

Background: Familial mutations in amyloid precursor protein (APP) increase amyloid-β peptide generation and aggregation leading to Alzheimer disease (AD).
Results: A protective A2T mutation impairs not only β-secretase-mediated APP cleavage but also, unexpectedly, Aβ aggregation.
Conclusion: The protective mutation modulates amyloid formation.
Significance: The interpretation that a lifelong suppression of β-secretase is sufficient to protect against AD is not supported by these novel data.
Keywords: Aggregation, Alzheimer Disease, Amyloid-β (Aβ), Kinetics, Thermodynamics
Abstract
Missense mutations in alanine 673 of the amyloid precursor protein (APP), which corresponds to the second alanine of the amyloid β (Aβ) sequence, have dramatic impact on the risk for Alzheimer disease; A2V is causative, and A2T is protective. Assuming a crucial role of amyloid-Aβ in neurodegeneration, we hypothesized that both A2V and A2T mutations cause distinct changes in Aβ properties that may at least partially explain these completely different phenotypes. Using human APP-overexpressing primary neurons, we observed significantly decreased Aβ production in the A2T mutant along with an enhanced Aβ generation in the A2V mutant confirming earlier data from non-neuronal cell lines. More importantly, thioflavin T fluorescence assays revealed that the mutations, while having little effect on Aβ42 peptide aggregation, dramatically change the properties of the Aβ40 pool with A2V accelerating and A2T delaying aggregation of the Aβ peptides. In line with the kinetic data, Aβ A2T demonstrated an increase in the solubility at equilibrium, an effect that was also observed in all mixtures of the A2T mutant with the wild type Aβ40. We propose that in addition to the reduced β-secretase cleavage of APP, the impaired propensity to aggregate may be part of the protective effect conferred by A2T substitution. The interpretation of the protective effect of this mutation is thus much more complicated than proposed previously.
Introduction
Familial forms of Alzheimer disease are caused by mutations in APP or in the PSEN1 and PSEN2 genes. All these mutations affect the generation of the Aβ2 peptide, which is generated from APP by consecutive proteolytic cleavages mediated by the β- and the γ-secretase. APP is the substrate and PSEN protein is the catalytic subunit of the γ-secretase, thus AD-causing mutations occur both in the substrate and protease. This is still a major argument for the hypothesis that abnormal Aβ generation is central to the disease process (1–3). However, it is important to notice that these mutations do not necessarily exert a purely quantitative effect on Aβ generation. Indeed, only the Swedish double mutation KM670/671NL (4), the recessive A2V mutation (5), and the putative AD mutation E11K (6) (Fig. 1) are known to increase Aβ generation. Other mutations in the C-terminal part of Aβ (amino acids 43–46) shift the initial ϵ-cut by γ-secretase from amino acids 50–51 to amino acids 49–50 and increase the relative amount of long, more aggregation-prone Aβ fragments versus shorter peptides, without increasing the total amount of Aβ (7–9). Other mutations, e.g. at amino acid residues 22–23 near the central hydrophobic domain of Aβ (Fig. 1), affect the intramolecular β-hairpin formation, thus altering Aβ peptide self-assembly (10, 11).
FIGURE 1.
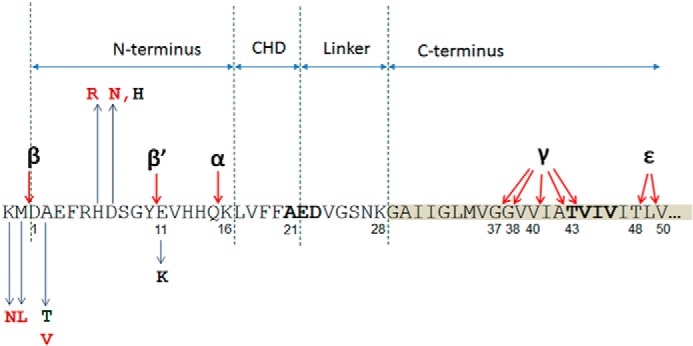
Aβ sequence of amyloid precursor protein. N-terminal part of Aβ fragment (amino acids 1–16) is followed by a central hydrophobic domain (CHD, amino acids 17–21), which is separated from the hydrophobic C-terminal part by a hydrophilic linker (amino acids 22–28). Main secretase cleavage sites (α, β, β′, γ, and ϵ) are indicated with arrows. Pathogenic N-terminal substitutions in or next to the Aβ sequence are shown in red and include KM670/671NL (Swedish), A2V and H6R (English), and D7N (Tottori). Green, the protective mutation A2T. Black, putative pathogenic mutations E11K and D7H. Familial forms of Alzheimer disease mutation-prone amino acid clusters in central hydrophobic domain and at the C terminus are shown in bold.
Aggregation propensity is an intrinsic property of the Aβ sequence and correlates with the length of the hydrophobic C terminus (12–14). Aggregation-prone Aβ42 and Aβ43 alloforms are usually more neurotoxic than the more abundantly generated Aβ40 in different experimental paradigms (14–17). A neurotoxic property of Aβ indeed emerges during the initial stages of its aggregation and is associated with the presence of oligomeric Aβ structures (15, 17, 18). The latter remain the subject of intense investigation (18–20). Compelling evidence, however, indicates that such aggregated Aβ peptides can directly affect memory formation, synaptic transmission, and neuron viability (18, 20).
The central role of APP misprocessing and amyloid generation in the pathogenesis of Alzheimer disease recently received considerable support from the finding of a protective mutation in the APP sequence in a large population-based study (21). This A673T allele, previously described as a natural APP polymorphism (22), was investigated in 41 elderly carriers, including three homozygous ones, and these carriers appeared to be protected against AD but also to perform significantly better in cognitive tests compared with a noncarrier population of the same age (21). These authors provided evidence that the Ala-to-Thr substitution decreased β-secretase cleavage of APP and therefore the generation of Aβ peptide with about 40% in a cell line (21).
A simple interpretation of the data suggests that this mutation provides a life-long and mild suppression (20% in heterozygous carriers) of Aβ generation by BACE-1. Obviously, this would provide a strong argument for the therapeutic development of BACE-1 inhibitors and its preventative use in persons at risk for AD. However, some caution with the interpretation of these data is indicated because little experimental work has been done to evaluate the effect of this mutation on aggregation or neurotoxic properties of the peptide (23).
Experimental studies of mutations at the N terminus, for instance the so-called English (H6R) and Tottori (D7N) mutations, show an increased oligomerization and/or fibrillogenesis of mutant Aβ peptides (24, 25). More intriguingly, apart from the protective Ala-to-Thr mutation (21), an Ala to Val mutation has also been identified in a single Italian kindred with early onset inherited AD. This disease is characterized by both increased Aβ production and increased fibrillogenesis (5, 26). In vitro studies of the A2V mutant Aβ peptides revealed that they were more toxic than wild type Aβ peptides and also that mixing mutant with wild type (WT) Aβ in an equimolar ratio decreased their toxicity, in line with the protected status of heterozygous carriers of this mutation (5). Thus, this mutation appears not only to affect the total production but also to have a profound effect on the biophysical and toxic properties of the Aβ peptide, in a way not explained by currently available models for Aβ toxicity.
We therefore set out to investigate the impact of the A2T and A2V mutations on Aβ production but also aggregation. We show that in addition to effects on β-cleavage, these mutations profoundly affect aggregation kinetics and the free energy of Aβ aggregation. Furthermore, both mutant peptides interact with wild type Aβ and thermodynamically destabilize its aggregation. These findings provide novel insights into the biophysical parameters that determine aggregation and toxicity of Aβ and reinforce the concept that “quality” of the Aβ mix is more important than absolute amounts of Aβ peptide in the causation of AD.
EXPERIMENTAL PROCEDURES
SFV Expression System and APP Mutagenesis
The plasmid pSFV1-huAPP695 has been described previously (27). APP mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The following primers were used to introduce the A673T mutation: 5′-ctctgaagtgaagatggatacggaattccgacatgactcag-3′ (forward) and 5′-ctgagtcatgtcggaattccgtatccatcttcacttcagag-3′ (reverse). To introduce the A673V mutation, the following primers were used: 5′-ctgaagtgaagatggatgtagaattccgacatgactc-3′ (forward) and 5′-gagtcatgtcggaattctacatccatcttcacttcag-3′ (reverse). The mutant constructs were transformed into the XL10 gold ultracompetent cells, and a restriction-grade DNA was isolated. After verification of the mutant sequence using 5′-GTCTTGGCCAACATGATTAGTG-3′ as primer, the APP-expressing and the helper plasmid were transformed into DH5α cells. The DNA was isolated, linearized at the SpeI site, purified using the PCR purification kit (Qiagen, catalog no. 28106), eluted in nuclease-free water, and transcribed using the Megascript SP6 kit (catalog no. AM1330M, Ambion/Invitrogen) according to the manufacturer's instructions in the presence of m7G(5′)ppp(5′)G (catalog no. AM8048, Ambion/Invitrogen). The viral vector was produced in baby hamster kidney cells as described (28), aliquoted, and stored at −80 °C.
Neuronal Culture Transduction
Primary neuronal cultures were prepared from C57Bl6 mouse brains at embryonic day 14 and plated on 6-cm dishes (Nunc) coated with poly-l-lysine (Sigma), one embryo brain per three dishes. Neurons were seeded in minimal essential medium (Invitrogen, catalog no. 31095-029) supplemented with horse serum, penicillin, and streptomycin (PenStrep, Invitrogen, catalog no. 15140-122). 4 h after plating, medium was replaced with Neurobasal medium (NB, Invitrogen, catalog no. 21103-049) supplemented with B27 (Invitrogen, catalog no. 17504-044), and PenStrep.
Neuronal transduction was performed at in vitro culture day 3. 112 μl of viral vector-containing supernatant was added to 1.2 ml of the culture medium for 45 min. Medium was replaced with 1.3 ml of NB+B27+PenStrep as above, and the neuron culture-conditioned medium was collected after 4.5 h. The cells were then washed two times with ice-cold PBS and lysed for 15 min in 300 μl of ice-cold radioimmunoprecipitation buffer (150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8) with complete protease inhibitors. The neurons were then scraped and centrifuged at 13,000 rpm for 15 min to remove debris, and the supernatants were snap-frozen and stored at −80 °C until further processing.
Antibodies to APP Fragments
The following antibodies were used: rabbit polyclonal B63 antibody raised against the C terminus of APP (29); mouse monoclonal 22C11 antibody recognizing the N terminus of APP and total sAPP, from Chemicon/Biognost; rabbit polyclonal antibody recognizing sAPPβ, from Covance; mouse monoclonal 6E10 antibody recognizing the N-terminal part of the Aβ fragment (amino acids 1–16), from Covance.
Western Blot
Neuronal lysates and supernatants were mixed with 4× sample buffer (450 mm Tris-HCl, pH 8.45, 12% glycerol, 4% SDS, 0.0025% Coomassie Blue, 0.0025% phenol red) plus β-mercaptoethanol at a final concentration of 2% and denatured for 15 min at 75 °C. 12 μl of each sample was loaded on precast 4–12% Novex BisTris gel and electrophoresed at 125 V, 400 mA for 1.5 h. Proteins were further transferred on 0.2-μm nitrocellulose membrane (Protran) at 25 V, 400 mA during 1 h and 40 min. The membrane was blocked in Tris-buffered saline (TBS) with 0.1% Tween 20 and 5% nonfat milk. The primary antibodies were applied overnight at 4 °C. Peroxidase-coupled secondary antibodies were applied for 1 h at room temperature. The blots were developed chemiluminescently using enhanced luminol and digitized by means of the ImageQuant LAS4000 mini reader. Quantification of bands was performed in AIDA Image Analyzer version 4.27.
Aβ ELISA
To detect soluble Aβ in neuronal cultures, a sandwich ELISA with end-specific monoclonal antibodies from Janssens Pharmaceutica and generously made available to us by Dr. Mark Mercken was used as described previously (7). JRFcAb40/28 and JRFcAb42/26 raised against the C terminus of Aβ(x-40) and Aβ(x-42), respectively, were used as capture antibodies, and JRFAbN/25 recognizing the N-terminal 1–7 amino acids of human Aβ was used as the detection antibody.
96-Well plates (Nunc MaxiSorp) were coated with Aβ40 or Aβ42 capture antibodies at 1.5 μg/ml. After overnight incubation at 4 °C, the plates were washed and blocked with casein buffer (0.1% in PBS). Human Aβ standards (Aβ40, Aβ40-A2T, and Aβ40-A2V; Aβ42, Aβ42-A2T, and Aβ42-A2V) in dimethyl sulfoxide (DMSO) were diluted in casein buffer and pre-mixed with the detection antibody at 1:2000. For Aβ42 detection, undiluted culture medium samples were used, and a 3× dilution in casein buffer was done for Aβ40 detection. Medium mix with the corresponding antibody was added to the coated ELISA plate and incubated overnight at 4 °C. The plates were developed using 3,3′,5,5′-tetramethylbenzidine plus H2O2 (Sigma). The reactions were stopped with 2 n H2SO4, and absorbance at 450 nm was read on PerkinElmer Life Sciences Envision 2103 multilabel reader. Optical signal versus log Aβ concentration was fitted in GraphPad Prism software with a sigmoid curve with variable slope.
Solubilization of Aβ Peptides
Aβ42 variants (wild type, A2T and A2V mutants, and purity >95% by HPLC/C18) were received from JPT Peptide Technologies GmbH. Aβ40 variants (wild type, A2T and A2V mutants, purity >95% by HPLC/C18) were received from Giotto Bioscience and stored at −20 °C. Prior to solubilization, the vials containing the peptides were brought to room temperature to avoid formation of condensate and briefly centrifuged to collect powder on the bottom. Peptides were dissolved in 2 ml of hexafluoroisopropanol (1,1,1,3,3,3-hexafluor-2-propanol, or HFIP, 99%, Aldrich catalog no. 10522-8) and briefly sonicated, and HFIP was evaporated under a nitrogen stream. HFIP was added for a second time to obtain a peptide concentration of 1 mg/ml. Mixtures of wild type and mutant (Aβ/Aβ mutant) peptides were prepared by mixing the HFIP stock solutions at ratios corresponding to 67, 50, and 33% of mutant peptide.
250 μl of HFIP-dissolved Aβ was dried under nitrogen flow to obtain a peptide film that was resolubilized in DMSO (>99.9%, Sigma catalog no. D41640) to a final peptide concentration of 5 mg/ml, vortexed, and diluted in Tris/EDTA buffer, pH 7.4 (50 mm Tris, 1 mm EDTA), to 100 μm peptide. Protein concentration was measured with the protein assay (catalog no. 500-0006, Bio-Rad).
Thioflavin T Fluorescence Assay of Aggregation Kinetics
90 μl of freshly solubilized Aβ peptide was loaded on black 96-well plate with a μ-clear bottom (catalog no. 655096, Greiner Bio One), mixed with 10 μl of 0.12 mm aqueous solution of thioflavin T, and sealed with transparent film to prevent evaporation. Aggregation kinetics of Aβ was followed in situ at 25 °C using a Fluostar OPTIMA plate reader at λex = 440 nm and λem = 480 nm. Gain was set to the 5% of signal intensity in blank probe (unbound dye). Orbital shaking (2 s) was applied before reading every 15 min during ≈63 h.
Critical Concentration and Gibbs Energy Evaluation
A series of Aβ peptide samples from 100 to 1.2 μm was incubated for 6 weeks in low adhesion plastic vials (catalog no. B74030, Bioplastics) at room temperature. The samples were centrifuged during 3 min at 14,000 rpm at 4 °C, and soluble peptide content was assessed by means of the protein assay (Bio-Rad). Insoluble peptide fraction was calculated by subtracting the soluble peptide fraction (C) from the total concentration (C0), and a critical concentration (Cc) was evaluated as described (31). Cc refers to a concentration above which no further increase in soluble peptide is observed and below which there is less than 10% of detectable insoluble material. The Gibbs free energy of aggregation relates to the critical concentration as described under “Results.”
Immune Dot Blot
50 μm Aβ solutions were aggregated for 2 h, and 3 μl were blotted on a nitrocellulose membrane (0.1 μm, Protran) and dried. The membrane was blocked in TBS + 0.1% Tween 20 + 5% nonfat milk for 1 h at room temperature, and the primary antibodies were applied at 1:1000 overnight. The following primary antibodies were used: mouse monoclonal antibody 4G8 recognizing a middle region of Aβ (amino acids 18–22), from Sigma; mouse monoclonal antibody 3D6 recognizing N terminus of Aβ (amino acids 1–5), from Elan Pharmaceuticals; rabbit polyclonal “conformation-specific” antibody A11 recognizing nonfibrillar oligomers, from Invitrogen; rabbit polyclonal conformation-specific antibody OC recognizing fibrillar oligomers and fibers, from Calbiochem/Millipore. The secondary peroxidase-coupled antibodies were applied for 1 h, and the dot blot was developed in the same way as the Western blots described above.
AFM Analysis of Aβ Oligomers
The 1 × 1 cm silicon/SiO2 substrates (silicon with 300 nm thermally grown SiO2) were cleaned in Piranha solution (1 volume of H2O2 and 3 volumes of H2SO4) for 15 min to make them hydrophilic, then rinsed thoroughly with ultrapure water (UPW, 18.2 megohms), and kept in UPW until use. 40 μl of 50 μm Aβ sample was deposited on the substrates, dried, then rinsed with UPW, and dried again. The AFM images were obtained in tapping mode in ambient conditions with JPK III AFM (The NanoWizard 3 BioScience AFM) using a silicon tip with an initial radius between 2 and 12 nm on a V shaped nitride cantilever (Brucker, MSNL-10, cantilever F). The nominal spring constant of the cantilever was 0.6 n/m and the frequency used was 120 ± 10 kHz. AFM images were analyzed by means of the WSxM software.
Transmission Electron Microscopy of Aβ Fibers
5 μl of aggregated Aβ were adsorbed to carbon-coated Formvar film on 400-mesh copper grids (Laborimpex, S162–4H) for 1 min. The grids were washed five times in UPW and stained with 2% w/v uranyl acetate for 10 s. Samples were then examined with a JEOL JEM-1400 microscope at 80 kV.
Fourier Transform Infrared Spectroscopy
The infrared spectra of aggregated Aβ samples were recorded using a Bruker Tensor 27 infrared spectrophotometer (Bruker Optik GmbH) equipped with a Bio-ATR II accessory. Spectra were recorded at a resolution of 4 cm−1, and 128 accumulations were performed per measurement, at a wave number range from 900 to 4000 cm−1. UPW was used to record a blank signal. The obtained spectra were vehicle-subtracted and rescaled in the amide I area (from 1600 to 1700 cm−1).
ANS Assay of Hydrophobic Exposure
20 μm pre-aggregated Aβ peptide was mixed with an aqueous solution of 8-anilinonaphthalene-1-sulfonate (ANS) dye at a 1:5 molar ratio and loaded on a 96-well plate. Fluorescent emission of ANS (λex = 355 nm) was recorded between 400 and 700 nm with a 2- or 5-nm step on the Tecan Infinite M1000 plate reader. Unbound dye has a maximum of emission at around 535 nm, and blue shift of bound dye emission indicates lower polarity of bound entities.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6 software. One-way analysis of variance with Tukey multiple comparison tests was used to analyze APP processing. Unpaired two-tailed t test was used for analysis of kinetic and thermodynamic parameters of aggregation, ELISAs, and dot blots. Pearson correlation coefficients were used to evaluate correlation between kinetic parameters. Statistical significance of the observations is indicated in the figure legends.
RESULTS
Effects of A2T and A2V Mutation on β-Secretase Processing of APP in Neurons
To confirm and expand previous findings in non-neuronal cell lines (5, 21), we evaluated the effect of the Ala-to-Thr (A673T and A2T) and Ala-to-Val (A673V and A2V) substitutions on APP processing in primary neurons. Site-directed mutagenesis was used to introduce either an A2T or an A2V mutation in the neuronal 695-amino acid-long isoform of human APP, which was then delivered to primary mouse neurons by means of the SFV-based expression system as before (27). Secreted and membrane-associated products of APP processing were analyzed by means of Western blot (Fig. 2A and Table 1) and sandwich ELISA (Fig. 2, B and C). In agreement with previous reports (5, 21), the disease-causative APP A2V mutation induced a robust increase in β-cleavage products sAPPβ and APP-C99 at the cost of α-cleavage (Fig. 2A and Table 1). This was accompanied by a significant increase in Aβ40 and Aβ42 secretion (Fig. 2C). The protective A2T APP mutant in contrast reduced β-cleavage significantly, but in contrast to the observations made in non-neuronal cells (21), no increment in sAPPα was observed (Fig. 2A and Table 1). The significant decrease in sAPPβ (Table 1) was coherent with the 2.3-fold reduction in soluble Aβ40 and an almost 3-fold reduction in soluble Aβ42 (Fig. 2C).
FIGURE 2.

Mutations in Ala-673 affect human APP processing in SFV-transduced primary neurons. A, fragments of human (h)APP(695) generated during β-, β′-, and γ-secretase-mediated cleavage in mouse primary neuronal culture. On the right, C-terminal fragments generated during APP proteolysis, resolved in 16% Tris/Tricine gel, and probed with B63 antibody against the C terminus of APP. C99 is CTFβ; C89 is CTFβ′; C83 is CTFα; and pCTFs are phosphorylated C-terminal fragments. Semi-quantification of band intensities is presented in Table 1. Note that A673T/A673V refers to the position of mutation in the longest APP isoform (APP(770)), and in APP(695) this corresponds to A598T/A598V and in the Aβ sequence to A2T/A2V. B, calibration curves for ELISA detection of Aβ mutants by using JRFAbN/25 antibody against N-terminal amino acids 1–7 of human Aβ, means ± S.D. C, ELISA quantification of soluble Aβ40 and Aβ42 released by mutant human APP(695)-overexpressing neurons, means + S.E. Statistical significance (unpaired two-tailed t test) is indicated by *, p < 0.05; **, p < 0.01; and ***, p < 0.001; n = 3.
TABLE 1.
Semi-quantification of human APP(695) proteolytic fragments generated in transduced mouse neurons
Corresponding Western blots are shown in Fig. 2A. Data are normalized to the human APP expression level and are presented as M ± S.D. from three independently transduced cultures in triplicate. Significance of difference between mutant and WT APP is indicated by *, p < 0.05; **, p < 0.01; ****, p < 0.0001, or NS, not significant; one-way analysis of variance ith Tukey multiple comparison's test, CI 99%, is shown.
| WT APP | A673V | A673T | |
|---|---|---|---|
| C99 | 0.48 ± 0.14 | 1.04 ± 0.02** | 0.27 ± 0.02, NS. |
| sAPPβ | 0.18 ± 0.07 | 1.02 ± 0.02**** | 0.001 ± 0.000* |
| sAPPα | 1.01 ± 0.04 | 0.31 ± 0.07* | 0.81 ± 0.21, NS. |
| sAPP | 0.91 ± 0.08 | 0.92 ± 0.12, NS | 0.65 ± 0.14, NS |
Therefore, causative and protective mutations modulate β-cleavage in neurons in distinct opposite directions. This confirms in neurons that these mutations affect β-secretase cleavage of the APP protein, but the question whether these mutations would also affect aggregation properties of Aβ has not been addressed before. More importantly, we also wondered how these mutant Aβ peptides would interact with their wild type Aβ counterparts as this is directly relevant for the situation in the heterozygote carriers of these mutations.
A2T and A2V Mutations Affect Aβ40 Aggregation
We used two independently produced batches of wild type Aβ42 and Aβ40 and of Aβ42 and Aβ40 containing either the A2T or the A2V mutations, all with >95% purity. The peptides were first dissolved in HFIP and then dried under a nitrogen stream and reconstituted in dimethyl sulfoxide (DMSO). DMSO solution was further diluted with Tris/EDTA buffer, pH 7.4, to reach a final concentration of peptide of 25 μm. The aggregation of Aβ peptides was followed using the β-sheet-sensitive fluorescent dye thioflavin T (ThT).
The A2V and A2T mutants either as a pure mixture or as a 1:1 mixture with wild type Aβ42 followed the similar fast aggregation course as pure wild type Aβ42 peptide, without a discernible lag phase (Fig. 3A) suggesting that nucleation of all Aβ42 samples occurred during the dead time of the experiment (≤30 min) (Fig. 3A).
FIGURE 3.
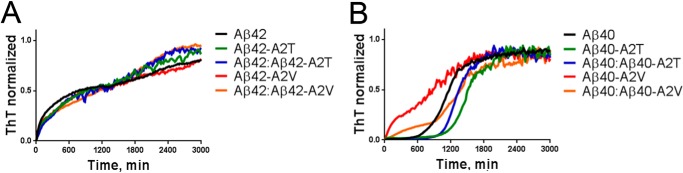
Effect of the A2T and A2V mutations on the aggregation course of Aβ peptides. A, aggregation kinetics of Aβ42 variants monitored in the ThT incorporation assay in Tris/EDTA buffer, pH 7.4. Peptide concentration is 25 μm. Data were normalized to the maximal ThT signal, averaged from four experiments with two independent preparations from two independently produced batches, and a mean value was plotted. B, aggregation kinetics of Aβ40 variants monitored in the ThT incorporation assay in Tris/EDTA buffer, pH 7.4. Peptide concentration is 25 μm. Data were normalized to the maximal ThT signal and averaged from four experiments with two independent preparations from two independently produced batches, and a mean value was plotted.
The Aβ40 peptide is intrinsically less aggregation-prone. In the context of Aβ40, a remarkable impact of the A2T and A2V mutations on aggregation became apparent. Aβ40-A2V on the one hand aggregated without a lag phase following a pattern that is more like wild type Aβ42 than Aβ40, and on the other hand, Aβ40-A2T showed a 1.5-fold increase in lag phase (Fig. 3B). Of note, the kinetic curves of the equimolar mixes of Aβ40 with the mutant variants represented an average between the wild type and the pure mutant suggesting little interaction of wild type and mutant Aβ in these mixes.
A2T and A2V Mutations Affect Kinetic Parameters of Aβ40 Aggregation
We next determined how the nucleation (tlag) and the fiber growth rate (k) are affected by changes in the initial peptide concentration. The rate of aggregation (k) was obtained by fitting a single exponential function to the growth phase of the aggregation curve, and lag time (tlag) was determined as the intersection of the linear part of the initial growth phase with the baseline (Fig. 4A) (32). A series of dilutions from 100 to 1.2 μm was prepared from Aβ40 (Fig. 4B), and its mutant variants (Fig. 4, C and D) and kinetic curves were obtained using the ThT incorporation assay.
FIGURE 4.

A2T and A2V mutations affect the kinetic parameters of Aβ40 aggregation. A, kinetic analysis of the aggregation curves. Straight line (b) was fitted to the baseline, and duration of nucleation or lag phase (tlag) was determined as the time point where line b intersected straight line a, a tangent to the steepest region of the elongation curve (32). Growth rate k was determined as the slope of the exponential part of the elongation curve (green). B–D, concentration-dependent aggregation of Aβ40 variants (B, wild type Aβ40; C, Aβ40-A2T; and D, Aβ40-A2V) in Tris/EDTA buffer, pH 7.4, monitored in the thioflavin T incorporation assay. Inset, aggregation of correspondent mutants at 1.2 and 3.7 μm. E–G, correlation between kinetic parameters of wild type Aβ40 (E), Aβ40-A2T (F), and Aβ40-A2V (G). Note that A2T mutation disrupts the inverse correlation between k and tlag. Significance of correlation was evaluated from the Pearson coefficients shown on the plots and is indicated by **, p < 0.01, means ± S.E., n = 3.
Increasing the peptide concentration typically leads to an acceleration of the elongation rate (k) and a shortening of the lag time (tlag). Because k and tlag are governed by similar physicochemical interactions, usually a (negative) correlation between k and tlag is observed for almost any peptide tested (33). Interestingly, the A2T mutant deviated from that rule (Fig. 4F), although the anti-correlation was observed for the wild type Aβ40 (Fig. 4E) and for Aβ40-A2V (Fig. 4G). The A2V mutation increases k dramatically, whereas tlag becomes shorter with increasing concentrations and is shorter compared with wild type (Fig. 4G). Conversely, the elongation rate for the A2T mutant is similar as for the wild type, but the tlag of the A2T mutant does not change much with increasing concentrations (Fig. 4F). At 100 μm, however, the aggregation kinetics of the A2T mutant becomes biphasic resembling the lag-free aggregation of the A2V mutant (Fig. 4G). The first plateau of the A2T aggregation curve includes only 19 ± 0.6% (n = 3) of the second plateau height and thus is three times less pronounced as compared with the A2V curve (63 ± 11%, n = 3).
We next wondered how mixtures of Aβ/Aβ-A2T would behave in this assay. Increasing Aβ-A2T in the Aβ/Aβ-A2T mixes revealed a small gradual increase in tlag (Fig. 5, A and B), although at 100 μm, the aggregation became biphasic and lag-free. However, the disease-causing Aβ-A2V inhibits nucleation when present at low amounts (33%, Fig. 5, C and D), but the presence of 50% or more of A2V in the mix led to a strong decrease of the lag phase (Fig. 5, C and D). The data thus implicate a concentration-dependent inhibitory effect of the protective A2T mutant and a complex behavior of the disease-causing A2V mutant on the nucleation propensity of wild type Aβ40.
FIGURE 5.

Aβ40-A2T and Aβ40-A2V mutants alter the nucleation of wild type Aβ40. A, shift of the lag phase duration in the mixes of wild type Aβ40 with Aβ40-A2T mutant. Maximum concentration of Aβ peptide is 100 μm. B, change in the nucleation phase duration (tlag) in the mixtures of the wild type Aβ40 with different percentage (0–100%) of Aβ40-A2T. M+S.E., n = 3. Note a change of the pure A2T kinetics indicative of the lag phase abrogation at a very high concentration. Significance of difference from the wild type peptide (black bars) was evaluated in unpaired t test and is indicated by **, p < 0.01, ***, p < 0.001. C, shift of the lag phase duration in the mixes of the wild type Aβ40 with Aβ40-A2V mutant. Maximum concentration of Aβ peptide is 100 μm. Note an increase in the lag phase at 33% of A2V in the mixture, not observable at a higher proportion of the mutant. D, change in the nucleation phase duration (tlag) in the mixtures of the wild type Aβ40 with different percentage (0–100%) of Aβ40-A2V. Means + S.E., n = 3. Significance of difference from the wild type peptide (black bars) was evaluated in the unpaired two-tailed t test and is indicated by ***, p < 0.001.
Effect of A2T and A2V Mutations on Aβ Oligomerization
We next asked whether differently aggregating A2T and A2V mutants form different oligomers, using both Aβ42 and Aβ40 peptides. The number of analytical tools to study such oligomers is limited, but AFM allows visualizing such species to a certain extent. We focused on early aggregates formed by the mutants and their equimolar mixtures with the WT Aβ42 and Aβ40 peptides, and we found a broad range of amorphous structures after 2 h of incubation (Fig. 6, A and D). All Aβ42 preparations formed larger aggregates compared with Aβ40, and mutant preparations formed smaller structures than corresponding wild type peptides. The A2T mutants and their mixes with WT Aβ40 and Aβ42 formed the smallest aggregates in all preparations, probably indicative of impaired nucleation, as observed in the kinetic assay (Fig. 4, C and F).
FIGURE 6.

Effect of the A2T and A2V mutations on morphology, immune reactivity, and hydrophobic exposure of Aβ42 and Aβ40 oligomers. A, atomic force micrographs of pre-aggregated Aβ42 mutants and their equimolar mixes with the wild type Aβ42. Images are obtained in the tapping mode under ambient conditions. On the right, Aβ42 oligomer size histograms obtained via the multiple Gaussian distribution fit of particle height. The peak height is 3.1 nm for WT Aβ42, 2.49 nm for Aβ40-A2T, 2.76 nm for Aβ40-A2V, 2.72 nm for Aβ40/Aβ40-A2T, and 2.9 nm for Aβ40/Aβ40-A2V. B, immune dot blot of Aβ42. Aβ was incubated in Tris/EDTA buffer, pH 7.4, for 2 h, blotted on a nitrocellulose membrane, and probed with conformation-specific antibodies A11 and OC. 4G8 antibody, which reacts with all Aβ forms, was used as a loading control. Right panel, densitometric analysis of Aβ42 dot blots. A11 and OC reactivity of mutant aggregates are significantly diminished compared with WT aggregates (unpaired two-tailed t test; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001, means+ S.E., n = 4). C, normalized fluorescent spectra of 8-anilinonaphthalene-1-sulfonate (ANS) dye upon its binding to pre-aggregated Aβ42 mutants and their 1:1 mixes with the wild type peptide. A mean value from n = 2 in duplicate is plotted. Increased magnitude and blue shifting of ANS spectra are indicative of increased hydrophobicity of the ANS-bound entities. D, atomic force micrographs of pre-aggregated Aβ40 mutants and their equimolar mixes with the wild type Aβ40. Images are obtained in the tapping mode under ambient conditions. On the right: Aβ40 oligomer size histograms obtained via the multiple Gaussian distribution fit of particle height. The peak height is as follows: 2.87 nm for WT Aβ40, 2.38 nm for Aβ40-A2T, 2.43 nm for Aβ40-A2V, 2.19 nm for Aβ40/Aβ40-A2T, and 2.47 nm for Aβ40/Aβ40-A2V. E, immune dot blot of Aβ40. Aβ was incubated in Tris/EDTA buffer, pH 7.4, for 2 h, blotted on a nitrocellulose membrane, and probed with A11 and OC antibodies. 4G8 antibody was used as a loading control. Right panel: densitometric analysis of Aβ40 dot blots. A11 and OC reactivities of mutant aggregates are significantly diminished compared with WT aggregates (unpaired two-tailed t test; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001, means + S.E., n = 4). F, normalized fluorescent spectra of the ANS dye upon its binding to pre-aggregated Aβ40 mutants and their 1:1 mixes with the wild type peptide. A mean value from n = 2 in duplicate is plotted. veh, vehicle; a.u., arbitrary units; Wavel, wave length.
We also made use of conformation-sensitive polyclonal antibodies; A11 and OC claimed to recognize nonfibrillar and fibrillar aggregates, respectively (34). A11 reactivity of oligomeric species is often regarded as a correlate of their toxic potencies (35, 36). Wild type Aβ42 and to a smaller extent Aβ40 peptide pre-aggregated for 2 h showed strong A11- and OC-positive staining (Fig. 6, B and E), implicating the co-existence of both the nonfibrillar and fibrillar oligomers in these preparations. Interestingly, both the A2T and A2V mutants of Aβ42 and Aβ40 were consistently A11- and OC-negative suggesting the involvement of the N terminus of these peptides in the formation of the tertiary structure recognized by these conformation-specific antibodies (Fig. 6, B and E). The equimolar mixes of WT peptides with either A2T or A2V variants were A11- and OC-positive, although the A11 signal was rather proportional to the amount of WT peptide in these samples.
We next probed the hydrophobic exposure of the putative oligomers using ANS binding assay (Fig. 6, C and F, and Table 2), considered to be another correlate to the toxic properties of oligomers (37, 38). In this assay, the fluorescence of ANS increases upon binding to the lipophilic entities, and the emission maximum undergoes a blue shift. The mixes of A2T and A2V with the wild type Aβ42 and Aβ40 demonstrated a lower magnitude and a slight red shift of ANS spectra compared with the WT and mutant peptide spectra (Fig. 6, C and F, and Table 2). Thus, both protective and causative mutants affect tertiary structure of the early aggregates, resulting in decreased exposed hydrophobic surface of both A2T and A2V variants.
TABLE 2.
Analysis of ANS spectra of Aβ peptides shown in Fig. 6, C and F
Data are presented as means ± S.D. from n = 2 in duplicate. a.u. is arbitrary unit.
| Aβ40 |
Aβ42 |
|||
|---|---|---|---|---|
| Emission peak | Relative peak magnitude | Emission peak | Relative peak magnitude | |
| nm | a.u. | nm | a.u. | |
| Vehicle | 532 ± 3 | 0.46 ± 0.00 | 535 ± 5 | 0.52 ± 0.02 |
| WT | 498 ± 3 | 1 ± 0.01 | 502 ± 6 | 1 ± 0.02 |
| A2T | 513 ± 2 | 0.73 ± 0.03 | 510 ± 8 | 0.9 ± 0.01 |
| WT/A2T | 527 ± 2 | 0.56 ± 0.02 | 512 ± 6 | 0.77 ± 0.01 |
| A2V | 507 ± 5 | 0.86 ± 0.04 | 510 ± 5 | 0.86 ± 0.02 |
| WT/A2V | 521 ± 2 | 0.61 ± 0.01 | 520 ± 8 | 0.73 ± 0.03 |
Effect of A2T and A2V Mutations on Aβ Fiber Structure
We next investigated how the two different mutations affected fibril formation. Aβ40 and Aβ42 mixtures were aggregated for 2 weeks, and their secondary structure was analyzed by means of FTIR (Fig. 7). Both Aβ42 and Aβ40 peptides showed a strong enrichment for β-sheet structure (peak at 1627 cm−1, Fig. 7, A and C, in line with previous observations (39, 40)). The mixtures of the wild type Aβ42 with A2T and A2V mutants were structurally different from the pure Aβ42 peptides. Additional absorbance was observed between 1657 and 1665 cm−1 indicative of unstructured or helical components, in addition to the β-sheet content (Fig. 7B). Aβ40 mutants and Aβ40-based mixtures were all enriched in β-sheet structures (Fig. 7, C and D).
FIGURE 7.

Secondary structure of Aβ mutants and their mixtures with the wild type peptide. A and B, Fourier transform infrared absorbance spectra of Aβ42 variants and their 1:1 mixes (B) with the wild type peptide aggregated for 2 weeks. Spectra are recorded in concentrated peptides (2 mg/ml) in Tris/EDTA buffer, pH 7.4. Nonconcentrated samples of Aβ42 (0.2 mg/ml) were used as a background. Peaks at 1627 cm−1 correspond to the β-sheet content; the peak at 1650 cm−1 is indicative of a mixed structure (α-helix plus random coil), and absorbance between 1657 and 1665 cm−1 (B) includes structures different from β-sheet (disordered or helical). A peak at 1690 cm−1 is assigned to antiparallel β-sheet structures. C and D, FTIR spectra of Aβ40 variants and their 1:1 mixes with the wild type peptide aggregated for 2 weeks. Spectra are recorded in concentrated peptides (2 mg/ml) in Tris/EDTA buffer, pH 7.4. IR absorbance at 1627 and 1690 cm−1 corresponds to the β-sheet content. The β-sheet peak of Aβ40 A2T is shifted to 1617 cm−1 (C).
We also analyzed the aggregated material using transmission electron microscopy. Aβ42-A2T and Aβ42-A2V fibers were co-occurring with amorphous aggregates in contrast to the net fibers of WT Aβ42 (Fig. 8A) and did not undergo further structural change after prolonged incubation (data not shown). The Aβ40 containing material formed predominantly amorphous aggregates after 2 weeks of incubation (Fig. 8C) that converted into mature fibers after 6 weeks of incubation (Fig. 8D). Aβ42 and Aβ40 variants became 85–90% insoluble after a few weeks of incubation except the A2T mutants, which still contained twice as much soluble peptide as the wild type and A2V mutants (Fig. 8, B and E).
FIGURE 8.

Impact of the A2T and A2V mutations on the morphology and residual soluble peptide content in aggregated Aβ variants. A, transmission electron micrographs of the fibers obtained after 2 weeks of quiescent incubation of Aβ42 variants and their equimolar mixes with the wild type Aβ42. B, residual soluble peptide content in Aβ42 variants incubated at concentrations above 50 μm for 2 weeks. Significance (unpaired two-tailed t test) is indicated by *, p < 0.05, means + S.E., n = 3. C, transmission electron micrographs of the fibers formed by Aβ40 variants and their equimolar mixes with the wild type Aβ40 after 2 weeks and 6 weeks (D) of quiescent incubation. E, residual soluble peptide content in Aβ40 mutants incubated at 100 μm for 6 weeks. Significance (unpaired two-tailed t test) is indicated by *, p < 0.05, means + S.E., n = 3. F, immune dot blot of Aβ42 fibers. Aβ was incubated in Tris/EDTA buffer, pH 7.4, for 2 weeks, blotted on a nitrocellulose membrane, and probed with conformation-specific antibodies A11 and OC. 4G8 antibody was used as a loading control. 3D6 antibody recognizing the first five amino acids of Aβ was used to probe N-terminal accessibility in mutant aggregates. Immune reactivity of Aβ40 fibers did not change between 2 and 6 weeks of aggregation and was identical to the one of Aβ42. Right panel: densitometric analysis of Aβ fiber dot blots. OC reactivity of the mutant fibers is significantly diminished compared with the WT fibers (unpaired two-tailed t test; **, p < 0.01, means + S.E., n = 3–4). A11 signal intensities of fibers can be considered negligible.
Immune reactivity of both Aβ42 and Aβ40 fibers with A11 or OC antibodies (Fig. 8F) was similar to what was observed with the shorter incubations of Aβ42 and Aβ40 (Fig. 6, B and E); the mutant fibers were not reactive with the OC antibody; the A11 reactivity in the fibrillar wild type peptides was negligible. Interestingly, the accessibility of N termini in aggregated samples did not change as staining with the N-terminal 3D6 antibody was maintained (Fig. 8F).
Effects on Gibbs Free Energy of Aggregation
The Gibbs free energy of aggregation relates to the critical concentration and can be calculated by Equation 1,
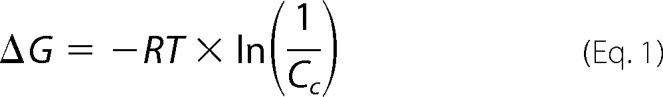 |
where R is the gas constant (R = 8.314 J/mol·K); T represents the absolute temperature, and Cc is the critical concentration of the peptide (31). All calculations were done for T = 298 K.
We thus first evaluated the critical concentrations of Aβ40 and mutant Aβ40 (Fig. 9A). A series of peptide dilutions from 100 to 1.2 μm was incubated for 6 weeks at room temperature, and the residual soluble peptide was monitored after 1, 3, and 6 weeks (Fig. 9A). The critical concentrations (Fig. 9A) were determined as described (31) after 6 weeks of incubation. Interestingly, the solubility of the A2V mutant drops faster as compared with the wild type Aβ40 (Fig. 9A), in line with kinetic data, but then further precipitation slows down, which results in comparable critical concentrations with WT peptide (Fig. 9A). These were used to calculate Gibbs free energy of aggregation (ΔG) using Equation 1, above (Fig. 9B). ΔG value provides a thermodynamic quantification for the tendency of the peptide mixture to populate the amyloid state, i.e. the lower ΔG, the more favorable the amyloid state. In good agreement with previous work (31), we found a ΔG for WT Aβ40 of −7 ± 0.23 kcal/mol, although under the same conditions the ΔG of A2T was significantly higher (ΔG = −6.29 ± 0.18 kcal/mol). The ΔG of A2V was within error the same (ΔG = −6.89 ± 0.13 kcal/mol) as the wild type Aβ40.
FIGURE 9.

Impact of the A2T and A2V mutations on the critical concentration and the Gibbs free energy of aggregation. A, soluble (solid lines) and insoluble (dashed lines) peptide fractions in Aβ40 variants (black, wild type; green, A2T mutant; red, A2Vmutant) measured by means of the Bio-Rad protein assay after 1, 3, and 6 weeks of incubation. Data are presented as means ± S.E., n = 3. Critical concentration (Cc) values are indicated at corresponding plots and refer to the concentration of soluble peptide above which the soluble content does not change and below which there is <10% of detectable insoluble material. Cc of Aβ40-A2T is significantly different (**, p < 0.01, unpaired two-tailed t test) from Cc of WT Aβ40 and Cc of Aβ40-A2V. B, Gibbs free energy (ΔG) of mutant and WT Aβ40 peptide estimated from the critical concentration values as described under “Experimental Procedures,” significance versus the wild type peptide and A2V mutant is indicated as **, p < 0.01, unpaired two-tailed t test; means + S.D., n = 3. C, changes in the Gibbs free energy (ΔΔG) in the mixtures of Aβ40 with either A2T or A2V mutants estimated according to the equation ΔΔG = ΔG(X) − ΔG(Ala), where X is A2T or A2V mutant present in a given mixture at indicated proportion (0–100%). ΔG values for each mixture are shown in Table 3.
We next assessed the impact of the A2T and A2V mutant on Gibbs energy of Aβ40 aggregation in a series of mixes (Table 3 and Fig. 9C). The changes in the Gibbs free energy (ΔΔG) in the mixtures of Aβ40 with either A2T or A2V mutants were calculated using the equation ΔΔG = ΔG(X) − ΔG(Ala), where X is the A2T or A2V mutant present in a given mixture. As can be seen from the positive increments in Gibbs energy ΔΔG conferred to the WT by the presence of the A2T or A2V mutant (Fig. 9C), both A2T and A2V mutant peptides solubilize WT Aβ40. A2T does this to a greater extent than A2V. When A2V mutant constituted 33% of the mix, both aggregation kinetics as well as amyloid stability decreased (Figs. 5, C and D, and 9C). For A2T, the difference in lag phase duration between mixes was less dramatic (Fig. 5, A and B), and ΔΔG values for all the mixes were comparably high, around 0.7–0.9 kcal/mol, indicating a strong destabilizing effect of A2T on Aβ40 aggregation. Changes in Gibbs energy thus followed the change in the nucleation phase of aggregation, in line with the literature (31).
TABLE 3.
Changes of Gibbs free energy of Aβ40 aggregation upon the titration of the wild type Aβ40 with mutant (A2X) peptides
ΔG is determined as described under “Experimental Procedures” and presented as means ± S.D., n = 3.
| A2X + WT Aβ40 |
X = Thr | X = Val | |
|---|---|---|---|
| % WT | % A2X | ||
| ΔG, kcal/mol | ΔG, kcal/mol | ||
| 100 | 0 | −7.0 ± 0.23 | −7.0 ± 0.23 |
| 67 | 33 | −6.09 ± 0.0 | −6.3 ± 0.37 |
| 50 | 50 | −6.1 ± 0.02 | −6.66 ± 0.1 |
| 33 | 67 | −6.05 ± 0.06 | −6.71 ± 0.04 |
| 0 | 100 | −6.29 ± 0.18 | −6.89 ± 0.13 |
DISCUSSION
The A2V and the A2T mutations in the APP sequence are intriguing as they have opposing effects on the risk to develop Alzheimer disease (5, 21). We confirm here that these mutations close to the β-secretase cleavage site in APP have indeed opposite effects on the proteolytic processing of APP toward Aβ peptides. This reinforces the amyloid hypothesis of AD, which suggests that abnormal amyloid generation is a triggering, initiating, or driving event in the disease cascade (1). The first conclusion of our work is, however, that the A2T mutation has a profound effect on a series of biophysical parameters of Aβ peptide aggregation. Thus, the effects of the protective A2T mutant with regard to AD pathobiology are not only quantitative (21) but also qualitative as suggested before (23). In fact, the A2V mutation exerts also complex interactions with the wild type peptide explaining why it is only disease-causing in the homozygous situation (5).
To compare the effects of the protective and causative mutations on Aβ peptide aggregation, we performed detailed kinetic analysis of Aβ self-assembly, and we also compared the behavior of the peptides at equilibrium. Interestingly, although the effects of the N-terminal mutations on the kinetic parameters measured for aggregation were masked by the high aggregation propensity of the Aβ42 peptide (Fig. 3A), the onset of dynamic equilibrium was differentially affected in Aβ42-A2T and Aβ42-A2V peptides (Fig. 8B). These observations are in line with the proposed critical role of the C terminus in the kinetics of amyloid formation and its minor role in Aβ peptide solubility at equilibrium (13). Study of the less hydrophobic and less aggregation-prone Aβ40 peptide, however, revealed not only the increased residual solubility of A2T (Fig. 8E) but also the delayed aggregation of the A2T mutant and enhanced aggregation of the A2V mutant when above the critical concentrations (Figs. 3B, 4, and 9A). Because nucleation time (tlag) inversely correlates with sequence hydrophobicity (41), the kinetic effect of the protective mutation on Aβ40 aggregation could originate from the subtle changes in hydrophobicity conferred by the polar threonine. This is congruent with the ANS binding data showing a reduced hydrophobicity of Aβ40-A2T aggregates (Fig. 6F, left panel). A delayed onset of elongation and increased residual solubility conferred by the A2T mutation are indicative of less productive interactions between the monomers and/or the larger building blocks above the critical concentrations. This is in line with the AFM data revealing the presence of small aggregates in A2T mutants pre-aggregated for 2 h (Fig. 6, A and D).
Experimental evidence suggests that readily aggregating sequences have lower critical concentrations; in other words, kinetic and thermodynamic aspects of aggregation are governed by the same physicochemical mechanisms (31), and this holds for Aβ mutations at different positions (41). In case of the A2V mutation, the relation between the speed and the energy of aggregation appears more complex. On the one hand, the accelerated kinetics (Figs. 3B and 4D), reminiscent of Aβ42 (Fig. 3A), pointed to a very efficient self-assembly, possibly fostered by interactions between the more hydrophobic A2V mutant N-terminal tails. On the other hand, at concentrations significantly exceeding the critical one (8.5 ± 0.9 μm), these interactions become less productive. Alternatively, a structural re-arrangement of aggregation nuclei occurs leading to an intermediate plateau stage during elongation that can be regarded as another lag phase (Fig. 4D). This rate-limiting process may explain why the Gibbs aggregation energy of Aβ40-A2V was, at the end, not lower than the energy of the wild type sequence (Fig. 9A).
Hortschansky et al. (31) compared changes in Gibbs energy conferred by substitution of residue 18 of Aβ40 with different amino acids. Of particular interest, substitution of Gly-18 with threonine caused a 0.11 kcal/mol increase of the Gibbs aggregation energy of Aβ peptide as compared with a thermodynamically more favorable glycine-to-valine substitution (G18V) that diminished the aggregation energy of Aβ40 by −0.03 kcal/mol. The magnitude of changes in ΔG between A2T and A2V mutants is larger than the difference between G18T and G18V energies suggesting a surprisingly pronounced effect of N-terminal mutations on fibrillization. Alternatively, small differences in methodology used to measure the critical concentrations should be considered. Additional evidence for the important effects of the N-terminal mutations on Aβ aggregation is the delayed onset of mutant fiber formation. The residual solubility of Aβ40-A2V decreases already after 1 week (Fig. 9A); however, the A2V mutants similarly to A2T form mature fibers only after prolonged incubation (Fig. 8, C and D). The transition of heterogeneous aggregates formed after 2 weeks of incubation (Fig. 8C) to a more mature structure (Fig. 8D) is delayed due to the kinetic and/or thermodynamic barriers that govern the aggregation of the mutant Aβ40.
It is true that the N terminus of the Aβ fragment does not take part in the amyloid fiber core formation and is usually regarded as unstructured. Recent crystallization studies brought forward an α-helical conformation of Aβ's N terminus in complex with the 3D6-based therapeutic antibody bapineuzumab (42). Experimental evidence nevertheless suggests that N-terminal substitutions can affect Aβ aggregation (5, 24, 25, 43). For instance, before being identified in the clinic, the A2V mutation itself was isolated as an aggregation-prone variant by screening mutant Aβ40-GFP fusions in Escherichia coli (44).
The second important conclusion from our work is that mixtures of Aβ peptides behave in a complex way. This is particularly relevant if one considers that in reality Aβ peptides are always heterogeneous, affecting their ultimate biophysical behaviors in various ways (14, 15, 20). In the mixtures of the wild type Aβ40 with the A2T mutant, the lag phase extended proportionally to the increase of A2T mutant in the mixture (Fig. 5, A and B), although these changes were overall modest. At the highest tested concentration (100 μm), the aggregation kinetics of pure Aβ40 was altered resembling the one of Aβ40-A2V, but this effect was not present at lower concentrations of the mutant (Fig. 4, C and D).
Changes in the Gibbs aggregation energy were comparably high in all A2T mixtures indicating a strong destabilizing effect of Aβ40-A2T on Aβ40 aggregation (Fig. 9C and Table 3). This was reflected in the lower hydrophobic exposure of Aβ/Aβ-A2T mixes (for both Aβ42 and Aβ40, Fig. 6, C and F, left panel) and in the presence of very small aggregates during the first few hours of aggregation (Fig. 6, A and D). FTIR spectra revealed non-β-sheet structures in Aβ42/Aβ42-A2T peptide mixtures supporting a destabilizing effect of the A2T mutation on amyloid fiber formation (Fig. 7B).
In the mixtures of the wild type Aβ40 with the A2V mutant, the lag phase duration inversely correlated with the amount of A2V mutant (Fig. 5C) and was coherent with the gradual decrease of the aggregation energy (Fig. 9B). The alanine-to-valine substitution was also thermodynamically destabilizing as judged from the positive increment in ΔG (Fig. 9C), and lower hydrophobic exposure of Aβ/Aβ-A2V mixes (Fig. 6, C and F, right panel) was observed.
Decreased aggregation propensity of A2T mutants and heterozygous mixtures of A2T/V with the wild type peptide render them more available for degradation. This is in line with the absence of cerebral amyloidosis in A2T carriers even at advanced age (45) and with the protected status of heterozygous carriers of A2V (5). These effects of A2T and A2V mutations on peptide aggregation are striking; however, the interpretation of Aβ aggregation studies in the context of any clinical phenotypes should take into account other aspects of Aβ and APP biology. A recent study (46) showed that A2T and A2V mutations rendered WT APP incapable of mediating TGFβ2-induced cell death. Moreover, the flexible N-terminal part of the Aβ fragment itself may be important for interaction with some “toxic Aβ receptors,” e.g. FcybII (30), and thus, N-terminal mutations in Aβ may affect receptor-driven synaptotoxic cascades. Furthermore, the changes in full-length Aβ production caused by the A2T and A2V mutations might also affect more indirectly the toxic or aggregation behaviors of the resulting Aβ mixes. Of note, non-neuronal cells expressing the A2V mutation showed an increased secretion of N-truncated species Aβ(11–40), Aβ(11–42), and AβN3pE-42 (5). It will be interesting to check whether A2T and A2V mutations affect the N-terminal truncated species generation in patients as well.
In conclusion, the complex effects of the A2T mutation link this mutation to various aspects of Aβ biophysics. The simple conclusion that the chronic and mild lowering of the amount of Aβ produced over a lifetime explains that the protective effect of this mutant against AD has therefore to be considered with sufficient caution (23). Reduced Aβ production by A2T mutants should not be considered as the “ultimate” argument in favor for BACE1 drug development as the effects of this mutation cannot only be explained in quantitative terms. We have previously already shown that Aβ pathobiology is not only driven by quantity but that Aβ peptide behavior is also strongly modulated by the quality and relative composition of the Aβ profile in a mixture (15). Basically any subtle alterations in the biophysical properties of the Aβ peptide can have major effects at the level of seeding and aggregation. The complexity of these processes should not be ignored when contemplating approaches to cure or prevent AD.
This work was supported by grants from VIB, University of Leuven, Funds for Scientific Research Flanders, Flanders Institute for Science and Technology, and the Federal Office for Scientific Affairs of Belgium (Belspo), Grant IUAP P7/16, and a Methusalem grant of the KULeuven/Flemish Government (to B. D. S.), and part of this work was done in the frame of ERA-NET NEURON “AβID.”
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- SFV
- Semliki Forest virus
- UPW
- ultrapure water
- ANS
- 8-anilinonaphtalene-1-sulfonic acid
- HFIP
- 1,1,1,3,3,3-hexafluor-2-propanol
- sAPP
- soluble APP
- AFM
- atomic force microscopy.
REFERENCES
- 1. Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712 [DOI] [PubMed] [Google Scholar]
- 2. Golde T. E., Streit W. J., Chakrabarty P. (2013) Alzheimer's disease risk alleles in TREM2 illuminate innate immunity in Alzheimer's disease. Alzheimers Res. Ther. 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardy J., Bogdanovic N., Winblad B., Portelius E., Andreasen N., Cedazo-Minguez A., Zetterberg H. (2014) Pathways to Alzheimer's disease. J. Int. Med. 275, 296–303 [DOI] [PubMed] [Google Scholar]
- 4. Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. (1992) A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N terminus of β-amyloid. Nat. Genet. 1, 345–347 [DOI] [PubMed] [Google Scholar]
- 5. Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A. R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., Manzoni C., Francescucci B., Spagnoli A., Cantù L., Del Favero E., Levy E., Salmona M., Tagliavini F. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou L., Brouwers N., Benilova I., Vandersteen A., Mercken M., Van Laere K., Van Damme P., Demedts D., Van Leuven F., Sleegers K., Broersen K., Van Broeckhoven C., Vandenberghe R., De Strooper B. (2011) Amyloid precursor protein mutation E682K at the alternative β-secretase cleavage β′-site increases Aβ generation. EMBO Mol. Med. 3, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chávez-Gutiérrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H., Wiltfang J., Serneels L., Karran E., Gijsen H., Schymkowitz J., Rousseau F., Broersen K., De Strooper B. (2012) The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eckman C. B., Mehta N. D., Crook R., Perez-tur J., Prihar G., Pfeiffer E., Graff-Radford N., Hinder P., Yager D., Zenk B., Refolo L. M., Prada C. M., Younkin S. G., Hutton M., Hardy J. (1997) A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of Aβ42(43). Hum. Mol. Genet. 6, 2087–2089 [DOI] [PubMed] [Google Scholar]
- 9. Goate A., Chartier-Harlin M. C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706 [DOI] [PubMed] [Google Scholar]
- 10. Kulic L., McAfoose J., Welt T., Tackenberg C., Späni C., Wirth F., Finder V., Konietzko U., Giese M., Eckert A., Noriaki K., Shimizu T., Murakami K., Irie K., Rasool S., Glabe C., Hock C., Nitsch R. M. (2012) Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation. Transl. Psychiatry 2, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murakami K., Masuda Y., Shirasawa T., Shimizu T., Irie K. (2010) The turn formation at positions 22 and 23 in the 42-mer amyloid β peptide: the emerging role in the pathogenesis of Alzheimer's disease. Geriatr. Gerontol. Int. 10, S169–S179 [DOI] [PubMed] [Google Scholar]
- 12. Jarrett J. T., Berger E. P., Lansbury P. T., Jr. (1993) The C terminus of the β protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 695, 144–148 [DOI] [PubMed] [Google Scholar]
- 13. Jarrett J. T., Berger E. P., Lansbury P. T., Jr. (1993) The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 [DOI] [PubMed] [Google Scholar]
- 14. Vandersteen A., Masman M. F., De Baets G., Jonckheere W., van der Werf K., Marrink S. J., Rozenski J., Benilova I., De Strooper B., Subramaniam V., Schymkowitz J., Rousseau F., Broersen K. (2012) Molecular plasticity regulates oligomerization and cytotoxicity of the multipeptide-length amyloid-β peptide pool. J. Biol. Chem. 287, 36732–36743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuperstein I., Broersen K., Benilova I., Rozenski J., Jonckheere W., Debulpaep M., Vandersteen A., Segers-Nolten I., Van Der Werf K., Subramaniam V., Braeken D., Callewaert G., Bartic C., D'Hooge R., Martins I. C., Rousseau F., Schymkowitz J., De Strooper B. (2010) Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito T., Suemoto T., Brouwers N., Sleegers K., Funamoto S., Mihira N., Matsuba Y., Yamada K., Nilsson P., Takano J., Nishimura M., Iwata N., Van Broeckhoven C., Ihara Y., Saido T. C. (2011) Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 14, 1023–1032 [DOI] [PubMed] [Google Scholar]
- 17. Dahlgren K. N., Manelli A. M., Stine W. B., Jr., Baker L. K., Krafft G. A., LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 18. Walsh D. M., Selkoe D. J. (2007) A β oligomers–a decade of discovery. J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 19. Teplow D. B. (2013) On the subject of rigor in the study of amyloid β-protein assembly. Alzheimers Res. Ther. 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 21. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R. R., Huttenlocher J., Bjornsdottir G., Andreassen O. A., Jönsson E. G., Palotie A., Behrens T. W., Magnusson O. T., Kong A., Thorsteinsdottir U., Watts R. J., Stefansson K. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 22. Peacock M. L., Warren J. T., Jr., Roses A. D., Fink J. K. (1993) Novel polymorphism in the A4 region of the amyloid precursor protein gene in a patient without Alzheimer's disease. Neurology 43, 1254–1256 [DOI] [PubMed] [Google Scholar]
- 23. De Strooper B., Voet T. (2012) Alzheimer's disease: A protective mutation. Nature 488, 38–39 [DOI] [PubMed] [Google Scholar]
- 24. Hori Y., Hashimoto T., Wakutani Y., Urakami K., Nakashima K., Condron M. M., Tsubuki S., Saido T. C., Teplow D. B., Iwatsubo T. (2007) The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 282, 4916–4923 [DOI] [PubMed] [Google Scholar]
- 25. Ono K., Condron M. M., Teplow D. B. (2010) Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 285, 23186–23197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giaccone G., Morbin M., Moda F., Botta M., Mazzoleni G., Uggetti A., Catania M., Moro M. L., Redaelli V., Spagnoli A., Rossi R. S., Salmona M., Di Fede G., Tagliavini F. (2010) Neuropathology of the recessive A673V APP mutation: Alzheimer disease with distinctive features. Acta Neuropathol. 120, 803–812 [DOI] [PubMed] [Google Scholar]
- 27. De Strooper B., Simons M., Multhaup G., Van Leuven F., Beyreuther K., Dotti C. G. (1995) Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 14, 4932–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Annaert W. G., Levesque L., Craessaerts K., Dierinck I., Snellings G., Westaway D., George-Hyslop P. S., Cordell B., Fraser P., De Strooper B. (1999) Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J. Cell Biol. 147, 277–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Annaert W. G., Esselens C., Baert V., Boeve C., Snellings G., Cupers P., Craessaerts K., De Strooper B. (2001) Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron 32, 579–589 [DOI] [PubMed] [Google Scholar]
- 30. Kam T. I., Song S., Gwon Y., Park H., Yan J. J., Im I., Choi J. W., Choi T. Y., Kim J., Song D. K., Takai T., Kim Y. C., Kim K. S., Choi S. Y., Choi S., Klein W. L., Yuan J., Jung Y. K. (2013) FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer's disease. J. Clin. Invest. 123, 2791–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hortschansky P., Christopeit T., Schroeckh V., Fändrich M. (2005) Thermodynamic analysis of the aggregation propensity of oxidized Alzheimer's β-amyloid variants. Protein Sci. 14, 2915–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hortschansky P., Schroeckh V., Christopeit T., Zandomeneghi G., Fändrich M. (2005) The aggregation kinetics of Alzheimer's β-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 14, 1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christopeit T., Hortschansky P., Schroeckh V., Gührs K., Zandomeneghi G., Fändrich M. (2005) Mutagenic analysis of the nucleation propensity of oxidized Alzheimer's β-amyloid peptide. Protein Sci. 14, 2125–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 36. Laganowsky A., Liu C., Sawaya M. R., Whitelegge J. P., Park J., Zhao M., Pensalfini A., Soriaga A. B., Landau M., Teng P. K., Cascio D., Glabe C., Eisenberg D. (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubnovitsky A., Sandberg A., Rahman M. M., Benilova I., Lendel C., Härd T. (2013) Amyloid-β protofibrils: size, morphology and synaptotoxicity of an engineered mimic. PloS one 8, e66101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bolognesi B., Kumita J. R., Barros T. P., Esbjorner E. K., Luheshi L. M., Crowther D. C., Wilson M. R., Dobson C. M., Favrin G., Yerbury J. J. (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–740 [DOI] [PubMed] [Google Scholar]
- 39. Lin S. Y., Chu H. L. (2003) Fourier transform infrared spectroscopy used to evidence the prevention of β-sheet formation of amyloid β(1–40) peptide by a short amyloid fragment. Int. J. Biol. Macromol. 32, 173–177 [DOI] [PubMed] [Google Scholar]
- 40. Broersen K., Jonckheere W., Rozenski J., Vandersteen A., Pauwels K., Pastore A., Rousseau F., Schymkowitz J. (2011) A standardized and biocompatible preparation of aggregate-free amyloid β peptide for biophysical and biological studies of Alzheimer's disease. Protein Eng. Des. Sel. 24, 743–750 [DOI] [PubMed] [Google Scholar]
- 41. Meinhardt J., Tartaglia G. G., Pawar A., Christopeit T., Hortschansky P., Schroeckh V., Dobson C. M., Vendruscolo M., Fändrich M. (2007) Similarities in the thermodynamics and kinetics of aggregation of disease-related Aβ(1–40) peptides. Protein Sci. 16, 1214–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miles L. A., Crespi G. A., Doughty L., Parker M. W. (2013) Bapineuzumab captures the N-terminus of the Alzheimer's disease amyloid-β peptide in a helical conformation. Sci. Rep. 3, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qahwash I., Weiland K. L., Lu Y., Sarver R. W., Kletzien R. F., Yan R. (2003) Identification of a mutant amyloid peptide that predominantly forms neurotoxic protofibrillar aggregates. J. Biol. Chem. 278, 23187–23195 [DOI] [PubMed] [Google Scholar]
- 44. Kim W., Hecht M. H. (2008) Mutations enhance the aggregation propensity of the Alzheimer's A β peptide. J. Mol. Biol. 377, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kero M., Paetau A., Polvikoski T., Tanskanen M., Sulkava R., Jansson L., Myllykangas L., Tienari P. J. (2013) Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol. Aging 2013. 34, 1518.e1–e3 [DOI] [PubMed] [Google Scholar]
- 46. Hashimoto Y., Matsuoka M. (2014) A mutation protective against Alzheimer's disease renders amyloid β precursor protein incapable of mediating neurotoxicity. J. Neurochem. 130, 291–300 [DOI] [PubMed] [Google Scholar]