resolving sequence locations and binding sites throughout most of the RyR structure, and thus should further our understanding of the structure-function correlations of this channel. Determining the binding locations of A-DPc10 and other peptide-based biosensors of the RyR functional state may provide a rational basis to design probes necessary for diagnostic and therapeutic discovery targeting the RyR pathological leak, which represents an important unmet health need (54).
Acknowledgments
Computational resources were provided by the University of Minnesota Supercomputing Institute. Fluorescence spectroscopy studies were in part carried out at the University of Minnesota Biophysical Spectroscopy Center. We are thankful to Octavian Cornea for outstanding technical assistance.
This work was supported by National Institutes of Health grant No. R01HL092097 (to D.M.B. and R.L.C.), grant No. R01AR059124 (to J.D.F.), and grant Nos. R01GM27906 and R37AG26160 (to D.D.T.).
Supporting Material
References
- 1.Fill M., Copello J.A. Ryanodine receptor calcium release channels. Physiol. Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 2.Lanner J.T., Georgiou D.K., Hamilton S.L. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bers D.M. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014;76:107–127. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 4.Bers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton S.L., Serysheva I.I. Ryanodine receptor structure: progress and challenges. J. Biol. Chem. 2009;284:4047–4051. doi: 10.1074/jbc.R800054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samsó M., Feng W., Allen P.D. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramachandran S., Chakraborty A., Meissner G. Structural determinants of skeletal muscle ryanodine receptor gating. J. Biol. Chem. 2013;288:6154–6165. doi: 10.1074/jbc.M112.433789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramachandran S., Serohijos A.W., Dokholyan N.V. A structural model of the pore-forming region of the skeletal muscle ryanodine receptor (RyR1) PLoS Comput. Biol. 2009;5:e1000367. doi: 10.1371/journal.pcbi.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tung C.C., Lobo P.A., van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 10.Yuchi Z., Lau K., van Petegem F. Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure. 2012;20:1201–1211. doi: 10.1016/j.str.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 11.Zhu L., Zhong X., Liu Z. Modeling a ryanodine receptor N-terminal domain connecting the central vestibule and the corner clamp region. J. Biol. Chem. 2013;288:903–914. doi: 10.1074/jbc.M112.429670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Petegem F. Ryanodine receptors: structure and function. J. Biol. Chem. 2012;287:31624–31632. doi: 10.1074/jbc.R112.349068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornea R.L., Nitu F., Fruen B.R. FRET-based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc. Natl. Acad. Sci. USA. 2009;106:6128–6133. doi: 10.1073/pnas.0813010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samsó M., Shen X., Allen P.D. Structural characterization of the RyR1-FKBP12 interaction. J. Mol. Biol. 2006;356:917–927. doi: 10.1016/j.jmb.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Cornea R.L., Nitu F.R., Fruen B.R. Mapping the ryanodine receptor FK506-binding protein subunit using fluorescence resonance energy transfer. J. Biol. Chem. 2010;285:19219–19226. doi: 10.1074/jbc.M109.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo T., Cornea R.L., Bers D.M. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto T., Ikemoto N. Spectroscopic monitoring of local conformational changes during the intramolecular domain-domain interaction of the ryanodine receptor. Biochemistry. 2002;41:1492–1501. doi: 10.1021/bi015581z. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto T., Ikemoto N. Peptide probe study of the critical regulatory domain of the cardiac ryanodine receptor. Biochem. Biophys. Res. Commun. 2002;291:1102–1108. doi: 10.1006/bbrc.2002.6569. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi S., Bannister M.L., Ikemoto N. Dantrolene stabilizes domain interactions within the ryanodine receptor. J. Biol. Chem. 2005;280:6580–6587. doi: 10.1074/jbc.M408375200. [DOI] [PubMed] [Google Scholar]
- 20.Uchinoumi H., Yano M., Matsuzaki M. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tateishi H., Yano M., Matsuzaki M. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc. Res. 2009;81:536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mochizuki M., Yano M., Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 23.Ono M., Yano M., Matsuzaki M. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi S., Yano M., Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J. Am. Coll. Cardiol. 2009;53:1993–2005. doi: 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto T., Yano M., Matsuzaki M. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–772. doi: 10.1161/CIRCULATIONAHA.107.718957. [DOI] [PubMed] [Google Scholar]
- 26.Oda T., Yang Y., Bers D.M. In cardiomyocytes, binding of unzipping peptide activates ryanodine receptor 2 and reciprocally inhibits calmodulin binding. Circ. Res. 2013;112:487–497. doi: 10.1161/CIRCRESAHA.111.300290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girgenrath T., Mahalingam M., Fessenden J.D. N-terminal and central segments of the type 1 ryanodine receptor mediate its interaction with FK506-binding proteins. J. Biol. Chem. 2013;288:16073–16084. doi: 10.1074/jbc.M113.463299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Söding J., Biegert A., Lupas A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo T., Fruen B.R., Bers D.M. FRET detection of calmodulin binding to the cardiac RyR2 calcium release channel. Biophys. J. 2011;101:2170–2177. doi: 10.1016/j.bpj.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haugland R.P., Spence M.T.Z., Basey A. Molecular Probes; Eugene, OR: 2005. The Handbook: A Guide to Fluorescent Probes and Labeling Technologies. [Google Scholar]
- 31.Burkhard P., Taylor P., Walkinshaw M.D. X-ray structures of small ligand-FKBP complexes provide an estimate for hydrophobic interaction energies. J. Mol. Biol. 2000;295:953–962. doi: 10.1006/jmbi.1999.3411. [DOI] [PubMed] [Google Scholar]
- 32.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF CHIMERA—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Pintilie G.D., Zhang J., Gossard D.C. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wriggers W. Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2010;2:21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleywegt G.J., Jones T.A. Databases in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1998;54:1119–1131. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- 36.Brunger A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 37.Brünger A.T., Adams P.D., Warren G.L. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Choi U.B., Strop P., Weninger K.R. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vrljic M., Strop P., Brunger A.T. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat. Struct. Mol. Biol. 2010;17:325–331. doi: 10.1038/nsmb.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brünger A.T., Krukowski A., Erickson J.W. Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr. A. 1990;46:585–593. doi: 10.1107/s0108767390002355. [DOI] [PubMed] [Google Scholar]
- 41.Voss N.R., Gerstein M., Moore P.B. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 2006;360:893–906. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 42.Muschielok A., Andrecka J., Michaelis J. A nano-positioning system for macromolecular structural analysis. Nat. Methods. 2008;5:965–971. doi: 10.1038/nmeth.1259. [DOI] [PubMed] [Google Scholar]
- 43.Muschielok A., Michaelis J. Application of the nano-positioning system to the analysis of fluorescence resonance energy transfer networks. J. Phys. Chem. B. 2011;115:11927–11937. doi: 10.1021/jp2060377. [DOI] [PubMed] [Google Scholar]
- 44.Rasnik I., Myong S., Ha T. DNA-binding orientation and domain conformation of the E. coli rep helicase monomer bound to a partial duplex junction: single-molecule studies of fluorescently labeled enzymes. J. Mol. Biol. 2004;336:395–408. doi: 10.1016/j.jmb.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 45.Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- 46.Fessenden J.D. Förster resonance energy transfer measurements of ryanodine receptor type 1 structure using a novel site-specific labeling method. PLoS ONE. 2009;4:e7338. doi: 10.1371/journal.pone.0007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radermacher M., Rao V., Wagenknecht T. Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J. Cell Biol. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma M.R., Penczek P., Wagenknecht T. Cryoelectron microscopy and image analysis of the cardiac ryanodine receptor. J. Biol. Chem. 1998;273:18429–18434. doi: 10.1074/jbc.273.29.18429. [DOI] [PubMed] [Google Scholar]
- 49.Lobo P.A., van Petegem F. Crystal structures of the N-terminal domains of cardiac and skeletal muscle ryanodine receptors: insights into disease mutations. Structure. 2009;17:1505–1514. doi: 10.1016/j.str.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 50.Huang X., Fruen B., Liu Z. Calmodulin-binding locations on the skeletal and cardiac ryanodine receptors. J. Biol. Chem. 2012;287:30328–30335. doi: 10.1074/jbc.M112.383109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Z., Wang R., Wagenknecht T. Dynamic, inter-subunit interactions between the N-terminal and central mutation regions of cardiac ryanodine receptor. J. Cell Sci. 2010;123:1775–1784. doi: 10.1242/jcs.064071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oda T., Yano M., Matsuzaki M. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 53.Samsó M., Wagenknecht T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- 54.Kushnir A., Marks A.R. Ryanodine receptor patents. Recent Pat. Biotechnol. 2012;6:157–166. doi: 10.2174/1872208311206030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.