

Abstract
AIM: To investigate the effects of N-acetylcysteine (NAC) on endoplasmic reticulum (ER) stress and tissue injury during liver ischemia reperfusion injury (IRI).
METHODS: Mice were injected with NAC (300 mg/kg) intraperitoneally 2 h before ischemia. Real-time polymerase chain reaction and western blotting determined ER stress molecules (GRP78, ATF4 and CHOP). To analyze the role of NAC in reactive oxygen species (ROS)-mediated ER stress and apoptosis, lactate dehydrogenase (LDH) was examined in cultured hepatocytes treated by H2O2 or thapsigargin (TG).
RESULTS: NAC treatment significantly reduced the level of ROS and attenuated ROS-induced liver injury after IRI, based on glutathione, malondialdehyde, serum alanine aminotransferase levels, and histopathology. ROS-mediated ER stress was significantly inhibited in NAC-treated mice. In addition, NAC treatment significantly reduced caspase-3 activity and apoptosis after reperfusion, which correlated with the protein expression of Bcl-2 and Bcl-xl. Similarly, NAC treatment significantly inhibited LDH release from hepatocytes treated by H2O2 or TG.
CONCLUSION: This study provides new evidence for the protective effects of NAC treatment on hepatocytes during IRI. Through inhibition of ROS-mediated ER stress, NAC may be critical to inhibit the ER-stress-related apoptosis pathway.
Keywords: N-acetylcysteine, Reactive oxygen species, Endoplasmic reticulum stress, Apoptosis, Liver ischemia-reperfusion
Core tip: The protective effect of N-acetylcysteine (NAC) treatment has been confirmed in IRI; however, its underlying mechanism remains unclear. Our previous data showed that endoplasmic reticulum (ER) stress is critical for the development of liver IRI. We have found new evidence for the protective effects of NAC on hepatocytes during liver IRI. Our data demonstrated that NAC modulates ER stress signaling and reduces ER-stress-mediated apoptosis during liver IRI. These findings provide further support for the usefulness of exploring NAC as a potential alternative drug for treating liver IRI.
INTRODUCTION
Ischemia reperfusion injury (IRI) is an important inducing factor in liver damage and failure in various clinical settings, such as hepatic trauma, resection, liver transplantation and circulatory shock[1-4]. ER stress mediates liver injury during liver IRI[5,6]. ER, as an intracellular organelle, plays a key role in protein synthesis, folding, assembly and transportation. Upset in any balances of the ER causes ER stress. Generally, ER stress can give rise to an unfolded protein response (UPR), which tries to reduce ER stress by inhibiting protein synthesis, increasing chaperones, and decomposing misfolded proteins[7-10]. However, excess or prolonged UPR results in apoptosis as a cytoprotective response in stressed cells[9,10].
Reactive oxygen species (ROS) play a prominent causative role in liver damage following IR[11,12]. ROS can start the cascade of cell damage, necrosis/apoptosis, and subsequent pro-inflammatory responses. Recently, studies have shown that ROS can trigger ER stress in vivo and in vitro[13,14]. Here, we hypothesized that ROS-induced ER stress results in cell death during liver IRI.
N-acetylcysteine (NAC), an antioxidant drug, is an inexpensive and simple water-soluble molecule that has been used as a treatment for congestive and obstructive lung diseases for nearly 50 years. Since the mid-1970s, it has also been used to treat paracetamol intoxication[15]. In addition, it has been used to treat hepatitis and acute liver failure, in which it protects the liver by increasing hepatic glutathione (GSH), which serves as a substrate to inactivate or combine with foreign substances[16,17]. Indeed, NAC has beneficial effects on IRI in the central nervous system, heart, kidney and liver[18-21]. To date, however, no data are available on the effects of NAC on ROS-induced ER stress during IRI. This study was designed to elucidate whether NAC inhibits ROS and ROS-induced ER stress during hepatic IRI.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice were purchased from the Laboratory Animal Resources of Nanjing Medical University (NJMU), China. The animals were fed a laboratory diet and kept under constant environmental conditions, with 12-h light-dark cycles. Procedures were carried out in accordance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee (IACUC) of NJMU (Protocol Number NJMU08-092) approved the animal protocol.
Surgical procedure and experimental design
We used a partial (70%) hepatic ischemia model. Midline laparotomy was performed under 10% chloral hydrate [0.3 g/kg, injected intraperitoneally (ip)] anesthesia in mice. In the IR group, all structures in the portal triad (hepatic artery, portal vein, and bile duct) to the left and median liver lobes were occluded with an atraumatic bulldog clamp for 90 min. The clamp was then removed to allow reperfusion. The abdomen was immediately closed with a continuous 4-0 silk suture. The mice were sacrificed 6 h after reperfusion, and blood, as well as liver tissue samples, were harvested for analysis. In the sham-operated group, mice were given anesthesia and subjected to laparotomy and exposure of the portal triad without hepatic ischemia. In the NAC treatment group, mice were injected ip with 300 mg/kg NAC for 2 h before the operation. NAC was purchased from Sigma-Aldrich (Shanghai, China) and diluted in saline to 3 mg/mL.
Serum biochemical analysis
Blood samples were centrifuged to obtain serum. The serum level of alanine aminotransferase (ALT) was measured to assess the extent of hepatocyte damage, using an automated chemical analyzer (Olympus Automated Chemistry Analyzer AU5400, Tokyo, Japan).
Histopathological study
Liver specimens were fixed with 10% neutral formaldehyde and embedded in paraffin. The specimens were sectioned at 4 μm and stained with hematoxylin and eosin. The sections were subjected to histopathological analysis by light microscopy. Sections were scored from 0 to 4 for sinusoidal congestion, vacuolization of hepatocyte cytoplasm, and parenchymal necrosis, as described by Suzuki et al[22].
GSH, malondialdehyde and caspase-3 activities
GSH, malondialdehyde (MDA) and caspase-3 activities were assessed in liver tissues. The activities were measured with a GSH, MDA and Caspase-3 assay kit (Jiancheng Biotechnology, Nanjing, China), according to the manufacturer’s instructions.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling staining
Paraffin sections (4 μm thick) were deparaffinized in toluene and dehydrated through a graded series of ethanol solutions. Transferase dUTP nick-end labeling (TUNEL) was used to stain the sections, using a commercially available kit (in situ Cell Death Detection Kit; Roche-Boehringer Mannheim, Germany).
Western blotting
Proteins were extracted from liver tissues subjected to ischemia or cell lysates, and the Bradford assay (Bio-Rad, Hercules, CA, United States) was used to determine the protein concentration. About 30 μg of the protein sample was resolved by SDS-PAGE and transferred to nitrocellulose membranes (Sunshine Biotechnology, China). These membranes were blocked in skimmed milk powder (5% w/v) with phosphate-buffered saline with 0.1% Tween 20 (PBS-T) at 4 °C overnight. Membranes were incubated with primary antibodies for GRP78, CHOP, BCL-2, BCL-xl, and β-actin (Cell Signaling Technology, Danvers, MA, United States), and ATF4 (Santa Cruz Biotechnology, Santa Cruz, CA, United States). Following three washes with PBS-T, the membranes were incubated for 1 h at room temperature with peroxidase-conjugated secondary antibody (Cell Signaling Technology). The final results were obtained by exposure to autoradiographic film (Kodak XAR film), and visualized via a chemiluminescent detection system (ECL Substrate Western Blot Detection System; Pierce, Rockford, IL, United States).
Quantitative real-time reverse transcriptase polymerase chain reaction
The Super-Script First-Strand Synthesis System (Invitrogen, Carlsbad, CA, United States) was used to perform the reverse transcription reactions. To determine the relative number of cDNA molecules in the reverse transcribed samples, real-time PCR analyses were performed using the Light-Cycler system (Roche, Indianapolis, IN, United States). PCR was performed according to the previously described procedures[23]. PCR was performed using 10 μL 2 × Master Mix SYBR Green I (Takara, Japan), 0.25 μmol/L of each 5’ and 3’ primer, and 2 μL samples or water to a final volume of 20 μL. Samples were denatured at 94 °C for 5 min. Amplification and fluorescence determination were carried out in three steps for 45 cycles: denaturation at 94 °C for 10 s, annealing at 60 °C for 15 s, and extension at 72 °C for 20 s. At the end of extension, SYBR green fluorescence was detected, which reflects the amount of double-stranded DNA. To discriminate specific from nonspecific cDNA products, a melting curve was obtained at the end of each run. Products were denatured at 95 °C for 3 s, and the temperature was then decreased to 58 °C for 15 s and raised slowly from 58 to 95 °C, using a temperature transition rate of 0.1 °C/s. Data were normalized with the hypoxanthine phosphoribosyltransferase (HPRT) levels in the samples. Oligo 6.0 was used to design the primers. Primer sets (sense sequence and antisense sequence, respectively) for the following genes were: HPRT forward, 5’-TCA ACG GGG GAC ATA AAA GT-3’, reverse, 5’-TGC ATT GTT TTA CCA GTG TCA A-3’; GRP78 forward, 5’-CTG AGG CGT ATT TGG GAA AG-3’, reverse, 5’-TCA TGA CAT TCA GTC CAG CAA-3’; ATF4 forward, 5’-ATG GCC GGC TAT GGA TGA T-3’, reverse, 5’-CGA AGT CAA ACT CTT TCA GAT CCA TT-3’; CHOP forward, 5’-CTT GAG CCT AAC ACG TCG ATT-3’, reverse, 5’-TGC ACT TCC TTC TGG AAC ACT-3’.
Cell culture and treatment
A two-step in situ collagenase perfusion procedure[24] was used to isolate mouse hepatocytes. Livers from the C57BL/6 mice were perfused in situ through the portal vein with EGTA buffer (0.5 mmol/L EGTA, 137 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 0.65 mmol/L MgSO4, and 10.07 mmol/L HEPES at pH 7.4) at a flow rate of 5 mL/min for 10 min, followed by collagenase buffer (67 mmol/L NaCl, 6.7 mmol/L KCl, 4.76 mmol/L CaCl2, 0.035% collagenase type II, and 10.07 mmol/L HEPES at pH 7.6) at a flow rate of 5 mL/min for 15 min. After centrifugation, the hepatocytes were collected and seeded in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were preincubated with 5 mmol/L NAC for 6 h, then 200 μmol/L H2O2 (3 h for PCR and western blotting, 24 h for LDH analysis) or 1 μM thapsigargin (TG), an ER stress activator, 24 h for LDH analysis) treatment to induce oxidative and ER stress.
Statistical analysis
Differences among groups were determined for statistical significance using one-way ANOVA or Student’s t test. All P values were two-sided, and P < 0.05 was considered statistically significant. Statistical calculations were performed using SPSS (Chicago, IL, United States). Data were presented as means ± SD from at least three independent experiments.
RESULTS
NAC treatment attenuates ROS-induced liver injury after IR
NAC was injected ip at 300 mg/kg 2 h before warm ischemia. Antioxidants like GSH are physiological countermeasures to free radicals such as ROS, and after IR, the hepatic GSH content is decreased. In the present study, IR livers had decreased GSH levels (13.50 ± 1.32 mg/g and 4.5 ± 0.65 mg/g protein, respectively, P < 0.001). The IR-induced GSH depletion was almost completely inhibited in the NAC-treated mice (4.5 ± 0.65 mg/g and 11.0 ± 1.08 mg/g protein, respectively, P < 0.005) (Figure 1A). MDA, an index of lipid peroxidation that indicates the levels of the oxygen free radicals, was significantly increased after reperfusion in the IR group compared with the sham group (39.0 ± 3.06 mmol/g and 129.8 ± 12.70 mmol/g weight tissue, respectively, P < 0.005). MDA was significantly decreased in the NAC-treated group compared with the IR group (129.8 ± 12.70 mmol/g and 51.75 ± 6.18 mmol/g weight tissue, respectively, P < 0.005) (Figure 1B). These data indicated that NAC treatment reduced oxidative stress in liver IR.
Figure 1.
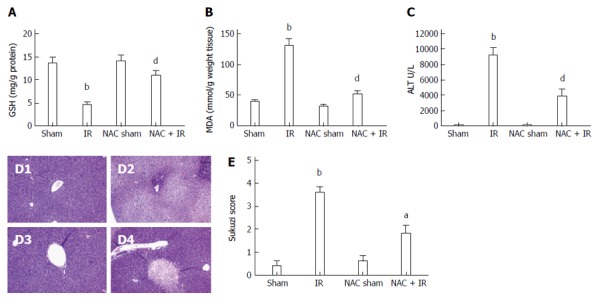
N-acetylcysteine treatment attenuates reactive oxygen species-induced liver injury after ischemia-reperfusion. Mice were subjected to 90 min partial liver ischemia, followed by 6 h reperfusion. A and B: reactive oxygen species levels evaluated by glutathione (mg/g protein) and malondialdehyde (nmol/g tissue); mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; dP < 0.01 vs IR group; C: hepatocellular function evaluated by alanine aminotransferase (U/L); mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; dP < 0.01 vs IR group; D: histopathological analysis of livers harvested 6 h after reperfusion: D1: sham group: normal hepatic architecture; D2: IR group: severe hepatic lobule distortion, sinusoidal congestion, apparent edema, vacuolization and massive necrosis; D3: NAC sham group: normal hepatic architecture; D4: NAC + IR group: mild vacuolization, punctate necrosis and edema; E: severity of liver IRI by Suzuki’s histological grading; mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; aP < 0.05 vs IR group. NAC: N-acetylcysteine; IR: Ischemia-reperfusion; ALT: Alanine aminotransferase.
We then assessed whether the inhibitory effect of NAC on IR-induced oxidative stress could reduce hepatocellular damage directly inflicted by ROS during liver IR. Figure 1C shows that NAC treatment significantly attenuated the increase of serum transaminases after reperfusion (9144 ± 1064 U/L and 3783 ± 1017 U/L, respectively, P < 0.05). Liver serum enzyme data were in line with liver pathological analyses. Histological analysis showed distinct areas of necrosis in the IR group, which was significantly improved in the NAC treatment group (Figure 1D). The histological parameters observed in the sham (0.40 ± 0.25), IR (3.6 ± 0.25), NAC sham (0.60 ± 0.30) and NAC IR (1.8 ± 0.37) groups were according to Suzuki et al[22] (Figure 1E). These data indicated that NAC treatment significantly attenuated ROS-induced liver injury after IR.
NAC treatment reduces hepatocellular apoptosis in liver after IR
Following reperfusion, the TUNEL assay assessed hepatocellular apoptosis. TUNEL-positive cells were markedly fewer in the NAC-pretreated IR liver compared with the IR group (Figure 2A). TUNEL-positive cells as a percent of total hepatocytes in the four groups were: Sham 1.5% ± 0.29%, IR 72.0% ± 5.08%, NAC sham 1.75% ± 0.48% and NAC + IR 20.5% ± 4.41%, indicating that hepatocellular apoptosis was significantly inhibited by NAC treatment after reperfusion (Figure 2B). Active caspase-3 directly triggered hepatocellular apoptosis following liver IR and indicated the levels of apoptosis. Figure 2C displays that the level of caspase-3 was significantly inhibited after NAC treatment in ischemic liver compared with the IR control (0.925 ± 0.11 U and 2.35 ± 0.25 U, respectively; P < 0.005).
Figure 2.
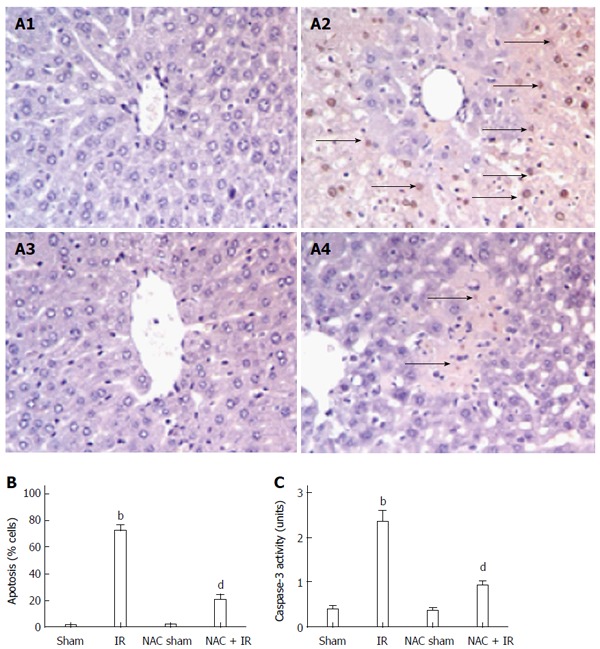
N-acetylcysteine treatment reduces hepatocellular apoptosis in liver after ischemia-reperfusion. A: Liver apoptosis by transferase dUTP nick-end labeling staining: A1: Sham group; A2: IR group; A3: NAC sham group; A4: NAC + IR group; B: apoptotic cells were quantified in six high-power fields (× 400), and expressed as percentages of apoptotic cells among total cells; mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; dP < 0.01 vs IR group; C: caspase-3 activity; mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; dP < 0.001 vs IR group. NAC: N-acetylcysteine; IR: Ischemia-reperfusion.
ROS-mediated ER stress is inhibited by NAC treatment in liver after IR
Previous studies have shown that ROS is an important inducer of the ER stress response, which mediates cell death[25-27]. The above data showed that NAC treatment significantly inhibited ROS generation during liver IR. We analyzed whether NAC treatment reduced ER stress responses mediated by ROS during liver IRI. We assessed gene expression and protein levels of ER stress molecules, including GRP78, ATF4 and CHOP. GRP78 is a central regulator of ER homeostasis, because of its multiple functional roles in protein folding, ER calcium binding, and controlling of the activation of transmembrane ER stress sensors. The ATF4-CHOP pathway, downstream of GRP78, is the main apoptotic pathway of ER stress[23]. These molecules are expressed at low levels under physiological conditions, but are induced strongly during severe ER stress. As shown in Figure 3A, GRP78, ATF4 and CHOP gene expressions were significantly elevated in the ischemic liver after reperfusion. These genes were significantly inhibited by NAC treatment after reperfusion. In addition, GRP78, ATF4 and CHOP proteins were further analyzed by western blotting, which agreed with the PCR data (Figure 3B and C). These data indicated that NAC treatment suppressed ROS-mediated ER stress during liver IRI.
Figure 3.
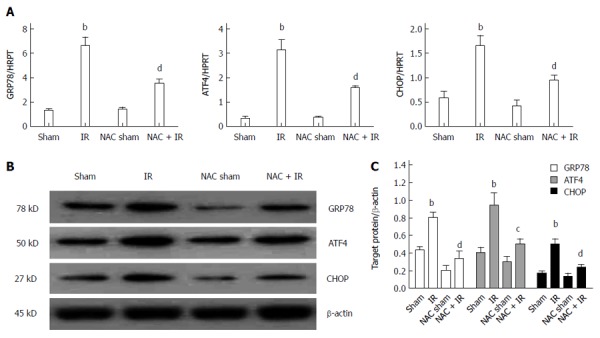
Reactive oxygen species-mediated endoplasmic reticulum stress is inhibited by N-acetylcysteine treatment in liver after ischemia-reperfusion. A: Endoplasmic reticulum stress related gene (GRP78, ATF4 and CHOP) expressions in livers harvested 6 h after reperfusion, by quantitative real-time reverse transcriptase polymerase chain reaction analysis; mean ± SD, n = 3-5/group, bP < 0.01 vs sham group; dP < 0.01 vs IR group; B: western blot-assisted analysis of GRP78, ATF4, CHOP and β-actin; C: Relative quantities of protein of GRP78, ATF4 and CHOP, mean ± SD, n= 3-5/group, bP < 0.01 vs sham group; dP < 0.01, cP < 0.05 vs IR group; representative of three experiments. NAC: N-acetylcysteine; IR: Ischemia-reperfusion.
ROS-mediated ER stress is inhibited by NAC treatment in vitro
To confirm the effects of NAC treatment on ROS-mediated ER stress, primary hepatocytes were isolated and cultured in vitro. Primary hepatocytes were initially treated with or without 5 mmol/L NAC for 6 h, and then with 200 μmol H2O2 for 3 h. RNA and proteins were harvested from treated cells. The gene expressions of GRP78, ATF4 and CHOP were analyzed by qRT-PCR, which showed that NAC treatment significantly repressed H2O2-induced GRP78 (1.24 ± 0.24 and 0.52 ± 0.08, respectively, P < 0.05), ATF4 (7.03 ± 1.09 and 2.59 ± 0.39, respectively, P < 0.005), and CHOP (18.55 ± 2.14 and 6.40 ± 1.32, respectively, P < 0.05) (Figure 4A). Furthermore, protein expression of GRP78, ATF4 and CHOP was further detected by western blotting, which agreed with the gene expression data (Figure 4B). These data indicated that NAC treatment suppressed ROS-mediated ER stress in hepatocytes.
Figure 4.
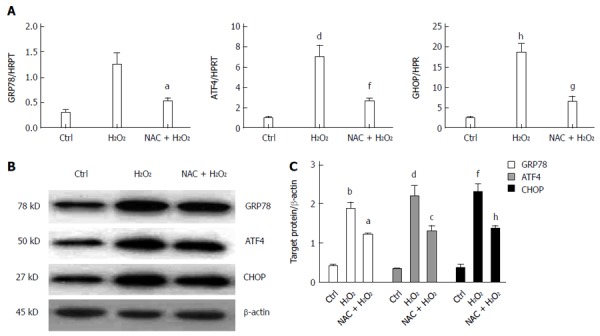
Reactive oxygen species-mediated endoplasmic reticulum stress is inhibited by N-acetylcysteine treatment in vitro. A: Endoplasmic reticulum stress related gene (GRP78, ATF4 and CHOP) expressions in primary hepatocytes harvested 3 h after H2O2 treatment by quantitative real-time reverse transcriptase polymerase chain reaction analysis; mean ± SD, n = 3-5/group, bP < 0.01, dP < 0.01, hP < 0.01 vs Ctrl group; aP < 0.05, fP < 0.01, gP < 0.05 vs NAC + H2O2 group; B: western blot-assisted analysis of GRP78, ATF4, CHOP and β-actin; C: Relative quantities of protein of GRP78, ATF4 and CHOP, mean ± SD, n = 3-5/group, bP < 0.01, dP < 0.01, fP < 0.01 vs Ctrl group; aP < 0.05, cP < 0.05, hP < 0.01 vs NAC + H2O2 group; representative of three experiments. NAC: N-acetylcysteine; IR: Ischemia-reperfusion; Ctrl: Control.
ER stress-related apoptosis is inhibited by NAC treatment
Previous studies have shown that Bcl-2 and other Bcl-2 family members are important molecules during ER-stress-induced apoptosis[28,29]. The present work examined the levels of antiapoptotic Bcl-2 family members Bcl-2 and Bcl-xl by western blotting. Figure 5A shows that liver IR significantly inhibited the levels of Bcl-2 and Bcl-xl, which could be restored by NAC treatment. To assess directly the roles of NAC treatment on ROS-mediated ER stress or ER stress induced cell death, we analyzed the effects of NAC treatment on H2O2- or TG-induced hepatocellular death by LDH in vitro. Figure 5B shows that NAC treatment significantly reduced H2O2-induced (30.71 ± 1.09 U/L and 15.36 ± 1.83 U/L, respectively, P < 0.005) and TG-induced (35.37 ± 3.20 U/L and 15.62 ± 2.41 U/L, respectively, P < 0.005) death in hepatocytes. These data indicated that NAC treatment inhibited ROS or the ER-stress-mediated apoptosis pathway.
Figure 5.
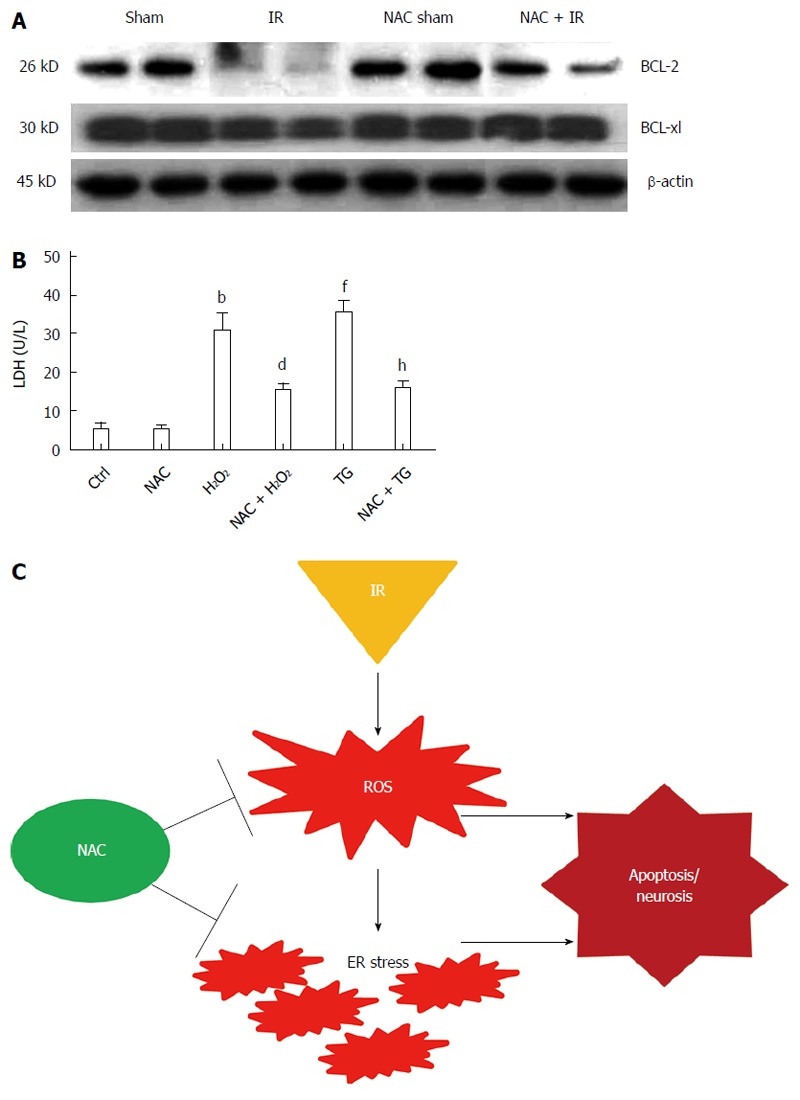
Reactive oxygen species or endoplasmic reticulum stress-induced hepatocyte death is inhibited by N-acetylcysteine treatment. A: Western blot-assisted analysis of BCL-2, BCL-xl and β-actin; B: released lactate dehydrogenase level of primary hepatocytes after H2O2 (200 μmol) or TG (1 µmol) treatment; mean ± SD, n = 4/group, bP < 0.01 vs Ctrl group; dP < 0.01 vs H2O2 group; fP < 0.01 vs Ctrl group; hP < 0.01 vs TG group; C: schematic illustration of NAC works in IR-stressed liver. NAC: N-acetylcysteine; IR: Ischemia-reperfusion; Ctrl: Control; ROS: Reactive oxygen species.
DISCUSSION
NAC is an antioxidant that provides resistance to tissue injury caused by ROS. It has been reported extensively to have excellent preventive and therapeutic effects on various disorders, including paracetamol intoxication, hepatitis, acute liver failure and IRI[16,21,30,31]. The mechanisms and molecules of NAC treatment in connection with tissue protection have been reported in various diseases. NAC has a number of different pharmacological effects, predominantly based on increased mitochondrial and cytosolic production of GSH, one of the principal free radical scavenging agents in humans[32]. In paracetamol toxicity, GSH repletion reduces the level of covalent binding of the toxic metabolite, making NAC an effective antidote[33]. The Stravitz RT group has shown that NAC treatment improves transplant-free survival in patients with non-paracetamol acute liver failure by ameliorating the production of IL-17[34]. Some recent studies have demonstrated that ROS can trigger ER stress in vivo and in vitro[13,14]. The present study aimed to elucidate whether the ROS-induced ER stress is inhibited by NAC treatment after liver IRI, and if yes, to investigate the effects of ER stress inhibition on apoptosis.
We first investigated the effects of NAC treatment on ROS during liver IRI. Mice were injected with 300 mg/kg NAC prior to induction of the liver IRI model. To asses ROS levels, GSH and MDA levels were determined. Our results showed that NAC treatment significantly decreased ROS levels after liver IRI, which was consistent with previous data[35,36]. We then examined the effects of NAC on hepatocellular damage by liver enzymes, which supported the view that NAC treatment had a significantly protective effect. Histological study of the ischemic liver showed that NAC treatment markedly improved liver damage after IRI, which was consistent with the biochemical findings. Suzuki scores were also significantly decreased in the ischemic liver by NAC treatment. Consistent with reported data on liver IRI models, NAC treatment significantly attenuated liver injury[35-37].
Apoptosis/necrosis plays crucial role in liver IRI and reflect liver tissue damage directly. The present study assessed apoptotic cells in liver sections after IRI and showed that the positive cells were markedly increased in the IR group. In addition, the mean number of positive cells detected in the NAC treatment group was 20.21% of that in the IR group. Caspase-3 levels, which indicate the extent of apoptosis in the liver tissue, were analyzed in ischemic liver tissue after reperfusion. NAC treatment effectively repressed caspase-3 levels upregulation following reperfusion, which was in line with the TUNEL results.
Some studies demonstrated the induction of ER stress during IR in different organs, including the liver[5,6,23]. Disruption of ER homeostasis triggers cellular stress responses, including the UPR, an ER-specific stress response. GRP78, ATF-4 and CHOP, which members of the UPR signaling pathway, are important for the maintenance of normal cellular homeostasis[38-40]. In addition, ROS has been shown to trigger ER stress[13,14]. Our previous studies demonstrated that the ATF4-CHOP pathway, downstream of GRP78, is the main apoptotic pathway of ER stress in the ischemic liver[23]. GRP78, ATF-4 and CHOP genes and proteins were analyzed to evaluate the effects of NAC treatment on ER stress during liver IR. Our data showed that IR significantly increased GRP78, ATF-4 and CHOP expression in the liver. Upregulations of GRP78, ATF-4 and CHOP were significantly inhibited by NAC treatment after IR. To determine further the role of NAC in ROS-mediated ER stress, a cell-death model was set up via treating primary hepatocytes with 200 μmol H2O2 in vitro. We determined whether NAC treatment inhibited expression of GRP78, ATF-4 and CHOP. Figure 4 shows that H2O2 treatment significantly enhanced expression of GRP78, ATF-4 and CHOP, which was significantly suppressed in the NAC treatment group. These data clearly imply that ER stress is involved in oxidative stress in cells. Mitochondrial dysfunction induced by ROS has a strict relation with ER stress[41]. Importantly, the ROS-induced ER stress was inhibited by NAC treatment in vivo and in vitro.
Prolonged and excessive ER stress can cause cell apoptosis or death. LDH activity correlates with cell membrane disruption and is thus regarded as a reliable indicator of cell damage and/or death[42]. In addition, LDH levels have often been used an index to assess hepatocellular death[43]. We measured LDH activity to reflect hepatocyte damage or death. Figure 5A shows that H2O2 treatment significantly increased LDH release, which was significantly inhibited by NAC pretreatment. To investigate further the effects of NAC on ER-stress-induced cell death, TG (an activator of ER stress) was used to treat hepatocytes. The results demonstrated that NAC treatment effectively reduced hepatocellular death induced by TG treatment. UPR activates c-JNK phosphorylation, which has been suggested as a pro-apoptotic link by direct phosphorylation of mitochondrial proteins, including members of the BCL-2 family[12,13,28,29]. Our present findings are consistent with other studies in which IR-induced ER stress inhibited expression of antiapoptotic protein (BCL-2 and BCL-xl) in the liver. Our results also indicate that NAC treatment recovers expression of BCL-2 and BCL-xl during liver IRI.
In conclusion, our research indicates that NAC treatment attenuates the liver injury induced by IR and reinforces the usefulness of exploring NAC as a potential alternative therapeutic drug during liver IRI. The protective effects of NAC not only reflect its confirmed antioxidant effects, but also are caused by regulation of ER stress molecules and prevention of related apoptosis development (Figure 5C). However, the application of our results has some limitations in the clinical setting.
ACKNOWLEDGMENTS
We are grateful to Yuan-Fang Fu at UCLA for revising the English text.
COMMENTS
Background
The protective effect of N-acetylcysteine (NAC) treatment has been confirmed during IRI in several organs, but its underlying mechanism remains unclear.
Research frontiers
IRI is an important inducing factor in liver damage and failure in various clinical settings, such as hepatic trauma, resection, liver transplantation and circulatory shock. Although many drugs or agents have been evaluated in preventing liver IRI, the outcomes were not satisfactory.
Innovations and breakthroughs
This study revealed new evidence for the protective effects of NAC treatment on hepatocytes during liver IRI. The data demonstrate that protection associated with NAC treatment is not only caused by the confirmed antioxidant effects, but also by regulation of endoplasmic reticulum (ER) stress molecules and inhibition of relative apoptosis during liver IRI.
Applications
This study provides evidence further supporting the usefulness of NAC as an alternative therapeutic drug during liver IRI.
Terminology
ER stress is an unfolded protein response reflecting the accumulation of unfolded and misfolded proteins in the ER lumen.
Peer review
The study reports experimental data that highlight the potential use of NAC in the treatment of IRI. The experimental techniques were appropriate, and the authors present their findings in an adequate way.
Footnotes
Supported by First Affiliated Hospital of Nanjing Medical University and the National Natural Science Foundation of China, Grant No. 81100270, No. 81070380, No. 81310108001, No. 81210108017 and No.81273261
P- Reviewer: Bjornsson B, Hummel R S- Editor: Ma YJ L- Editor: Stewart G E- Editor: Liu XM
References
- 1.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–G26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 2.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 3.Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg. 2001;181:160–166. doi: 10.1016/s0002-9610(00)00573-0. [DOI] [PubMed] [Google Scholar]
- 4.Que X, Debonera F, Xie J, Furth EE, Aldeguer X, Gelman AE, Olthoff KM. Pattern of ischemia reperfusion injury in a mouse orthotopic liver transplant model. J Surg Res. 2004;116:262–268. doi: 10.1016/j.jss.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, Eckhoff DE, Contreras JL. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138:342–351. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Ren F, Cheng Q, Bai L, Shen X, Gao F, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Endoplasmic reticulum stress modulates liver inflammatory immune response in the pathogenesis of liver ischemia and reperfusion injury. Transplantation. 2012;94:211–217. doi: 10.1097/TP.0b013e318259d38e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 8.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 10.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao J, Zhang C, Wang P, Lu L, Zhang F. All-trans retinoic acid alleviates hepatic ischemia/reperfusion injury by enhancing manganese superoxide dismutase in rats. Biol Pharm Bull. 2010;33:869–875. doi: 10.1248/bpb.33.869. [DOI] [PubMed] [Google Scholar]
- 12.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol. 2011;26 Suppl 1:173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 13.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu K, Shen NY, Xu XS, Su HB, Wei JC, Tai MH, Meng FD, Zhou L, Zhang YL, Liu C. Emodin induces human T cell apoptosis in vitro by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction. Acta Pharmacol Sin. 2013;34:1217–1228. doi: 10.1038/aps.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- 16.Gunduz H, Karabay O, Tamer A, Ozaras R, Mert A, Tabak OF. N-acetyl cysteine therapy in acute viral hepatitis. World J Gastroenterol. 2003;9:2698–2700. doi: 10.3748/wjg.v9.i12.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squires RH, Dhawan A, Alonso E, Narkewicz MR, Shneider BL, Rodriguez-Baez N, Olio DD, Karpen S, Bucuvalas J, Lobritto S, et al. Intravenous N-acetylcysteine in pediatric patients with nonacetaminophen acute liver failure: a placebo-controlled clinical trial. Hepatology. 2013;57:1542–1549. doi: 10.1002/hep.26001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian H, Zhang Q, Li H, Zhang G. Antioxidant N-acetylcysteine and AMPA/KA receptor antagonist DNQX inhibited mixed lineage kinase-3 activation following cerebral ischemia in rat hippocampus. Neurosci Res. 2003;47:47–53. doi: 10.1016/s0168-0102(03)00186-x. [DOI] [PubMed] [Google Scholar]
- 19.Oksuz H, Senoglu N, Yasim A, Turut H, Tolun F, Ciralik H, Bilge F. Propofol with N-acetylcysteine reduces global myocardial ischemic reperfusion injury more than ketamine in a rat model. J Invest Surg. 2009;22:348–352. doi: 10.1080/08941930903214750. [DOI] [PubMed] [Google Scholar]
- 20.Seguro AC, Poli de Figueiredo LF, Shimizu MH. N-acetylcysteine (NAC) protects against acute kidney injury (AKI) following prolonged pneumoperitoneum in the rat. J Surg Res. 2012;175:312–315. doi: 10.1016/j.jss.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 21.Smyrniotis V, Arkadopoulos N, Kostopanagiotou G, Theodoropoulos T, Theodoraki K, Farantos C, Kairi E, Paphiti A. Attenuation of ischemic injury by N-acetylcysteine preconditioning of the liver. J Surg Res. 2005;129:31–37. doi: 10.1016/j.jss.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki S, Nakamura S, Koizumi T, Sakaguchi S, Baba S, Muro H, Fujise Y. The beneficial effect of a prostaglandin I2 analog on ischemic rat liver. Transplantation. 1991;52:979–983. doi: 10.1097/00007890-199112000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Rao J, Qin J, Qian X, Lu L, Wang P, Wu Z, Zhai Y, Zhang F, Li G, Wang X. Lipopolysaccharide preconditioning protects hepatocytes from ischemia/reperfusion injury (IRI) through inhibiting ATF4-CHOP pathway in mice. PLoS One. 2013;8:e65568. doi: 10.1371/journal.pone.0065568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatano E, Brenner DA. Akt protects mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis through NK-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1357–G1368. doi: 10.1152/ajpgi.2001.281.6.G1357. [DOI] [PubMed] [Google Scholar]
- 25.Pan C, Giraldo GS, Prentice H, Wu JY. Taurine protection of PC12 cells against endoplasmic reticulum stress induced by oxidative stress. J Biomed Sci. 2010;17 Suppl 1:S17. doi: 10.1186/1423-0127-17-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 27.Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin Immunol. 2009;21:156–163. doi: 10.1016/j.smim.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zingarelli B, Hake PW, Burroughs TJ, Piraino G, O’connor M, Denenberg A. Activator protein-1 signalling pathway and apoptosis are modulated by poly(ADP-ribose) polymerase-1 in experimental colitis. Immunology. 2004;113:509–517. doi: 10.1111/j.1365-2567.2004.01991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alipour M, Buonocore C, Omri A, Szabo M, Pucaj K, Suntres ZE. Therapeutic effect of liposomal-N-acetylcysteine against acetaminophen-induced hepatotoxicity. J Drug Target. 2013;21:466–473. doi: 10.3109/1061186X.2013.765443. [DOI] [PubMed] [Google Scholar]
- 31.Hadžić N. Challenging the dogmas; the NAC tie. Hepatology. 2013;57:1297–1300. doi: 10.1002/hep.26044. [DOI] [PubMed] [Google Scholar]
- 32.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray KM, Carpenter MJ, Baker NL, DeSantis SM, Kryway E, Hartwell KJ, McRae-Clark AL, Brady KT. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169:805–812. doi: 10.1176/appi.ajp.2012.12010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stravitz RT, Sanyal AJ, Reisch J, Bajaj JS, Mirshahi F, Cheng J, Lee WM. Effects of N-acetylcysteine on cytokines in non-acetaminophen acute liver failure: potential mechanism of improvement in transplant-free survival. Liver Int. 2013;33:1324–1331. doi: 10.1111/liv.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demir S, Inal-Erden M. Pentoxifylline and N-acetylcysteine in hepatic ischemia/reperfusion injury. Clin Chim Acta. 1998;275:127–135. doi: 10.1016/s0009-8981(98)00078-3. [DOI] [PubMed] [Google Scholar]
- 36.Seifi B, Kadkhodaee M, Delavari F, Mikaeili S, Shams S, Ostad SN. Pretreatment with pentoxifylline and N-acetylcysteine in liver ischemia reperfusion-induced renal injury. Ren Fail. 2012;34:610–615. doi: 10.3109/0886022X.2012.660827. [DOI] [PubMed] [Google Scholar]
- 37.Sener G, Tosun O, Sehirli AO, Kaçmaz A, Arbak S, Ersoy Y, Ayanoğlu-Dülger G. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003;72:2707–2718. doi: 10.1016/s0024-3205(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 38.Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- 39.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 40.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim JH, Lee HJ, Ho Jung M, Song J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell Signal. 2009;21:169–177. doi: 10.1016/j.cellsig.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Jauregui HO, Hayner NT, Driscoll JL, Williams-Holland R, Lipsky MH, Galletti PM. Trypan blue dye uptake and lactate dehydrogenase in adult rat hepatocytes--freshly isolated cells, cell suspensions, and primary monolayer cultures. In Vitro. 1981;17:1100–1110. doi: 10.1007/BF02618612. [DOI] [PubMed] [Google Scholar]
- 43.Sanders SW, Dukes GE, Gray P, Tolman KG. Toxicity of heparin in isolated rat hepatocytes. Biochem Pharmacol. 1984;33:2223–2226. doi: 10.1016/0006-2952(84)90658-0. [DOI] [PubMed] [Google Scholar]