

Introduction
Huntington's disease is a neurodegenerative disorder that affects muscle coordination. It is the most common genetic cause of abnormal involuntary writhing movements called chorea and becomes noticeable in the middle age. The disease is caused by an autosomal dominant mutation on either of an individual's two copies of genes called Huntington. We present neuroimaging findings in a middle aged case of Huntington's disease.
Case report
A 43-year-old male patient, symptomatic for 5 yrs with involuntary, irregular, non-patterned movements presented to the Neurology Department of a service hospital. These movements were not associated with posturing. On physical examination, the movements were low amplitude affecting distal part of the limbs, face and tongue. Non-contiguous involvement was noted. No associated looseness, imbalance or weakness of limbs were observed. There was no cognitive decline or change in behaviour. His laboratory parameters were unremarkable.
MRI examination of the brain was performed for further evaluation of the patient. Axial T1 and T2W images of the brain showed reduction in the size of putamen and caudate nuclei bilaterally (putaminal involvement more than the caudate nuclei). Partial loss of normal caudate impression on the frontal horns with resultant dilatation of the frontal horns was observed. No abnormal signal was however noted in the region of affection. These findings are suggestive of Huntington's disease (Fig. 1a and b). Compare these findings with normal brain at similar level (Fig. 2a and b). In our case on further questioning it was revealed that the individual's father had similar disease with onset at the age of 50 yrs.
Fig. 1.
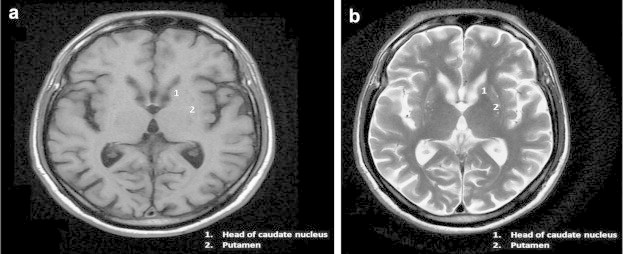
Symmetrical partial atrophy of head of caudate nucleus, gross atrophy of putamen. (a) Axial T1W and (b) Axial T2W images of the patient at the level of basal ganglia.
Fig. 2.
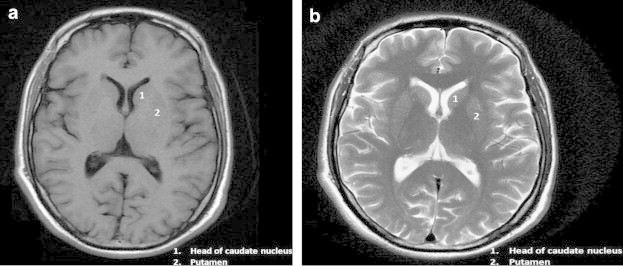
Normal brain at the level of basal ganglia. (a) Axial T1W and (b) Axial T2W images.
Discussion
Huntington's disease is an autosomal dominant neurodegenerative disorder affecting four to five persons per million. The disease shows complete penetrance and is determined by a gene located on short arm of chromosome 4. Patients present with movement disorder in form of choreoathetosis, rigidity (Westphal variant), dementia and emotional disturbances.1,2
On neuroimaging atrophy of the corpus striatum involving the caudate and putamen is seen. This change generally proceeds from medial to lateral and dorsal to ventral. These changes are better appreciated on MRI than on CT. The atrophy of caudate results in loss of normal bulge of these nuclei on the frontal horns with resultant focal dilatation of the frontal horns. Signal changes in form of either a hyper or hypointensity may be seen in the striatum on T2W images. The hyperintensity is attributed to neuronal loss and gliosis whereas the iron accumulation accounts for the hypointense signal. Two groups have reported T2W hyperintensity present in all patients with rigid form but only occasionally with the classical hyperkinetic form of disease.1–3
Volumetric studies reveal marked reduction in the volumes of striatal structures as well as some reduction in thalamus and hippocampus. Greater magnitude of atrophic change was noted in the putamen compared to caudate in a study of mildly affected patients.4
MR spectroscopy in these patients reveals a reduction in NAA and creatine in the basal ganglia of 60% in symptomatic and of 30% in the presymptomatic patients.5 In advanced cases other parts of the brain such as olives, pons, cerebellum, temporal lobes and white matter tracts may get affected. Cerebral cortical atrophy in such cases correlates well with the neuropsychological symptoms.6 Moreover in vivo functional MRI and diffusion tensor imaging (DTI) have shown promise in detection of preclinical HD mutation carrier years before the onset of symptoms.7
To conclude neuroimaging, particularly MRI, remains a cornerstone in the diagnosis and assessing the severity of Huntington's disease. Genetic testing can be used to confirm the diagnosis if the family history is not forthcoming. Genetic counselling to those affected may provide guidance on the implications of the disease.
Conflicts of interest
All authors have none to declare.
References
- 1.Holondy Andrei I., George Ajax E., de Leon Mony J., Karimi Sassan, Golomb James. Neurodegenerative disorders. In: Haaga John R., editor. CT and MRI of the Whole Body. 5th ed. Mosby Elsevier; Philadelphia: 2009. pp. 352–353. [Google Scholar]
- 2.Jack C.R., Jr., Lexa F.J., Tojanowski J.Q., Braffman B.H., Atlas S.W. Normal aging, dementia and neurodegenerative disease. In: Atlas S.W., editor. Magnetic Resonance Imaging of the Brain and Spine. 3rd ed. Lippincott Williams & Wilkins; Philadelphia: 2002. pp. 1208–1210. [Google Scholar]
- 3.Oliva D., Carella F., Savoiardo M. Clinical and magnetic resonance features of the classic and akinetic-rigid variants of Huntington's disease. Arch Neurol. 1993;50:17–19. doi: 10.1001/archneur.1993.00540010013010. [DOI] [PubMed] [Google Scholar]
- 4.Harris G.J., Pearlson G.D., Peyser C.E. Putamen volume reduction on magnetic resonance imaging exceeds caudate changes in mild Huntington's disease. Ann Neurol. 1992;31:69–75. doi: 10.1002/ana.410310113. [DOI] [PubMed] [Google Scholar]
- 5.Squire L.R., Amaral D.G., Press G.A. Magnetic resonance imaging of hippocampal formation and mammillary nuclei distinguish medial temporal lobe and diencephalic amnesia. J Neurosci. 1990;10:3106–3117. doi: 10.1523/JNEUROSCI.10-09-03106.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starkstein S.E., Brandt J., Bylsma F., Peyser C., Folstein M., Folstein S.E. Neuropsychological correlates of brain atrophy in Huntington's disease: a magnetic resonance imaging study. Neuroradiology. 1992;34:487–489. doi: 10.1007/BF00598956. [DOI] [PubMed] [Google Scholar]
- 7.Bohanna I., Georgiou-Karistianis, Hannan A.J., Egan G.F. Magnetic resonance imaging as an approach towards identifying neurpathological biomarkers for Huntington's disease. Brain Res Rev. 2008;58:209–225. doi: 10.1016/j.brainresrev.2008.04.001. [DOI] [PubMed] [Google Scholar]