

Background: Cancer cells activate their microenvironment through secreting several cytokines.
Results: The cytokine IL-6 activates breast stromal fibroblasts through STAT3-dependent up-regulation of the RNA binding protein AUF1, which stabilizes the SDF-1, α-SMA, TGF-β1, and IL-6 mRNAs.
Conclusion: Up-regulation of AUF1 enhances the procarcinogenic effects of breast stromal fibroblasts.
Significance: The IL-6/STAT3/AUF1 pathway could constitute an important prognostic and/or therapeutic target.
Keywords: Breast Cancer, Gene Expression, Interleukin 6 (IL-6), mRNA Decay, Myofibroblast, RNA-binding Protein, STAT3, Tumor Microenvironment, AUF1
Abstract
The development and spread of mammary carcinomas require synergetic interplay between tumor cells and their microenvironment through paracrine secretions, which are still not well defined. We have shown here that interleukin-6 (IL-6), either recombinant or secreted from highly invasive breast cancer cells, down-regulates the tumor suppressor proteins p16INK4A, p21WAF1, and p53 and activates breast stromal fibroblasts in a paracrine manner. The formation of myofibroblasts requires p16INK4A down-regulation and the activation of the JAK2/STAT3 pathway. Indeed, the transcription factor STAT3 positively controls the expression of the three major myofibroblast markers, SDF-1, α-smooth muscle actin (α-SMA), and TGF-β1, and mediates IL-6-related down-regulation of p16INK4A, p21WAF1, and p53 as well as the activation of stromal fibroblasts. Importantly, these effects were mediated through STAT3-dependent up-regulation of the mRNA-binding protein AUF1, whose promoter contains three canonical STAT3 binding sites. AUF1 binds the SDF-1, α-SMA, TGF-β1, and IL-6 mRNAs and reduces their turnover. Consequently, specific AUF1 down-regulation inhibits IL-6-dependent activation of breast stromal fibroblasts, whereas AUF1 ectopic expression of p37AUF1 activated these cells and enhanced their paracrine induction of epithelial-to-mesenchymal transition in breast cancer cells, which shows a non-cell-autonomous oncogenic function of AUF1. Together, these results demonstrate a major role of IL-6 in activating breast stromal fibroblasts through STAT3-dependent AUF1 induction.
Introduction
Breast stroma-carcinoma interactions play major roles in the development and spread of tumor cells. Stroma is composed of various types of cells, including fibroblasts, adipocytes, and immune cells. Recent studies have made it increasingly apparent that tumor microenvironment does not exist simply as a passive support structure, but it is rather a hallmark of cancer that plays an active and crucial role in the onset and progression of tumor cells, through dynamic and interdependent interactions (1, 2). This functional cross-talk is mediated through different molecules that not only foster the growth of the initial tumor cells but empower them to invade and metastasize (3, 4).
Three-dimensional co-culture experiments have shown that fibroblasts were converted into a graded pattern of myogenic differentiation (myofibroblasts) when confronted with tumor cells, with a strong effect on cells that were in the immediate vicinity of tumor cells (5). Recently, conversion of resident fibroblasts into myofibroblasts during the course of tumor progression was also observed in a co-implantation breast tumor xenograft model (6). These results indicate the possible transformation of breast stromal fibroblasts to myofibroblasts under cancer-related paracrine effects. In fact, TGF-β can induce the production of the major myofibroblast marker α-smooth muscle actin (α-SMA)2 in mammary fibroblasts in vitro and consequently transdifferentiates these cells into active fibroblasts (7).
Interleukin-6 (IL-6) is among those soluble factors that are secreted from both cancer cells and stromal fibroblasts. IL-6 is a multifunctional cytokine that plays a key role in both innate and acquired immune responses, hematopoiesis, inflammation, and the regulation of growth and differentiation of cancer cells (8). Breast cancer tissues express high levels of IL-6 as compared with matched normal tissues, and these levels increase with tumor grade (9, 10). Furthermore, IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland (11). Several studies have shown a significant role of IL-6 in the migration and invasion of breast cancer cells as well as their epithelial to mesenchymal transition (12–15). Recently, Chang et al. (16) have shown increased levels of IL-6 at the leading edge of invasive human breast tumors.
Importantly, IL-6 is also an activator of signal transducer and activator of transcription 3 (STAT3) in various cancer cells, including breast cancer cell lines (17–19). STAT3 activation can take place through both autocrine expression of IL-6 and paracrine activation by IL-6 from stroma (20, 21). STAT3 is an oncogene that has been found persistently phosphorylated/active in different human cancer cell lines and primary tumors, including breast cancers (22). Indeed, phosphorylated STAT3 is expressed in about 40% of all breast cancers (23, 24). STAT3 controls the expression of several cancer-related genes, both tumor suppressors and oncogenes. Most of these genes are common to both wound healing and cancer (25). Therefore, STAT3 activation constitutes an important link between inflammation and cancer (26).
Recent findings have shown that the transformation of breast stromal fibroblasts to myofibroblasts is under the control of tumor suppressor proteins, such as p21WAF (p21), p53, PTEN, and CAV-1 (27). Furthermore, we have recently shown that the expression of the cyclin-dependent kinase inhibitor p16INK4A (hereafter referred to as p16) is reduced in active cancer-associated fibroblasts and that p16 down-regulation plays a major role in the activation of breast stromal fibroblasts (28). In the present report, we sought to determine the molecules and pathways that underlie the paracrine effects of breast cancer cells on stromal fibroblasts, leading to their activation and the down-regulation of p16, p21, and p53. We have clearly shown that IL-6 plays a major role in these processes through the activation of the RNA binding protein AUF1 in a STAT3-dependent manner.
EXPERIMENTAL PROCEDURES
Cells and Cell Culture
Breast fibroblast cells NBF-6 and TCF-64 were obtained and used as described previously (29). MDA-MB-231 and MCF-10A cells were obtained from ATCC and were cultured following their instructions. All supplements were obtained from Sigma except for antibiotic and antimycotic solutions, which were obtained from Invitrogen. Human IL-6 recombinant protein (hBA-184) (Santa Cruz Biotechnology, Inc.).
Cellular Lysate Preparation and Immunoblotting
This has been performed as previously described (30). Antibodies directed against α-SMA, TGF-β1 (2AR2), stromal-derived factor-1 (SDF-1), Twist-1, vimentin (RV202), AUF1 (ab50692), and IL-6 were purchased from Abcam (Cambridge, MA); those against STAT3, phospho-STAT3-Tyr705 (D3A7), Snail (C15D3), E-cadherin (24E10), EpCam (UV1D9), GP-130, JAK-2 (D2E12), and phospho-JAK-2 (Tyr1007/1008) were from Cell Signaling (Danvers, MA); antibody against p16INK4A was from BD Biosciences; antibody against ZEB-1 (4C4) was from Abnova (Taipei, Taiwan); and antibodies against p21 (F-5), p53 (DO-1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; FL-335) were purchased from Santa Cruz Biotechnology.
Chromatin Immunoprecipitation (ChIP) Assay
Cells (106) were treated with 2% formaldehyde for 10 min at room temperature to cross-link the transcription factor to DNA. The cross-linking was terminated by the addition of glycine (0.125 m). After washing with PBS, cells were collected and resuspended in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris, pH 8.1) with protease inhibitors. The sonicated lysate was processed using the ChIP assay kit following the manufacturer's instructions (Cell Signaling). ChIP experiments were performed using antibody against STAT3. Anti-IgG and anti-H3 antibodies were used as negative and positive controls, respectively. Input is the supernatant of the negative control (mock), and served as a positive PCR control, which represents the starting genomic DNA that was used for normalization. Immunoprecipitated chromatin was analyzed by PCR and qPCR using GAPDH as an unlinked locus control. The sequences of the primers used were as follows: BS1, 5′-CCAAACCCGAACACAAACTC-3′ and 5′-TCCTAAAACGTACCGTGATTCC-3′; BS2, 5′-CAAAAGCAAAGCAAAAACAATG-3′ and 5′-GGGTGACATTGAGGTTTTCG-3′; BS3, 5′-CCCTCTAGCCGCTACTTCG-3′ and 5′-GCCCTCTCGCTACCCTTTATG-3′; GAPDH, 5′-TACTAGCGGTTTTACGGGCG-3′ and 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′.
RNA Purification, RT-PCR, and Quantitative RT-PCR (qRT-PCR)
Total RNA was purified using the TRI reagent (Sigma) according to the manufacturer's instructions and was treated with RNase-free DNase before cDNA synthesis using the RT-PCR kit (Clontech) for both miRNAs and mRNAs. For RT-PCR, cDNA was amplified using the Platinum® Taq DNA polymerase (Invitrogen). After electrophoresis on ethidium bromide-stained 2% agarose gels, the intensity of the PCR products was determined with the Quantity One program (Bio-Rad) and was normalized against GAPDH. For qRT-PCR, the RT2 Real-TimeTM SYBR Green qPCR master mix (Roche Applied Science) was used, and the amplifications were performed utilizing a Light Cycler 480 (Roche Applied Science). The melting curve data were collected to check PCR specificity, and the amount of PCR products was measured by threshold cycle (Ct) values, and the relative ratio of specific genes to GAPDH for each sample was then calculated. The respective primers are as follows: CDKN2A, 5′-GAGGCCGATCCAGGTCATGA-3′ and 5′-GCACGGGTCGGGTGAGA-3′; CDKN1A, 5′-GAGACTCTCAGGGTCGAAAACG-3′ and 5′-GATTAGGGCTTCCTCTTGGAGAA-3′; TP53, 5′-CAGTCTACCTCCCGCCATAA-3′ and 5′-CCACAACAAAACACCAGTGC-3′; C-MYC, 5′-TGCCAGGACCCGCTTCT-3′ and 5′-GAAAAAAATCCAGCGTCTAAGCA-3′; AUF1, 5′-GATCAAGGGGTTTTGGCTTT-3′ and 5′-GTTGTCCATGGGGACCTCTA-3′; GAPDH, 5′-GAGTCCACTGGCGTCTTC-3′ and 5′-GGGGTGCTAAGCAGTTGGT-3′; α-SMA, 5′-CCGACCGAATGCAGAAGGA-3′ and 5′-ACAGAGTATTTGCGCTCCGAA-3′; SDF1, 5′-TAGTCAAGTGCGTCCACGA-3′ and 5′-GGACACACCACAGCACAAAC-3′; TGF-β1, 5′-TGTGTGCTGAAGCCATCGTTG-3′ and 5′-CCGGCTTGTCTGAAAAGGTCA-3′; IL-6, 5′-GACAAAGCCAGAGTCCTTCAGAGA-3′ and 5′-CTAGGTTTGCCGAGTAGATCT-3′.
Immunoprecipitation and RT-PCR
Cell lysates were prepared from confluent cells; 3 mg were incubated in the lysis buffer (50 mm Tris (pH 8), 100 mm NaCl, 10% glycerol, protease inhibitors, 5 mm DTT, and 2 units/ml RNasin); and 5 mg of AUF1 mouse monoclonal antibody (mouse IgG was used as control) were added and mixed at 4 °C for 4 h. An equal volume of protein A-agarose was added per immunoprecipitation and mixed overnight at 4 °C. After centrifugation, the pellet was resuspended in 1 ml of TRI reagent used for RNA extraction. qRT-PCRs were performed as described above.
siRNA Transfection
IL-6 siRNA, STAT3 siRNA, and control siRNA were obtained from Qiagen. AUF1 siRNA (pSILENCER-AUF15), which targets all four AUF1 isoforms (31), was a generous gift from Dr. Gorospe. The transfections were carried out using the RNAi Fect reagent (Qiagen) as recommended by the manufacturer.
Analysis of mRNA Stability
Cells were challenged with actinomycin D (5 μg/ml) for various periods of time (0–6 h), and then total RNA was purified and assessed using qRT-PCR. One-phase exponential decay curve analysis (SigmaPlot) was used to assess the mRNA decay kinetics, considering the values at time 0 as 100%. The time corresponding to 50% remaining mRNA was considered as mRNA half- life.
Viral Infection
Lentivirus-based vectors bearing CDKN2A-ORF (pCDH) or p37AUF1-ORF as well as their respective controls (Origene) were used to prepare the lentiviral supernatant from 293FT cells. Lentiviral supernatants were collected 48 h post-transfection, filtered, and used for infection. 24 h later, media were replaced with complete media, and cells were grown for 3 days.
Luciferase Reporter Assay
Cells were plated at 1 × 105 cells/well on 6-well plates and transfected with 3 μg of the Gaussia luciferase reporter vector containing either human wild type AUF1 promoter or mutated sequence of one of the three STAT3 binding sites. A non-promoter-containing plasmid was used as a control (GeneCopoeia). Transfections were carried out using Lipofectamine 2000 as recommended by the manufacturer (Invitrogen). At 24 h post-transfection, cells were recultured in fresh media and reincubated for 72 h. Subsequently, medium was collected and used to measure the secreted Gaussia luciferase in a 96-well plates in triplicate as recommended by the manufacturer (GeneCopoeia). The mean and S.E. were calculated from three wells for each promoter activity and presented as a percentage of the non-stimulated control.
ELISAs
Supernatants from 24-h fibroblast cell cultures were harvested, and ELISA was performed according to the manufacturer's instructions (R&D Systems). The OD was used at 450 nm on a standard ELISA plate reader. These experiments were performed in triplicate.
Cytokine Array
Serum-free conditioned media (SFCM) were applied to RayBio human cytokine antibody array membrane-5 (RayBiotech, Inc.) as recommended by the manufacturer. Briefly, the membranes were incubated with 1 ml of SFCM overnight and then were washed and reincubated with the Biotin-conjugated anti-cytokines. Subsequently, the cytokines were visualized by incubating the membranes with HRP-conjugated streptavidin secondary antibodies, and then the membranes were exposed to x-ray films.
Cell Migration, Invasion, and Proliferation
These assays were performed in a real-time and label-free manner using the xCELLigence RTCA technology (Roche Applied Science), which measures impedance changes in a meshwork of interdigitated gold microelectrodes located at the bottom well (E-plate) or at the bottom side of a microporous membrane (CIM-Plate 16). Cell migration and invasion were assessed as per the manufacturer's instructions. In brief, 2 × 104 cells in serum-free medium were added to the upper wells of the CIM-Plate with a thin layer of Matrigel basement membrane matrix (for invasion) or without (for migration), and a complete medium was added to the lower chamber wells used as a chemoattractant. Subsequently, the plates were incubated in the RTCA for 24 h, and the impedance value of each well was automatically monitored by the xCELLigence system and expressed as a cell index value, which represents cell status based on the measured electrical impedance change divided by a background value. Each assay was performed in triplicate.
For the proliferation assay, exponentially growing cells (2 × 104) were seeded in E-plate with complete medium as per the manufacturer's instructions. All data were recorded and analyzed by the RTCA software. Cell index was used to measure the change in the electrical impedance divided by the background value to represent cell status. Each assay was performed in triplicate.
Conditioned Media
Cells were cultured in medium without serum for 24 h, and then media were collected and briefly centrifuged. The resulting supernatants were either used immediately or frozen at −80 °C until needed.
Statistical Analysis
Statistical analysis was performed by Student's t test, and p values of 0.05 and less were considered as statistically significant.
RESULTS
Breast Cancer Cells Down-regulate Stromal Fibroblast p16, p21, and p53 in a Paracrine Manner
Do breast cancer cells affect the expression of breast stromal fibroblast tumor suppressor proteins? To address this question, normal breast stromal fibroblasts (TCF-64; tumor counterpart fibroblast, isolated from a histologically normal part of a breast harboring a tumor), and NBF-6 (normal breast fibroblast, obtained from a mammoplasty) were indirectly co-cultured with SFCM derived from the non-carcinogenic epithelial cells MCF-10A (used as control) and the highly invasive breast cancer cells MDA-MB-231. Serum-free medium (SFM) was also used as negative control. Cells were harvested after 24 h, whole cell lysates were prepared and used for immunoblotting analysis utilizing specific antibodies, and GAPDH was used as an internal control. Fig. 1A shows that whereas SFCM from MCF-10A cells had only a marginal effect on the level of p16, SFCM from MDA-MB-231 cells (MDA-SFCM) strongly decreased p16 level in both TCF-64 and NBF-6 as compared with the basal level (SFM). Like the p16 level, p21 and p53 levels were also markedly reduced upon treatment of stromal fibroblasts with MDA-SFCM (Fig. 1A). Interestingly, a similar effect was obtained at the mRNA level of the three genes (Fig. 1B). This indicates that breast cancer cells can reduce the expression of the three important tumor suppressor proteins p16, p21, and p53 in breast stromal fibroblasts in a paracrine manner.
FIGURE 1.
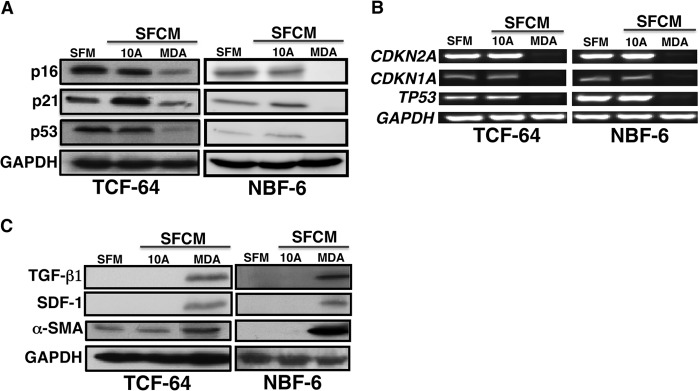
Breast cancer cells down-regulate stromal fibroblast p16, p21 and p53 in a paracrine manner. SFM as well as SFCM collected from MCF-10A (10A) and MDA-MB-231 (MDA) cells were used to treat fibroblast cells for 24 h. A and C, cell lysates were prepared and used for immunoblotting analysis using antibodies for the indicated proteins, and GAPDH was used as an internal control. B, total RNA was prepared and used for RT-PCR utilizing primers specific for the indicated genes. The amplified products were electrophoresed on 2% ethidium bromide-stained agarose gels. These results are typical of at least three independent experiments.
To test whether MDA-SFCM activated breast stromal fibroblasts, we assessed the level of three important markers of active fibroblasts, α-SMA, SDF-1, and TGF-β1. Fig. 1C shows that the level of these three proteins increased markedly following treatment of fibroblasts with MDA-SFCM as compared with SFM and SFCM from MCF-10A cells, which suggests the activation of these fibroblasts.
Paracrine IL-6 Down-regulates p16, p21, and p53 in Breast Stromal Fibroblasts
To identify the secreted factor(s) responsible for p16, p21, and p53 down-regulation in breast stromal fibroblasts, we performed cytokine array using media conditioned with MDA-MB-231 or MCF-10A cells. Fig. 2A shows great differential expression of several cytokines, such as MCP-1, GM-CSF, IGFBP-4, and IL-6. However, IL-6 showed the strongest differential expression, with a very high level in MDA-MB-231 cells as compared with MCF-10A cells. This result was confirmed by ELISA, which showed the secretion of 35 ng/ml IL-6 from MDA-MB-231 cells (Fig. 2B). Therefore, we decided to investigate the possible implication of IL-6 in the transformation of stromal fibroblasts to myofibroblasts as well as the down-regulation of p16, p21, and p53. To this end, IL-6 was first inhibited in MDA-SFCM using anti-IL-6 inhibitory antibody, whereas IgG antibody was used as a control. Fig. 2C shows that IL-6 inhibition suppressed the effect of MDA-SFCM on the expression of p16, p21, and p53 in breast stromal fibroblasts. On the other hand, adding pure IL-6 protein (5, 10, and 35 ng/ml) to serum-free culturing medium decreased the levels of the p16 protein in a dose-dependent manner, with the maximum effect reached at 35 ng/ml IL-6. The effect on p21 and p53 was much stronger, and both proteins disappeared upon treatment with only 5 ng of IL-6 (Fig. 2C). Interestingly, the effect of 35 ng/ml IL-6 was similar to the exposure of cells to MDA-SFCM (Fig. 2C); therefore, this concentration was used to test the effect of IL-6 on the mRNA level using qRT-PCR.
FIGURE 2.

Exogenous IL-6 down-regulates p16, p21, and p53 in breast stromal fibroblasts. A, SFCM from the indicated cells were collected and applied onto a human cytokine antibody array membrane. The circles indicate the spots corresponding to IL-6, whereas the arrows indicate other cytokines with higher levels in MDA-MB-231 cells as compared with MCF-10A cells. B, SFM and SFCM from the indicated cells were used to assess the level of IL-6 by ELISA. Error bars, S.D. C, breast stromal fibroblast NBF-6 cells were treated with the indicated combinations, and then cell lysates were prepared and were used for immunoblotting analysis. These experiments were repeated several times, and representative blots are shown. D and G, total RNA was prepared form NBF-6 cells treated with the indicated media, and then the mRNA levels of the indicated genes were assessed by qRT-PCR. Error bars, S.D. E, MDA-MB-231 cells were transfected with IL-6 siRNA or control siRNA (Ctrl), and then cell lysates were prepared and used for immunoblotting. F, NBF-6 cells were exposed to SFM and SFCM from MDA-MB-231 cells harboring control siRNA or IL-6 siRNA, and then cell lysates were prepared and used for immunoblotting analysis.
Adding IL-6 to SFM strongly reduced the mRNA levels of p16, p21, and p53 in breast stromal fibroblasts (Fig. 2D). In order to confirm the role of IL-6 in mediating the paracrine effect of MDA-MB-231 cells on breast stromal fibroblasts, IL-6 was knocked down in MDA-MB-231 cells using specific siRNA (Fig. 2E). Then SFCM were collected from these cells and their corresponding controls expressing control siRNA, and then they were used to treat fibroblast cells. Fig. 2F shows that, whereas SFCM from control cells markedly suppressed the expression of the three proteins, SFCM from IL-6-deficient cells had only a marginal effect. This indicates that IL-6 down-regulation inhibited the paracrine effect of MDA-MB-231 cells on the expression of p16, p21, and p53 proteins in breast stromal fibroblasts. Together, these results demonstrate that paracrine IL-6 is responsible for p16, p21, and p53 down-regulation in breast stromal fibroblasts.
Paracrine IL-6 Activates Breast Stromal Fibroblasts in a p16-dependent Manner
After showing the role of IL-6 in down-regulating important tumor suppressor genes, we sought to test whether IL-6 treatment activates stromal fibroblasts. Therefore, breast stromal fibroblast cells (NBF-6) were incubated with SFM containing different concentrations of the IL-6 protein for 24 h, and then the levels of IL-6, α-SMA, SDF-1, and TGF-β1 were assessed by immunoblotting. SFM and MDA-SFCM were used as negative and positive controls, respectively. Fig. 2C (right) shows that the levels of α-SMA increased markedly in the presence of 5 ng/ml IL-6. On the other hand, the levels of SDF-1 and TGF-β1 increased in a dose-dependent manner, reaching the maximum in response to 35 ng/ml IL-6 (Fig. 2C, right). On the contrary, specific inhibition of IL-6 in MDA-SFCM abolished the effect of MDA-MB-231 cells on the expression of these proteins (Fig. 2C, left). Furthermore, IL-6 knockdown in MDA-MB-231 cells abolished the effect of the MDA-SFCM on the levels of the IL-6, α-SMA, SDF-1, and TGF-β1 proteins in stromal fibroblasts (Fig. 2F). Interestingly, IL-6 (35 ng/ml) and MDA-SFCM strongly increased the mRNA levels of IL-6, α-SMA, SDF-1, and TGF-β1 (Fig. 2G), indicating that the IL-6 paracrine effect on these genes is at the mRNA level. These results indicate that IL-6 can reactivate breast stromal fibroblast cells.
To confirm these results, we assessed the migration/invasion abilities of breast stromal fibroblasts in the presence and absence of IL-6 as described above, using the RTCA-DP xCELLigence system. Fig. 3A shows that although breast stromal fibroblasts (NBF-6) treated with MDA-SFCM exhibited high migration and invasion abilities, IL-6 inhibition in this SFCM suppressed these abilities. On the other hand, adding IL-6 (35 ng/ml) to SFM markedly increased the migratory and the invasiveness capacities of these fibroblasts (Fig. 3A). However, although this increase reached the level obtained with MDA-SFCM for the invasion, it was 50% lower as to the migration (Fig. 3A). This indicates that paracrine IL-6 increases the migration/invasion abilities of breast stromal fibroblasts.
FIGURE 3.
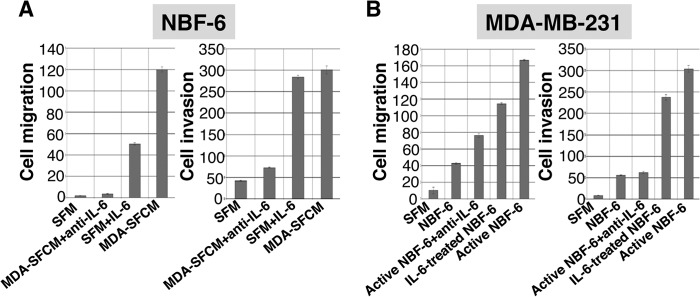
IL-6 activates breast stromal fibroblasts. A, exponentially growing NBF-6 cells (2 × 104) were exposed to SFM, SFM containing IL-6 (35 ng/ml), SFM conditioned with MDA-MB-231 cells (MDA-SFCM), and MDA-SFCM containing anti-IL-6 inhibitory antibody (25 ng/ml). B, MDA-MB-231 cells (2 × 104) were incubated with SFM or SFCM obtained from the indicated cells in the presence of anti-IL-6 inhibitor antibody (25 ng/ml) or in the absence of the inhibitor. Cells were added to the upper wells of CIM-Plates, and then the migration and invasion abilities of both NBF-6 and MDA-MB-231 cells were assessed for 24 h by the real-time RTCA-DP xCELLigence system. Error bars, S.D.
To further show the IL-6-dependent reactivation of stromal fibroblasts, we assessed the ability of NBF-6 preincubated for 24 h with IL-6 or MDA-SFCM (active NBF-6) in enhancing the migration/invasion abilities of breast cancer cells in a paracrine manner. Therefore, SFCM from these cells were collected and used to treat MDA-MB-231 cells. Fig. 3B shows that SFCM from IL-6-treated NBF-6 and active NBF-6 enhanced the migration/invasion abilities of MDA-MB-231 cells as compared with those treated with SFM or SFCM from non-treated NBF-6 cells. Interestingly, the paracrine pro-invasive/migratory effects of active NBF-6 were inhibited when SFCM from these cells were pretreated with anti-IL-6 antibody (Fig. 3B). This confirms that IL-6 is responsible for breast stromal fibroblast reactivation.
Next, we sought to elucidate the role of p16 down-regulation in the MDA-MB-231/IL-6-dependent paracrine activation of breast stromal fibroblasts. Therefore, NBF-6 cells were first infected either with a plasmid bearing the open reading frame (ORF) of the p16-coding gene CDKN2A, CDKN2A-ORF (NBF6.ORF cells), or a control plasmid (NBF6C cells). Fig. 4A shows a clear increase in the p16 protein level in cells expressing the CDKN2A-ORF as compared with their corresponding controls. Interestingly, this increase in p16 expression had only a slight inhibitory effect on the cell cycle and cell proliferation (Fig. 4B). Subsequently, these cells were incubated in the presence of MDA-SFCM or IL-6 for 24 h, and then cell lysates were prepared and used to assess protein levels by immunoblotting. Fig. 4C shows that whereas MDA-SFCM and IL-6 reduced the levels of p16, p21, and p53 in NBF6C as compared with cells treated with SFM, this reduction was abolished in cells expressing the CDKN2A-ORF. Furthermore, the increase in the expression of TGF-β1, SDF-1, IL-6, and α-SMA observed in NBF6C was suppressed in NBF6.ORF (Fig. 4C). This indicates that p16 down-regulation is an important step toward IL-6-dependent activation of breast stromal fibroblasts.
FIGURE 4.

IL-6 activates breast stromal fibroblasts in a p16-dependent manner. NBF-6 cells were infected with a control vector (NBF6C) or a vector bearing the CDKN2A-ORF (NBF6.ORF). A, immunoblotting analysis. B, cell proliferation and cell cycle were analyzed 3 days postinfection. Left, cell proliferation analysis of exponentially growing cells (2 × 104) using the RTCA-DP xCELLigence system. Right, cell cycle analysis using flow cytometry. The numbers indicate the proportion of cells in different phases of the cell cycle. C, cells were treated with the indicated media, and the levels of the indicated proteins were assessed by immunoblotting.
STAT3 Plays a Major Role in IL-6-dependent Reactivation of Breast Stromal Fibroblasts
The obtained data raised an important question as to how paracrine IL-6 down-regulates p16, p21, and p53. It is well known that IL-6 activates the JAK2/STAT3 pathway and that active STAT3 represses p21 and p53 (26, 32). Therefore, we first investigated the possible STAT3 activation in breast stromal fibroblasts upon exposure to MDA-SFCM or IL-6. To this end, TCF-64 and NBF-6 cells were incubated for 24 h with SFM, SFM containing IL-6, or MDA-SFCM. Fig. 5A shows that MDA-SFCM or the presence of IL-6 in SFM increased the level of the IL-6 receptor (GP-130) and IL-6 and consequently activated the JAK2 and STAT3 proteins in these cells. On the other hand, IL-6 knockdown in MDA-MB-231 cells or IL-6 inhibition by specific antibody in MDA-SFCM abolished the activation of the JAK2/STAT3 pathway (Fig. 5A). Thereby, we decided to investigate the possible implication of the STAT3 pathway in the IL-6-dependent down-regulation of p16, p21, and p53 proteins. Therefore, STAT3 was first knocked down in NBF-6 cells (NBF-6.STAT3si) (Fig. 5B). Fig. 5C shows that STAT3 down-regulation increased the level of p16, p21, and p53, whereas it reduced the expression of IL-6, TGF-β1, SDF-1, and α-SMA. This indicates that STAT3 modulates the expression of all of these genes in breast stromal fibroblasts. Subsequently, NBF-6.STAT3si and NBF-6.controlsi cells were treated with IL-6 (35 ng/ml) for 24 h. Treatment of NBF-6.controlsi with SFM was used as a control. Fig. 5D shows that STAT3 knockdown abolished IL-6-dependent down-regulation of p16, p21, and p53. Concomitantly, no effect was observed on the levels of IL-6, TGF-β1, SDF-1, and α-SMA as compared with SFM (Fig. 5D). This suggests that STAT3 activation is essential for IL-6-dependent reactivation of breast stromal fibroblasts.
FIGURE 5.

STAT3 controls the expression of p16, p21, and p53 and the reactivation of breast stromal fibroblasts. A, stromal fibroblasts were treated with the indicated media for 24 h, and then cell lysates were prepared and utilized for immunoblotting analysis using antibodies against the indicated proteins. B, NBF-6 cells were transfected with STAT3 siRNA (STAT3si) or control siRNA (Ctrl), and the STAT3 level was assessed by immunoblotting. C and D, control and STAT3si cells were either not treated or exposed to the indicated media, and then the expression of the indicated proteins was assessed by immunoblotting using specific antibodies. E, control and STAT3si cells were either not treated or challenged with the indicated media, and then the migration/invasion abilities were assessed using the RTCA-DP xCELLigence system.
To confirm the major role of STAT3 in activating stromal fibroblasts, we evaluated the effect of this transcription factor on the IL-6-dependent increase in the migration/invasion abilities of NBF-6 cells. To this end, NBF-6.STAT3si and NBF-6.control-si cells were treated with MDA-SFCM or SFM containing IL-6 for 24 h, and then invasion and migration were assessed using the RTCA-DP xCELLigence system. Fig. 5E shows a potent increase in the invasion/migration abilities of NBF-6.controlsi cells treated with MDA-SFCM or SFM containing IL-6 as compared with cells treated with SFM. On the other hand, no effect was observed on NBF-6.STAT3si cells wherein STAT3 was specifically down-regulated (Fig. 5E). These results indicate that STAT3 plays a major role in the IL-6-dependent activation of breast stromal fibroblasts.
STAT3 Represses p16, p21, and p53 through Positive Regulation of AUF1
Next, we sought to elucidate the molecular mechanism that underlies the STAT3-dependent repression of p16, p21, and p53 in response to IL-6. In fact, the three tumor suppressor genes are post-transcriptionally regulated by the RNA-binding protein AUF1, which is a family of four proteins generated by alternative pre-mRNA splicing (31, 33, 34). Therefore, we hypothesized that STAT3 may negatively regulate these genes through the activation of their negative regulator AUF1. To test this possibility, we first assessed the expression of AUF1 in NBF-6.STAT3si cells as compared with their controls. Indeed, STAT3 down-regulation reduced the level of both bands corresponding to AUF1, as compared with the basal level observed in control cells (Fig. 6A, right). Furthermore, IL-6- and MDA-SFCM-dependent activation of STAT3 increased the level of AUF1 (Fig. 6A, left). However, this effect was abolished in STAT3-knocked down cells (Fig. 6A, left). This indicates that AUF1 expression is modulated in a STAT3-dependent manner.
FIGURE 6.

STAT3 binds AUF1 promoter and positively regulates its expression. A, immunoblotting, as described in the legend to Fig. 5, C and D. B, total RNA was purified from NBF-6 cells harboring either control siRNA (dark bars) or STAT3 siRNA (open bars), and then specific primers were used to amplify the indicated genes by qRT-PCR. Error bars, S.D. (*, p < 0.01; **, p < 0.001). C, RNAs bound to the AUF1 protein were isolated by immunoprecipitation from Ctrl and STAT3si cells using anti-AUF1 antibody or anti-IgG (used for control cells), and then the mRNAs of the CDKN2A, CDKN1A, and TP53 genes were amplified by qRT-PCR. Data were normalized to the levels of the highly abundant GAPDH mRNA in each IP sample and represented as the enrichment of each mRNA. Error bars, S.E. of three different experiments. *, p ≤ 0.001. D, AUF1 promoter region with the three STAT3 binding sites and their positions. E, ChIP assay. Chromatin was purified from the indicated cells and then immunoprecipitated using anti-STAT3 antibody, and anti-H3 and anti-IgG antibodies were used as positive and negative controls, respectively. Subsequently, three different STAT3 binding sites were amplified by PCR (top) and qPCR (bottom) using specific primers, and their abundance was plotted relative to the input. These experiments were performed in triplicate. Error bars, S.D. (*, p < 0.05). F, top, schematic representation of the luciferase reporter vector. Bottom (histogram), control as well as STAT3si cells were stably transfected with a luciferase reporter vector bearing the wild-type AUF1 promoter segment containing either the three wild-type STAT3 binding sites or the same segment mutated in only one of these sites. The reporter activity was assessed at 72 h post-transfection. Data were presented as percentage change in reporter activity as compared with the negative control cells. Error bars, S.D. (#, difference between the wild-type and the mutated sequence in the control cells) (***, p < 0.001).
To confirm these findings, we tested the effect of STAT3 down-regulation on the level of the AUF1 mRNA by qRT-PCR. Fig. 6B shows that the AUF1 mRNA level was strongly reduced in NBF-6.STAT3si as compared with NBF-6.controlsi. Concomitantly, the levels of CDKN2A, CDKN1A, and TP53 mRNAs were markedly up-regulated in NBF-6.STAT3si cells (Fig. 6B). Next, we checked the effect of STAT3 down-regulation on the binding of AUF1 to its targets. To this end, AUF1-mRNA ribonucleoprotein complexes were obtained from NBF-6.STAT3si cells and their controls by immunoprecipitation (IP) using anti-AUF1 antibody (IgG was used for control cells only), and the level of AUF1-bound mRNAs was assessed upon qRT-PCR amplification using specific primers. Fig. 6C shows amplification of the CDKN1A, CDKN2A, and TP53 mRNAs following immunoprecipitation with anti-AUF1 but not with the IgG antibody, indicating the binding of the AUF1 protein to these transcripts. Importantly, the levels of these mRNAs that were bound to AUF1 decreased in NBF-6.STAT3si cells as compared with their control cells (Fig. 6C). This indicates that STAT3 down-regulation reduces the level of AUF1 binding to its targets. This suggests that STAT3 controls AUF1 expression, and because STAT3 is a transcription factor, this effect could be at the transcriptional level. Therefore, we searched for the possible presence of STAT3 binding sites in the promoter of the AUF1 gene. Fig. 6D shows the presence of three STAT3 cognate binding sites (BS1, BS2, and BS3).
Next, we sought to investigate the binding of STAT3 to these sites. To this end, chromatin was prepared from NBF-6.STAT3si and NBF-6.controlsi cells, and STAT3-DNA and H3-DNA complexes were pulled down using anti-STAT3 or anti-H3 antibodies, respectively, while IgG was used as a negative control. Subsequently, the promoter regions encompassing BS1, BS2, BS3, and the GAPDH promoter were amplified by PCR and qPCR. Fig. 6E shows strong amplification of these AUF1 promoter regions from chromatin immunoprecipitated with anti-STAT3 antibody and also with anti-H3 antibody used as positive control but not with IgG. On the other hand, the GAPDH promoter was only marginally amplified from chromatin immunoprecipitated with anti-STAT3 and anti-IgG antibodies, whereas it was strongly amplified from chromatin immunoprecipitated with the anti-H3 antibody (Fig. 6E). Interestingly, STAT3 binding to its cognate binding sites was reduced in NBF-6.STAT3si cells (Fig. 6E). This indicates the binding of STAT3 to the AUF1 promoter in vivo.
To further confirm the implication of STAT3 in the transcriptional regulation of AUF1, a fragment of the AUF1 promoter encompassing the three STAT3 binding sites was cloned in a Gaussia luciferase reporter vector, as shown in Fig. 6F (top), and then was introduced into NBF-6.STAT3si cells as well as their respective control cells. Interestingly, the luciferase activity was markedly reduced in NBF-6.STAT3si cells as compared with control cells (Fig. 6F, bottom). This indicates that STAT3 plays a major role in AUF1 transcription. Similarly, the use of one mutated STAT3 binding site strongly affected the luciferase activity, which was further reduced in STAT3-defective cells (Fig. 6F, bottom). This shows that the three STAT3 binding sites are important for effective STAT3-dependent activation of the AUF1 promoter, suggesting that the three sites are required for efficient binding of STAT3.
IL-6-related Down-regulation of p16, p21, and p53 and the Activation of Stromal Fibroblasts is AUF1-dependent
Next, we investigated whether paracrine IL-6 modulates the expression of breast stromal AUF1. To this end, cells were treated as described in Fig. 5A, and protein levels were assessed by immunoblotting. Fig. 7A shows that MDA-SFCM and SFM containing IL-6 increased the level of AUF1 in both NBF-6 and TCF-64 cells. Consequently, the levels of p16, p21, and p53 were strongly reduced as compared with control cells treated with SFM (Fig. 7A). However, the inhibition of IL-6 in MDA-SFCM or in MDA-MB-231 cells abolished the effect on AUF1 and its targets (Fig. 7A). This indicates that IL-6 up-regulates AUF1 in breast stromal fibroblasts in a paracrine manner.
FIGURE 7.

IL-6 effects on stromal fibroblasts are AUF1-dependent. A, cells were exposed to the indicated media for 24 h, and then cell lysates were prepared and used for immunoblotting analysis utilizing specific antibodies as indicated. B, NBF-6 cells containing control siRNA or AUF1 siRNA were treated as indicated for 24 h, and then the levels of the indicated proteins were assessed by immunoblotting using specific antibodies. C, RNAs bound to the AUF1 protein were immunoprecipitated from NBF-6 cells treated either with SFM (control) or SFM containing IL-6 using anti-AUF1 antibody or anti-IgG (used for control cells), and then the mRNAs of the indicated genes were amplified by qRT-PCR. Data were normalized to the levels of the highly abundant GAPDH mRNA in each IP sample and represented as the enrichment of each mRNA. Error bars, S.D.; *, p ≤ 0.001. D, NBF-6 cells bearing either ctrl.siRNA or AUF1si were first incubated in SFM containing IL-6 for 24 h and then were treated with actinomycin D for the indicated periods of time. Total RNA was extracted, and the remaining amounts of the mRNAs were assessed using qRT-PCR. The values at time 0 were considered as 100%. The dashed lines reveal the half-life (50% mRNA remaining) for each mRNA analyzed, and the corresponding values are indicated in boxes. Error bars, S.D. E, Ctrl.siRNA and AUF1.siRNA NBF-6 cells were incubated in SFM containing IL-6 for 24 h, and then their migration and invasion abilities were assessed by the real-time RTCA-DP xCELLigence system for 24 h. F, NBF-6 cells bearing control siRNA or AUF1 siRNA were incubated in SFM containing IL-6 for 24 h and then were reincubated in IL-6-free SFM for another 24 h. The resulting SFCM were used to challenge MDA-MB-231 cells, and then their migration/invasion abilities were assessed as described above. G, whole cell lysates were prepared from MDA-MB-231 cells that were previously cultured in SFM or SFM conditioned with NBF-6.ctrl.siRNA and NBF-6.AUF1.siRNA cells that were pretreated with IL-6. The levels of the indicated proteins were assessed by immunoblotting using specific antibodies.
We have next asked whether AUF1 plays a role in IL-6-dependent activation of breast stromal fibroblasts. To this end, AUF1 was knocked down using specific siRNA, and scrambled siRNA was used as control. Fig. 7B shows AUF1 siRNA-dependent down-regulation of AUF1. To assess the IL-6 effect, cells were incubated for 24 h in the presence of SFM or SFM containing IL-6. Interestingly, AUF1 knockdown suppressed IL-6-dependent down-regulation of p16, p21, and p53 (Fig. 7B). Concomitantly, the IL-6-dependent increase in the expression of IL-6, SDF-1, α-SMA, and TGF-β1 was also inhibited in AUF1-defective IL-6-treated cells as compared with their corresponding controls (Fig. 7B). This indicates that AUF1 is a key player in the IL-6-dependent down-regulation of p16, p21, and p53 and also the activation of breast stromal fibroblasts.
To confirm this, we investigated the effect of paracrine IL-6 on the AUF1 binding to the CDKN1A, CDKN2A, and TP53 mRNAs. To this end, NBF-6 cells were treated either with SFM or SFM containing IL-6 for 24 h, and then AUF1-mRNA ribonucleoprotein complexes were obtained by IP using anti-AUF1 antibody (IgG was used for control cells) and were used for qRT-PCR amplification utilizing specific primers. Fig. 7C shows higher the amplification of the CDKN1A, CDKN2A, and TP53 mRNAs when cells where cultured in media containing IL-6 than in the absence of this cytokine. Similar results were obtained for the IL-6, SDF-1, α-SMA, and TGF-β1 mRNAs. However, the IL-6-dependent increase in the AUF1 binding to SDF-1, α-SMA, and TGF-β1 was less important (Fig. 7C). This indicates that paracrine IL-6 increases the level of AUF1 and consequently its binding to its targets. This also shows that AUF1 may post-transcriptionally regulate the expression of IL-6, SDF-1, α-SMA, and TGF-β1.
To further show the role of AUF1 in modulating the expression of these genes in response to paracrine IL-6, we studied the stability of the corresponding mRNAs in NBF-6 cells bearing control or AUF1 siRNA. These cells were incubated in SFM containing IL-6 (35 ng/ml) for 24 h, and then they were treated with actinomycin D for various periods of time (0, 0.5, 1, and 2 h). Fig. 7D shows that whereas AUF1 down-regulation protected the CDKN1A, CDKN2A, and TP53 mRNAs from IL-6-induced turnover, it instead enhanced the turnover of IL-6, SDF-1, and α-SMA. However, no effect was observed on the stability of the TGF-β1 mRNA (Fig. 7D). Therefore, we decided to investigate the role of AUF1 on the stability of the TGF-β1 mRNA for a longer period of time. Fig. 7D shows that although the TGF-β1 mRNA half-life is 11 h in control cells, it is only 2 h 20 min in AUF1-defective cells. This indicates that the TGF-β1 mRNA turnover is also under the control of AUF1.
To demonstrate that IL-6-dependent up-regulation of AUF1 is an important step toward the activation of breast stromal fibroblasts, we first studied the effect of AUF1 down-regulation in NBF-6 cells on their migration/invasion abilities upon exposure to IL-6. Therefore, NBF-6.AUF1si cells and their control cells were incubated in SFM containing IL-6 for 24 h, and then their migration/invasion abilities were assessed as described above. Fig. 7E shows that AUF1 down-regulation inhibited the migration/invasion capabilities of breast fibroblasts exposed to IL-6. Next, we investigated the ability of these fibroblasts to enhance the migration/invasion of breast cancer cells in a paracrine manner. Therefore, SFM were conditioned with NBF-6.AUF1si and their control cells for 24 h, and then the resulting SFCM as well as SFM (used as negative control) were used to test the effect on the migration/invasion of MDA-MB-231 cells as described above. Fig. 7F shows that AUF1 down-regulation strongly inhibited the paracrine pro-migratory/invasive capabilities of IL-6-treated stromal fibroblasts.
Finally, we investigated the effect of IL-6-treated AUF1-defective breast stromal fibroblasts in inducing epithelial-to-mesenchymal transition (EMT) in breast cancer cells. To this end, we checked the expression levels of epithelial and mesenchymal markers in MDA-MB-231 cells exposed to SFM or SFCM from NBF6.AUF1.siRNA cells and their control cells, which were primarily exposed to IL-6. Fig. 7G shows that IL-6 treatment of control cells, but not AUF1-deficient cells, induced the EMT process in breast cancer cells in a paracrine fashion. Indeed, SFCM from IL-6-exposed control cells decreased the level of E-cadherin and EpCAM (two important epithelial markers) and up-regulated the mesenchymal markers Snail1, N-cadherin, vimentin, ZEB1, and TWIST1 (Fig. 7G). On the other hand, AUF1 down-regulation inhibited this paracrine induction of EMT (Fig. 7G). Together, these results clearly show that IL-6-dependent activation of breast stromal fibroblasts is AUF1-related.
AUF1 Up-regulation Activates Breast Stromal Fibroblasts
To further show the role of AUF1 in the activation of breast stromal fibroblasts, we introduced AUF1-ORF corresponding to the p37 isoform (p37AUF1) or an empty vector into NBF-6 cells (NBF6.AUF1-ORF or NBF6.ctr, respectively), and then we assessed the levels of p16, p21, and p53 by immunoblotting. It is noteworthy that p37AUF1 has the strongest affinity for AU-rich elements in vitro (34). Fig. 8, A and B, shows that the introduction of p37AUF1 increased the level of AUF1 protein and mRNA, respectively. Interestingly, AUF1 up-regulation reduced the levels of the p16, p21, and p53 proteins and mRNAs (Fig. 8, A and B). Concomitantly, the protein and mRNA levels of IL-6, SDF-1, TGF-β1, and α-SMA were strongly up-regulated in p37AUF1-expressing cells as compared with control cells (Fig. 8, A and B). This indicates that AUF1 up-regulation in breast stromal fibroblasts plays a major role in their activation. To confirm this, we tested the effect of AUF1 up-regulation on the invasion/migration abilities of breast stromal fibroblasts. To this end, we assessed the migration/invasion abilities of NBF-6 cells expressing p37AUF1 and their controls as described in the legend to Fig. 5E. Fig. 8C shows that AUF1 up-regulation markedly increased both the invasion and the migration capacities of breast stromal fibroblasts as compared with control cells.
FIGURE 8.

AUF1 up-regulation activates breast stromal fibroblasts and enhances their paracrine procarcinogenic effects. A, whole cell lysates were prepared from NBF-6 cells expressing either p37AUF1-ORF or a control plasmid. The levels of the indicated proteins were assessed by immunoblotting. B, total RNA was purified and used to assess the mRNA levels of the indicated genes by qRT-PCR. Error bars, S.D. C, the migration/invasion abilities of NBF-6 cells expressing either p37AUF1-ORF or a control plasmid were assessed using the RTCA-DP xCELLigence system. D, SFM and SFCM prepared from NBF-6 cells expressing either p37AUF1-ORF (NBF6-AUF1.ORF) or a control plasmid (NBF6-AUF1C) were used to challenge MDA-MB-231 cells. The invasion/migration abilities of MDA-MB-231 cells were assessed using the RTCA-DP xCELLigence system. E, whole cell lysates were prepared from MDA-MB-231 cells that were previously cultured in SFM or SFM conditioned with NBF-6 cells pretransfected with a control plasmid (Ctrl) or a plasmid bearing p37AUF1 (AUF1). The levels of the indicated proteins were assessed by immunoblotting using specific antibodies.
AUF1 Up-regulation in Breast Stromal Fibroblasts Triggers EMT in Breast Cancer Cells
To further confirm the AUF1-dependent activation of breast stromal fibroblasts, we tested the paracrine effect of these cells on the invasion/migration abilities of breast cancer cells. To this end, SFM and SFCM from NBF6.AUF1-ORF and NBF6.ctr cells were used to treat MDA-MB-231 cells, and their invasion/migration were assessed over 24 h using the RTCA-DP xCELLigence system. Fig. 8D shows that the expression of AUF1 in stromal fibroblasts strongly enhanced the migration and invasion abilities of breast cancer cells in a paracrine manner. Next, we checked the expression levels of epithelial and mesenchymal markers in MDA-MB-231 cells exposed to SFCM from NBF6.AUF1-ORF and NBF6.ctr cells. Fig. 8E shows that SFCM from stromal fibroblasts expressing high level of AUF1 decreased the level of E-cadherin and EpCAM and up-regulated vimentin and N-cadherin (two important markers of mesenchymal cells). To shed light on the molecular mechanism underlying ectopic AUF1-dependent induction of EMT, we tested the effect of SFCM from NBF6.AUF1-ORF on the expression of the EMT inducers, Snail1, ZEB-1, and Twist-1, which suppress the expression of E-cadherin (35–38). Interestingly, SFCM from stromal fibroblasts expressing a high level of AUF1 increased the expression level of the three repressors as compared with control cells (Fig. 8E). Together, these results indicate that AUF1 up-regulation in breast stromal fibroblasts triggers EMT in breast cancer cells in a paracrine manner, which demonstrates their active status.
DISCUSSION
It has become clear that tumors evolve through accumulation of cellular and molecular changes of both cell-autonomous and paracrine origins. In addition, tumor cells activate stromal cells, which fuel cancer growth and spread. It is therefore of the utmost importance to define the molecules and pathways that control these carcinogenic cross-talks. It has been previously reported that breast cancer cells can activate stromal fibroblasts and that TGF-β plays an important role in this process (7, 39). Recently, Xu et al. have also shown the role of breast cancer cells CD147 in the transformation of quiescent fibroblasts to myofibroblasts (40). It is also clear that the STAT3/IL-6 pathway plays a major role in this paracrine effect of cancer cells on their microenvironment (22, 26). However, the role of this pathway in transactivating breast stromal fibroblasts is still unclear. In the present report, we have demonstrated that IL-6 is a major messenger that breast cancer cells utilize to activate their adjacent stromal fibroblasts. Indeed, the highly invasive breast cancer cells MDA-MB-231 activated normal breast stromal fibroblasts and down-regulated the three important tumor suppressor proteins p16, p21, and p53 in an IL-6-dependent manner. These cells secrete several other cytokines, but the effect of IL-6 was predominant. Indeed, whereas IL-6 inhibition abolished the paracrine effect on breast fibroblasts, adding IL-6 to the culture medium yielded an effect similar to that of the medium conditioned with the MDA-MB-231 cells (Fig. 2).
At the molecular level, it has been reported that cancer cells can suppress p53 in adjacent fibroblasts in a paracrine manner. In some combinations of tumor cells and fibroblasts, the inhibitory effect was at the p53 basal level, whereas in other cases, the inhibition was observed only upon exposure to genotoxic agents (41). Therefore, to the best of our knowledge, the present report provides the first indication that the paracrine effect of breast cancer cells leads to the down-regulation of the three tumor suppressor proteins p16, p21, and p53 and that IL-6 is a major activator of breast stromal fibroblasts. Indeed, IL-6 down-regulated these three major tumor suppressor proteins and up-regulated several myofibroblast markers, such as SDF-1 and α-SMA (Fig. 9).
FIGURE 9.
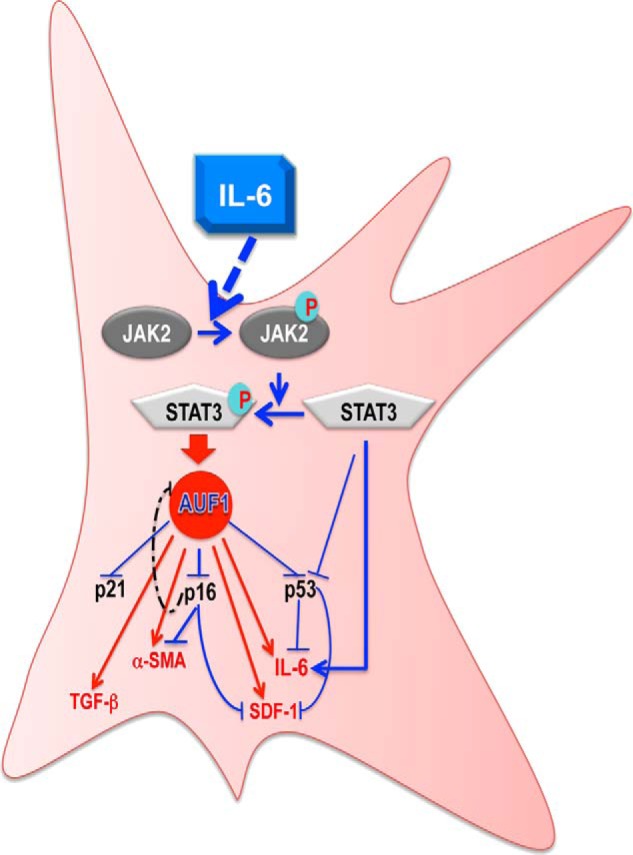
Schematic representation of the role of IL-6 in transactivating breast stromal fibroblasts. Paracrine IL-6 activates the JAK2/STAT3 pathway, which transactivates AUF1 through STAT3 binding to the AUF1 promoter. This leads to down-regulation of the tumor suppressor proteins p16, p21, and p53 and also the up-regulation of α-SMA, SDF-1, IL-6, and TGF-β1 via direct binding of AUF1 to their mRNAs. STAT3 can also suppress the expression of p53/p21 and activates IL-6 in an AUF1-independent manner. For more details, see “Discussion.”
Furthermore, we have shown that p16 down-regulation is important for such activation. This corroborates our recent findings showing that most human cancer-associated fibroblasts have reduced levels of p16 and that specific p16 down-regulation increases the expression of α-SMA and SDF-1 and activates breast stromal fibroblasts (28). Similar findings were reported for p53 and p21 (29, 42–44), indicating that down-regulation of these tumor suppressor genes is an important step toward the activation of stromal fibroblasts. Therefore, we addressed the molecular mechanism underlying IL-6-dependent down-regulation of these tumor suppressor proteins and have shown that STAT3 activation is important for this down-regulation and the activation of stromal fibroblasts upon exposure to IL-6. Furthermore, STAT3 down-regulation, in the absence of IL-6 treatment, increased the levels of p16, p21, and p53 and reduced the expression of several markers of active fibroblasts, such as SDF-1, TGF-β1, and α-SMA. In fact, it has been previously shown that active STAT3 represses p21 and p53 (26, 32). Therefore, we present here the first indication that although STAT3 is also a negative regulator of p16, it positively controls SDF-1, TGF-β1, and α-SMA, which leads to an increase in the procarcinogenic effects of breast stromal fibroblasts. This indicates that STAT3 activation plays a major role in the transactivation of breast stromal fibroblasts and that STAT3 also has a non-cell-autonomous tumor-promoting activity. This shows that STAT3 activation plays a tremendous role in carcinogenesis, not only in cancer cells but also in stromal fibroblasts, and coordinates the reciprocal cross-talk between cancer cells and these mesenchymal cells, which promotes tumor growth and spread. Furthermore, STAT3 promotes angiogenesis through activating angiogenic factors, such as VEGF-A and HIF-1α, and also modulates the immune system through various cytokines, such as IL-6, IL-10, and TGF-β (22, 26).
Importantly, we have shown that these STAT3 effects are mediated through the transactivation of the RNA-binding protein AUF1, which plays a major role in the activation of breast stromal fibroblasts. Indeed, in addition to its known role in down-regulating the three major tumor suppressor proteins p16, p21, and p53 (31, 33), we have shown here that AUF1 binds and stabilizes IL-6, SDF-1, TGF-β1, and α-SMA mRNAs, which is a major feature of active fibroblasts. Regarding IL-6, it has been shown previously that ectopic expression of AUF1 stabilizes IL-6, which corroborates our findings. However, IL-6 was also stabilized by specific AUF1 down-regulation (45), which is in disagreement with the present results. This discrepancy could be due to the fact that Paschoud et al. (45) used mouse NIH 3T3 cells, whereas our findings were in breast stromal fibroblasts.
Several of the AUF1 targets, such as p21, p53, and IL-6, are known to be under the control of STAT3 (26), indicating the possible coexistence of both direct and indirect STAT3-dependent regulation of these genes, which demonstrates the capital role of this transcription factor (Fig. 9). The fact that STAT3 and AUF1 control the expression of a plethora of other important autocrine and paracrine cancer-related genes (26, 33) indicates that this STAT3/AUF1 axis plays a major role in the activation of breast stromal fibroblasts and also their paracrine prometastatic effects. Notably, there is a negative feedback loop between p16 and AUF1 (Fig. 9). Indeed, p16 also negatively regulates AUF1 post-transcriptionally (46), which could explain the key role of p16 in the activation of stromal fibroblasts. Therefore, either AUF1 up-regulation or p16 down-regulation will both lead to stromal fibroblast transactivation and the consequent promotion of metastasis. This effect on p16/AUF1 could be through STAT3 activation as shown here or through other pathways. These results also show that AUF1 has a non-cell-autonomous oncogene function, which enhances carcinogenesis in a paracrine manner.
In addition, several recent lines of evidence indicate that AUF1 has autocrine procarcinogenic effects in several solid tumors (47). Indeed, mice engineered to overexpress p37AUF1 developed undifferentiated sarcomas expressing high levels of cyclin D1, c-Myc, and c-Fos, with high vascularization and cellularity that progressed to a late stage and resulted in animal death (48). Furthermore, cytoplasmic expression of AUF1 was higher in malignant thyroid tissues as compared with benign tissues, and total or exon-selective knockdown of AUF1 led to growth inhibition accompanied by the up-regulation of cell cycle inhibitors (49). Moreover, Abdelmohsen et al. (50) have recently reported that AUF1 represses Dicer and consequently influences miRNA levels. They have also shown low expression of AUF1 in normal tissues but high levels in cancer tissues from colon, stomach, breast, kidney, liver, and pancreas (50). Together, these results indicate a wide ranging mode of oncogenic action for AUF1 (47).
In summary, the present results provide clear evidence that IL-6 activates breast stromal fibroblasts through STAT3-dependent induction of AUF1, which leads to the down-regulation of p16, p21, and p53 and the up-regulation of IL-6, SDF-1, TGF-β1, and α-SMA. This shows the major role of IL-6, STAT3, and AUF1 in the synergetic interplay between breast carcinomas and their stromal fibroblasts. Therefore, targeting AUF1 could have a potent antimetastatic effect and therefore may be of great therapeutic value.
Acknowledgments
We are grateful to Dr. Myriam Gorospe for kindly providing the pSILENCER-AUF15 plasmid and Dr. H. Ghebeh for help.
This work was performed under Research Advisory Council Proposal 2120034 and was supported in part by a King Abdulaziz City for Science and Technology Grant 11-BIO1435-20, under National Comprehensive Plan for Science and Technology.
- α-SMA
- α-smooth muscle actin
- qPCR
- quantitative PCR
- qRT-PCR
- quantitative RT-PCR
- SFCM
- serum-free conditioned medium/media
- SFM
- serum-free medium/media
- IP
- immunoprecipitation
- EMT
- epithelial-to-mesenchymal transition.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Pietras K., Ostman A. (2010) Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. 316, 1324–1331 [DOI] [PubMed] [Google Scholar]
- 3. Albini A., Mirisola V., Pfeffer U. (2008) Metastasis signatures: genes regulating tumor-microenvironment interactions predict metastatic behavior. Cancer Metastasis Rev. 27, 75–83 [DOI] [PubMed] [Google Scholar]
- 4. Allen M., Louise Jones J. (2011) Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J. Pathol. 223, 162–176 [DOI] [PubMed] [Google Scholar]
- 5. Rønnov-Jessen L., Petersen O. W., Koteliansky V. E., Bissell M. J. (1995) The origin of the myofibroblasts in breast cancer: recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J. Clin. Invest. 95, 859–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kojima Y., Acar A., Eaton E. N., Mellody K. T., Scheel C., Ben-Porath I., Onder T. T., Wang Z. C., Richardson A. L., Weinberg R. A., Orimo A. (2010) Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. U.S.A. 107, 20009–20014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaughan M. B., Howard E. W., Tomasek J. J. (2000) Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 257, 180–189 [DOI] [PubMed] [Google Scholar]
- 8. Knüpfer H., Preiss R. (2007) Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 102, 129–135 [DOI] [PubMed] [Google Scholar]
- 9. Chavey C., Bibeau F., Gourgou-Bourgade S., Burlinchon S., Boissière F., Laune D., Roques S., Lazennec G. (2007) Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 9, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schafer Z. T., Brugge J. S. (2007) IL-6 involvement in epithelial cancers. J. Clin. Invest. 117, 3660–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sansone P., Storci G., Tavolari S., Guarnieri T., Giovannini C., Taffurelli M., Ceccarelli C., Santini D., Paterini P., Marcu K. B., Chieco P., Bonafè M. (2007) IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 117, 3988–4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasser A. K., Sullivan N. J., Studebaker A. W., Hendey L. F., Axel A. E., Hall B. M. (2007) Interleukin-6 is a potent growth factor for ER-α-positive human breast cancer. FASEB J. 21, 3763–3770 [DOI] [PubMed] [Google Scholar]
- 13. Spaeth E. L., Dembinski J. L., Sasser A. K., Watson K., Klopp A., Hall B., Andreeff M., Marini F. (2009) Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 4, e4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tamm I., Cardinale I., Krueger J., Murphy J. S., May L. T., Sehgal P. B. (1989) Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. J. Exp. Med. 170, 1649–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sullivan N. J., Sasser A. K., Axel A. E., Vesuna F., Raman V., Ramirez N., Oberyszyn T. M., Hall B. M. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang Q., Bournazou E., Sansone P., Berishaj M., Gao S. P., Daly L., Wels J., Theilen T., Granitto S., Zhang X., Cotari J., Alpaugh M. L., de Stanchina E., Manova K., Li M., Bonafe M., Ceccarelli C., Taffurelli M., Santini D., Altan-Bonnet G., Kaplan R., Norton L., Nishimoto N., Huszar D., Lyden D., Bromberg J. (2013) The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 15, 848–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Akira S., Nishio Y., Inoue M., Wang X. J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71 [DOI] [PubMed] [Google Scholar]
- 18. Gao S. P., Mark K. G., Leslie K., Pao W., Motoi N., Gerald W. L., Travis W. D., Bornmann W., Veach D., Clarkson B., Bromberg J. F. (2007) Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Invest. 117, 3846–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dethlefsen C., Højfeldt G., Hojman P. (2013) The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 138, 657–664 [DOI] [PubMed] [Google Scholar]
- 20. Lieblein J. C., Ball S., Hutzen B., Sasser A. K., Lin H. J., Huang T. H., Hall B. M., Lin J. (2008) STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 8, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sriuranpong V., Park J. I., Amornphimoltham P., Patel V., Nelkin B. D., Gutkind J. S. (2003) Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 63, 2948–2956 [PubMed] [Google Scholar]
- 22. Groner B., Lucks P., Borghouts C. (2008) The function of Stat3 in tumor cells and their microenvironment. Semin. Cell Dev. Biol. 19, 341–350 [DOI] [PubMed] [Google Scholar]
- 23. Dolled-Filhart M., Camp R. L., Kowalski D. P., Smith B. L., Rimm D. L. (2003) Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 9, 594–600 [PubMed] [Google Scholar]
- 24. Berishaj M., Gao S. P., Ahmed S., Leslie K., Al-Ahmadie H., Gerald W. L., Bornmann W., Bromberg J. F. (2007) Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 9, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dauer D. J., Ferraro B., Song L., Yu B., Mora L., Buettner R., Enkemann S., Jove R., Haura E. B. (2005) Stat3 regulates genes common to both wound healing and cancer. Oncogene 24, 3397–3408 [DOI] [PubMed] [Google Scholar]
- 26. Yu H., Kortylewski M., Pardoll D. (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51 [DOI] [PubMed] [Google Scholar]
- 27. Aboussekhra A. (2011) Role of cancer-associated fibroblasts in breast cancer development and prognosis. Int. J. Dev. Biol. 55, 841–849 [DOI] [PubMed] [Google Scholar]
- 28. Al-Ansari M. M., Hendrayani S. F., Shehata A. I., Aboussekhra A. (2013) p16(INK4A) Represses the paracrine tumor-promoting effects of breast stromal fibroblasts. Oncogene 32, 2356–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hawsawi N. M., Ghebeh H., Hendrayani S. F., Tulbah A., Al-Eid M., Al-Tweigeri T., Ajarim D., Alaiya A., Dermime S., Aboussekhra A. (2008) Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 68, 2717–2725 [DOI] [PubMed] [Google Scholar]
- 30. Al-Mohanna M. A., Al-Khalaf H. H., Al-Yousef N., Aboussekhra A. (2007) The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. Nucleic Acids Res. 35, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang W., Martindale J. L., Yang X., Chrest F. J., Gorospe M. (2005) Increased stability of the p16 mRNA with replicative senescence. EMBO Rep. 6, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu G., Wright K. L., Ma Y., Wright G. M., Huang M., Irby R., Briggs J., Karras J., Cress W. D., Pardoll D., Jove R., Chen J., Yu H. (2005) Role of Stat3 in regulating p53 expression and function. Mol. Cell Biol. 25, 7432–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lal A., Mazan-Mamczarz K., Kawai T., Yang X., Martindale J. L., Gorospe M. (2004) Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 23, 3092–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wagner B. J., DeMaria C. T., Sun Y., Wilson G. M., Brewer G. (1998) Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics 48, 195–202 [DOI] [PubMed] [Google Scholar]
- 35. Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 [DOI] [PubMed] [Google Scholar]
- 36. Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Savagner P., Gitelman I., Richardson A., Weinberg R. A. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 [DOI] [PubMed] [Google Scholar]
- 37. Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 38. Jablonka S., Bandilla M., Wiese S., Bühler D., Wirth B., Sendtner M., Fischer U. (2001) Co-regulation of survival of motor neuron (SMN) protein and its interactor SIP1 during development and in spinal muscular atrophy. Hum. Mol. Genet. 10, 497–505 [DOI] [PubMed] [Google Scholar]
- 39. Rønnov-Jessen L., Petersen O. W. (1993) Induction of α-smooth muscle actin by transforming growth factor-β1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 68, 696–707 [PubMed] [Google Scholar]
- 40. Xu J., Lu Y., Qiu S., Chen Z. N., Fan Z. (2013) A novel role of EMMPRIN/CD147 in transformation of quiescent fibroblasts to cancer-associated fibroblasts by breast cancer cells. Cancer Lett. 335, 380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bar J., Feniger-Barish R., Lukashchuk N., Shaham H., Moskovits N., Goldfinger N., Simansky D., Perlman M., Papa M., Yosepovich A., Rechavi G., Rotter V., Oren M. (2009) Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 28, 933–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kiaris H., Chatzistamou I., Trimis G., Frangou-Plemmenou M., Pafiti-Kondi A., Kalofoutis A. (2005) Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 65, 1627–1630 [DOI] [PubMed] [Google Scholar]
- 43. Moskovits N., Kalinkovich A., Bar J., Lapidot T., Oren M. (2006) p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 66, 10671–10676 [DOI] [PubMed] [Google Scholar]
- 44. Trimis G., Chatzistamou I., Politi K., Kiaris H., Papavassiliou A. G. (2008) Expression of p21waf1/Cip1 in stromal fibroblasts of primary breast tumors. Hum. Mol. Genet. 17, 3596–3600 [DOI] [PubMed] [Google Scholar]
- 45. Paschoud S., Dogar A. M., Kuntz C., Grisoni-Neupert B., Richman L., Kühn L. C. (2006) Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell Biol. 26, 8228–8241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Al-Khalaf H. H., Colak D., Al-Saif M., Al-Bakheet A., Hendrayani S. F., Al-Yousef N., Kaya N., Khabar K. S., Aboussekhra A. (2011) p16(INK4a) positively regulates cyclin D1 and E2F1 through negative control of AUF1. PLoS One 6, e21111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zucconi B. E., Wilson G. M. (2011) Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Front. Biosci. (Landmark Ed.) 16, 2307–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gouble A., Grazide S., Meggetto F., Mercier P., Delsol G., Morello D. (2002) A new player in oncogenesis: AUF1/hnRNPD overexpression leads to tumorigenesis in transgenic mice. Cancer Res. 62, 1489–1495 [PubMed] [Google Scholar]
- 49. Trojanowicz B., Brodauf L., Sekulla C., Lorenz K., Finke R., Dralle H., Hoang-Vu C. (2009) The role of AUF1 in thyroid carcinoma progression. Endocr. Relat. Cancer 16, 857–871 [DOI] [PubMed] [Google Scholar]
- 50. Abdelmohsen K., Tominaga-Yamanaka K., Srikantan S., Yoon J. H., Kang M. J., Gorospe M. (2012) RNA-binding protein AUF1 represses Dicer expression. Nucleic Acids Res. 40, 11531–11544 [DOI] [PMC free article] [PubMed] [Google Scholar]