

Background: The A673T variant of the amyloid precursor protein (APP) protects against Alzheimer disease (AD).
Results: A673T reduces BACE1 processing of APP by decreasing catalytic turnover and reduces amyloid-β(1–42) aggregation.
Conclusion: A673T APP protects against AD primarily by reducing Aβ production and also by reducing aggregation.
Significance: The biochemical nature of the A673T protective mutation provides insight into AD development.
Keywords: Alzheimer Disease, Amyloid Precursor Protein (APP), Amyloid-β (AB), β-Secretase 1 (BACE1), Neurobiology, Proteolytic Enzyme
Abstract
Pathogenic mutations in the amyloid precursor protein (APP) gene have been described as causing early onset familial Alzheimer disease (AD). We recently identified a rare APP variant encoding an alanine-to-threonine substitution at residue 673 (A673T) that confers protection against development of AD (Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., Stefansson, H., Sulem, P., Gudbjartsson, D., Maloney, J., Hoyte, K., Gustafson, A., Liu, Y., Lu, Y., Bhangale, T., Graham, R. R., Huttenlocher, J., Bjornsdottir, G., Andreassen, O. A., Jönsson, E. G., Palotie, A., Behrens, T. W., Magnusson, O. T., Kong, A., Thorsteinsdottir, U., Watts, R. J., and Stefansson, K. (2012) Nature 488, 96–99). The Ala-673 residue lies within the β-secretase recognition sequence and is part of the amyloid-β (Aβ) peptide cleavage product (position 2 of Aβ). We previously demonstrated that the A673T substitution makes APP a less favorable substrate for cleavage by BACE1. In follow-up studies, we confirm that A673T APP shows reduced cleavage by BACE1 in transfected mouse primary neurons and in isogenic human induced pluripotent stem cell-derived neurons. Using a biochemical approach, we show that the A673T substitution modulates the catalytic turnover rate (Vmax) of APP by the BACE1 enzyme, without affecting the affinity (Km) of the APP substrate for BACE1. We also show a reduced level of Aβ(1–42) aggregation with A2T Aβ peptides, an observation not conserved in Aβ(1–40) peptides. When combined in a ratio of 1:9 Aβ(1–42)/Aβ(1–40) to mimic physiologically relevant mixtures, A2T retains a trend toward slowed aggregation kinetics. Microglial uptake of the mutant Aβ(1–42) peptides correlated with their aggregation level. Cytotoxicity of the mutant Aβ peptides was not dramatically altered. Taken together, our findings demonstrate that A673T, a protective allele of APP, reproducibly reduces amyloidogenic processing of APP and also mildly decreases Aβ aggregation. These effects could together have an additive or even synergistic impact on the risk of developing AD.
Introduction
Genetic studies of Alzheimer disease (AD)5 have helped shape our current understanding of disease etiology. For example, mutations in presenilin1 (PSEN1) and presenilin 2 (PSEN2), enzymes involved in the processing of APP, have been found in autosomal dominant familial cases of AD (2–4). More recently, PSEN1 mutations have also been identified in some sporadic cases of late onset AD (5). Numerous disease-associated mutations in the APP gene itself have also been identified. Many of these mutations cluster at or near the β- or γ-proteolytic sites, favoring either the overproduction of total amyloid-β (Aβ) (6–8) or an increased ratio of the pro-aggregating Aβ(1–42) species relative to Aβ(1–40) (9–12). In other instances, mutations within the Aβ peptide promote an increased propensity for aggregation (7, 13, 14). Together, these genetic findings provide strong support for the amyloid hypothesis of AD, which postulates that an imbalance in the production and clearance of Aβ initiates a cascade of amyloid accumulation, neurotoxicity, and neurodegeneration (15).
Identification of genetic variants that protect from AD can be equally informative in revealing the mechanisms underlying disease biology, and they could guide therapeutic approaches to block disease development or progression. Recently, we reported a low frequency variant in the APP gene (rs63750847-A) that confers significant protection against AD (1). This variant results in an alanine-to-threonine substitution at position 673 in the APP gene (A673T), adjacent to the β-cleavage site. Our primary characterization of the A673T variant demonstrated that this mutation made APP a less favorable substrate for BACE1, resulting in reduced levels of Aβ production (1). However, the A673T substitution lies within the Aβ peptide and thus could also alter the properties of the Aβ peptide itself. Here, we examine the effects of this variant on the kinetics of APP processing by BACE1, and additionally, we evaluate the impact of this substitution on Aβ aggregation and biological activity, including microglial uptake and cellular toxicity. Our results provide further support for reduced β-site cleavage of the A673T APP variant and additionally reveal modest alterations in Aβ(1–42) peptide aggregation, as assessed using synthetic peptides.
EXPERIMENTAL PROCEDURES
Cellular APP Cleavage Assays
Human APP695 cDNAs encoding WT APP, APP A673T, or APP A673V were transfected into 293T cells maintained in DMEM + 10% FBS as described previously (1). After 4 days in vitro (DIV), cells were lysed using RIPA buffer with Complete Protease Inhibitor Mixture (Roche Applied Science) to assay for β-CTFs. The cell supernatant was also collected to confirm reduced Aβ production. Dissociated cortical neuron cultures (plated at 2.5 × 104 cells/well in a 96-well plate) were prepared from E16.5 C57BL/6J mice. Prior to plating, cells were nucleofected with Amaxa (Lonza, Basel, Switzerland) with cDNAs encoding the same human APP695 constructs described above or β-galactosidase (control). After 6 DIV, cells were incubated with 50 μl/well fresh cell medium for 24 h. The medium was collected and used for Aβ ELISA measurements (see below). For both cell types, the efficiency of transfection across different constructs was compared by lysing a separate well of cells with Ambion Cells-to-CTTM (Invitrogen) to assess APP RNA levels by quantitative PCR. Human cortical neurons (catalogue no. DDP-NC-1.0) derived from control donor iPS cells (clone ID 01279.107), the isogenic APP-A673V iPS cells (01279.A32), and the isogenic APP-A673T iPS cells (01279.A27) were ordered from Cellular Dynamics International Inc. (Madison, WI). Human iPSC-derived neurons were plated in 8-well chamber slide (BD 354688) at a density of 1 × 105 cells/well. The culture slides were prepared with poly-d-lysine and laminin following the Cellular Dynamics International user's guide. Half of the conditioned media was replaced with fresh warm media every 2 days. To confirm APP expression, cells were fixed with 4% paraformaldehyde, 4% sucrose, permeabilized with 0.1% Triton X-100, and blocked in 2% BSA/PBS. Cells were stained with rabbit monoclonal anti-APP clone Y188 (Abcam, Cambridge, MA) and mouse anti-MAP2 clone 5F9 (Abcam, Cambridge, MA) in 2% BSA/PBS for 2 h. After washing, the secondary goat anti-mouse IgG AlexaFluor®488 antibody A10680 (Invitrogen) and goat anti-rabbit IgG AlexaFluor®568-conjugated antibody A11036 (Invitrogen) were applied for 30 min at 1:200 in 2% BSA/PBS. Media were harvested after 4 DIV for sAPP analysis and after 14 DIV for Aβ measurements. Experiments were performed at least three times, with technical triplicates in each experiment.
β-CTF Analysis
Cell lysates from transfected 293T cells were run on 4–12% Novex BisTris gels (Invitrogen) for SDS-PAGE analysis. β-CTFs were detected by incubating blots with the C-terminal polyclonal APP antibody A8717 (Sigma) or the monoclonal mid-domain APP antibody 6E10 (Covance, Dedham, MA). Imaging was performed on the Bio-Rad VersaDoc gel imaging system. β-CTFs were quantified using an ELISA from IBL International (catalogue no. 27776, Hamburg, Germany) following the manufacturer's guidelines.
Aβ and sAPP Immunoassays
Aβ40 and Aβ42 peptides were measured from transfected HEK 293 and primary neuron cell supernatants by sandwich ELISAs as described previously (1). Briefly, rabbit polyclonal antibody specific for the C terminus of Aβ40 or Aβ42 (Millipore, Bedford, MA) was coated onto plates, and biotinylated anti-Aβ monoclonal antibody 6E10 (Covance, Dedham, MA) was used for detection. The assays had a lower limit of quantification values of 6.24 pg/ml for Aβ40 and 18.76 pg/ml for Aβ42. For iPSC-derived neurons, Aβ40 and Aβ42 peptides were measured from cell supernatants using a sandwich electro-chemiluminescence immunoassay (Meso Scale Discovery catalogue no. K15148E, Gaithersburg, MD). Cell supernatants from all cell types were also assayed for sAPP cleavage products by using a sAPPα/sAPPβ multiplex kit (Meso Scale Discovery catalogue no. K15120E).
TR-FRET Cleavage of WT and Swedish APP(662–688) Substrates
N-terminally biotinylated peptide substrates containing either WT (biotin-KTEEISEVKMDAEFRHDSGYEVHHQKL), A673T (biotin-KTEEISEVKMDTEFRHDSGYEVHHQKL), or A673V (biotin-KTEEISEVKMDVEFRHDSGYEVHHQKL) and similar peptides with the Swedish mutation in combination (biotin-KTEEISEVNLDAEFRHDSGYEVHHQKL, biotin-KTEEISEVNLDTEFRHDSGYEVHHQKL, and biotin-KTEEISEVNLDVEFRHDSGYEVHHQKL) were custom-synthesized by American Peptide Co. Streptavidin-d2 (SA-d2) and anti-Aβ monoclonal antibody 6E10 (Covance, Dedham, MA) labeled with europium cryptate (6E10-Eu) were purchased from CisBio (Bedford, MA). We previously demonstrated that the A673T mutation does not interfere with detection by the 6E10 antibody (1). Enzyme reactions were carried out in 384-well black low volume proxiplate (PerkinElmer Life Sciences) with 50 nm recombinant human BACE1 ECD (Genentech) and 5 μm peptide substrate in the reaction buffer (50 mm sodium acetate, pH 4.4, 0.1% CHAPS, 0.1% BSA) at ambient temperature for up to 48 h for non-Swedish peptides and up to 30 min for peptides containing the Swedish mutation. The enzyme reactions were quenched with the addition of equal volume stop buffer (200 mm Tris, pH 8, 0.1% CHAPS, 0.1% BSA). Peptides in the reactions were then further diluted to 300 nm prior to the addition of an equal volume of detection reagent (10 nm anti-Aβ 6E10-Eu, 75 nm SA-d2, 800 mm KF, 200 mm Tris, pH 8, 0.1% CHAPS, 0.1% BSA), and the mixture was incubated at ambient temperature for 1 h. The plates were measured in an EnVision (PerkinElmer Life Sciences) reader, and the TR-FRET ratio with 320 nm excitation, 615 nm donor emission, and 665 nm acceptor emission was calculated as (em665/em615)·Z, where Z = 5000. The reaction progress curves were determined by subtracting background signal at time 0 for each reaction, and reaction rates (min−1) were calculated from linear fits using GraphPad Prism (Sunnyvale, CA).
FRET Km Determination with Short APP(668–675) Peptides
Three FRET peptide substrates, containing WT APP amino acid sequence 668–675 (E(EDANS)EVKMDAEFK(Dabcyl)NH2), A673T (E(EDANS)EVKMDTEFK(Dabcyl) NH2), or A673V (E(EDANS)EVKMDVEFK(Dabcyl)NH2) were custom-synthesized by American Peptide Co. (Sunnyvale, CA). Each peptide was conjugated with a fluorescent donor molecule EDANS at the N terminus and a quencher molecule Dabcyl at the C terminus. The peptide substrates were titrated up to 100 μm in reaction buffer (50 mm sodium acetate, pH 4.4, 0.1% CHAPS, 0.1% BSA), and the proteolytic reaction was carried out using 50 nm BACE1 at ambient temperature. The enzyme reactions were quenched at various time points up to 48 h with the addition of equal volume stop buffer (200 mm Tris, pH 8, 0.1% CHAPS, 0.1% BSA). Fluorescence intensity at 490 nm with excitation at 340 nm was measured using Tecan M1000 fluorescence reader (Tecan, Männedorf, Switzerland), and the total fluorescence intensity gain was determined by subtracting background fluorescence at time 0 from the fluorescence intensity of each reaction mixture at different time points. Reaction rates (min−1) were calculated from linear fits of the reaction progress curves, and the Michaelis-Menten equation was applied to determine Km and Vmax values using GraphPad Prism (Sunnyvale, CA).
IC50 Determination of APP Peptide A673 Variants in FRET Cleavage Assay
FRET peptide substrate with the Swedish mutation (Rh-EVNLDAEFK-Quencher) was obtained from Invitrogen. Enzyme reactions were carried out with 50 nm BACE1, 150 nm FRET peptide substrate with Swedish mutation, and up to 100 μm biotinylated peptide in reaction buffer (50 mm sodium acetate, pH 4.4, 0.1% CHAPS, 0.1% BSA) at ambient temperature. Fluorescence intensity (530 nm excitation, 590 nm emission) was continuously monitored for 85 min on a Tecan M1000 fluorescence reader. Initial background signal at each peptide concentration was subtracted from each reaction time. Reaction rates (min−1) were calculated from linear fits, and peptide IC50 values were generated using a four-parameter nonlinear fit in GraphPad Prism.
Aggregation Kinetics as Measured by Thioflavin-T Fluorescence
The synthetic peptides (AnaSpec, Fremont, CA) were disaggregated by dissolution in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), followed by evaporation under a stream of nitrogen. Immediately before analysis, the peptide film was thoroughly dissolved in 10 mm NaOH, neutralized in an equal volume of 100 mm sodium phosphate buffer, pH 7.4, and filtered with a 0.2-μm syringe filter. UV absorbance of the solution at 280 nm was measured, and the peptide concentration was determined using the respective theoretical extinction coefficient and adjusted to 1.0 mg/ml with 5 mm NaOH, 50 mm sodium phosphate buffer, pH 7.4. The redissolved and adjusted peptide solution was transferred into a well of the 96-well plate containing an equal volume of 56 μm thioflavin-T (ThT) in 2 mm NaN3, 50 mm phosphate buffer, pH 7.4. All data were recorded with a Tecan Safire II reader and black, clear-bottom, and sealed 96-well plates (Corning Glass) in bottom-reading mode at 37 °C. Kinetic reads were taken every 15 min (Aβ(1–40)) or every 10 min (Aβ(1–42) and Aβ(1–42)/Aβ(1–40) mixtures). ThT was excited at 450 nm, and the fluorescence emission signal was read at 490 nm. For each experiment, traces of the mean of three technical replicates were plotted using GraphPad Prism 5, and error bars of the final point indicate standard deviation.
Microglial Aβ Uptake Assay and Immunocytochemistry
Primary microglia were prepared from P2 C57BL/6J mice at a density of 6 × 104 cells per well in 8-well poly-d-lysine-coated chamber slides, and the Aβ uptake assay was performed 48 h later. Aβ(1–40) and Aβ(1–42) peptides were custom-synthesized by rPeptide (Bogart, GA) and prepared as oligomers according to Stine et al. (16) with the following modifications: peptides were resuspended in 1:10 v/v 1% NH4OH/HFIP to 20 μm and lyophilized overnight. Lyophilized peptides were resuspended in anhydrous dimethyl sulfoxide (DMSO) D2650 (Sigma) to 5 mm and immediately diluted to 100 μm in ice-cold cell culture medium (phenol red-free Ham's F-12), vortexed for 30 s, and incubated at 4 °C for 24 h. Peptides were aliquoted and stored at −80 °C. 1 μm of Aβ(1–40) or Aβ(1–42) peptides was added to primary microglial cultures. After 90 min, microglia were washed three times with cold PBS, immediately fixed in 4% paraformaldehyde, 4% sucrose. Cells were then blocked in 2% BSA/PBS and permeabilized in 0.1% Triton X-100 (Sigma catalogue no. 93426-250 ml) for 20 min and stained with mouse anti-Aβ (Covance, Dedham, MA, catalogue no. SIG-39320) at 1:500 and rabbit anti-Iba1 019–19741 (Wako, Osaka, Japan) at 1:200 in 2% BSA/PBS for 2 h. After washing, the secondary goat anti-mouse IgG AlexaFluor®488 antibody A10680 (Invitrogen) and goat anti-rabbit IgG AlexaFluor®568-conjugated antibody A11036 (Invitrogen) were applied for 30 min at 1:200 in 2% BSA/PBS. Immunostained cells were analyzed on a Zeiss 200 M confocal microscope (Thornwood, NY) and processed with Photoshop CS5. To quantify intracellular Aβ uptake by microglia, the area corresponding to intracellular Aβ was delineated using Iba1 staining, and the raw integrated signal intensity was determined per microglia. The quantification was performed using ImageJ software, and values are represented in arbitrary units. 10–20 cells from each condition were quantified in each of three independent experiments, and data from the three experiments were then averaged.
Cellular Toxicity Assays
Cortical neurons were isolated from E16.5 C57BL/6J mice and plated at a density of 3.5 × 104 cells per well in 96-well poly-d-lysine-coated plates (BD Biosciences). At 6 DIV, cells were treated with freshly prepared Aβ(1–40) or Aβ(1–42) WT, A2T, or A2V synthetic peptides (rPeptide, Bogart, GA) or vehicle. The peptides were initially resuspended in NH4OH/HFIP and lyophilized as described for microglial experiments. Lyophilized peptides were then dissolved in DMSO to generate a 5 mm stock and immediately diluted in culture medium and incubated with cells. After 72 h, cell viability was assessed using the Cell Titer Glo assay, following the manufacturer's instructions (Promega, Madison, WI). Experiments were performed three times, with technical triplicates per construct in each experiment.
RESULTS
A673T Variant Reduces BACE1 Cleavage of APP in Primary Neurons
We previously reported that the A673T variant of APP decreases production of the BACE1 cleavage product sAPPβ and both of the amyloidogenic peptides Aβ40 and Aβ42 by ∼40% when overexpressed in HEK 293 cells (1). To extend these findings, we looked at the production of another APP BACE1 cleavage product, the β-CTF, C99. We transfected HEK 293 cells with APP WT or the protective A673T variant. For comparison, we also examined two pathogenic APP variants, A673V and K670N/M671L (Swedish mutation), which have both been shown to increase Aβ and β-CTF production. We first examined β-CTF production by Western blot analysis of cell lysates using two antibodies as follows: A8717, which detects both α- and β-CTF, and 6E10, which detects only β-CTF (Fig. 1A). CTFs were readily detected in APP-transfected cells and absent in control GFP-transfected cells. As expected, higher levels of β-CTF were seen in cells expressing the pathogenic APP variants A673V or K670N/M671L; conversely, APP A673T reduced β-CTF levels. To quantify these changes, we used a β-CTF ELISA to analyze these cell lysates. A673T reduced β-CTF levels to ∼25% that of WT APP levels (Fig. 1B). This reduction is consistent with the effects of A673T on sAPPβ and Aβ production as we reported previously (1).
FIGURE 1.

Decreased cellular production of β-secretase cleavage products from APP A673T. A, representative Western blot analysis of cell lysates from HEK 293 cells transfected with GFP control, WT, A673T, A673V, or K670N/M671L APP. α- and β-CTF fragments were detected using an antibody specific for the C terminus of APP (A8717), and β-CTF fragments were specifically detected using an antibody just distal to the β-secretase cleavage site (6E10). An antibody to actin was used as a loading control for both blots. B, ELISA quantification of β-CTF cleavage product from these same cell lysates. C and D, ELISA quantification of Aβ40 (C) and Aβ42 (D) from supernatants of cultured mouse cortical neurons transfected with WT, A673T, or A673V human APP. E, ratio of sAPPβ/sAPPα as determined by immunoassay analysis of cell supernatants from the same cortical neurons. Values represent mean ± S.D. of three independent experiments, each with technical replicates. Two-tailed t test was compared with WT APP; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Our findings thus far have focused on APP processing in the HEK 293 cell line. To confirm that A673T had similar effects on APP processing by BACE1 in neurons, we performed experiments in cultured primary mouse cortical neurons. Cultured neurons were transfected with cDNA encoding human APP WT, the protective A673T variant, the pathogenic APP A673V variant, or a control vector expressing β-gal. All constructs expressed APP at similar levels, as confirmed by quantitative PCR (data not shown). We assessed the formation of APP cleavage products Aβ40, Aβ42, sAPPβ, and sAPPα in the media of transfected cells. Our assays were designed to specifically detect cleavage products from transfected human APP constructs and did not detect endogenous mouse APP cleavage products. Similar to what was observed in HEK 293 cells, we found that the A673T APP variant generated less Aβ40 and Aβ42 peptides as compared with WT APP (Fig. 1, C and D). Aβ40 was reduced by 38 (±25)% and Aβ42 by 35 (±10)%. APP A673V increased Aβ40 and Aβ42 levels by ∼2.5-fold. Immunoassay analysis for sAPPβ and sAPPα in cell supernatants (Fig. 1E) showed that the A673T APP variant reduced the ratio of sAPPβ relative to sAPPα when compared with WT APP by 49 (±5)%. Conversely, the pathogenic APP A673V variant robustly increased the sAPPβ/sAPPα ratio.
Overexpression of APP by transfection may result in nonphysiological processing if overexpressed substrate overwhelms endogenous proteases. Thus, we next set out to investigate APP processing of these variants when expressed at normal endogenous cellular levels. To do this, we used human iPSC lines that have been engineered via TALEN-mediated SNP alteration to carry A673T- or A673V-specific changes in APP (17). The isogenic cell lines were differentiated into cortical neurons. The differentiated cells from the three isogenic lines appeared morphologically similar to each other and uniformly expressed APP (Fig. 2A). Similar to transfected mouse neurons, we observed that the A673T APP iPSC human neurons generated less Aβ40 and Aβ42 peptides as compared with WT APP (Fig. 2, B and C). Aβ40 was reduced by 33 (±7)% and Aβ42 by 36 (±12)%. In contrast, APP A673V increased both Aβ40 and Aβ42 levels by ∼2-fold. The reduction in β-secretase cleavage products was confirmed by measuring the ratio of sAPPβ relative to sAPPα by immunoassay (Fig. 2D). The A673T APP variant reduced the sAPPβ/sAPPα ratio by 32 (±18)% compared with WT APP. Conversely, the pathogenic APP A673V variant increased the sAPPβ/sAPPα ratio ∼4-fold.
FIGURE 2.

Decreased β-secretase cleavage products from APP A673T in human iPSC-derived neurons. A, representative immunofluorescence images of differentiated cortical control (WT), isogenic APP-A673T, or isogenic APP-A673V iPS cells. Cells were stained with antibodies to MAP2 (red) and APP (green). B and C, ELISA quantification of Aβ40 (B) and Aβ42 (C) from cell supernatants collected from the different cell lines. D, ratio of sAPPβ/sAPPα as determined by immunoassay analysis of cell supernatants from the same iPSC-derived neurons. Values represent mean ± S.D. of three independent experiments, each with technical replicates. Two-tailed t test was compared with WT APP; *, p < 0.05; **, p < 0.01.
The consistency of our results across transfected HEK 293 cells and primary neurons and isogenic human iPSC-derived neurons confirms the strong influence of the residue at position 673 on BACE1 cleavage. The reproducible decreases in β-secretase cleavage of APP across these different cell types suggest that similar alterations in APP processing likely occur in neurons in the human brain.
A673T Decreases the Vmax of APP Processing by BACE1
Previously, we demonstrated that the A673T mutation decreased catalytic cleavage of an APP substrate by BACE1 in a reconstituted in vitro enzyme assay (1). Here, we investigated the mechanism by which the mutation at this residue modulated BACE1 enzymatic activity. First, by utilizing TR-FRET detection against long (27 amino acids) APP peptide substrates, we demonstrated that the A673T variant displayed ∼50% reduced cleavage activity relative to the WT substrate (Fig. 3A), consistent with our previous results determined by mass spectrometric detection. In comparison, the A673V mutation appeared to be a better substrate for BACE1 with an estimated 3-fold increase in cleavage rate relative to WT APP peptides at 5 μm substrate concentration. When the mutations at Ala-673 were combined with the Swedish APP mutation (K670N/M671L, Swe), the same differential modulations of BACE1 activity by threonine and valine were observed (Fig. 3B), however with much more rapid kinetics as a result of the Swe mutations.
FIGURE 3.

BACE1 processing of APP substrates in a reconstituted in vitro enzyme assay. A and B, TR-FRET analysis of cleavage rates for A673T and A673V peptides in a WT context (A) or in combination with the Swedish (K670N/M671L) mutation (B). C, kinetic analysis of FRET cleavage data. A673T and A673V mutations decreased and increased the rate of catalytic cleavage (Vmax) of the short APP substrate, respectively, without affecting the Km value. D, competitive inhibition of the catalytically efficient Swedish peptide substrate by peptide substrates without Swedish mutation, having a slow cleavage rate. WT, A637T and A673V competitive peptides yielded comparable IC50, suggesting similar binding affinity. Values represent mean ± S.D. of three independent experiments, each with technical replicates.
Utilizing a fluorogenic FRET substrate that spanned from P4 to P5′ of the APP BACE1 cleavage sequence, the kinetic parameters of WT APP were compared with the A673T and A673V mutations. The results showed that A673T decreased and A673V increased the Vmax, but neither altered the Km value, suggesting that mutations at this residue have an impact on the catalytic turnover of the substrate instead of substrate binding affinity (Fig. 3C).
To confirm that the mutations at Ala-673 do not modulate the binding affinity of peptide substrates to BACE1, we performed a substrate competition assay. We used the FRET peptide substrate containing the Swedish mutation as a detection method for BACE1 activity, and we titrated non-Swedish long peptides (WT, A673T, and A673V) into the BACE1 reaction. At a reaction time of 1–2 h, although the Swe FRET peptide substrate is efficiently cleaved by BACE1, the catalytic turnover of the unlabeled non-Swedish peptides by BACE1 is negligible. In other words, the non-Swedish peptides act as competitive binders to inhibit cleavage of the Swe FRET substrate in this assay setup, and their titration should yield IC50 curves that reflect their respective binding affinities. The IC50 values of WT, A673T, and A673V peptides were comparable with each other (ranging between 26 and 33 μm), further supporting the hypothesis that the mutations at residue Ala-673 of APP do not alter their binding affinity to the BACE1-active site (Fig. 3D).
Aggregation Level of A2T Aβ Variant Peptides Is Reduced in the Context of Aβ(1–42) but Not Aβ(1–40)
Our results indicate that the APP A673T variant is a less favorable substrate for BACE1 cleavage. However, the A673T variant also results in a single amino acid substitution (A2T) in Aβ peptides, and thus it could alter the properties of the Aβ peptide itself. Therefore, we compared the aggregation kinetics of synthetic WT, A2T, and A2V Aβ peptides, as both Aβ(1–40) and Aβ(1–42) species, by ThT incorporation and fluorescence.
As shown in Fig. 4, Aβ(1–40) peptides generally aggregate very slowly relative to Aβ(1–42). However, the A2V substitution promotes a significant increase in both the aggregation kinetics and the extent of aggregation in Aβ(1–40) (Fig. 4A). This result is similar to observations reported by Di Fede et al. (7). By contrast, aggregation kinetics for WT and A2T Aβ(1–40) were indistinguishable from each other. When A2V Aβ(1–40) peptides were combined with WT Aβ(1–40) or A2T Aβ(1–40) peptides in a 1:1 ratio, we observed intermediate rates and levels of aggregation.
FIGURE 4.

Aggregation kinetics of Aβ peptides measured by ThT fluorescence. A–C, process of Aβ aggregation was monitored over time using synthetic WT, A2T, or A2V Aβ peptides of pure Aβ(1–40) (A), pure Aβ(1–42) (B), or a 1:9 ratio of Aβ(1–42) to Aβ(1–40) (C). Mixtures of WT, A2T, and A2V are one-to-one molar ratios. Three separate experiments are shown for each set of peptides. Traces represent the mean of three replicate measurements, with ± S.D. shown for the final point. a.u., arbitrary units.
When assessing Aβ(1–42) aggregation kinetics (Fig. 4B), we observed similar profiles between WT Aβ(1–42) and A2V Aβ(1–42) peptides. In comparison, A2T Aβ(1–42) showed a reproducibly lower level of aggregation than WT Aβ(1–42). Mixtures of A2T Aβ(1–42) with either WT Aβ(1–42) or A2V Aβ(1–42) showed intermediate levels of aggregation.
In vivo, Aβ peptides exist as a heterogeneous mixture. As an approximate representation of this heterogeneity, we looked at aggregation kinetics of a mixture of Aβ(1–42) and Aβ(1–40) in a 1:9 ratio, representative of what has been observed in human CSF (18). When mixed together in this manner, we observed aggregation kinetics intermediate between those with pure Aβ(1–40) or Aβ(1–42) peptides (Fig. 4C). The A2V peptide mixture again showed the most rapid kinetics. The A2T mixture showed slower kinetics than the WT mixed peptides in two of three experiments, and similar kinetics in the third. Mixing A2T with WT Aβ in this context results in an aggregation profile similar to WT Aβ alone.
Taken together, these results suggest that substitutions at position 2 in Aβ peptides can impact peptide aggregation properties. The most dramatic effect is with A2V substitution in the context of Aβ(1–40). This mutation appears to convert Aβ(1–40) into a much more aggregation-prone molecule with Aβ(1–42)-like properties. However, A2V did not alter aggregation in the context of Aβ(1–42), likely because this longer peptide is already very aggregation-prone. In contrast, A2T does not appear to alter Aβ(1–40) aggregation, but it does reduce the aggregation level of Aβ(1–42). When Aβ(1–42) and Aβ(1–40) are mixed together in a physiologically relevant ratio of 1:9, the A2T variant overall has slightly slower aggregation kinetics compared with WT. However, the resulting aggregation of A2T remains substantial, making it difficult to predict the impact of this mutation on pathological amyloid accumulation, particularly in heterozygous carriers.
Microglial Uptake of A2T Aβ(1–42) Synthetic Peptides Is Reduced in Comparison with WT Aβ Peptides
Many recent Alzheimer genetic hits, including TREM2 and CD33, highlight the importance of microglial biology in Alzheimer disease (19–22). Microglial uptake of soluble Aβ is believed to be an important clearance mechanism in the brain (23). We wondered whether the different Aβ variants we have examined might be differentially taken up by microglia. We used enriched primary microglial cultures to assess uptake of synthetic WT, A2T, and A2V Aβ peptides. Again, we assessed both Aβ(1–40) and Aβ(1–42) peptides prepared as oligomers. Microglia were incubated with 1 μm Aβ oligomers for 90 min, after which cells were fixed, and internalized Aβ was visualized by immunocytochemistry.
As shown in Fig. 5A, Aβ could be visualized inside microglia under all conditions. However, when quantified (Fig. 5B), it was clear that WT Aβ(1–42) was internalized to a much greater extent than WT Aβ(1–40). This likely reflects the higher level of aggregation with WT Aβ(1–42) compared with WT Aβ(1–40), because aggregation stimulates microglial internalization of extracellular debris (including Aβ). A similar trend is seen when comparing the A2T peptides (Fig. 5A, middle panels); low levels of A2T Aβ(1–40) are internalized, although significantly higher levels of A2T Aβ(1–42) are taken up. However, the average Aβ fluorescence in cells treated with A2T Aβ(1–42) is approximately half that of cells treated with WT Aβ(1–42). This correlates with the reduced overall aggregation of A2T Aβ(1–42) relative to WT (Fig. 4B). Interestingly the A2V peptides were internalized at high levels both as Aβ(1–40) and Aβ(1–42) species. This likely reflects the higher aggregation state of A2V Aβ(1–40) (see Fig. 4A), thereby resulting in behavior more like an Aβ(1–42) peptide.
FIGURE 5.
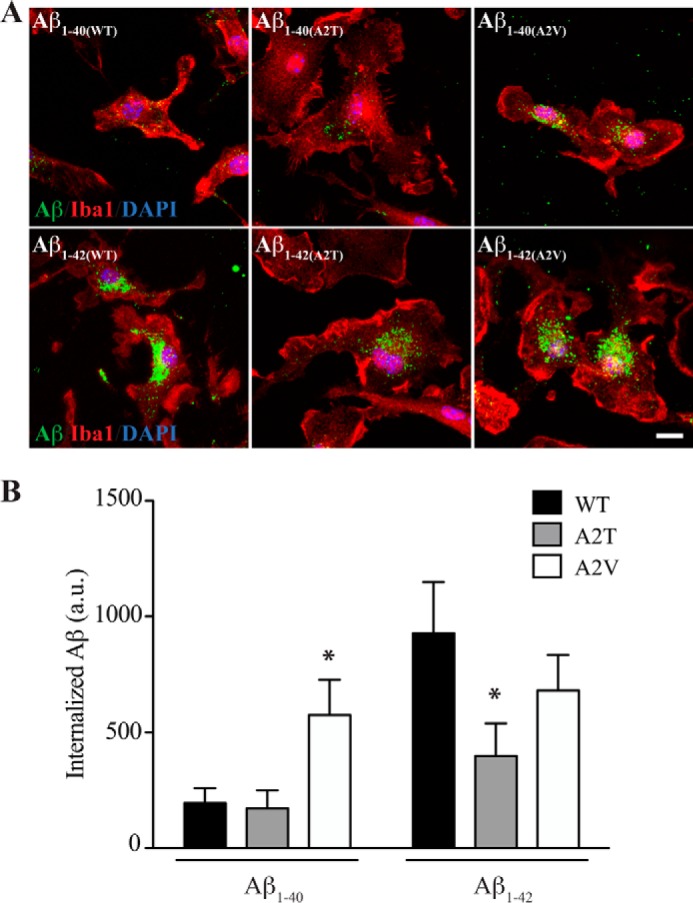
Microglial uptake of WT, A2T, or A2V Aβ peptides. A, primary microglial cultures were incubated with WT, A2T, or A2V mutants of either Aβ(1–40) (upper panels) or Aβ(1–42) (lower panels) for 90 min. Microglia were then fixed and stained for Iba1 (red), DAPI (blue), and Aβ (green). Representative confocal images for each condition show that Aβ(1–42) peptides accumulate in microglia to a greater extent compared with Aβ(1–40). Scale bar, 10 μm. B, quantification of internalized Aβ as measured by average immunofluorescence signal intensity. Values represent mean ± S.D. of three independent experiments, each analyzing >10 cells per condition). Two-tailed t test, compared with WT Aβ; *, p < 0.05. a.u., arbitrary units.
In conclusion, our results suggest that microglial internalization of the different Aβ variants we assayed is highly correlated with the aggregation properties of that variant. There do not appear to be any further inherent differences between the ability of microglia to internalize the Aβ variants.
Cellular Toxicity of A2T Aβ Synthetic Peptides Does Not Differ from That of WT Aβ Peptides
Aβ peptides are believed to be toxic to neurons, and at high concentrations can induce cell death in vitro. To investigate the impact of the A2T substitution on Aβ toxicity, we treated neuronal cultures with synthetic WT, A2T, and A2V Aβ peptides, both in the context of Aβ(1–40) and Aβ(1–42). Mixed primary cortical cultures were incubated with the different synthetic peptides ranging in concentration from 0.1 to 10 μm for 3 days, at which time cellular toxicity was assessed using a luminescent viability assay. For both Aβ(1–40) and Aβ(1–42), only slight toxicity was observed after treatment with the WT and A2T peptides at concentrations up to 3 μm (Fig. 6). Notably, A2V peptides showed no toxicity at these concentrations. At 10 μm, all Aβ(1–40) and Aβ(1–42) peptides were somewhat toxic, reducing cell viability by ∼20–40%, regardless of the nature of the variant at position 2 of Aβ.
FIGURE 6.

Cortical toxicity by WT, A2T, or A2V Aβ peptides. A and B, cultured mouse cortical neurons (E16.5) were treated with synthetic WT, A2T, or A2V Aβ(1–40) peptides (A) or Aβ(1–42) peptides (B) for 3 days at a concentration ranging from 0.1 to 10 μm. Cell viability was assessed by a luminescent assay. Results are normalized to vehicle-treated controls. Values represent mean ± S.D. of three independent experiments, each with technical replicates.
Comparing across the full concentration range, toxicity from A2T peptides was not significantly different from WT peptides. However, A2V peptides, both Aβ(1–40) and Aβ(1–42), were overall less toxic than WT peptides across the concentration range, and this difference was significant (p < 0.05 for Aβ(1–40) and p < 0.001 for Aβ(1–42), two-way analysis of variance). However, the effect size was small. These results seem somewhat counterintuitive, as the pathogenic peptide might be expected to be more toxic. However, it has been proposed that low molecular weight oligomers are cytotoxic to cultured neurons. It is possible that fewer of these smaller oligomers are present in the rapidly aggregating A2V peptide preparations. Furthermore, an experimental system such as ours that requires application of high concentrations of synthetic peptides to cultured neurons to elicit any toxicity may not be a good model of the disease state (24). Nonetheless, these results suggest that the Aβ peptides produced by the A673T APP variant are no less or no more toxic than WT Aβ.
DISCUSSION
The results from our biochemical and cellular analyses of the A673T APP variant support the conclusion that a likely mechanism for protection is via reduced APP cleavage, leading to decreased Aβ production over the lifetime of an A673T carrier. We demonstrate that the reduction in amyloidogenic processing of APP can be replicated across transfected cell lines and primary neurons, as well as in isogenic human iPSC-derived neurons that express APP at endogenous levels. Mechanistically, we show that A673T reduces the catalytic turnover of APP by BACE1, whereas an alternative residue, A673V, at the same site increases the catalytic turnover rate by BACE1. Neither residue alters the affinity of APP for BACE1. Therefore, the residue at the Ala-673 site is a critical determinant of amyloidogenic processing of APP at both the enzyme and cellular level.
An alternative model, consistent with reduction in APP processing, is that protection is conferred by reduced production of β-CTFs and/or soluble APPβ. We observe that both β-CTFs and soluble APPβ are reduced in A673T-expressing cells (Figs. 1 and 2). This possibility is supported by reports that suggest a role for these APP products in mediating neuronal toxicity (25–27). Nevertheless, because of the observation that mutations in APP center on the production and/or aggregation potential of Aβ (for example, the Dutch APP mutation E693Q causes Aβ to aggregate on blood vessels causing hereditary cerebral hemorrhage with amyloidosis (28–30)), combined with the paucity of mutations in APP directly in the cytoplasmic (β-CTFs) or N-terminal (soluble APPβ) region of APP that alter the risk for Alzheimer disease, the most plausible hypothesis is that Aβ mediates disease.
We next investigated the aggregation properties of Aβ, comparing the Ala-673 variants. We observed the greatest effect with A2V in Aβ(1–40), leading to enhanced aggregation, similar to previous reports (7). However, A2V Aβ(1–42) aggregation is not altered, likely because Aβ(1–42) aggregates so readily even in WT Aβ(1–42) form. Conversion of Aβ(1–40) to Aβ(1–42) aggregation-prone behavior could easily explain pathology in A673V carriers, because Aβ(1–40) is ∼10–20-fold more abundant than Aβ(1–42) in human CSF (18). Indeed, a 1:9 mixture of Aβ(1–42)/Aβ(1–40) showed increased aggregation kinetics for A2V peptides relative to WT peptides. Our results with A2V peptides confirm previously reported findings relative to Aβ(1–40) (7); however, we were not able to replicate the “heterozygous protection” proposed upon mixing WT and A2V peptides.
Of note, we also observed a consistent reduction in aggregation level of A2T peptides compared with WT peptides for Aβ(1–42) but not for Aβ(1–40). When comparing a 1:9 mixture of Aβ(1–42)/Aβ(1–40), the A2T peptides display slightly slower aggregation kinetics compared with WT peptides. An equimolar mixture of WT and A2T peptides prepared in a 1:9 ratio of Aβ(1–42)/Aβ(1–40) behaved identically to WT peptides (Fig. 4C). This in vitro experiment is our closest approximation of the Aβ species that might be present in an A673T heterozygous individual. As such, the reduced aggregation of the A2T Aβ(1–42) peptides likely translates to a small impact on amyloid deposition in vivo. Nevertheless, this may contribute in part to genetic protection by the A673T variant, in combination with the substantial reduction in Aβ generation.
Interestingly, the results of our microglial uptake assay correlated directly with the aggregation results. Because microglia are known to internalize aggregated proteins, this outcome is not surprising. These data confirm our findings that A2V increases aggregation in the context of Aβ(1–40), and A2T reduces aggregation in the context of Aβ(1–42), functionally leading to increased and decreased microglial internalization, respectively. However, we failed to detect any other fundamental differences in how microglia recognize and engulf the variant peptides. Thus, microglial uptake of these different variants in vivo is likely to be driven purely by Aβ levels and extent of aggregation.
Our cell toxicity data suggest that the A673T variant does not confer protection against AD by generating an Aβ peptide that is inherently less toxic than WT Aβ. However, toxicity assays involving Aβ are highly artificial and must be interpreted with caution (24). As an illustration of the potential inconsistencies of these types of experiments, previous studies investigating the toxicity of the A673V variant suggested that A2V showed increased cellular toxicity (7); in contrast, we show that A2V may be slightly less toxic in vitro. Rather than draw spurious conclusions from these results, we simply suggest caution in over-interpreting Aβ-induced cellular toxicity results.
We previously reported that the A673T variant is equally protective in apoE4 carriers and noncarriers (1); thus, the relative protective effect of the A673T variant is substantially more robust in apoE4 carriers. Taking into consideration that apoE4 carriers are considerably more likely to accumulate amyloid, and thus generate amyloid plaques and develop AD (31–33), the findings reported herein further support the hypothesis that apoE4 is mediating its effects via Aβ. Specifically, the protective A673T APP variant that reduces amyloid production and may also reduce Aβ aggregation results in a determining factor for the risk of developing AD, negating any apoE genotype effect.
We investigated several mechanisms by which A673T may confer protection against AD. The most compelling mechanism supported by our studies is that this variant reduces BACE1-mediated processing of APP. We observed this result reproducibly across multiple cell models. In addition, we found that A2T peptides are less prone to aggregation in the context of Aβ(1–42) but not in the context of Aβ(1–40). In terms of translating these findings to humans, our results predict that a heterozygote A673T carrier will have a significant, but relatively moderate, reduction in Aβ production (∼20%). The rare nature of this mutation has made it difficult to examine endogenous Aβ levels in carriers, although attempts to do so are ongoing. In heterozygote A673T carriers, we predict that Aβ aggregation and amyloid formation will be similar or slightly reduced when compared with WT individuals. However, any resulting reduction in Aβ aggregation may work additively, or even synergistically, with reduced Aβ levels to lessen AD risk in carriers. Although it is unclear how early Aβ reduction is needed to provide protection from AD, these mechanistic studies of human APP variants further validate efforts to reduce Aβ production and aggregation as therapeutic approaches to treat Alzheimer disease.
Acknowledgments
We thank Bill Meilandt and Kristin Wildsmith for technical advice on assays, Coby Carlson at Cellular Dynamics International for technical discussions on iPS cell lines, and Gai Ayalon and David Hansen for critical reading of the manuscript.
Footnotes
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- APP
- amyloid precursor protein
- ThT
- thioflavin-T
- β-CTF
- β-C-terminal fragment
- DIV
- days in vitro
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- iPSC
- induced pluripotent stem cell
- Swe
- Swedish
- HFIP
- 1,1,1,3,3,3-hexafluoro-2-propanol
- sAPP
- soluble APP
- TR-FRET
- time-resolved FRET.
REFERENCES
- 1. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R. R., Huttenlocher J., Bjornsdottir G., Andreassen O. A., Jönsson E. G., Palotie A., Behrens T. W., Magnusson O. T., Kong A., Thorsteinsdottir U., Watts R. J., Stefansson K. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 2. Cruts M., van Duijn C. M., Backhovens H., Van den Broeck M., Wehnert A., Serneels S., Sherrington R., Hutton M., Hardy J., St George-Hyslop P. H., Hofman A., Van Broeckhoven C. (1998) Estimation of the genetic contribution of presenilin-1 and −2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 7, 43–51 [DOI] [PubMed] [Google Scholar]
- 3. Hardy J., Crook R. (2001) Presenilin mutations line up along transmembrane α-helices. Neurosci. Lett. 306, 203–205 [DOI] [PubMed] [Google Scholar]
- 4. Kauwe J. S., Jacquart S., Chakraverty S., Wang J., Mayo K., Fagan A. M., Holtzman D. M., Morris J. C., Goate A. M. (2007) Extreme cerebrospinal fluid amyloid β levels identify family with late-onset Alzheimer's disease presenilin 1 mutation. Ann. Neurol. 61, 446–453 [DOI] [PubMed] [Google Scholar]
- 5. Cruchaga C., Haller G., Chakraverty S., Mayo K., Vallania F. L., Mitra R. D., Faber K., Williamson J., Bird T., Diaz-Arrastia R., Foroud T. M., Boeve B. F., Graff-Radford N. R., St Jean P., Lawson M., Ehm M. G., Mayeux R., Goate A. M. (2012) Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS One 7, e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Citron M., Oltersdorf T., Haass C., McConlogue L., Hung A. Y., Seubert P., Vigo-Pelfrey C., Lieberburg I., Selkoe D. J. (1992) Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature 360, 672–674 [DOI] [PubMed] [Google Scholar]
- 7. Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A. R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., Manzoni C., Francescucci B., Spagnoli A., Cantù L., Del Favero E., Levy E., Salmona M., Tagliavini F. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou L., Brouwers N., Benilova I., Vandersteen A., Mercken M., Van Laere K., Van Damme P., Demedts D., Van Leuven F., Sleegers K., Broersen K., Van Broeckhoven C., Vandenberghe R., De Strooper B. (2011) Amyloid precursor protein mutation E682K at the alternative β-secretase cleavage β′-site increases Aβ generation. EMBO Mol. Med. 3, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Jonghe C., Esselens C., Kumar-Singh S., Craessaerts K., Serneels S., Checler F., Annaert W., Van Broeckhoven C., De Strooper B. (2001) Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 10, 1665–1671 [DOI] [PubMed] [Google Scholar]
- 10. Kwok J. B., Li Q. X., Hallupp M., Whyte S., Ames D., Beyreuther K., Masters C. L., Schofield P. R. (2000) Novel Leu723Pro amyloid precursor protein mutation increases amyloid β42(43) peptide levels and induces apoptosis. Ann. Neurol. 47, 249–253 [DOI] [PubMed] [Google Scholar]
- 11. Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T. D., Hardy J., Hutton M., Kukull W., Larson E., Levy-Lahad E., Viitanen M., Peskind E., Poorkaj P., Schellenberg G., Tanzi R., Wasco W., Lannfelt L., Selkoe D., Younkin S. (1996) Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 2, 864–870 [DOI] [PubMed] [Google Scholar]
- 12. Suzuki N., Cheung T. T., Cai X. D., Odaka A., Otvos L., Jr., Eckman C., Golde T. E., Younkin S. G. (1994) An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (β APP717) mutants. Science 264, 1336–1340 [DOI] [PubMed] [Google Scholar]
- 13. Nilsberth C., Westlind-Danielsson A., Eckman C. B., Condron M. M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D. B., Younkin S. G., Näslund J., Lannfelt L. (2001) The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat. Neurosci. 4, 887–893 [DOI] [PubMed] [Google Scholar]
- 14. Tomiyama T., Nagata T., Shimada H., Teraoka R., Fukushima A., Kanemitsu H., Takuma H., Kuwano R., Imagawa M., Ataka S., Wada Y., Yoshioka E., Nishizaki T., Watanabe Y., Mori H. (2008) A new amyloid β variant favoring oligomerization in Alzheimer's-type dementia. Ann. Neurol. 63, 377–387 [DOI] [PubMed] [Google Scholar]
- 15. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 16. Stine W. B., Jungbauer L., Yu C., LaDu M. J. (2011) Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 670, 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carlson C. B., Mangan K. P., Wang J., Aoyama N., McLachlan M., Burke T., Delaura S., Jones E. (2014) Society for Neuroscience Annual Meeting, November 15–19, 2014, Washington, D. C. [Google Scholar]
- 18. Fagan A. M., Younkin L. H., Morris J. C., Fryer J. D., Cole T. G., Younkin S. G., Holtzman D. M. (2000) Differences in the Aβ40/Aβ42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann. Neurol. 48, 201–210 [PubMed] [Google Scholar]
- 19. Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J. C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J. (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., Abraham R., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Jones N., Stretton A., Thomas C., Richards A., Ivanov D., Widdowson C., Chapman J., Lovestone S., Powell J., Proitsi P., Lupton M. K., Brayne C., Rubinsztein D. C., Gill M., Lawlor B., Lynch A., Brown K. S., Passmore P. A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A. D., Beaumont H., Warden D., Wilcock G., Love S., Kehoe P. G., Hooper N. M., Vardy E. R., Hardy J., Mead S., Fox N. C., Rossor M., Collinge J., Maier W., Jessen F., Rüther E., Schürmann B., Heun R., Kolsch H., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frolich L., Hampel H., Gallacher J., Hull M., Rujescu D., Giegling I., Goate A. M., Kauwe J. S., Cruchaga C., Nowotny P., Morris J. C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P. P., Van Broeckhoven C., Livingston G., Bass N. J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C. E., Tsolaki M., Singleton A. B., Guerreiro R., Muhleisen T. W., Nothen M. M., Moebus S., Jockel K. H., Klopp N., Wichmann H. E., Pankratz V. S., Sando S. B., Aasly J. O., Barcikowska M., Wszolek Z. K., Dickson D. W., Graff-Radford N. R., Petersen R. C., van Duijn C. M., Breteler M. M., Ikram M. A., DeStefano A. L., Fitzpatrick A. L., Lopez O., Launer L. J., Seshadri S., Berr C., Campion D., Epelbaum J., Dartigues J. F., Tzourio C., Alperovitch A., Lathrop M., Feulner T. M., Friedrich P., Riehle C., Krawczak M., Schreiber S., Mayhaus M., Nicolhaus S., Wagenpfeil S., Steinberg S., Stefansson H., Stefansson K., Snaedal J., Bjornsson S., Jonsson P. V., Chouraki V., Genier-Boley B., Hiltunen M., Soininen H., Combarros O., Zelenika D., Delepine M., Bullido M. J., Pasquier F., Mateo I., Frank-Garcia A., Porcellini E., Hanon O., Coto E., Alvarez V., Bosco P., Siciliano G., Mancuso M., Panza F., Solfrizzi V., Nacmias B., Sorbi S., Bossu P., Piccardi P., Arosio B., Annoni G., Seripa D., Pilotto A., Scarpini E., Galimberti D., Brice A., Hannequin D., Licastro F., Jones L., Holmans P. A., Jonsson T., Riemenschneider M., Morgan K., Younkin S. G., Owen M. J., O'Donovan M., Amouyel P., Williams J. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M. A., van Duijn C. M., Thorsteinsdottir U., Kong A., Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., Gallins P. J., Buxbaum J. D., Jarvik G. P., Crane P. K., Larson E. B., Bird T. D., Boeve B. F., Graff-Radford N. R., De Jager P. L., Evans D., Schneider J. A., Carrasquillo M. M., Ertekin-Taner N., Younkin S. G., Cruchaga C., Kauwe J. S., Nowotny P., Kramer P., Hardy J., Huentelman M. J., Myers A. J., Barmada M. M., Demirci F. Y., Baldwin C. T., Green R. C., Rogaeva E., St George-Hyslop P., Arnold S. E., Barber R., Beach T., Bigio E. H., Bowen J. D., Boxer A., Burke J. R., Cairns N. J., Carlson C. S., Carney R. M., Carroll S. L., Chui H. C., Clark D. G., Corneveaux J., Cotman C. W., Cummings J. L., DeCarli C., DeKosky S. T., Diaz-Arrastia R., Dick M., Dickson D. W., Ellis W. G., Faber K. M., Fallon K. B., Farlow M. R., Ferris S., Frosch M. P., Galasko D. R., Ganguli M., Gearing M., Geschwind D. H., Ghetti B., Gilbert J. R., Gilman S., Giordani B., Glass J. D., Growdon J. H., Hamilton R. L., Harrell L. E., Head E., Honig L. S., Hulette C. M., Hyman B. T., Jicha G. A., Jin L. W., Johnson N., Karlawish J., Karydas A., Kaye J. A., Kim R., Koo E. H., Kowall N. W., Lah J. J., Levey A. I., Lieberman A. P., Lopez O. L., Mack W. J., Marson D. C., Martiniuk F., Mash D. C., Masliah E., McCormick W. C., McCurry S. M., McDavid A. N., McKee A. C., Mesulam M., Miller B. L., Miller C. A., Miller J. W., Parisi J. E., Perl D. P., Peskind E., Petersen R. C., Poon W. W., Quinn J. F., Rajbhandary R. A., Raskind M., Reisberg B., Ringman J. M., Roberson E. D., Rosenberg R. N., Sano M., Schneider L. S., Seeley W., Shelanski M. L., Slifer M. A., Smith C. D., Sonnen J. A., Spina S., Stern R. A., Tanzi R. E., Trojanowski J. Q., Troncoso J. C., Van Deerlin V. M., Vinters H. V., Vonsattel J. P., Weintraub S., Welsh-Bohmer K. A., Williamson J., Woltjer R. L., Cantwell L. B., Dombroski B. A., Beekly D., Lunetta K. L., Martin E. R., Kamboh M. I., Saykin A. J., Reiman E. M., Bennett D. A., Morris J. C., Montine T. J., Goate A. M., Blacker D., Tsuang D. W., Hakonarson H., Kukull W. A., Foroud T. M., Haines J. L., Mayeux R., Pericak-Vance M. A., Farrer L. A., Schellenberg G. D. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee C. Y., Landreth G. E. (2010) The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 25. Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Tamayev R., D'Adamio L. (2012) Inhibition of γ-secretase worsens memory deficits in a genetically congruous mouse model of Danish dementia. Mol. Neurodegener. 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tamayev R., Matsuda S., Arancio O., D'Adamio L. (2012) β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol. Med. 4, 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandez-Madrid I., Levy E., Marder K., Frangione B. (1991) Codon 618 variant of Alzheimer amyloid gene associated with inherited cerebral hemorrhage. Ann. Neurol. 30, 730–733 [DOI] [PubMed] [Google Scholar]
- 29. Levy E., Carman M. D., Fernandez-Madrid I. J., Power M. D., Lieberburg I., van Duinen S. G., Bots G. T., Luyendijk W., Frangione B. (1990) Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126 [DOI] [PubMed] [Google Scholar]
- 30. Van Broeckhoven C., Haan J., Bakker E., Hardy J. A., Van Hul W., Wehnert A., Vegter-Van der Vlis M., Roos R. A. (1990) Amyloid β protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248, 1120–1122 [DOI] [PubMed] [Google Scholar]
- 31. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fleisher A. S., Chen K., Liu X., Ayutyanont N., Roontiva A., Thiyyagura P., Protas H., Joshi A. D., Sabbagh M., Sadowsky C. H., Sperling R. A., Clark C. M., Mintun M. A., Pontecorvo M. J., Coleman R. E., Doraiswamy P. M., Johnson K. A., Carpenter A. P., Skovronsky D. M., Reiman E. M. (2013) Apolipoprotein E ϵ4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol. Aging 34, 1–12 [DOI] [PubMed] [Google Scholar]
- 33. Reiman E. M., Chen K., Liu X., Bandy D., Yu M., Lee W., Ayutyanont N., Keppler J., Reeder S. A., Langbaum J. B., Alexander G. E., Klunk W. E., Mathis C. A., Price J. C., Aizenstein H. J., DeKosky S. T., Caselli R. J. (2009) Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]