

Abstract
The SNARE complex plays a vital role in vesicle fusion arising during neuronal exocytosis. Key components in the regulation of SNARE complex formation, and ultimately fusion, are the transmembrane and linker regions of the vesicle-associated protein, synaptobrevin. However, the membrane-embedded structure of synaptobrevin in its prefusion state, which determines its interaction with other SNARE proteins during fusion, is largely unknown. This study reports all-atom molecular-dynamics simulations of the prefusion configuration of synaptobrevin in a lipid bilayer, aimed at characterizing the insertion depth and the orientation of the protein in the membrane, as well as the nature of the amino acids involved in determining these properties. By characterizing the structural properties of both wild-type and mutant synaptobrevin, the effects of C-terminal additions on tilt and insertion depth of membrane-embedded synaptobrevin are determined. The simulations suggest a robust, highly tilted state for membrane-embedded synaptobrevin with a helical connection between the transmembrane and linker regions, leading to an apparently new characterization of structural elements in prefusion synaptobrevin and providing a framework for interpreting past mutation experiments.
Introduction
Communication across neural synapses is accomplished by exocytosis, whereby Ca2+ influx into the presynaptic neuron, normally induced by an action potential, causes rapid synaptic vesicle fusion and the release of neurotransmitter into the synaptic cleft (1–4). Key to the cellular machinery responsible for vesicle fusion is the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Formation of this complex is responsible for bringing synaptic vesicles into close proximity with the cellular membrane and providing at least part of the energy necessary for fusion of the two membranes (1,2,5,6). The vital role played by the SNARE proteins in neuronal exocytosis is clearly evidenced by the effects of clostridial neurotoxin light-chain proteases, which disrupt proper SNARE function and result in the extremely lethal pathologies of botulism and tetanus (3,7). Thus, determining the key structural elements of SNARE proteins that drive membrane fusion is key to the continued development of an adequate model for neuronal communication in both healthy and diseased states.
The neuronal SNARE complex is composed of synaptobrevin, residing on the vesicle membrane, along with syntaxin and SNAP25, embedded in the plasma membrane. Syntaxin and SNAP25 apparently form a transient structure before full SNARE complex formation is accomplished by the addition of synaptobrevin (7). The full complex is a heterotrimer, composed of synaptobrevin, syntaxin, and SNAP25 (1,7) (Fig. 1 a). Formation of the complex is thought to proceed from the N-terminal (cytosolic) region of the proteins toward the C-terminal (membrane proximal) region (3,4). To promote a single membrane fusion event, several SNARE complexes are likely required to form (8).
Figure 1.
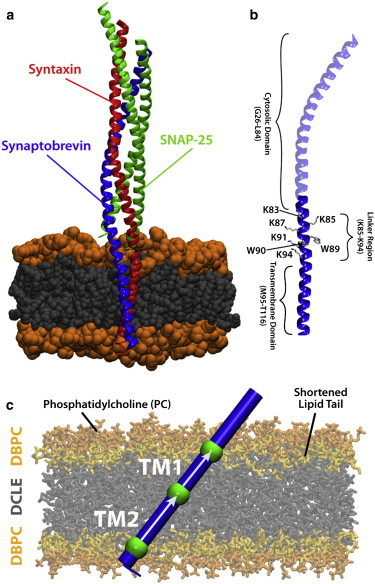
Synaptobrevin and the SNARE complex. (a) After membrane fusion occurs, synaptobrevin, which is located initially in the vesicle membrane, forms a helical bundle with syntaxin and SNAP25 in the cell membrane. (b) Synaptobrevin is composed of a cytosolic domain (residues 1–84), a so-called linker region (residues 85–94), and a transmembrane domain (residues 95–116). (c) Simulation system used to investigate the tilt of different regions of truncated synaptobrevin (residues 78–116) with respect to the membrane normal. Regions TM1 and TM2, respectively representing the N- and C-terminal halves of the synaptobrevin TM and linker regions, and the vectors used to characterize the tilt angles, are shown. The highly mobile membrane mimetic model (HMMM) (9) was employed to enable increased sampling of protein configurations. To see this figure in color, go online.
After fusion, the soluble portion of the SNARE complex adopts a highly twisted, parallel four-helix bundle conformation extending ∼120 Å (4,6,7). The interior of the helical bundle consists mostly of hydrophobic residues along with a single polar layer, which is highly conserved across SNARE proteins (7,10). In the postfusion state, the transmembrane (TM) regions of syntaxin and synaptobrevin also interact, with helical continuity being maintained from the cytosolic domain into both the linker and TM regions of syntaxin and synaptobrevin, which may help to drive fusion (7,11). The full complex with TM domains has been shown to adopt a nearly parallel orientation with respect to the membrane normal (11) (Fig. 1 a).
Although the postfusion SNARE complex, including the TM domains of syntaxin and synaptobrevin, has been well characterized (11), the configuration of synaptobrevin before complex formation (prefusion state) has been the source of much debate. Prefusion synaptobrevin consists of 116 residues, which have historically been grouped into three regions: the cytosolic (residues 1–84), the linker (residues 85–94), and the TM (residues 95–116) (11–14) (Fig. 1 b). The linker region contains the conserved WWKNLK motif (15). We note that the “linker region” has been termed as such based on the assumption that it acts as a connection between the TM and cytosolic regions and was assumed not to be involved in membrane insertion of synaptobrevin. As discussed later, we will show that the linker region plays a key role not only in membrane insertion of the protein, but also in determining the tilt angle of the TM region, and therefore, it should probably be named differently. Complex formation is believed to induce a structural transition in the cytosolic domain of synaptobrevin from a disordered coil to an α-helix (1,3,5,10). Changes in the TM and linker regions of synaptobrevin during complex formation are less understood due to competing interpretations of experimental data available for the prefusion configuration (12,15).
Measurements (12,15–17) for the insertion depth and tilt angle of the linker region relative to the membrane have yielded relatively consistent results but with vastly different structural interpretations. Experiments have shown that the tryptophan residues of the linker region are inserted well below the phosphate layer of the membrane (12) and that the linker region has a large tilt with respect to the membrane normal (15,17). The results can be interpreted through either a continuous helix or a disordered connection between the TM and linker regions (12,15). Circular dichroism (CD) experiments suggest the existence of a continuous helix (15), whereas coarse-grained simulations have demonstrated a disordered connection (16). A helical connection between the TM and linker regions would invalidate the currently proposed structural segments of synaptobrevin (11). More importantly, the flexibility of the connection between the TM and linker regions could determine the mechanism by which SNARE complex formation induces fusion (7). Thus, we cannot establish an adequate model for the regulatory role of synaptobrevin during fusion without determining its membrane-embedded prefusion structure.
Experimental results have shown that the TM and linker regions of synaptobrevin play key roles in the regulation of both SNARE complex formation and full fusion (17–20). Specifically, fusion is highly sensitive to alterations, both additions and deletions, of the C-terminus of synaptobrevin (19,20). It is not clear, however, whether these mutations affect the structure of isolated synaptobrevin or its interactions within the SNARE complex during fusion. Thus, by characterizing the structure of the TM and linker regions, the effect of mutations can be classified as altering either the prefusion configuration or the dynamics of the fusion process.
The composition of the lipid environment has also been shown to affect the conformation of synaptobrevin in the membrane (21,22). Thus, comparisons between experiments that differ in both mutations to synaptobrevin and the lipid composition are difficult due to the coupling of the two effects (12,20,23). However, characterizing the prefusion structure of both wild-type and mutant synaptobrevin in a consistent lipid environment permits one to disentangle the effects of mutations and membrane composition on synaptobrevin during SNARE complex formation.
In the following, we use molecular-dynamics (MD) simulations to develop a model for the native configuration of synaptobrevin in a phosphatidylcholine (PC) lipid bilayer utilizing a novel membrane mimetic in conjunction with full membrane simulations. Furthermore, we examine the effects of key mutations on tilt and insertion depth of synaptobrevin. Our results demonstrate that the TM and linker regions in prefusion synaptobrevin form a continuous helix (residues 85–99), which adopts a highly tilted configuration with a well-defined orientation in the membrane due to the alignment of several tryptophan and lysine residues in the linker region. The C-terminal part (residues 101–116) of the TM helix is found to be mechanically decoupled from the rest of the TM and linker region by a flexible glycine kink (residue 100). Thus, our results suggest a different interpretation of structural elements for prefusion synaptobrevin than proposed for the postfusion SNARE complex (11). The tilted state is robust against both the addition of residues to the C-terminus and a large range of different initial conditions, including different initial tilt and insertion depth relative to the membrane.
Methods
Constructing the highly mobile membrane mimetic
Due to the entangled and disordered structures that lipid tails form, the dynamics of membrane-associated phenomena is often difficult to capture on timescales accessible to atomistic MD simulations. A believed-novel membrane mimetic developed in our lab (9,24), which greatly expedites the dynamics of lipids without compromising the atomistic details of the headgroups, was used in this study to explore the lipid-protein interactions and native tilt of synaptobrevin and its mutants (Fig. 1 c). In the highly mobile membrane mimetic (HMMM) model, the membrane core is replaced by the organic solvent DCLE (1,1-dichloroethane) and the headgroup region is represented by short-tailed lipids, such as DBPC (1,2-dibutyl-sn-glycero-3-phosphatidylcholine). A more detailed description and discussion of the HMMM model and its properties can be found in Ohkubo et al. (9).
A layer of DCLE measuring 98 × 98 × 38 Å3 and containing 2666 molecules (giving a density of 1.2 g/mL) was constructed using the PACKMOL software (25). Following this construction, 300 copies of DBPC were placed (150 per leaflet) such that the phosphorous atom of each short-tailed lipid coincided with the edge of the DCLE layer, giving an area per lipid of 64 Å2, which closely matches the physiological area per lipid for POPC (26). The lipids were each randomly rotated around their principal axis to introduce additional structural disorder and to decrease the time needed to equilibrate the system. DCLE molecules overlapping short-tailed lipids were removed. The system was then solvated and ionized to 150 mM NaCl using the SOLVATE and AUTOIONIZE plugins of VMD (27), respectively, giving the system final dimensions of 98 × 98 × 83 Å3 with ∼72,000 atoms. Because proper membrane width is integral to this study, mild harmonic constraints (k = 0.05 kcal/mol⋅Å2) were applied to each phosphorus atom in the direction of the membrane normal to restrain each headgroup to its original z position, thereby maintaining the thickness of the membrane at the physiologically relevant value for POPC (39 Å) (26). These constraints were applied for the duration of each simulation and allowed for tight regulation of the membrane thickness. The membrane was then energy-minimized for 5000 steps and equilibrated in an NPnT ensemble (constant pressure and temperature, fixed area in the xy plane, algorithm provided by the MD code NAMD) for 2.5 ns. The resulting system was used in all membrane simulations of synaptobrevin.
Building and equilibrating synaptobrevin
A recent crystal structure of the postfusion SNARE complex (PDB:3IPD) (11) was used as the starting point for simulations of synaptobrevin. The coordinates of synaptobrevin were taken from the complex, capped with a C-terminal carboxy group and an N-terminal ammonium group, and placed in a full POPC membrane measuring 85 × 85 Å2, generated using the MEMBRANE BUILDER plugin of VMD. The system was energy-minimized for 3000 steps and equilibrated for 4 ns, including 2 ns with the protein backbone harmonically restrained (k = 5.0 kcal/mol ⋅ Å2) in all directions and 2 ns with the protein backbone harmonically restrained in the xy plane (i.e., allowed to move in the z direction).
Following this, synaptobrevin was truncated to only include the TM and linker regions (residues 78–116), while discarding the rest of the protein. In the crystal structure (11), the TM and linker regions form a continuous helix, a conformation which has been verified experimentally for isolated synaptobrevin (15). Using the PSFGEN plugin of VMD, the N-terminus was then capped with an N-acetyl group and the C-terminus was capped with a methyl group (unless otherwise noted) to neutralize the ends of the helix and avoid nonphysiological interactions with the membrane.
The truncated synaptobrevin was then placed in a PC HMMM and all overlapping molecules were removed, resulting in a system size of ∼70,000 atoms. Moreover, the helix was placed such that the depth of synaptobrevin in the DBPC membrane matched that of the depth in the full lipid membrane. The resulting system was energy-minimized for 3000 steps and the protein held fixed for 3 ns allowing the short-tailed lipids to relax around the protein. The resulting truncated synaptobrevin structure was then rotated to create a distribution of starting tilts for our simulations; overlapping molecules were again removed. Initial tilts with the lysines in the linker region (residues 87, 91, and 94) angled toward the center of the membrane are denoted with a prime (′).
Although the initial configuration of synaptobrevin in our simulations is constructed from a postfusion crystal structure (11), our initial model incorporates two key experimentally verified structural properties of the prefusion state. Experimental results show that the TM and linker regions form a continuous helix (15) and that the cytosolic region is disordered (3,15). Although the CD data that show a continuous helix was measured in β-octyl glucoside (15), similar measurements in lipid-like detergents (dodecylphosphocholine) showed no significant difference in CD spectra. The disordered cytosolic region is not expected to significantly affect membrane structure of prefusion synaptobrevin and, therefore, the cytosolic domain has been truncated in our simulations. Thus, in the absence of a prefusion crystal structure, our model provides a reasonable starting point for investigation of the properties of prefusion synaptobrevin.
Addition of residues to C-terminus
Previous biochemical studies have shown that extending synaptobrevin with charged residues hinders the fusion process (20). Therefore, to test whether the additional residues affect the native configuration of synaptobrevin in the membrane, we created three structures (GG, KK, and EE) in which synaptobrevin was extended by two residues. The new C-terminus was capped with a methyl group as discussed above. The lengthened synaptobrevin was then fixed except for the two additional residues and energy-minimized for 4000 steps followed by simulation in vacuo for 100 ps. The resulting structure was placed in the DBPC membrane with the same initial configuration as the crystal structure (i.e., 10′°) and simulated using an NPnT ensemble.
Conversion of HMMM model to conventional membrane
To test whether the tilted model of synaptobrevin generated in the HMMM membrane was physiologically relevant, we replaced the HMMM membrane with a conventional POPC membrane using the final snapshot in the WT40 simulation. The main goal in this transformation was to perturb the headgroup-protein interaction as little as possible while allowing the POPC membrane to relax around the convergent, highly tilted state. To accomplish this, a single POPC lipid was superimposed onto each DBPC molecule in the system such that the headgroup orientation and positioning was preserved. Once all lipids were placed, the DBPC and DCLE that make up the HMMM membrane were removed from the system. To minimize perturbation to the lipid-protein interactions, we allowed the POPC membrane to slowly relax around the protein. The lipid tails were allowed to melt for 2 ns while harmonically restraining both the lipid headgroups and the protein (k = 5.0 kcal/mol⋅Å2). Because the HMMM membrane was slightly thicker than a POPC, we continued to restrain the cis-leaflet headgroups, while releasing the restraints on the trans-leaflet for 2 ns. Next, the restraints on the cis-leaflet, as well as the constant area constraint, were lifted and the system equilibrated for 2 ns. This allowed the membrane to equilibrate to the correct AL as well as equilibrate around the protein, which was still harmonically restrained. Following this, all restraints were released and the system simulated for 100 ns in an NPT ensemble (P = 1.0 atm, T = 310 K).
Simulation protocols
All simulations employed NAMD2 (28) utilizing the CHARMM27 set of force-field parameters with ϕ/ψ cross-term map corrections (29) for proteins, the CHARMM36 set of parameters for the short-tailed lipids (30), and the CHARMM general force field for the DCLE molecules (31). Parameters were developed for DBPC based on previously available parameters for POPC by simply shortening the fatty acid tails (9). The TIP3P model for water was used across all simulations (32). Constant pressure was maintained at a target of 1 atm using the Nosé-Hoover-Langevin piston method (33,34). Constant temperature was kept at 310 K using a Langevin damping coefficient of 1 ps−1. Nonbonded interactions were cut off beyond 12 Å with a smoothing function applied beyond 10 Å. Long-range electrostatics were calculated using the particle-mesh Ewald method (35) with a grid density of >1 Å−3. A time step of 2 fs was used with bonded and nonbonded forces calculated every time step and particle-mesh Ewald forces calculated every other time step. System setup, visualization, and analysis were carried out using VMD (27).
Analysis
We observed a decoupling between the top (TM1: 85–99) and bottom (TM2: 101–116) sections of the TM helix, which we attribute to the flexibility of synaptobrevin at G100. Therefore, it is more meaningful to measure and report separately the tilt angles of TM1 and TM2 (Fig. 1 c) with respect to the membrane normal (z axis in this study). To characterize the tilt of the separate regions of synaptobrevin (TM1 and TM2), we determined the center of mass of the backbone atoms of three groups of atoms along the helix, namely residues 89–94, 98–103, and 110–115. The TM1 vector was designated to be the vector from residues 98–103 to residues 89–94, and the TM2 vector from residues 110–115 to residues 98–103. We then calculated the angle between each TM vector and the membrane normal. We also calculated the helix vector using the method of Åqvist (36), which uses a modified least-squares fit of the Cα atoms to determine the best-fit vector through the helix. The results from this method are very similar to the center-of-mass method and are presented in Fig. S1 in the Supporting Material. Because the position of W89/W90 is important for putting previous studies in context of a structural model, we also monitored the insertion depth of these residues in the simulations (12,17). This was done by measuring the distance between the midpoint of the bilayer and the average position of the α-carbons of W89/W90.
Results and Discussion
Our simulations demonstrate that the synaptobrevin transmembrane (TM) and linker regions form a continuous helix (residues 85–99), which adopts a highly tilted configuration with a well-defined orientation in the membrane due to the alignment of several lysine residues in the linker region. The C-terminal half (residues 101–116) of the TM helix forms a continuous helix, which is mechanically decoupled from the rest of the TM region by a flexible glycine kink (residue 100). Thus, our results suggest a different interpretation of structural elements for prefusion synaptobrevin than proposed for the postfusion SNARE complex (11). The tilted state is robust against both the addition of residues to the C-terminus and a large range of different initial conditions, including different initial tilts and insertion depths relative to the membrane.
Prefusion synaptobrevin has a well-defined structure in the membrane
Prior results from experiments and simulations have yielded two conflicting models for membrane-embedded synaptobrevin (12,15,16). One model proposes that the TM (residues 95–116) and linker (residues 85–94) regions are connected by a disordered segment, with little or no tilt in the TM domain, but a large tilt with regard to the membrane in the linker region (12,16); the second model proposes that the TM and linker regions form a continuous helix with a large tilt on the basis of CD and infrared spectroscopic measurements (15). To distinguish between the two models, we employed MD simulations with a wide range of initial orientations to establish a well-defined structure for the TM and linker regions of synaptobrevin in the presence of a membrane. Determining the membrane-bound structure is key to both understanding the fusogenic properties of synaptobrevin and offering a correct interpretation of mutagenesis experiments.
To establish a robust and convergent membrane-embedded state, we have employed simulations with initial tilts of 20′°, 10′°, 0°, 20°, and 40° with respect to the membrane normal (Fig. 2 a). We note that in all of the simulations, the so-called linker region is initially out of the membrane, that is, consistent with the general assumption that this region merely links the TM region to the rest of the protein and is not involved in membrane insertion of synaptobrevin. By considering the results from simulations with multiple initial conditions, we can characterize the fluctuations of the TM and linker regions about a robust prefusion structure. The results are summarized in Table 1.
Figure 2.
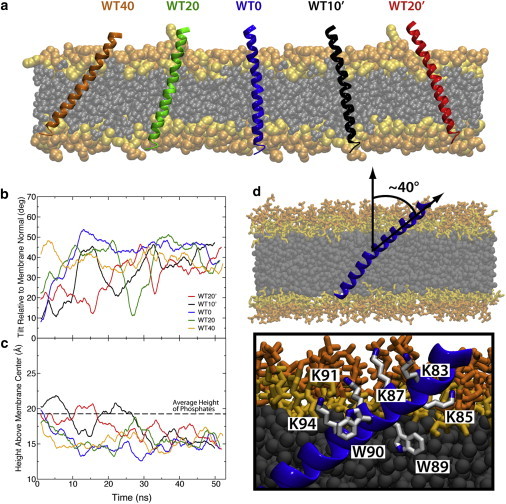
Membrane-embedded synaptobrevin. (a) Initial conditions for simulations with wild-type synaptobrevin varied in both initial insertion depth and tilt. The simulations are labeled according to starting tilt with respect to the membrane normal. Initial tilts with the lysines in the linker region (residues 87, 91, and 94) angled toward the center of the membrane are denoted with a prime (′). (b) Running average of the tilt angle between TM1 and the membrane normal. The tilt angle converges after ∼30 ns. (c) Running average of the height of the tryptophan residues above the midpoint of the membrane. The distance below the phosphate layer of the membrane (dashed line) for the tryptophan residues converges to a value of ∼5 Å. (d) The highly tilted state resulting in all cases is characterized by lysine residues aligned along one side of the synaptobrevin helix and tryptophan residues inserted into the membrane on the other side of the helix. To see this figure in color, go online.
Table 1.
Membrane tilt and insertion depth in various simulations of wild-type synaptobrevin
| System | Initial tilt (°) | TM1 tilt (°)a | Initial insertion (Å) | Insertion depth (Å)b |
|---|---|---|---|---|
| WT20′ | 22.2 | 44.4 ± 5.0 | 19.6 | 15.5 ± 1.2 |
| WT10′ | 13.2 | 41.0 ± 5.7 | 19.4 | 16.4 ± 1.5 |
| WT0 | 3.3 | 35.5 ± 6.7 | 19.0 | 14.7 ± 1.2 |
| WT20 | 16.8 | 41.4 ± 8.4 | 18.2 | 15.5 ± 1.3 |
| WT40 | 36.8 | 38.4 ± 5.5 | 17.3 | 15.2 ± 1.4 |
| Average | — | 40.1 ± 3.4 | — | 15.5 ± 0.6 |
Average tilt over the last 20 ns of the simulation. The tilt is measured for residues 91–100 relative to the membrane normal.
Average insertion depth over the last 20 ns of the simulation. The insertion depth is defined as the distance of the center of mass for the Cα of tryptophan residues 89–90 above the midpoint of the membrane.
Simulations across all initial tilt angles yielded a robust structure requiring <30 ns of simulation in all cases. The first significant finding is that in all cases, the TM/linker region exhibits a deeper insertion into the membrane (∼5 Å) with regard to its initial position (Fig. 2 c). Across all five simulations, we observe an average insertion depth of ∼5 Å below the phosphate layer for W89/W90 (Fig. 2 c), which is consistent with the electron paramagnetic resonance data of Kweon et al. (12). Additionally, in our simulations, we observe significant insertion into the acyl-chain region of the bilayer below K85, consistent with experimental accessibility data (12). The observed translocation of the protein into the membrane is primarily due to the insertion of the residues in the linker region, which, based on our results, appears to directly participate in determining the insertion depth of the TM region into the membrane. In fact, based on the convergent behavior of this region as observed in our simulations, at least for the prefusion state, this linker should be best regarded as a part of a single TM helix of synaptobrevin.
The membrane-embedded state is characterized by a continuous helical segment formed by the first half of the TM helix and the linker (residues 85–99, TM1), followed by a flexible glycine kink (residue 100), which is conserved across multiple species (15,37), and a C-terminal helix (residues 101–116, TM2). The TM1 region displayed a high tilt angle (∼40°) relative to the membrane normal and insertion of the tryptophan residues below the phosphate region of the bilayer (Fig. 2, b–d) in agreement with experimental studies (12,15). In fact, both EPR and IR experiments suggest that synaptobrevin makes an angle of ∼33–39° with the membrane normal (12,15), in complete agreement with what we observe in our simulations (Table 1). The TM2 region, however, displayed great flexibility, as is evident from the large fluctuations in tilt (Fig. 3, a and b).
Figure 3.
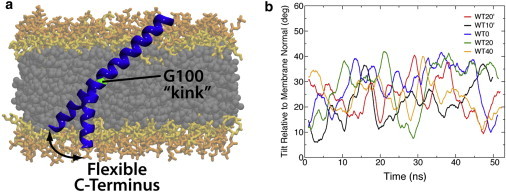
The glycine kink in the synaptobrevin TM domain. (a) Representative snapshots showing the kink of the synaptobrevin TM domain at G100. (b) Running average of the tilt angle between TM2 and the membrane normal, indicating high flexibility for the C-terminal half. To see this figure in color, go online.
The decoupling of TM2 from TM1 is consistent with Fourier transform infrared spectroscopy data from Bowen and Brünger (15), in which they measured an extremely low peptide order parameter for the synaptobrevin transmembrane construct in POPC bilayers. It was proposed that the low order parameter could be consistent with a kink at G100 causing independent motion of the N- and C-terminal halves of the transmembrane construct, which is indeed what we observe in our simulations. The robust nature of the membrane-embedded prefusion structure is clearly displayed by the convergence of simulations with starting configurations that range over 50° in tilt (Fig. 2, Table 1). Previous coarse-grained simulations have not produced a similar convergent state, most likely due to imposed secondary structure restraints on the TM and linker connection (16).
We attribute the highly tilted state observed in our simulations to the special arrangement of lysine and tryptophan residues in the linker region of synaptobrevin (12,15) (Fig. 2 d). The organization of residues in the linker region is highly conserved across synaptobrevin homologs (15), suggesting that the tilt observed in our simulations is a robust property of the entire class of SNARE proteins. Furthermore, our results suggest that the high tilt for the linker region is transmitted into the TM domain, which is likely capable of disordering lipid packing and of promoting fusion in a mechanism similar to viral fusion peptides (38–40).
The structural elements of synaptobrevin observed in our simulations suggest a new characterization of the TM domain, namely that the TM domain for membrane-embedded synaptobrevin actually includes residues 85–116. This apparently newly characterized TM domain consists of two helical sections, TM1 (residues 85–99) and TM2 (residues 101–116), respectively, which are mechanically decoupled by a glycine kink (G100) proposed in previous FTIR studies (15). The kink in the synaptobrevin TM helix is due to the weakened interaction between the amide of G100 and the carbonyl of M96 (see Fig. S2).
The weakened interaction allows for more mobility in the TM2 portion of the helix and an increase in the interactions between the C-terminus and the membrane (see Fig. S3, Fig. S4, Fig. S5, and Fig. S6). In our simulations, TM1 adopts a robust highly tilted state, whereas TM2 remains flexible and pivots around the G100 kink. Indeed, over the timecourse of the simulations the TM2 region adopts an average angle of 25.7 ± 2.7° (see Table S1 and Fig. S7 in the Supporting Material), which is significantly different from the average angle of 40.1 ± 3.4° for the TM1 region, clearly demonstrating that the two regions are decoupled. The previous characterization of synaptobrevin including distinct TM and linker regions (11–14) is inadequate for two reasons:
-
1.
The continuous helix (residues 85–99) makes the linker region and the beginning of the TM domain structurally indistinguishable; and
-
2.
The term “linker” implies a flexible connection between two domains, which is unsuitable to describe a highly coupled α-helical segment.
Addition of C-terminal residues does not affect the prefusion state
Experimental results have shown that SNARE-mediated fusion can be modulated by the introduction of additional residues to the C-terminus of synaptobrevin (20). The role of the additional amino acids could be to alter either the prefusion configuration of synaptobrevin, its interactions with other SNARE proteins during fusion, or both. By comparing the configuration of synaptobrevin both with and without additional C-terminal residues, one can isolate the effects on the prefusion structure.
We constructed three mutants (GG, KK, and EE), each with two additional amino acids attached to the C-terminus of synaptobrevin. The sequences chosen and the simulation results are compared in Fig. 4; simulation results are also listed in Table 2. The same simulation conditions were used as for the wild-type, and the initial tilt was set at 10′°. Interestingly, addition of two glycines, two lysines, or two glutamates did not affect the observed membrane-embedded structure; all the simulations converged to a state with a highly tilted TM and linker region, a continuous helical connection, and tryptophan residues inserted below the phosphate layer of the membrane, matching the wild-type configuration. For all mutants tested, the C-terminal region displayed large fluctuations in tilt, suggesting flexibility. In addition, measuring the contact probabilities of these mutants shows striking similarity to the wild-type simulation, with one exception: namely, the additional residues make contacts with the headgroup that are absent in the wild-type simulations (see Fig. S3, Fig. S4, Fig. S5, and Fig. S6). We conclude that the wild-type structure of the synaptobrevin TM and linker regions is robust against additions to the C-terminus.
Figure 4.
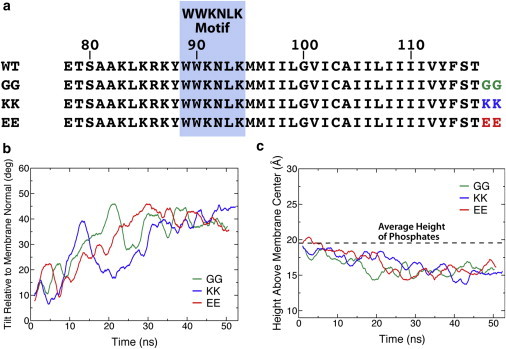
Additions to the synaptobrevin C-terminus. (a) Aminoacid sequence comparison of wild-type and mutants used in the simulations. (Blue box) Highly conserved WWKNLK motif. (b) Running average of the tilt angle between TM1 and the membrane normal. The plots converge to the wild-type value in ∼30 ns for all mutants. (c) Running average of the height of the tryptophan residues above the center of the membrane. The distance below the phosphate layer of the membrane for the tryptophan residues converges to a value of ∼5 Å. To see this figure in color, go online.
Table 2.
Membrane tilt and insertion depth in various simulations of mutant synaptobrevin
| System | Initial tilt (°) | TM1 tilt (°)a | Initial insertion (Å) | Insertion depth (Å)b |
|---|---|---|---|---|
| GG | 13.2 | 39.1 ± 5.1 | 19.3 | 15.7 ± 1.0 |
| KK | 13.2 | 40.2 ± 4.9 | 19.4 | 15.4 ± 1.2 |
| EE | 13.2 | 40.3 ± 4.9 | 19.4 | 15.7 ± 1.1 |
| Average | — | 39.9 ± 0.7 | — | 15.6 ± 0.2 |
Average tilt over the last 20 ns of the simulation. The tilt is measured for residues 91–100 relative to the membrane normal.
Average insertion depth over the last 20 ns of the simulation. The insertion depth is defined as the distance of the center of mass for the Cα of tryptophan residues 89–90 above the midpoint of the membrane.
Experiments have shown that SNARE-mediated fusion is negatively regulated by the addition of charged amino acids to the C-terminal end of synaptobrevin (20). Our results show that the addition of charged amino acids, such as lysine and glutamate, does not affect the orientation of synaptobrevin in the membrane. Thus, it is likely that the C-terminal end plays some further role during the fusion process that is not compatible with the addition of charged amino acids.
In the case that glycine residues are added, the entire C-terminus is still capable of inserting into the membrane. Glycine residues were experimentally observed to insignificantly alter SNARE-mediated fusion (20). Charged residues, which are detrimental to fusion, prevent full insertion, but provide enough flexibility for the protein to highly tilt. Thus, the major difference between the two cases involves the anchoring of the C-terminus to the phosphate layer of the membrane, a fact that provides support for a mechanism (20) in which synaptobrevin is pulled partially through the membrane during fusion.
The highly tilted state of synaptobrevin is conserved in the transformation of HMMM to conventional membrane
Although the HMMM membrane is successful in its goal of expediting lipid dynamics, the model itself does not necessarily claim to reproduce the mechanical and electrical properties of conventional membranes (i.e., DOPS, POPC, and DPPE). Therefore, to ensure that the highly tilted model of synaptobrevin generated using the HMMM membrane is a physiological state relevant to the biology of the SNARE complex, we tested whether the structure adopted in the HMMM membrane was stable in a conventional POPC membrane. Following a previously utilized scheme (41,42), we equilibrated a POPC membrane around the final snapshot of the WT40 system. After equilibration and relaxation of the membrane, we simulated the synaptobrevin/POPC system for 100 ns. In the full membrane, the tilted state of synaptobrevin remains extremely stable in the POPC membrane (Fig. 5).
Figure 5.
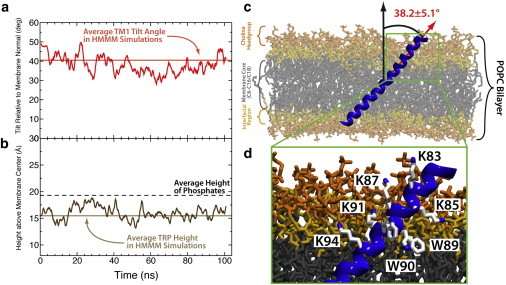
Structure of synaptobrevin in a POPC membrane. (a) Plot of the TM1 angle of synaptobrevin as a function of simulation time. (Light red line) Average TM1 angle obtained across the five wild-type simulations of synaptobrevin in the PC HMMM. (b) Plot of the height of the tryptophans above the membrane center. (Light-brown line) Average height of the tryptophans above the membrane center in the five wild-type simulations of synaptobrevin in the PC HMMM. (c) Snapshot of synaptobrevin in a conventional POPC membrane demonstrating that the highly tilted state observed in the HMMM simulations is indeed stable in the full membrane simulations. (Red arrow) Helical axis used to measure the TM1 angle (corresponds to the red trace shown in panel a). Moreover, the coloring scheme applied to the POPC bilayer in this figure is carried over from Fig. 2 for ease of comparison. (d) Closeup of the TM1 showing the position of the WWKNLK motif as well as additional lysine residues. This arrangement mimics that found in HMMM simulations and shown in Fig. 2. To see this figure in color, go online.
There is very little perturbation to either the average tilt angle of TM1 (38.2 ± 5.1°) or the average insertion depth of the tryptophan residues (16.0 ± 1.3 Å above membrane center) and the values obtained from the full membrane simulation agree very well with those found from HMMM simulations (Table 1). By plotting the TM2 tilt angle, we observe that, although the dynamics are slowed drastically compared to the HMMM simulations, TM2 remains decoupled from TM1 in the POPC simulations due to the presence of G100 and does not adopt a well-defined tilt within the membrane (Fig. 5). The PC HMMM was also able to capture the particular arrangement of lysine and tryptophan residues that is observed in the POPC membrane simulations (Fig. 5 d). Therefore, not only was the PC HMMM membrane able to capture physiologically relevant structures and dynamics observed in a conventional POPC membrane, but the HMMM membrane was able to capture these phenomena on a much faster timescale, allowing for more robust sampling and for the generation of statistical measures that would be prohibitively costly in conventional membrane simulations.
Conclusions
Through atomistic MD simulations we have demonstrated that the TM and linker regions of synaptobrevin adopt a robust configuration in the membrane. The configuration, as illustrated in Fig. 6, is characterized by two helical segments, TM1 and TM2. TM1, which includes the formerly-called linker region, has lysine residues aligned along a single face of the helix and tryptophan residues inserted below the phosphate layer of the membrane, which results in a highly tilted state (∼40°). TM2 is mechanically decoupled from TM1 due to a flexible glycine kink (G100). It is important to note that these structural dynamics do not appear to be an effect of the HMMM model used. The structure observed in the HMMM model is conserved in a conventional, full-tailed membrane simulation in addition to independent experimental results verifying the depth of penetration of the tryptophans (W89/W90) (12) and the formation of the mechanically decoupling glycine kink (G100) (15). The previous distinction between the TM and linker regions of synaptobrevin is inadequate to describe the robust, highly tilted helical TM1 segment observed in our simulations. The identification of a well-defined prefusion structure allowed us to analyze the effects of mutagenesis experiments, and, in the future, will permit a quantitative determination of lipid environment effects on synaptobrevin orientation.
Figure 6.
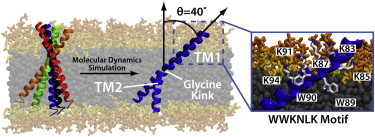
Conformation of membrane-embedded synaptobrevin. The membrane-embedded configuration is characterized by a highly tilted TM1 region, lysine residues aligned along a single face of the TM1 helix, and tryptophan residues inserted below the phosphate layer of the membrane. The TM2 helix is mechanically decoupled from TM1 by a kink at G100. The previously assigned linker region forms a continuous helix together with TM1 and is largely inserted into the membrane. To see this figure in color, go online.
We have further demonstrated that the addition of residues to the C-terminus of synaptobrevin does not alter the membrane-embedded structure. Both helical segments (TM1 and TM2) remain intact, and the TM1 segment adopts a highly tilted state similar to wild-type synaptobrevin. Extensive measurements of protein-lipid contacts between mutant synaptobrevin transmembrane helices and the membrane shows little to no change from the wild-type measurements. Therefore, structurally, there appears to be no change to synaptobrevin upon addition of residues to the C-terminus, and it is unclear what the cause for an increased energy barrier to fusion might be. Because the native state is not altered by the addition of residues to the C-terminus, the extra residues most likely affect the stability of fusion intermediates; that is, the asymmetry between SNARE-mediated fusion with the addition of glycine versus charged residues provides support for a mechanism in which the C-terminus is pulled into the membrane during fusion (20).
Although the cytosolic domains of individual SNARE proteins play a pivotal role in bringing two membranes into close proximity, the TM domains of syntaxin and synaptobrevin likely play an equally important role in facilitating fusion pore formation and subsequently full fusion (20,38,43). Thus, characterizing the structural transition that occurs in the synaptobrevin TM domain during SNARE-mediated fusion is pivotal for developing an understanding of exocytosis. We have presented here a well-defined model for the orientation of isolated synaptobrevin in a PC bilayer, which can be compared with future structural studies during the different stages of fusion to characterize the regulatory role of synaptobrevin.
Acknowledgments
This work was supported in part by the National Institutes of Health (grants No. U54-GM087519 and No. P41-GM104601 to E.T. and K.S., and grants No. R01-GM101048 and No. R01-GM086749 to E.T.) and the National Science Foundation (Graduate Research Fellowship to M.J.A.). All simulations have been performed at XSEDE resources (grant No. MCA06N060).
Footnotes
Andrew E. Blanchard and Mark J. Arcario contributed equally to this article.
Supporting Material
References
- 1.Fasshauer D., Otto H., Brünger A.T. Structural changes are associated with soluble n-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J. Biol. Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 2.Südhof T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 3.Brunger A.T. Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- 4.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 5.Südhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martens S., McMahon H.T. Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- 7.Sutton R.B., Fasshauer D., Brunger A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 8.Yersin A., Hirling H., Kasas S. Interactions between synaptic vesicle fusion proteins explored by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 2003;100:8736–8741. doi: 10.1073/pnas.1533137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohkubo Y.Z., Pogorelov T.V., Tajkhorshid E. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunger A.T., Weninger K., Chu S. Single-molecule studies of the neuronal SNARE fusion machinery. Annu. Rev. Biochem. 2009;78:903–928. doi: 10.1146/annurev.biochem.77.070306.103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stein A., Weber G., Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kweon D.-H., Kim C.S., Shin Y.-K. Regulation of neuronal SNARE assembly by the membrane. Nat. Struct. Biol. 2003;10:440–447. doi: 10.1038/nsb928. [DOI] [PubMed] [Google Scholar]
- 13.McNew J.A., Weber T., Rothman J.E. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell. 1999;4:415–421. doi: 10.1016/s1097-2765(00)80343-3. [DOI] [PubMed] [Google Scholar]
- 14.Südhof T.C., Baumert M., Jahn R. A synaptic vesicle membrane protein is conserved from mammals to Drosophila. Neuron. 1989;2:1475–1481. doi: 10.1016/0896-6273(89)90193-1. [DOI] [PubMed] [Google Scholar]
- 15.Bowen M., Brunger A.T. Conformation of the synaptobrevin transmembrane domain. Proc. Natl. Acad. Sci. USA. 2006;103:8378–8383. doi: 10.1073/pnas.0602644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durrieu M.-P., Bond P.J., Baaden M. Coarse-grain simulations of the R-SNARE fusion protein in its membrane environment detect long-lived conformational sub-states. ChemPhysChem. 2009;10:1548–1552. doi: 10.1002/cphc.200900216. [DOI] [PubMed] [Google Scholar]
- 17.Kweon D.-H., Kim C.S., Shin Y.-K. Insertion of the membrane-proximal region of the neuronal SNARE coiled coil into the membrane. J. Biol. Chem. 2003;278:12367–12373. doi: 10.1074/jbc.M211123200. [DOI] [PubMed] [Google Scholar]
- 18.Maximov A., Tang J., Südhof T.C. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fdez E., Martínez-Salvador M., Hilfiker S. Transmembrane-domain determinants for SNARE-mediated membrane fusion. J. Cell Sci. 2010;123:2473–2480. doi: 10.1242/jcs.061325. [DOI] [PubMed] [Google Scholar]
- 20.Ngatchou A.N., Kisler K., Lindau M. Role of the synaptobrevin C terminus in fusion pore formation. Proc. Natl. Acad. Sci. USA. 2010;107:18463–18468. doi: 10.1073/pnas.1006727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong J., Borbat P.P., Shin Y.-K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. USA. 2009;106:5141–5146. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellena J.F., Liang B., Tamm L.K. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc. Natl. Acad. Sci. USA. 2009;106:20306–20311. doi: 10.1073/pnas.0908317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddiqui T.J., Vites O., Fasshauer D. Determinants of synaptobrevin regulation in membranes. Mol. Biol. Cell. 2007;18:2037–2046. doi: 10.1091/mbc.E07-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arcario M.J., Ohkubo Y.Z., Tajkhorshid E. Capturing spontaneous partitioning of peripheral proteins using a biphasic membrane-mimetic model. J. Phys. Chem. B. 2011;115:7029–7037. doi: 10.1021/jp109631y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martínez L., Andrade R., Martínez J.M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 26.Kucerka N., Tristram-Nagle S., Nagle J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 27.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 28.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacKerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 30.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanommeslaeghe K., Hatcher E., MacKerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 33.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 34.Feller S.E., Zhang Y., Pastor R.W. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 35.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 36.Åqvist J. A simple way to calculate the axis of an α-helix. Comput. Chem. 1986;10:97–99. [Google Scholar]
- 37.Nonet M.L., Saifee O., Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J. Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Risselada H.J., Kutzner C., Grubmüller H. Caught in the act: visualization of SNARE-mediated fusion events in molecular detail. ChemBioChem. 2011;12:1049–1055. doi: 10.1002/cbic.201100020. [DOI] [PubMed] [Google Scholar]
- 39.Martin I., Ruysschaert J., Epand R.M. Role of the N-terminal peptides of viral envelope proteins in membrane fusion. Adv. Drug Deliv. Rev. 1999;38:233–255. doi: 10.1016/s0169-409x(99)00031-9. [DOI] [PubMed] [Google Scholar]
- 40.Légaré S., Lagüe P. The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. Proteins. 2014;82:2118–2127. doi: 10.1002/prot.24568. [DOI] [PubMed] [Google Scholar]
- 41.Baylon J.L., Lenov I.L., Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J. Am. Chem. Soc. 2013;135:8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arcario M.J., Tajkhorshid E. Membrane-induced subdomain rearrangement of talin subdomains providing a molecular mechanism for inside–out integrin activation. Biophys. J. 2014;107:2059–2069. doi: 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson M.B. SNARE complex zipping as a driving force in the dilation of proteinaceous fusion pores. J. Membr. Biol. 2010;235:89–100. doi: 10.1007/s00232-010-9258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.