

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a progressive disease of increasing public health concern. In western populations the disease has an estimated prevalence of 20%-40%, rising to 70%-90% in obese and type II diabetic individuals. Simplistically, NAFLD is the macroscopic accumulation of lipid in the liver, and is viewed as the hepatic manifestation of the metabolic syndrome. However, the molecular mechanisms mediating both the initial development of steatosis and its progression through non-alcoholic steatohepatitis to debilitating and potentially fatal fibrosis and cirrhosis are only partially understood. Despite increased research in this field, the development of non-invasive clinical diagnostic tools and the discovery of novel therapeutic targets has been frustratingly slow. We note that, to date, NAFLD research has been dominated by in vivo experiments in animal models and human clinical studies. Systems biology tools and novel computational simulation techniques allow the study of large-scale metabolic networks and the impact of their dysregulation on health. Here we review current systems biology tools and discuss the benefits to their application to the study of NAFLD. We propose that a systems approach utilising novel in silico modelling and simulation techniques is key to a more comprehensive, better targeted NAFLD research strategy. Such an approach will accelerate the progress of research and vital translation into clinic.
Keywords: Non-alcoholic fatty liver disease, Network, Metabolism, Systems biology, Modelling, Regulation, Simulation
Core tip: Research into non-alcoholic fatty liver disease (NAFLD) is dominated by human clinical studies and the use of animal models. We postulate that the wider use of systems biology approaches, incorporating novel modelling and simulation strategies, will yield greater insights into the mechanisms underlying NAFLD progression. Such insights are essential to the development of non-invasive diagnostic tools and novel therapies.
NON-ALCOHOLIC FATTY LIVER DISEASE
Non-alcoholic fatty liver disease (NAFLD) is considered to be the hepatic manifestation of the metabolic syndrome and a leading cause of liver-related morbidity and mortality in both adult and paediatric patients[1,2]. The metabolic syndrome is associated with abdominal obesity, high blood pressure, hypertriglyceridemia, hyperglycaemia and low high-density lipoprotein cholesterol levels[3]. Both the metabolic syndrome and NAFLD are associated with increased risk of cardiovascular disease (CVD) and type II diabetes[4], leading to increased morbidity and mortality. Importantly, NAFLD is a growing public health issue, with diagnosis increasing dramatically in the last two decades. NAFLD is now estimated to affect 20%-30% of adults in western populations, with an increased occurrence (70%-90%) in obese individuals, type II diabetics and has also been linked with type I diabetes[6,7].
NAFLD is a progressive inflammatory disease beginning with the macroscopic accumulation of fat in the liver (> 5%-10% by weight) in the absence of high alcohol intake or hepatitis[5,8], termed simple steatosis (SS) (Figure 1). SS causes no significant increase in liver related complications, and is widely accepted as a ‘benign’ adaptation to lipid loading in the liver. However, approximately 47% of individuals with SS will progress to non-alcoholic steatohepatitis (NASH), characterised by inflammatory infiltration of the liver and low-level fibrosis[9]. NASH is associated with a significantly increased risk of liver-related complications, hepatocellular carcinoma (HCC) and is an independent risk factor of CVD. Both SS and NASH are considered reversible through weight-loss, changes in diet and increased physical activity. However, approximately 25%-30% of individuals with NASH will develop irreversible fibrosis leading to cirrhosis, severe liver-related morbidity, high risk of HCC and the need for liver transplantation[10].
Figure 1.
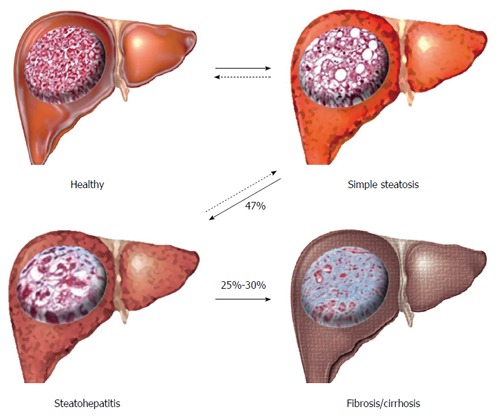
Progression and stages of non-alcoholic fatty liver disease. Livers, representing macroscopic changes with inserted micrographs of histological sections; collagen fibres stained with Masson’s trichrome stain (blue). Arrows represent disease progression/regression (dashed).
In addition to the increased morbidity and mortality, there is a high financial cost associated with overweight/obesity related diseases and their clinical management, estimated at £3.2bn per annum in the United Kingdom alone[11].
A major clinical challenge and research focus is the lack of robust non-invasive diagnostic tools for NAFLD. The population prevalence of the disease can still only be estimated since the only reliable method of disease staging and monitoring is though invasive liver biopsy. Although considered the gold standard, biopsy is prone to variations in both sampling and in evaluation by pathologists[12]. Ultrasonography and magnetic resonance imaging techniques are being developed for disease staging, but are presently unable to monitor/stage the inflammatory stages of the disease or require validation[7,13]. This lack of a reliable, non-invasive method for diagnosing, staging or monitoring NAFLD, means individuals are often only diagnosed incidentally, presenting with either elevated liver enzymes or a fatty liver on ultrasound in combination with the associated metabolic risk factors (e.g., abdominal obesity, type II diabetes). As such, diagnosis may be delayed, complicating treatment and damaging prognosis. In addition, a major concern to both patients and clinicians is the fact that following diagnosis, the most successful treatment options are limited to lifestyle changes. While statins, insulin-sensitizing drugs and bile acids have been investigated in NAFLD treatment, no effective, targeted drug therapies are currently available[14,15].
To develop novel biomarkers and effective therapeutic interventions, it is imperative to establish a better understanding of the mechanisms that underlie the pathology and progression of NAFLD. To this end, there has been an increasing research effort focused on this disease, with a greater than seven fold increase in the number of publications specifically investigating NAFLD and NASH in the last decade (Figure 2). Despite this increase however, our understanding of the molecular mechanisms underlying NAFLD pathogenesis remains modest.
Figure 2.
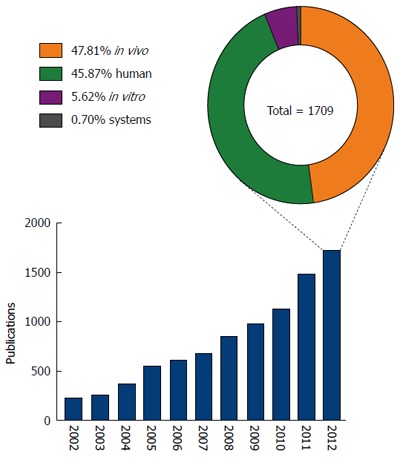
Non-alcoholic fatty liver disease publications and distribution of research. Graph shows the number of journal articles published (as listed on PubMed) between 2002 and 2012 focused on non-alcoholic fatty liver disease; pie chart shows the distribution of models/approaches (human, in vivo, in vitro, systems approach) used in research published during 2012.
Systems approaches have been widely adopted in many fields of biological research, but nutritional sciences does still not exploit the full potential of the tools available[16,17]. Currently, NAFLD research is dominated by the use of human patient studies and in vivo animal models, predominantly mouse and rat (Figure 2). Irrespective of the species used, in vivo animal models can be divided into three categories; feeding models, gene deficient/knockout models and combinations models; these have been extensively reviewed in the literature[18,19]. Research using in vitro models and/or systems biology approaches represented less than 7% of published NAFLD research in 2012. The relatively low usage of in vitro models may reflect a concern that in vitro approaches cannot accurately reproduce complex disease phenotypes such as NAFLD. However, such a belief ignores the recent advances in cell culture techniques allowing the enhancement of the ‘in vivo like’ phenotypes in vitro. Improved culturing of hepatocytes, and the other cell types in the liver, continues to be discussed in the literature and is an active area of method development[18,20,21]. For example, three-dimensional cell culture strategies can now greatly enhance the way that cells polarise in culture, a feature vital to the function of hepatocytes in vivo[22-24]. The use of co-culture experiments permits analyses of cell-cell signalling and metabolic networks,in particular between the different cell types that comprise the liver, other tissues or even with the gut microflora[18,25,26]. In addition to a limited use of in vitro techniques to study NAFLD, the negligible use of systems biology tools grossly under represents the ever-increasing array of tools for studying such cell networks. Within recent years there has been an explosion in systems biology tools for the study of complex biological networks[27-34]. Applied to the human organism, it has already proven to be an invaluable tool in understanding disease etiology, drug discovery and toxicology[35-39]. Indeed, systems tools have already begun to offer insight into the mechanisms of fatty liver disease; Sookoian et al[40] used a combination of data mining and network analysis tools to identify shared mechanistic pathways between non-alcoholic and alcoholic-fatty liver disease. While this generated novel insights, it only begins to scratch the surface of available in silico modelling and network simulation tools.
Given the impact that systems approaches have made in the understanding and treatment of other diseases, we posit that an increased application of these approaches towards NAFLD would reap similar rewards.
Systems biology approaches
Systems biology should not be considered as a discipline in and of itself, or merely as the computational branch of molecular biology. Systems biology is an approach to studying biological systems as a whole. Under pinning this is the principle that the key to understanding and predicting the behaviour of any biological systems and its response to the environment is the understanding of the networks that determine these responses[16,41,42]. Understanding how all the different cellular and tissue networks are interconnected in the context of the whole (human) organism and be able to predict network behaviours, is the ultimate goal of systems biology. However, work in this burgeoning field has typically considered either metabolic, signalling or gene regulatory networks independently.
Biological research has classically adopted a reductionist approach to the study of biological networks, identifying their component parts and the interactions between them that account for the observed properties of the sub-system under study. This ‘bottom-up’ approach is based on the assumption that a system, regardless of its size and complexity, is the sum of its component parts. The emergence of the high throughput, ‘‘omics’’ technologies, has generated the data needed to describe networks on a much larger scale than previously possible. This has led to the development of “top-down” modelling approaches that infer relationships between network components that change in the same way following perturbation of the system of interest.
The issue with the ‘bottom-up’ approach is that in order to simulate a network, intimate knowledge of its components, their connectivity and interaction is required. To model quantitatively, kinetic parameters must be painstakingly determined for each reaction and interaction in a given system. In many cases, such data is unavailable and often impossible to determine accurately with current experimental technologies. Furthermore, to determine such parameters for large-scale networks would be exorbitantly costly in terms of time and resources. On the other hand, the difficulty with the ‘top down’ approach is that while it allows us to study networks at a much larger scale, it does so at the cost of quantitative detail. While coverage of a given network is superior in terms of network coverage and data points generated, any insight into the mechanisms mediating the observed behaviour of the system is purely inferential. The precise nature of any mechanistic interactions would need to be confirmed through more targeted experimental, ‘bottom-up’ methods. Considering the limitations of these two approaches, a more comprehensive strategy is to combine both into a ‘‘middle-out’’ approach[43,44]. In such an approach, the best ‘‘bottom-up’’ models would be integrated with larger ‘‘top-down’’ models generated through high throughput ‘‘-omic’’ datasets, generating a global model with improved predictivity due to the constraints imposed through highly detailed quantitative information.
Strategies for modelling the liver in silico
Second to the brain, the liver is, arguably, the most complicated organ in the human body, with an array of diverse and essential functions. Macroscopically, the human liver is described as four lobes; the right, left, quadrate and caudate. Despite the livers diverse functionality, the microscopic architecture of the liver is remarkably uniform, regardless of the direction from which the tissue is sectioned. This uniformity of structure makes understanding and modelling the architecture of the liver, relatively, easier. Indeed, there has been some excellent progress in modelling the structural architecture and microvasculature in silico[45]. Although the microscopic architecture of the liver may be uniform, the functional behaviour across the liver is not[46]. This varies greatly with blood/flow, oxygenation, cell polarity and is dynamically regulated in response to extracellular and intracellular cues. The hepatocyte is the primary cell type of the liver, accounting for 60% of the cells in the liver, 80% of the parenchymal volume and responsible for the synthesis of 90% of expressed liver proteins[24,47]. However, it is important to remember that there are many other cell types within the liver (e.g., hepatic stellate cells, kupffer cells), all of which contribute to the overall functional behaviour of the liver. Thus, to fully understand liver function (and dysfunction) it is important to study the interrelationship of these cells types.
Modelling of liver functionality at either the macroscopic and microscopic scale is the aim of several groups around the world, with perhaps the largest consortium being the German-based virtual liver network (VLN), which aims to model the liver at all spatial, temporal, metabolic and regulatory levels utilising a range of experimental and computational approaches[22,48]. The potential impact of such tissue-level models can be seen in the successes of similar projects, such as the virtual heart that feed into the virtual physiological human (VPH) initiative[41,49].
Genome-scale networks
The sequencing and functional annotation of genomes has provided the foundation for ‘reconstruction’ of biological networks at the genome-scale. This reconstruction process has been described as a both a ‘top-down’ and a ‘bottom-up’ approach, however, it is more accurately described as a ‘middle-out’ process, incorporating data from multiple sources to reconstruct the best possible network model. In principle, genome-scale network reconstruction is possible for all biological networks; signalling, gene-regulatory or metabolic[50]. However, the overwhelming majority of large-scale reconstructions that have been generated have been genome scale metabolic networks (GSMN). This has been due, in part, to the fact that large datasets generated from high-throughput techniques in transcriptomics, proteomics and metabolomics can all be applied to the reconstruction of metabolic networks, as well as the extensive historical biochemical data generated through the study of metabolic reactions[31,51].
Essentially, GSMN reconstructions are comprehensive structured databases of the metabolic reactions and metabolites of a given organism. The database is first built based on the annotated genome sequence, but this is then supplemented with knowledge of biochemical reactions, physiological data and high-throughput datasets. Until relatively recently, metabolic network reconstruction has been predominately of single-cell organisms due to the availability and relative simplicity of their genomes; the Escherichia coli (K12-strain) genome was first published in 1997 and contains approximately 4000 genes[52], while the human genome sequence wasn’t completed until 2003 and encodes approximately 21000 protein coding genes[53]. The process of network reconstruction is both labour and resource intensive, the time required increasing with the size and complexity of the target organism’s genome. For instance, Recon1, one of the first published reconstructions of the human metabolic network, took a team of 6-8 individuals two years to complete[31,54]. Currently, it is not possible to completely automate the reconstruction of high quality networks. Moreover, since reconstructions begin with an annotated genome sequence of the target organism, it is important to note these annotated genome sequences use a combination of manual and automated curation, are incomplete and are continuously updated[55]. Indeed, our understanding of the function of much of the genome is evolving and an area of continued intensive research. The result of this is that GSMN reconstruction is an iterative process requiring extensive manual curation.
Metabolism is of particular interest because its disruption may be the cause or as a result of a given disease state. As such, metabolites are commonly used as biomarkers for disease diagnosis and monitoring; for example high blood glucose levels in diabetes or cholesterol as a marker of increased CVD risk[42,51]. Ironically no such biomarkers have been validated for NAFLD despite its extensive disruption of metabolism however, promising metabolomics research is on-going[56]. Two major global reconstructions of human metabolism were published in 2007; Recon1 and The Edinburgh model[54,57,58]. Recently, Recon1 has been extensively updated, Recon2, through a collaborative effort to generate the best possible human GSMN[59]. There has also been reconstruction of a number of mouse GSMNs[60-62], important for the translation of data from this widely exploited model species back into humans. The obvious issue with these models is that they are based upon the entire genome and are not tissue-specific. Automated computational methods have been proposed using transcriptomic datasets to tailor these whole-organism models into tissue-specific GSMN reconstructions[63-65]. While this approach can generate working models, manual curation remains essential to reconstruct high quality networks. Table 1 shows a summary of published human GSMNs and tissue specific metabolic networks; this is not intended as an exhaustive list and does not include work by groups generating numerous cell and tissue specific networks using computational reconstruction methods[59,64,66].
Table 1.
Published human and tissue/cell specific metabolic network reconstructions
| Human metabolic networks | Ref. |
| Recon1 | [54] |
| EHMN (Edinburgh human metabolic network) | [57] |
| HumanCyc | [67] |
| Recon2 | [59] |
| KEGG | [68] |
| Tissue/cell type | |
| Liver/hepatocyte | [69,70] |
| Brain | [71,72] |
| Kidney | [72-74] |
| Heart | [72,75,76] |
| Erythrocytes | [77] |
| Alveolar macrophages | [78] |
| Adipocyte | [79] |
| Muscle | [72] |
| Placenta | [72] |
| Lung | [72] |
| Pancreas | [72] |
| Testis | [72] |
| Spleen | [72] |
| Ovary | [72] |
| Prostate | [72] |
| Colon | [72] |
| Small intestine | [72] |
| Thymus | [72] |
| Skeletal muscle | [65] |
Two liver/hepatocyte specific GSMNs were published concurrently: one employing an automated reconstruction strategy, the other an exhaustive manual curation of transcript, protein, biochemical and physiological data, HepatoNet1; a comprehensive reconstruction of hepatocyte metabolism. Comprised of 2539 reactions and 777 individual metabolites, this model has the potential to be a tremendous resource for NAFLD research. As well as the liver itself, a number of other tissues of particular relevance to NAFLD (i.e., heart, adipocyte, pancreas, intestine, kidney) have been published increasing the potential for GSMNs to impact NAFLD research. Methods allowing the interconnectivity of metabolism between tissues to be explored have already been developed[71]. Bordbar et al[80] integrated the metabolic networks of the human adipocyte, hepatocyte and myocytes integrating data from obese and obese type II diabetics.
Analysis of metabolic network reconstructions
Although reconstruction itself can provide insight into the properties of a network, the biologist wants to be able to understand and ultimately predict, the impact of perturbations on the system through simulations. Typically for GSMNs this has been done through constraint-based flux balance analysis (FBA), a fundamental tool for interrogating metabolic networks. In short, FBA solves the mathematical, stoichiometrically balanced, expression of a metabolic network reconstruction constrained based on experimental data sets[81]. For example, a GSMN reconstruction may be constrained using “omics” data to reflect the gene-expression profile of a specific tissue or of a specific gene knockout (i.e., the reaction is constrained to 0). However, as a direct result of its underlying assumptions FBA has some inherent disadvantages that are of critical importance if we are to gain insight into liver disease. The development of FBA has been in the context of studying single cell organisms; mathematically, FBA defines an ‘‘objective function’’, essentially a sum of reactions that is solved to optimise flux towards a biological output, characteristic of the organism under study. For microorganisms this is commonly defined as an increase in ‘‘biomass’’ (i.e., growth). However, healthy mammalian adult organs typically maintain tissue size and density; although cells divide and turnover ‘‘biomass’’ is maintained. Moreover, definition of a single objective function for a tissue as functionally diverse as the liver is almost impossible. Furthermore, basic FBA assumes that gene expression is at steady-state and is unable to account for dynamic gene regulatory responses to environmental perturbation. Given the numerous functions of the liver and the fact that these multiple functions occur simultaneously and are dynamically regulated in response to the environment, the scope of standard FBA for NAFLD research is limited.
Modelling regulation of the metabolic network
Broadly, the modelling of signalling networks can be divided into two classes; the first analyses the structure and connectivity of the network to provide insight into its dynamics. The second uses experimentally determined kinetic parameters as well as network connectivity to model dynamic properties of the network[28,50]. It has been suggested that the only possible way to successfully model the regulatory mechanisms of human whole cell metabolism is through the use of fully mechanistic/kinetic models[22]. While a number of kinetic models have been published, these have been of small sub-networks of metabolism. The quantity of data that would be required, to generate such a model on a whole-cell scale would require an untenable amount of computational and financial resources.
A number of modified FBA approaches have been published attempting to address some of its limitations and enhance its suitability for simulating dynamic mammalian networks. Currently, qualitative models of regulatory processes can be integrated with FBA of metabolic networks using a quasi-steady-state simulation method. This ‘regulatory FBA’ (rFBA) uses Boolean rules to express the regulatory relationships between the genes of a network[82]. This allows representation of gene expression/repression, but does not represent transcription and translation processes or mechanistic details about the nature of regulatory molecules, such as protein, RNA, protein complexes, post translational modification. Although rFBA allows the simulation of gene regulation, this simulation is synchronous with all events in the regulatory network occurring at the same rate. Furthermore, the results of rFBA, as with conventional FBA are deterministic, producing a deterministic network solution at every time-step of the simulation. Two additional approaches, integrated FBA (iFBA)[83] and integrated dynamic FBA (idFBA)[84], utilize ordinary differential equations for simulation of sub-networks (e.g., signalling pathways) where kinetic quantitative data are available. However the use of these tools for human systems is limited given both the increased scale of genome complexity and the lack of such quantitative data.
We have recently published a novel strategy quasi steady state Petri nets (QSSPN)[29]. This approach uses Petri nets (PNs)[85] to represent gene regulatory and signalling networks and couples this to FBA of GSMNs, extending the size and the detail at which models that can be qualitatively simulated. Although PNs have previously been used to model cell signalling[86], this is the first time that it has been coupled to FBA allowing dynamic regulation of metabolism at the genome scale. The use of PNs readily allows the representation of gene expression at the level of the genome, transcript and protein level, using existing conventions, and permits incorporation of kinetic parameters across the model as they become available. Our approach also addresses the issue of single deterministic results. The QSSPN method incorporates stochastic elements that allow non-deterministic simulation and Monte Carlo sampling of multiple simulation outcomes. Simply put, this allows the researcher to predict the probability of a particular behaviour or whether a particular behaviour is possible within the model. Unlike rFBA, QSSPN also permits multiple activation levels and variable rates of activation across the signalling network. As a result, the signalling network is modelled asynchronously, a fundamental property of biological systems and key to the modelling of oscillations, activation and repression of constitutively expressed genes. We validated the QSSPN strategy modelling regulation of bile acid metabolism and are currently modelling the network response to lipid loading in hepatocytes. The ability to computationally examine genotype-phenotype relationships and generate predict probable system behaviours resulting from network dysregulation, promises novel insight into NAFLD pathogenesis in the future.
CONCLUSION
GSMNs represent current state of the art in modelling large-scale networks and studying complex interactions associated with disease. Efforts by ourselves and others aim to model the liver at all spatial, temporal, metabolic and regulatory levels utilising a range of experimental and computational approaches in an overall systems approach. Increasingly the ‘low hanging fruit’ of biomarker and therapeutic target discovery has been picked and the identification of future targets will require a more comprehensive understanding of the underlying molecular mechanisms. This can only be achieved through the use of advanced strategies combining novel computational approaches and experimental techniques. While systems biologists aim to have computationally modelled 90% of the human organism by 2038[38], NAFLD represents a complex, multifactorial and urgent disease burden, the elucidation of which would benefit from existing systems tools rather than a solely reductionist approach. We believe that a ‘middle-out’ approach incorporating an iterative cycle of model reconstruction, simulation, experimental design and model refinement represents the best strategy to research in NAFLD and metabolic disease. We would further argue that such a strategy is best suited, not to purely kinetic modelling strategies, but to novel in silico modelling approaches that can model metabolism, gene expression, regulation and signalling at a qualitative level, but are capable of integrating kinetic parameters as available. NAFLD and the metabolic syndrome emerge as the product of a myriad of genetic, dietary and environmental factors and understanding the relative and mechanistic contribution of these is vital. These are the diseases of the 21st century and we will require 21st century approaches to develop our understanding and subsequent treatments.
Footnotes
Supported by The Biotechnology and Biological Sciences Research Council, No. BB/I008195/1
P- Reviewer: He FC S- Editor: Zhai HH L- Editor: A E- Editor: Ma S
References
- 1.Pacifico L, Nobili V, Anania C, Verdecchia P, Chiesa C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J Gastroenterol. 2011;17:3082–3091. doi: 10.3748/wjg.v17.i26.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore JB. Non-alcoholic fatty liver disease: the hepatic consequence of obesity and the metabolic syndrome. Proc Nutr Soc. 2010;69:211–220. doi: 10.1017/S0029665110000030. [DOI] [PubMed] [Google Scholar]
- 3.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 4.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Järvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 5.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 6.Targher G, Bertolini L, Padovani R, Rodella S, Zoppini G, Pichiri I, Sorgato C, Zenari L, Bonora E. Prevalence of non-alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. J Hepatol. 2010;53:713–718. doi: 10.1016/j.jhep.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 7.al-Majed SA, al-Momen AK, al-Kassimi FA, al-Zeer A, Kambal AM, Baaqil H. Tuberculosis presenting as immune thrombocytopenic purpura. Acta Haematol. 1995;94:135–138. doi: 10.1159/000203995. [DOI] [PubMed] [Google Scholar]
- 8.Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:E462–E468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 9.Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, Ratziu V. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol. 2013;59:550–556. doi: 10.1016/j.jhep.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 10.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allender S, Rayner M. The burden of overweight and obesity-related ill health in the UK. Obes Rev. 2007;8:467–473. doi: 10.1111/j.1467-789X.2007.00394.x. [DOI] [PubMed] [Google Scholar]
- 12.Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. Liver biopsy. Hepatology. 2009;49:1017–1044. doi: 10.1002/hep.22742. [DOI] [PubMed] [Google Scholar]
- 13.Fierbinteanu-Braticevici C, Dina I, Petrisor A, Tribus L, Negreanu L, Carstoiu C. Noninvasive investigations for non alcoholic fatty liver disease and liver fibrosis. World J Gastroenterol. 2010;16:4784–4791. doi: 10.3748/wjg.v16.i38.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao J, Guo R, Fung ML, Liong EC, Tipoe GL. Therapeutic approaches to non-alcoholic fatty liver disease: past achievements and future challenges. Hepatobiliary Pancreat Dis Int. 2013;12:125–135. doi: 10.1016/s1499-3872(13)60021-1. [DOI] [PubMed] [Google Scholar]
- 15.Dowman JK, Armstrong MJ, Tomlinson JW, Newsome PN. Current therapeutic strategies in non-alcoholic fatty liver disease. Diabetes Obes Metab. 2011;13:692–702. doi: 10.1111/j.1463-1326.2011.01403.x. [DOI] [PubMed] [Google Scholar]
- 16.de Graaf AA, Freidig AP, De Roos B, Jamshidi N, Heinemann M, Rullmann JA, Hall KD, Adiels M, van Ommen B. Nutritional systems biology modeling: from molecular mechanisms to physiology. PLoS Comput Biol. 2009;5:e1000554. doi: 10.1371/journal.pcbi.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore JB, Weeks ME. Proteomics and systems biology: current and future applications in the nutritional sciences. Adv Nutr. 2011;2:355–364. doi: 10.3945/an.111.000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozkalemkas F, Ali R, Ozkan A, Ozcelik T, Ozkocaman V, Kunt-Uzaslan E, Bahadir-Erdogan B, Akalin H. Tuberculosis presenting as immune thrombocytopenic purpura. Ann Clin Microbiol Antimicrob. 2004;3:16. doi: 10.1186/1476-0711-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klingmüller U, Bauer A, Bohl S, Nickel PJ, Breitkopf K, Dooley S, Zellmer S, Kern C, Merfort I, Sparna T, et al. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst Biol (Stevenage) 2006;153:433–447. doi: 10.1049/ip-syb:20050067. [DOI] [PubMed] [Google Scholar]
- 21.Pan C, Kumar C, Bohl S, Klingmueller U, Mann M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol Cell Proteomics. 2009;8:443–450. doi: 10.1074/mcp.M800258-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Böttger J, et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol. 2013;87:1315–1530. doi: 10.1007/s00204-013-1078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selden C, Shariat A, McCloskey P, Ryder T, Roberts E, Hodgson H. Three-dimensional in vitro cell culture leads to a marked upregulation of cell function in human hepatocyte cell lines--an important tool for the development of a bioartificial liver machine. Ann N Y Acad Sci. 1999;875:353–363. doi: 10.1111/j.1749-6632.1999.tb08517.x. [DOI] [PubMed] [Google Scholar]
- 24.Decaens C, Durand M, Grosse B, Cassio D. Which in vitro models could be best used to study hepatocyte polarity? Biol Cell. 2008;100:387–398. doi: 10.1042/BC20070127. [DOI] [PubMed] [Google Scholar]
- 25.Gebhardt R, Hengstler JG, Müller D, Glöckner R, Buenning P, Laube B, Schmelzer E, Ullrich M, Utesch D, Hewitt N, et al. New hepatocyte in vitro systems for drug metabolism: metabolic capacity and recommendations for application in basic research and drug development, standard operation procedures. Drug Metab Rev. 2003;35:145–213. doi: 10.1081/dmr-120023684. [DOI] [PubMed] [Google Scholar]
- 26.Krause P, Saghatolislam F, Koenig S, Unthan-Fechner K, Probst I. Maintaining hepatocyte differentiation in vitro through co-culture with hepatic stellate cells. In Vitro Cell Dev Biol Anim. 2009;45:205–212. doi: 10.1007/s11626-008-9166-1. [DOI] [PubMed] [Google Scholar]
- 27.Gevorgyan A, Bushell ME, Avignone-Rossa C, Kierzek AM. SurreyFBA: a command line tool and graphics user interface for constraint-based modeling of genome-scale metabolic reaction networks. Bioinformatics. 2011;27:433–434. doi: 10.1093/bioinformatics/btq679. [DOI] [PubMed] [Google Scholar]
- 28.Ruths D, Nakhleh L, Ram PT. Rapidly exploring structural and dynamic properties of signaling networks using PathwayOracle. BMC Syst Biol. 2008;2:76. doi: 10.1186/1752-0509-2-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher CP, Plant NJ, Moore JB, Kierzek AM. QSSPN: dynamic simulation of molecular interaction networks describing gene regulation, signalling and whole-cell metabolism in human cells. Bioinformatics. 2013;29:3181–3190. doi: 10.1093/bioinformatics/btt552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao YC, Tsai MH, Chen FC, Hsiung CA. GEMSiRV: a software platform for GEnome-scale metabolic model simulation, reconstruction and visualization. Bioinformatics. 2012;28:1752–1758. doi: 10.1093/bioinformatics/bts267. [DOI] [PubMed] [Google Scholar]
- 31.Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Zhu X, Chen JY. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput Biol. 2009;5:e1000450. doi: 10.1371/journal.pcbi.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 34.Bois FY. GNU MCSim: Bayesian statistical inference for SBML-coded systems biology models. Bioinformatics. 2009;25:1453–1454. doi: 10.1093/bioinformatics/btp162. [DOI] [PubMed] [Google Scholar]
- 35.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Côté S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486:361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolodkin A, Sahin N, Phillips A, Hood SR, Bruggeman FJ, Westerhoff HV, Plant N. Optimization of stress response through the nuclear receptor-mediated cortisol signalling network. Nat Commun. 2013;4:1792. doi: 10.1038/ncomms2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutta B, Pusztai L, Qi Y, André F, Lazar V, Bianchini G, Ueno N, Agarwal R, Wang B, Shiang CY, et al. A network-based, integrative study to identify core biological pathways that drive breast cancer clinical subtypes. Br J Cancer. 2012;106:1107–1116. doi: 10.1038/bjc.2011.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolodkin A, Boogerd FC, Plant N, Bruggeman FJ, Goncharuk V, Lunshof J, Moreno-Sanchez R, Yilmaz N, Bakker BM, Snoep JL, et al. Emergence of the silicon human and network targeting drugs. Eur J Pharm Sci. 2012;46:190–197. doi: 10.1016/j.ejps.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Xie L, Wang J, Bourne PE. In silico elucidation of the molecular mechanism defining the adverse effect of selective estrogen receptor modulators. PLoS Comput Biol. 2007;3:e217. doi: 10.1371/journal.pcbi.0030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sookoian S, Pirola CJ. Systems biology elucidates common pathogenic mechanisms between nonalcoholic and alcoholic-fatty liver disease. PLoS One. 2013;8:e58895. doi: 10.1371/journal.pone.0058895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohl P, Noble D. Systems biology and the virtual physiological human. Mol Syst Biol. 2009;5:292. doi: 10.1038/msb.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panagiotou G, Nielsen J. Nutritional systems biology: definitions and approaches. Annu Rev Nutr. 2009;29:329–339. doi: 10.1146/annurev-nutr-080508-141138. [DOI] [PubMed] [Google Scholar]
- 43.Walker DC, Southgate J. The virtual cell--a candidate co-ordinator for ‘middle-out’ modelling of biological systems. Brief Bioinform. 2009;10:450–461. doi: 10.1093/bib/bbp010. [DOI] [PubMed] [Google Scholar]
- 44.Dobson PD, Lanthaler K, Oliver SG, Kell DB. Implications of the dominant role of transporters in drug uptake by cells. Curr Top Med Chem. 2009;9:163–181. doi: 10.2174/156802609787521616. [DOI] [PubMed] [Google Scholar]
- 45.Hoehme S, Brulport M, Bauer A, Bedawy E, Schormann W, Hermes M, Puppe V, Gebhardt R, Zellmer S, Schwarz M, et al. Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc Natl Acad Sci USA. 2010;107:10371–10376. doi: 10.1073/pnas.0909374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braeuning A, Ittrich C, Köhle C, Hailfinger S, Bonin M, Buchmann A, Schwarz M. Differential gene expression in periportal and perivenous mouse hepatocytes. FEBS J. 2006;273:5051–5061. doi: 10.1111/j.1742-4658.2006.05503.x. [DOI] [PubMed] [Google Scholar]
- 47.Malarkey DE, Johnson K, Ryan L, Boorman G, Maronpot RR. New insights into functional aspects of liver morphology. Toxicol Pathol. 2005;33:27–34. doi: 10.1080/01926230590881826. [DOI] [PubMed] [Google Scholar]
- 48.Holzhütter HG, Drasdo D, Preusser T, Lippert J, Henney AM. The virtual liver: a multidisciplinary, multilevel challenge for systems biology. Wiley Interdiscip Rev Syst Biol Med. 2012;4:221–235. doi: 10.1002/wsbm.1158. [DOI] [PubMed] [Google Scholar]
- 49.Noble D. Modelling the heart: insights, failures and progress. Bioessays. 2002;24:1155–1163. doi: 10.1002/bies.10186. [DOI] [PubMed] [Google Scholar]
- 50.Papin JA, Hunter T, Palsson BO, Subramaniam S. Reconstruction of cellular signalling networks and analysis of their properties. Nat Rev Mol Cell Biol. 2005;6:99–111. doi: 10.1038/nrm1570. [DOI] [PubMed] [Google Scholar]
- 51.Ma H, Goryanin I. Human metabolic network reconstruction and its impact on drug discovery and development. Drug Discov Today. 2008;13:402–408. doi: 10.1016/j.drudis.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 53.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 54.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BØ. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siegert S, Yu Z, Wang-Sattler R, Illig T, Adamski J, Hampe J, Nikolaus S, Schreiber S, Krawczak M, Nothnagel M, et al. Diagnosing fatty liver disease: a comparative evaluation of metabolic markers, phenotypes, genotypes and established biomarkers. PLoS One. 2013;8:e76813. doi: 10.1371/journal.pone.0076813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma H, Sorokin A, Mazein A, Selkov A, Selkov E, Demin O, Goryanin I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol. 2007;3:135. doi: 10.1038/msb4100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hao T, Ma HW, Zhao XM, Goryanin I. Compartmentalization of the Edinburgh Human Metabolic Network. BMC Bioinformatics. 2010;11:393. doi: 10.1186/1471-2105-11-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quek LE, Nielsen LK. On the reconstruction of the Mus musculus genome-scale metabolic network model. Genome Inform. 2008;21:89–100. [PubMed] [Google Scholar]
- 61.Selvarasu S, Karimi IA, Ghim GH, Lee DY. Genome-scale modeling and in silico analysis of mouse cell metabolic network. Mol Biosyst. 2010;6:152–161. doi: 10.1039/b912865d. [DOI] [PubMed] [Google Scholar]
- 62.Sigurdsson MI, Jamshidi N, Steingrimsson E, Thiele I, Palsson BØ. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shlomi T, Cabili MN, Herrgård MJ, Palsson BØ, Ruppin E. Network-based prediction of human tissue-specific metabolism. Nat Biotechnol. 2008;26:1003–1010. doi: 10.1038/nbt.1487. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Eddy JA, Price ND. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst Biol. 2012;6:153. doi: 10.1186/1752-0509-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008;4:e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agren R, Bordel S, Mardinoglu A, Pornputtapong N, Nookaew I, Nielsen J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput Biol. 2012;8:e1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005;6:R2. doi: 10.1186/gb-2004-6-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jerby L, Shlomi T, Ruppin E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol Syst Biol. 2010;6:401. doi: 10.1038/msb.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gille C, Bölling C, Hoppe A, Bulik S, Hoffmann S, Hübner K, Karlstädt A, Ganeshan R, König M, Rother K, et al. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol Syst Biol. 2010;6:411. doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lewis NE, Schramm G, Bordbar A, Schellenberger J, Andersen MP, Cheng JK, Patel N, Yee A, Lewis RA, Eils R, et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat Biotechnol. 2010;28:1279–1285. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hao T, Ma HW, Zhao XM, Goryanin I. The reconstruction and analysis of tissue specific human metabolic networks. Mol Biosyst. 2012;8:663–670. doi: 10.1039/c1mb05369h. [DOI] [PubMed] [Google Scholar]
- 73.Chang RL, Xie L, Xie L, Bourne PE, Palsson BØ. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol. 2010;6:e1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang AD, Dai SX, Huang JF. Reconstruction and analysis of human kidney-specific metabolic network based on omics data. Biomed Res Int. 2013;2013:187509. doi: 10.1155/2013/187509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karlstädt A, Fliegner D, Kararigas G, Ruderisch HS, Regitz-Zagrosek V, Holzhütter HG. CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst Biol. 2012;6:114. doi: 10.1186/1752-0509-6-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Y, Huang J. Reconstruction and analysis of human heart-specific metabolic network based on transcriptome and proteome data. Biochem Biophys Res Commun. 2011;415:450–454. doi: 10.1016/j.bbrc.2011.10.090. [DOI] [PubMed] [Google Scholar]
- 77.Bordbar A, Jamshidi N, Palsson BO. iAB-RBC-283: A proteomically derived knowledge-base of erythrocyte metabolism that can be used to simulate its physiological and patho-physiological states. BMC Syst Biol. 2011;5:110. doi: 10.1186/1752-0509-5-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bordbar A, Lewis NE, Schellenberger J, Palsson BØ, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mardinoglu A, Agren R, Kampf C, Asplund A, Nookaew I, Jacobson P, Walley AJ, Froguel P, Carlsson LM, Uhlen M, et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol Syst Biol. 2013;9:649. doi: 10.1038/msb.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bordbar A, Feist AM, Usaite-Black R, Woodcock J, Palsson BO, Famili I. A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst Biol. 2011;5:180. doi: 10.1186/1752-0509-5-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Covert MW, Schilling CH, Palsson B. Regulation of gene expression in flux balance models of metabolism. J Theor Biol. 2001;213:73–88. doi: 10.1006/jtbi.2001.2405. [DOI] [PubMed] [Google Scholar]
- 83.Covert MW, Xiao N, Chen TJ, Karr JR. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24:2044–2050. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee JM, Gianchandani EP, Eddy JA, Papin JA. Dynamic analysis of integrated signaling, metabolic, and regulatory networks. PLoS Comput Biol. 2008;4:e1000086. doi: 10.1371/journal.pcbi.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rohr C, Marwan W, Heiner M. Snoopy--a unifying Petri net framework to investigate biomolecular networks. Bioinformatics. 2010;26:974–975. doi: 10.1093/bioinformatics/btq050. [DOI] [PubMed] [Google Scholar]
- 86.Ruths D, Muller M, Tseng JT, Nakhleh L, Ram PT. The signaling petri net-based simulator: a non-parametric strategy for characterizing the dynamics of cell-specific signaling networks. PLoS Comput Biol. 2008;4:e1000005. doi: 10.1371/journal.pcbi.1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]