

Background: FlgJ is required for flagella formation in Salmonella enterica.
Results: The lytic activity of FlgJ was determined to be that of a β-N-acetylglucosaminidase using a novel assay.
Conclusion: FlgJ is a hydrolase and not a lytic transglycosylase.
Significance: This information is essential for the search and rational design of inhibitors that may serve as leads to a new class of antibiotics.
Keywords: Bacterial Pathogenesis, Cell Wall, Enzyme Catalysis, Peptidoglycan, Virulence Factor, N-Acetylglycosaminidase, Autolysin, Flagella, Lytic Transglycosylase, Muramidase
Abstract
The flagellum is a major virulence factor of motile pathogenic bacteria. This structure requires more than 50 proteins for its biogenesis and function, one of which is FlgJ. Homologs of FlgJ produced by the β- and γ-proteobacteria, such as Salmonella enterica, Vibrio spp., and both Sphingomonas sp. and Pseudomonas spp. are bifunctional, possessing an N-terminal domain responsible for proper rod assembly and a C-terminal domain possessing peptidoglycan lytic activity. Despite the amount of research conducted on FlgJ from these and other bacteria over the past 15 years, no biochemical analysis had been conducted on any FlgJ and consequently confusion exists as to whether the enzyme is a peptidoglycan hydrolase or a lytic transglycosylase. In this study, we present the development of a novel assay for glycoside lytic enzymes and its use to provide the first enzymatic characterization of the lytic domain of FlgJ from S. enterica as the model enzyme. Surprisingly, FlgJ functions as neither a muramidase nor a lytic transglycosylases but rather as a β-N-acetylglucosaminidase. As such, FlgJ represents the first autolysin with this activity to be characterized from a Gram-negative bacterium. At its optimal pH of 4.0, the Michaelis-Menten parameters of Km and kcat for FlgJ from S. enterica were determined to be 0.64 ± 0.18 mg ml−1 and 0.13 ± 0.016 s−1, respectively, using purified PG as substrate. Its catalytic residues were identified as Glu184 and Glu223.
Introduction
The flagellum is recognized as an important virulence factor of motile pathogenic bacteria allowing them to reach their specific target infection site(s), in addition to avoiding hostile environments, and accessing nutrients (1). The biogenesis of the flagellar system is best understood for members of Enterobacteriaceae, in particular Salmonella enterica. Salmonella serotypes are of interest because adherence of flagella to surfaces contributes to food contamination (e.g. 2) and its strong antigenic properties can provoke inflammatory responses during host infections (e.g. Ref. 3).
More than 50 genes are associated with flagellar formation and function in S. enterica (reviewed in Ref. 4), which involves 20 different proteins. These proteins form a hetero-oligomeric structure comprised of three major substructures: a basal body that spans the cell wall, a connecting hook, and a thin helical filament that extends into the extracellular environment (5). The basal body consists of an inner ring (MS ring) and two outer rings (L and P rings) connected to a central rod that crosses the cytoplasmic membrane, periplasm, and outer membrane. The rod component proteins are synthesized in the cytosol and transported to the periplasm via the flagellum-specific type III export pathway. This specialized export apparatus exists within a central pore in the MS ring (6–8). During flagella formation, the rod needs to penetrate through the peptidoglycan (PG)3 layer. At approximately 4 nm (9), the diameter of the rod is larger than the estimated pore size of the PG sacculus (10) and thus its insertion requires the localized and controlled lysis of PG strands.
PG is a heteropolymer of alternating glycan strands and peptide chains forming a rigid network that completely surrounds bacterial cells to maintain the integrity of their cytoplasmic membranes. The glycan strands are composed of repeating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) linked residues β-(1→4) and are cross-linked together through amide linkages between the stem peptides attached to the lactyl groups of MurNAc residues. Once formed, the PG sacculus is not a static structure as it requires constant biosynthesis and reinforcement to permit cellular growth and division. This continuous biosynthesis and turnover are maintained by the concerted action of the extra-cytoplasmic PG-synthesizing enzymes (the penicillin-binding proteins) and PG-lytic enzymes, which provide new sites for the insertion of the PG precursors, in addition to facilitating the separation of daughter cells (reviewed in Ref. 11). Different classes of lytic enzymes exist for the cleavage of each of the various linkages that exist within PG. Thus, with Gram-negative bacteria, peptidases (both carboxy- and endopeptidases) hydrolyze the peptide bonds linking the amino acids of stem peptides, whereas glycolytic enzymes cleave the β-(1→4) linkage between the amino sugars of the carbohydrate chains (recently reviewed in Refs. 12 and 13). Those enzymes that lead to cellular lysis if left uncontrolled are referred to as autolysins.
The predominant site of cleavage within the glycan chains of PG in Gram-negative bacteria is the linkage between MurNAc and GlcNAc residues and this is catalyzed by muralytic enzymes (Fig. 1). Two classes of muralytic enzymes exist, muramidases (EC 3.2.1.17; lysozymes) and lytic transglycosylases (EC 4.2.2.n1/n2; LTs), which differ in the nature of their reaction products. As hydrolases, the muramidases add water across the β-(1→4)-glycosidic bond to be cleaved and thereby generate a reducing MurNAc product. Although possessing the structural “lysozyme-fold,” the LTs, on the other hand, are not hydrolases. Instead, the LTs catalyze an intramolecular rearrangement involving the C-6 hydroxyl group of the MurNAc at the site of cleavage to effect lysis with the concomitant formation of a 1,6-anhydromuramoyl reaction product (14). The LTs represent a major class of autolysins in Gram-negative bacteria as they play essential roles in the biosynthesis, maintenance, and turnover of the PG sacculus (reviewed in Ref. 15).
FIGURE 1.

Glycolytic autolysins of bacteria. The β-N-acetylglucosaminidases hydrolyze the β-1,4 linkage between GlcNAc and MurNAc residues in PG, whereas the muralytic enzymes, muramidases and LTs, cleave between MurNAc and GlcNAc residues. The muramidases are hydrolytic but the LTs are not; instead they release GlcNAc residues with the concomitant formation of 1,6-anhydroMurNAc. The open and closed arrows denote exo- and endo-activity, respectively.
The other site of cleavage within the carbohydrate backbone of PG is the β-(1→4)-glycosidic linkage between GlcNAc and MurNAc residues and this is catalyzed by β-N-acetylglucosaminidases (EC 3.2.1.14; chitinase) (Fig. 1). These enzymes play a more predominant role as autolysins in the metabolism of PG within Gram-positive bacteria (13). However, to date, their autolytic activity has not been detected in any Gram-negative bacterium (11), but a related enzyme, β-N-acetylhexosaminidase (EC 3.2.1.52; NagZ), participates in the cytoplasmic events of PG turnover (13).
FlgJ of the flagella operon has been shown to be required for the proper assembly of the flagellum (16). The FlgJs produced by the β- and γ-proteobacteria, such as S. enterica, Vibrio spp., and both Sphingomonas sp. and Pseudomonas spp. are bifunctional, possessing an N-terminal domain responsible for proper rod assembly and a C-terminal domain possessing PG lytic activity (16–21). Despite the amount of research conducted on FlgJ from these and other bacteria, no biochemical analysis has been conducted on any FlgJ and consequently confusion exists as to whether they function as a hydrolase or an LT.
The crystal structure of FlgJ from Sphingomonas sp. strain A1, the only one solved, contains a lysozyme-like fold (20, 21) and was classified as a member the glycoside hydrolase family GH73 (20) of the CAZy database thereby reinforcing earlier suggestions that FlgJ is muralytic (16, 19–21, 23). However, the GH73 family additionally contains N-acetylglucosaminidases (EC 3.2.1.14), which also possess the lysozyme-fold (CAZy database). To further complicate the issue, the monofunctional FlgJs of Rhodobacter sphaeroides (25) and Caulobacter crescentus (26) lack lytic activity and this is compensated for by the production of putative LTs, which for the former bacterium is encoded within its Flg operon. Like other LTs, they too would possess the lysozyme-fold (15).
In this study, we present the development of a novel assay for glycoside lytic enzymes and its use to provide the first enzymatic characterization of the lytic domain of FlgJ from S. enterica serovar typhimurium (S. typhimurium) as the model enzyme. Surprisingly, FlgJ functions as neither a muramidase nor an LT but rather as a β-N-acetylglucosaminidase. Thus, it represents the first autolysin with this activity to be characterized from a Gram-negative bacterium.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
DNase I, RNase A, Pronase, isopropyl β-d-1-thiogalactopyranoside, and EDTA-free protease inhibitor tablets were purchased from Roche Diagnostics (Laval, QC, ON). T4 DNA ligase and restriction enzymes were from New England Biolabs (Mississauga, ON). Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose was obtained from Qiagen (Valencia, CA), whereas Source 15S resin was purchased from GE Healthcare. Fischer (Nepean, ON) provided acrylamide, glycerol, and Luria-Bertani (LB) growth medium. Unless otherwise stated, all other chemicals and reagents were from Sigma, and growth media from Difco (Detroit, MI).
Bacterial Strains and Growth
The source of plasmids and bacterial strains used in this study, together with their genotypic description are listed in Table 1. Escherichia coli strains DH5α and BL21(λDE3)pLysS were maintained on LB broth or agar at 37 °C, which were supplemented with kanamycin sulfate (50 μg/ml) and chloramphenicol (34 μg/ml) in the case of strains harboring plasmids pET28a(+) and pLysS, respectively. For overexpression studies and protein production, E. coli BL21(λDE3)pLysS was grown in SuperBroth (5 g of sodium chloride, 20 g of yeast extract, and 32 g of tryptone) at 30 °C with agitation. All strains were stored at −80 °C in 25% glycerol.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strains | Genotype | Source or Ref. |
|---|---|---|
| E. coli BL21(λDE3)pLysS | F- ompT hsdSB (rB−mB−) gal dcm rne131 (DE3)pLysS(Cmr) | Novagen |
| E. coli DH5α | fhuA2 lac(del)U169 phoA glnV44 Φ80′ lacZ(del)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | Invitrogen |
| S. enterica serovar typhimurium LT2 | Genome sequence strain | |
| Plasmids | Description | |
| pET28a(+) | IPTG-inducible T7 expersion vector; N- and C-terminal His6 tag, Kmr | Novagen |
| pACFH4 | pET28a (+) derivative containing flgJ from LT2 encoding FlgJ truncated by its N-terminal 151 amino acids with an N-terminal His6 tag on an NdeI/XhoI fragment; Kmr | This study |
| pACFH5 | pACFH4 derivative encoding FlgJ truncated by its C-terminal 10 amino acids | This study |
| pACFH6 | pACFH5 derivative encoding FlgJ with a Glu184 → Gln replacement | This study |
| pACFH8 | pACFH5 derivative encoding FlgJ with a Glu223 → Gln replacement | This study |
Isolation and Purification of PG
Samples of insoluble PG were isolated from S. typhimurium strain LT2 using the boiling SDS protocol and purified by enzyme treatment (amylase, DNase, RNase, and Pronase) as described by Clarke (27).
Conditions for PCR
Chromosomal DNA template for PCR was isolated from S. typhimurium LT2. All oligonucleotide primers used in this study are listed in Table 2 and were acquired from Integrated DNA Technologies (Coralville, IA). PCR amplifications were achieved in 50-μl volumes using a Bio-Rad Mycycler Thermal Cycler system. Purification of PCR products was performed using the MinElute PCR Purification kit (Qiagen) or the High Pure PCR Product Purification kit (Roche).
TABLE 2.
Oligonucleotide primers used in this study
Nucleotides underlined denote restriction sites and those in bold denote the replacements for site-directed mutagenesis (SDM).
| Primer name | Oligonucleotide sequence (5′-3′) | Target/final vector | Description | |
|---|---|---|---|---|
| 1 | FlgJFwd | ATCGCATATGAGTAAAGACTTTCTGGCCCG | pET28a (+)/pACFH4 | Forward primer for FlgJC |
| 2 | FlgJRev | CGATCTCGAGTTAAAAGAGATTGTCGAGATTCG | pET28a (+)/pACFH4 | Reverse primer for FlgJC |
| 3 | FlgJCFwd | CGATCTCGAGTTATTTGCTGACCTTTTCAC | pACFH4/pACFH5 | Reverse primer for FlgJC truncated by 30 nucleotides |
| 4 | FlgJE184QFwd | GATTCTGGCGCAGGCGGCACTG[b]CAGTCCGGCTGGG | pACFH5/pACFH6 | Forward primer for SDM of E184Q |
| 5 | FlgJE184QRev | GGCGGCACTG[b]CTGTCCGGCTGGGGGCAGCG | pACFH5/pACFH6 | Reverse primer for SDM of E184Q |
| 6 | FlgJE223QFwd | TGACGGAGATCACCACCACT[b]CAATACGAAA | pACFH5/pACFH8 | Forward primer for SDM of E223Q |
| 7 | FlgJE223QRev | GCTTCGCCATTTTCGTA[b]TTGAGTGGTGGT | pACFH5/pACFH8 | Reverse primer for SDM of E223Q |
Engineering of flgJ
The flgJ ORF was amplified by PCR using the primers that contained NdeI and XhoI sites to facilitate cloning of its 3′-end encoding the C-terminal lytic domain of FlgJ (henceforth termed FlgJC) and the incorporation of His6 tag from pET28a(+) at its N terminus. Amplified ORFs were cleaned using a PCR clean up kit, digested with the respective restriction enzymes, and ligated with appropriately digested pET28a(+) plasmid to yield pACFH4, which was then transformed into E. coli DH5α. Individual constructs were isolated from transformants, screened for the correct size insert, and sequenced to confirm nucleotide identity (Genomics Facility within the Advanced Analysis Centre, University of Guelph). Initial expression trials demonstrated that the recombinant protein was not stable in its E. coli host as it was partially digested. The major fragment was determined to lack its C-terminal 10 amino acids. Hence, a further truncation of flgJ was made by removing 30 nucleotides from its 3′ end. PCR products and pET28a(+) were purified and digested with the appropriate restriction enzymes prior to ligation yielding pACFH5. The final protein product, FlgJC, lacked its 151 N-terminal amino acids and 10 C-terminal amino acids.
Replacement of amino acids for enzymatic activity analysis was achieved using the QuikChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA) with pACFH5 as template. Following PCR amplification of plasmid constructs, products were digested overnight with DpnI and then transformed into E. coli DH5α. Selected colonies were sequenced to confirm nucleotide identity and a description of the resulting constructs is presented in Table 1.
Overproduction and Purification of FlgJC
E. coli BL21(λDE3)pLysS transformed with plasmid DNA was inoculated into SuperBroth supplemented with kanamycin sulfate (50 μg ml−1) and chloramphenicol (34 μg ml−1) and incubated at 37 °C until early exponential phase (A600 nm ∼ 0.6). Freshly prepared isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 1 mm and expression was induced for 4 h at 30 °C. Cells were harvested by centrifugation (10,000 × g, 10 min, 4 °C) and frozen at −20 °C. Thawed cell pellets were resuspended in lysis buffer (50 mm sodium phosphate buffer, pH 8.0, 500 mm NaCl, and 10 mm imidazole) containing Complete EDTA-free protease inhibitor mixture tablets, 10 μg ml−1 RNase A, and 5 μg ml−1 DNase I, and incubated on ice for 15 min prior to disruption with an Ultrasonic Liquid Processor (Heat Systems Inc., Toronto, Ontario, Canada) fitted with a macroprobe. The resulting cell lysate was clarified by centrifugation (5,000 × g, 10 min, 4 °C), and the collected soluble cell fractions were mixed with Ni2+-NTA agarose (2 ml liter−1 starting culture) on a Nutator for 1 h at 4 °C. The resin slurry was poured into a disposable column and the flow-through fractions were collected. Contaminating proteins were removed from the resin by washing with 3 column volumes of lysis buffer followed by 3 column volumes of wash buffer (lysis buffer containing 20 mm imidazole). Purified FlgJC was eluted in 5–10 ml elution buffer (lysis buffer containing 500 mm NaCl and 250 mm imidazole) and then dialyzed at 4 °C against 25 mm sodium phosphate buffer, pH 8.0.
FlgJC was further purified by cation-exchange chromatography on Source 15S. Protein samples were applied in dialysis buffer at a flow rate of 0.7 ml min−1 and separation of FlgJC from contaminants was achieved with the application of a linear gradient to 1 m NaCl. Under these conditions, FlgJC eluted in ∼80 mm NaCl.
Enzyme Activity Assays
Turbidometry
The turbidometric assay of Hash (28) was used to monitor the time course of PG solubilization by FlgJC. Micrococcus luteus whole cells were suspended in 25 mm citric acid, HEPES, and CHES (CCH) buffer containing 100 mm NaCl, pH 4.0, and sonicated to provide homogenous suspensions. Purified FlgJC was added to 200-μl aliquots of substrate suspension and the decrease in turbidity of the reaction mixtures was monitored continuously at A595 nm for 5 min to 1 h. For the determination of optimal pH, suspensions of PG were prepared in 25 mm CCH buffer with 100 mm NaCl, pH 3.0–8.0.
Muropeptide-based LT Assay
The HPLC-based LT assay developed by Blackburn and Clarke (29) was used to quantify the soluble muropeptides possessing 1,6-anhydromuramoyl residues released from insoluble substrate after enzyme treatment. A typical assay reaction mixture consisted of freeze-dried S. typhimurium PG suspended in 200 μl of 25 mm CCH buffer with 100 mm NaCl, pH 4.0, at a final concentration of 1.2 mg ml−1. Reactions were initiated by the addition of enzyme followed by incubation at 37 °C with gentle shaking. Reaction mixtures lacking the addition of enzyme served as negative controls. At appropriate intervals of time, individual reactions were stopped by rapid freezing using a dry ice/ethanol bath. When required for analysis, samples were thawed and subjected to centrifugation at 13,000 × g for 15 min at 4 °C. To determine kinetic parameters, the soluble muropeptides recovered in the supernatants were digested with 1.1 μm mutanolysin overnight with gentle shaking at 37 °C and then reduced with 135 mm sodium borohydride for 30 min at ambient temperature. The samples were hydrolyzed to their constituent amino sugars by incubation in 6 m HCl for 1 h and 30 min at 95 °C. The hydrolyzed samples were dried in vacuo and resuspended in water for amino sugar analysis by HPAEC and for LC-ESI-MS analysis.
[18O]H2O-based Assay
The [18O]H2O-based peptidase assay of Lood et al. (30) was adapted for the assay of muropeptides that permitted differentiation between hydrolytic and LT activity, as well as facilitate direct identification of any hydrolytic reaction products associated with both soluble and insoluble fractions of PG. Freeze-dried S. typhimurium PG was suspended in [18O]H2O to a final concentration of 1.7 mg ml−1 and briefly sonicated to provide homogenous suspensions. Reactions were initiated with the addition of enzyme followed by incubation at 37 °C for 30 min with gentle shaking. Mutanolysin and Pseudomonas aeruginosa soluble lytic transglycosylase (Slt) 70 served as positive hydrolase (muramidase) and LT controls, respectively. Reactions were stopped by rapid freezing. They were then thawed and solubilized reaction products were separated from insoluble PG by centrifugation (15,000 × g for 15 min at 4 °C) prior to analysis by LC-Q-TOF-MS. The insoluble fraction was washed four to five times with 200 μl of H2O and recovered each time by centrifugation (15,000 × g, 6 min, ambient temperature). The washed PG pellet was resuspended in 0.1 mm potassium phosphate buffer, pH 6.5, and solubilized by mutanolysin prior to LC-Q-TOF-MS analysis.
Identification of Reaction Products
The soluble portion of the products of a reaction of 10 mg ml−1 of S. typhimurium PG in 25 mm CCH buffer with 100 mm NaCl, pH 4.0, treated with 6.0 μm FlgJC for 30 min at 37 °C was divided into two aliquots. One of the aliquots was further solubilized by mutanolysin and then both were reduced with NaBH4 as described above. The reduced soluble reaction products were separated by RP-HPLC using a C18 column (ODS Hypersil 4-μm particle size, 250 × 4.6 mm, Thermo Scientific) at 45 °C previously equilibrated in 10 mm ammonium phosphate buffer, pH 5.6, containing 2% methanol at a flow rate of 0.5 ml min−1. Separation was affected by application of a linear gradient to 28% methanol in the same buffer over 175 min. Eluting muropeptides were detected by monitoring A210 nm and fractions collected manually were analyzed by LC-ESI-MS.
Mass Spectrometry
All MS analyses were conducted using instruments at the Mass Spectrometry Facility of the University of Guelph. LC-ESI-MS was performed using a Dionex UHPLC UltiMate 3000 liquid chromatograph interfaced to an amaZon SL ion trap mass spectrometer. Samples were injected onto a Poroshell 120 EC-C18 (2.7-μm particle size, 150 × 4.6 mm) column (Agilent) previously equilibrated at 0.2 ml/min with 0.1% formic acid in 2% acetonitrile. Separation of muropetides was achieved by application of a linear gradient to 0.1% formic acid in 100% acetonitrile over 30 min. The mass spectrometer electrospray capillary voltage was 4.5 kV and the drying temperature at 250 °C with a drying gas flow rate of 10 liters/min. Nebulizer pressure was 40 p.s.i. Nitrogen was used as both nebulizing and drying gas, and helium was used as collision gas at 60 p.s.i. The mass spectrometer scanning range was 100–2200 mass to charge in auto-MS/MS positive-ion mode.
LC-Q-TOF-MS was performed by injecting samples into an Agilent 1260 Infinity liquid chromatograph interfaced to an Agilent 6540 UHD accurate Mass Q-TOF mass spectrometer. An AdvanceBio Peptide Map C18 (2.1 × 100 mm) column (Agilent) was previously equilibrated at 0.2 ml min−1 with 0.1% formic acid containing 2% acetonitrile and separation of muropetides was achieved by application of a multistep gradient to 60% acetonitrile over 38 min and then linearly to 100% acetonitrile over 50 min. The mass spectrometer electrospray capillary voltage was maintained at 4.0 kV and the drying temperature at 350 °C with a flow rate of 5 liters min−1. Nebulizer pressure was 15 p.s.i. Nitrogen was used as nebulizing and drying gas as well as collision gas. The mass-to-charge ratio was scanned across the 300–2000 range and the MS/MS mass range was scanned from 50 to 3000 m/z in positive-ion auto MS/MS mode. The auto MS/MS mode was setup to fragment 3 precursor ions per cycle (1 spectrum/s) with collision energy set 2.5 eV offset and linearly increased to 100 eV for 3000 m/z.
Other Analytical Techniques
Identification of ORFs, protein translations, and isoelectric points (pI) were performed using ApE version 2.0.47 and ProtParam (31). BlastP searches of the NCBI database for FlgJ homologs were performed using the amino acid sequence of S. enterica FlgJ as the probe. Analyses of sequence data were performed using ClustalW2 software (32), whereas both secondary and tertiary structure predictions were made using Phyre2 (33, 34). Nucleotide sequencing of PCR products as well as plasmids was performed by the Genomic Facility of the Advanced Analysis Center, University of Guelph. Protein concentrations were determined using the Pierce BCA protein assay kit with BSA serving as the standard (Pierce Biotechnology, Rockford, IL). SDS-PAGE on 15% acrylamide gels was conducted by the method of Laemmli (35) with Coomassie Brilliant Blue staining and Western immunoblot analysis as previously described (36). Circular dichroism (CD) spectrometry was performed as described previously (37) using a 0.1-cm path length cell at an internal temperature of 25 °C. The spectra were recorded as an average of four data accumulations, with a scan speed of 50 nm min−1, bandwidth of 1 nm, 1 s, data pitch 1 nm, and range of 190–250 nm. Analyses of the spectra were performed using DichroWeb with the Selecon 3 program (38) and protein reference set 4 (39). Data were further analyzed using Contin (40).
RESULTS
In Silico Analysis of FlgJ
Multiple alignment of the amino acid sequence of S. typhimurium LT2 FlgJ with the known or hypothetical homologs of 61 members of the Enterobacteriaceae found in the genome database led to the identification of nine consensus motifs (supplemental Fig. S1). The majority of these (five) are associated with the C-terminal region of the protein, the domain that has been demonstrated to possess lytic activity (16). No structure is known for any of these enzymes, but the x-ray crystal structure of the C-terminal domain of Sphingomonas FlgJ (FlgJC) involving residues Gln154-Asn310 has been determined at high resolution (20, 21).
The amino acid sequence of the putative FlgJC of S. typhimurium FlgJ, involving residues Lys153-His307, is 43.5% identical and 72.7% similar to Sphingomonas sp. FlgJC (Fig. 2). Included in this identity is Glu185, the predicted catalytic residue of Sphingomonas FlgJC, which comprises consensus motif IV of the aligned Enterobacteriaceae sequences (supplemental Fig. S1). Not surprisingly given this overall similarity, the secondary structure of S. typhimurium FlgJC, as predicted by Phyre, is very similar to the known structure of Sphinogomonas FlgJC (compare α 51.9%; β 6.4% to α 49.0%; β 9.0%, respectively) (Fig. 2). The structure of S. typhimurium FlgJC predicted by Phyre using Sphingomonas sp. FlgJC (Protein Data Bank (PDB) code 2ZYC) as the template (E-value, 2.3e−43; score, 253.76) consists of two lobes, α and β, separated by a deep cleft that is proposed to serve as the active site. Glu184 of motif IV located on the α lobe is positioned appropriately within the middle of this active site cleft to potentially serve as a catalytic acid/base.
FIGURE 2.

Predicted structure of S. enterica FlgJC. A, amino acid alignment and comparison of the known secondary structure Sphingomonas sp. (Ssp) FlgJC with those of the S. enterica (Se) enzyme. The red and orange boxes denote the invariant Glu and Asp residues, respectively. The schematic (B) and surface (C) presentations of the predicted three-dimensional structure of S. enterica FlgJC. Residues 152 to 299 of the S. enterica FlgJC were threaded by Phyre2 onto FlgJC from Sphingomonas sp. (PDB 2ZYC). The positions of invariant Glu184, Glu223, Asp248, and Asp283 are depicted in yellow.
Glu184 also aligns with the identified catalytic acid/base residue of S. typhimurium MltE. Examination of the S. typhimurium genome led to our identification of seven hypothetical LTs, MltA, MltB, MltC, MltD, MltE, MltF, and Slt. A phylogenic analysis of these sequences with S. typhimurium FlgJC using ClustalW2 suggested that FlgJC is most closely related to MltC or MltE (Fig. 3). Further analysis indicated that FlgJC is more similar to MltE; MltC could not be aligned with FlgJC using the LALIGN server, whereas FlgJC was found to have 10.8% identity (25% similarity) to S. typhimurium MltE (Fig. 3). Although this level of identity is not high, it did involve a number of residues associated with motif IV of FlgJC, including Glu184. The homologous residue in MltE, Glu64, comprises the signature Glu-Ser motif of the LTs and represents the sole catalytic acid/base residue. Based on these data, and that it possess the Glu-Ser LT motif, FlgJC was proposed to function as an LT.
FIGURE 3.
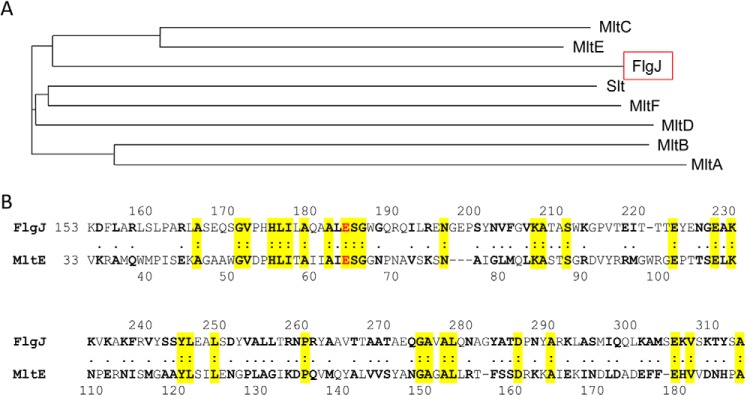
Comparison of S. enterica FlgJC primary structure with LTs. A, phylogram depicting relationship of FlgJC with the LTs identified in S. enterica. B, amino acid sequence alignment of FlgJC with S. enterica MltE. Residues highlighted in yellow denote identity and the Glu residues in red represent the predicted catalytic acid/base residues in the respective enzymes.
Overproduction and Purification of FlgJC
As observed by others previously (e.g. Ref. 16), attempts to prepare full-length S. typhimurium FlgJ failed because almost all of the protein precipitated from solution during its isolation and preliminary purification. Again as found earlier, altering buffer conditions or including detergents such as Triton X-100 did not alleviate this situation. Consequently, a truncated form of FlgJ had to be engineered to provide it in soluble form for its biochemical analysis.
The S. typhimurium flgJ gene has been engineered previously to produce recombinant FlgJC involving residues Ser152-Phe316 possessing an N-terminal His6 tag (16). We repeated this cloning by PCR amplification of the 3′ end of flgJ using genomic DNA of the same strain as template and ligating it into pET28a(+) giving pACFH4. Expression of this construct in E. coli BL21(λDE3)pLysS led to the overproduction of the protein product that was purified to apparent homogeneity (as determined by SDS-PAGE analysis; data not shown) by a combination of affinity chromatography on Ni2+-NTA-agarose and cation-exchange chromatography on Source 15S. The protein remained soluble but appeared to be susceptible to degradation into a major and several minor fragments. The major fragment, which retained the N-terminal His6 tag as observed by Western blot analysis, was found to have a molecular mass of 19.7 kDa by MALDI-TOF MS analysis (data not shown). These data indicated that cleavage was occurring at the C-terminal end of the protein to release a decapeptide. As this C-terminal decapeptide does not contain any residues of a consensus motif (supplemental Fig. S1), and hence unlikely to influence catalytic activity, a new vector (pACFH5) was generated using pACFH4 as a template, which provided a further truncation of flgJC by 30 codons. The protein product, now lacking the 151 N-terminal and 10 C-terminal amino acid residues of FlgJ, was again purified by a combination of affinity and cation-exchange chromatographies with routine yields of 7 mg/liter of cell culture. Importantly, this form of FlgJ was found to be stable and, for convenience, it will still be referred to as FlgJC.
The secondary structure of FlgJC was determined by CD spectroscopy involving a constrained least squares analysis. This analysis indicated the protein to be comprised of 30.8% α- and 18.6% β-structure (data not shown), which is similar to that predicted by Phyre and thus enhancing confidence in the modeled structure.
Lytic Activity of FlgJC
The turbidometric assay of Hash (28) was used to confirm the lytic activity of purified FlgJC with M. luteus whole cells as substrate. As seen from the representative plot presented in Fig. 4A, incubation of the whole cells with FlgJC led to the time-dependent loss of turbidity expected with the cleavage of the cell wall into soluble products by lytic enzymes. This loss of turbidity was continuous with time such that prolonged incubation led to eventual clearing of the cells, activity indicative of endo-acting lytic enzymes (15).
FIGURE 4.

PG lytic activity of FlgJC and its engineered variants. Enzyme was incubated at ambient temperature with cells of M. luteus suspended in 25 mm CCH buffer, pH 4.0, containing 100 mm NaCl and the progress of lysis was monitored turbidometrically at A595 nm. A, representative lytic activity of FlgJC at varying concentrations. ○, 0 μm; ■, 1.5 μm; ▴, 3.0 μm; and ●, 6.0 μm FlgJC. B, activity of engineered FlgJC variants at final concentrations of 6 μm. ○, negative control, no enzyme added; ●, wild-type FlgJC; ■, (Glu233 → Gln)FlgJC; and (▴) (Glu184 → Gln)FlgJC. The error bars denote S.D. (n = 3).
The dependence of this FlgJC activity on pH was assessed by plotting initial reaction rates of turbidity loss as a function of pH using a tripartite buffer system with pH ranging from 3 to 8. Under the conditions employed, the pH-activity profile was bell shaped with a maximal lytic activity occurring at pH 4.0 (Fig. 5). The specific activity of FlgJC at pH 4.0 was determined to be 0.33 ΔA600 units min−1 mg of protein−1.
FIGURE 5.

Dependence of FlgJC turbidometric activity on pH. The turbidometric activity of 6.0 μm FlgJC was determined using suspensions of purified insoluble PG as substrate in 25 mm CCH buffer with 100 mm NaCl, pH 3.0–8.0. The error bars denote S.D. (n ≥ 3).
MS Analysis of FlgJC Reaction Products
In an initial attempt to determine the reaction specificity of FlgJC, soluble reaction products from digests of S. typhimurium PG at pH 4.0 were recovered by centrifugation and subjected to LC-MS analysis as used previously for the analysis of muralytic reactions (e.g. 41). Very little soluble material was resolved by reverse-phase chromatography (Fig. 6, trace b) further suggesting endo-type activity of the enzyme leading to the production of larger muropeptides that either remained associated with insoluble PG and/or were not resolved by the reverse-phase HPLC column. Subsequent treatment of the soluble products with the muramidase mutanolysin did result in the detection of a number of muropeptides (trace d) that were distinct from PG digests with mutanolysin alone (trace c). However, given the complexity of the PG substrate in terms of its heterogeneous composition (involving chains of GlcNAc-MurNAc with varying stem peptide composition and extent of cross-linking, and with each terminating in 1,6-anhydro-MurNAc residue), it was impossible to determine what the initial reaction product of FlgJC was using this method.
FIGURE 6.
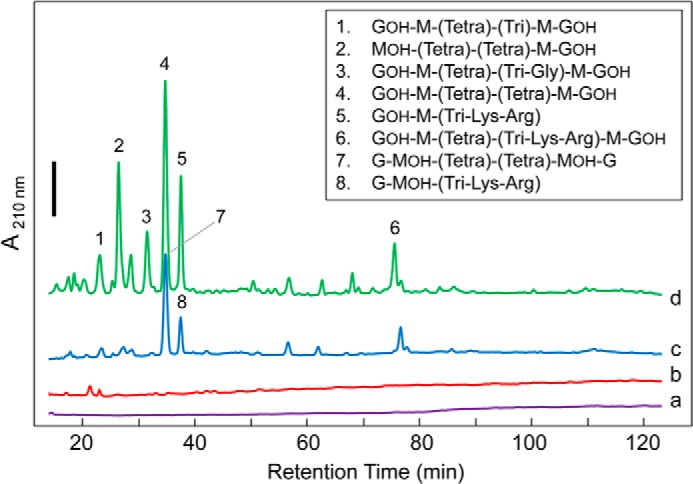
RP-HPLC-based analysis of FlgJC reaction products. S. typhimurium PG (10 mg ml−1) suspended in 25 mm CCH buffer, pH 4.0, containing 100 mm NaCl was treated with (a) no added enzyme; (b) 6.0 μm FlgJC; (c) 1.1 μm mutanolysin; and (d) 6.0 μm FlgJC followed by 1.1 μm mutanolysin. After incubation for 30 min at 37 °C, the soluble fractions were recovered by centrifugation and then reduced with NaBH4 prior to separation by RP-HPLC at 45 °C. The C18 column was equilibrated in 10 mm ammonium phosphate buffer, pH 5.6, containing 0.001% azide and 2% methanol at a flow rate of 0.5 ml min−1 and separation was affected by application of a linear gradient to 28% methanol in the same buffer over 175 min. Eluting muropeptides were detected by monitoring at A210 nm; the solid black bar denotes 0.02 absorbance units. See Table 4 for MS-based identification of muropeptides. G, GlcNAc; GOH, N-acetylglucosaminitol; M, MurNAc; MOH, N-acetylmuramitol; Tri, l-Ala-d-Glu-DAP; Tetra, l-Ala-d-Glu-DAP-d-Ala.
FlgJC as an LT
The HPLC-based assay developed for LTs (29) was employed to determine whether FlgJC does indeed function as an LT. With this assay, soluble reaction products liberated from the insoluble PG substrate are reduced with NaBH4, thereby converting any reducing GlcNAc and MurNAc associated with the muropeptides into their corresponding alditols, N-acetylglucosaminitol or N-acetylmuramitol, whereas any 1,6-anhydromuramic acid is left unaltered. Strong acid (6 m HCl) is then used to hydrolyze the muropeptides into their corresponding free amino sugars and amino acids, which are quantified by high pH anion-exchange chromatography-pulsed amperometric detection (HPAEC-PAD). Any 1,6-anhydromuramoyl residues generated by LT activity would be converted into muramic acid, whereas both glucosaminitol and muramitol would remain unaltered. Increasing concentrations with time of one of these products relative to the others that were intrinsic to the PG substrate defines the reaction specificity of the enzyme.
Using this method, no muramitol was detected in PG treated with FlgJC regardless of the length of incubation indicating that FlgJC is not a muramidase. Although muramic acid was detected in these samples (which would have originated from 1,6-anhydro-MurNAc), surprisingly its concentration did not increase significantly over time suggesting no LT activity. However, closer examination of the amino sugar analyses revealed that glucosaminitol production did increase over time. The FlgJC-catalyzed rates of glucosaminitol and muramic acid release from purified S. typhimurium PG as substrate at pH 4.0 were calculated to be 6.5 and 4.8 nmol min−1, respectively. The faster rate of glucosaminitol production relative to that of muramic acid indicates that FlgJC appears to function as a hydrolase and specifically as an N-acetylglucosaminidase.
The Michaelis-Menten parameters of FlgJC as a β-N-acetylglucosaminidase were determined by assaying for glucosaminitol production following NaBH4 reduction and acid hydrolysis of released reaction products from concentrations of PG ranging from 0.2 to 1.2 mg ml−1 in 25 mm CCH buffer, pH 4.0, at 37 °C (Fig. 7). Under these conditions, values of Km and kcat were determined to be 0.64 ± 0.18 mg ml−1 and 0.13 ± 0.016 s−1, respectively.
FIGURE 7.

Determination of the Michaelis-Menten parameters for FlgJC as a β-N-acetylglucosaminidase. S. typhimurium PG (0.2–1.2 mg ml−1) suspended in 25 mm CCH buffer, pH 4.0, containing 100 mm NaCl was treated with 6.0 μm FlgJC and the rates of glucosaminitol production were determined by HPAEC-based amino sugar analysis following NaBH4 reduction and acid hydrolysis of muropeptide products as described under “Experimental Procedures.” The error bars denote S.D. (n = 3).
Novel Assay for PG Hydrolytic Enzymes
A novel assay was developed to show unequivocally that FlgJC functions as a muramidase (hydrolase). This assay involves the labeling of hydrolytic reaction products with the stable isotope of oxygen, 18O, through the use of [18O]H2O, followed by MS analysis of reaction products. Using this heavy water, [18O]OH− would be incorporated as the C-1 hydroxyl of the MurNAc or GlcNAc product following muramidase- or β-N-acetylglucosaminidase-catalyzed hydrolysis of glycosidic linkages, respectively, which would cause an increase in the abundance of the third isotope peak of the reaction products as observed by MS. As the LT-catalyzed reaction pathway does not involve H2O, no 18O could be incorporated into reaction products and hence the isotopic distribution would not be altered. To validate this assay, S. typhimurium PG was digested with either mutanolysin (an N-acetylmuramidase) or P. aeruginosa Slt70 in the presence of [18O]H2O to serve as positive and negative controls, respectively. Soluble reaction products were collected by centrifugation and then subjected to LC-MS using a Q-TOF mass spectrometer as described under “Experimental Procedures.” As expected, only the muropeptide products generated by mutanolysin contained 18O. As seen in Fig. 8, the abundance of the third (heaviest) isotope peak of the muropetides from the mutanolysin digest were significantly increased relative to the natural abundance present in Slt70 reaction products.
FIGURE 8.

Q-TOF MS analysis of isotopic distribution of reaction products from PG lytic enzymes. S. typhimurium PG was suspended in [18O]H2O to a final concentration of 1.7 mg ml−1 and treated with: A, 1.1 μm mutanolysin; B, 1.0 μm P. aeruginosa Slt70; and C, 6.0 μm FlgJC. After incubation at 37 °C for 30 min, solubilized reaction products were separated from insoluble PG by centrifugation and the soluble products of each were subjected to LC-Q-TOF-MS analysis (left panels). For the FlgJC reaction, the PG pellet was washed exhaustively with H2O and then resuspended in 100 mm potassium phosphate buffer, pH 6.5, for solubilization by mutanolysin. The soluble muropeptides of this secondary digestion were recovered by centrifugation prior to LC-Q-TOF-MS analysis. The red spectral lines in the MS spectra denote the 18O-containing isotope of the respective muropeptides. Identification of select muropeptides (as indicated) was achieved by subsequent Q-TOF MS/MS analysis (right panels). The monoisotopic masses (M+H)+ are presented for each of the fragments detected.
Muropeptides containing high proportions of 18O were detected in both the soluble and (original) insoluble reaction products after digestion of S. typhimurium PG in 25 mm CCH buffer, pH 4.0, prepared in [18O]H2O (Fig. 8). Tandem MS analysis of these reaction products confirmed that the muropeptides enriched with 18O consisted exclusively of MurNAc(peptides)-GlcNAc, both cross- and uncross-linked. The complete list of reaction products liberated by FlgJC is presented in Table 3. With this knowledge, the major fractions obtained earlier by RP-HPLC (Fig. 6) that had been reduced with NaBH4 were re-examined by ESI-MS and then subjected to MS/MS analysis, which revealed muropeptides possessing glucosaminitol (Table 4). These data thus confirm that FlgJC functions as a hydrolase, and specifically as a β-N-acetylglucosaminidase.
TABLE 3.
Q-TOF MS/MS analysis of muropeptides released from insoluble PG by FlgJC in the presence of [18O]H2O
| Annotationa |
m/zb |
z | ||
|---|---|---|---|---|
| Expected | Observed | Δ | ||
| M-(tetra)-G | 471.6950 | 471.7043 | −0.0093 | 2 |
| M-(tri-Lys-Arg)-G | 578.2750 | 578.2833 | −0.0083 | 2 |
| [M-(tetra)-G]-[(tetra)-M-G] | 621.9233 | 621.9323 | −0.0090 | 3 |
| [M-(tetra)-G]-[(tri-Lys-Arg)M-G] | 692.9730 | 692.9859 | −0.0126 | 3 |
| [M-(tetra)-G]-[(tetra)M-G]-(tetra)-M-G | 929.0567 | 929.0594 | −0.0033 | 3 |
a G, GlcNAc; M, MurNAc; tri, l-Ala-d-Glu-DAP; tetra, l-Ala-d-Glu-DAP-d-Ala.
b The m/z data presented correspond to the 18O isotope-containing muropeptides.
TABLE 4.
Electrospray ionization-MS analysis of muropeptides released from insoluble PG by FlgJC
| Fraction | Annotationb |
m/z |
z | ||
|---|---|---|---|---|---|
| No.a | Expected | Observed | Δ | ||
| 1 | GOH-M-(tetra)-(tri)-M-GOH | 897.8850 | 897.8993 | −0.0143 | 2 |
| 2 | MOH-(tetra)-(tetra))-M-GOH | 831.8450 | 831.8759 | −0.0309 | 2 |
| 3 | GOH-M-(tetra)-(tri-Gly)-M-GOH | 617.9233 | 617.9461 | −0.0228 | 3 |
| 4 | GOH-M-(tetra)-(tetra)-M-GOH | 933.4000 | 933.4129 | −0.0129 | 2 |
| 5 | GOH-M- (tri-Lys-Arg) | 578.2850 | 578.2947 | −0.0097 | 2 |
| 6 | GOH-M-(tetra)-(tri-Lys-Arg)-M-GOH | 1039.9800 | 1039.9914 | −0.0114 | 2 |
| 7 | G-MOH-(tetra)-(tetra)-MOH-G | 933.4000 | 933.4171 | −0.0171 | 2 |
| 8 | G-MOH- (tri-Lys-Arg) | 578.2850 | 578.2905 | −0.0055 | 2 |
a The muropeptide fractions correspond to those of the RP-HPLC separation presented in Fig. 6.
b Identification of each muropeptide was made by MS/MS analysis of each parent ion (data not shown). G, GlcNAc; GOH, N-acetylglucosaminitol; M, MurNAc; MOH, N-acetylmuramitol; tri, l-Ala-d-Glu-DAP; tetra, l-Ala-d-Glu-DAP-d-Ala.
Identification of Catalytic Residues FlgJC
Typically, the mechanism of action of glycoside hydrolases involves two acidic amino acid residues that are opposed to each other across the active site cleft/pocket. Glu223 had been identified earlier as an essential residue in FlgJC from both S. typhimurium (16) and Sphingomonas sp. (20, 21), and this invariant residue comprises motif VI of the aligned sequences of identified Enterobacteriaceae FlgJs (supplemental Fig. S1). However, these earlier studies report different potential second catalytic residues. Asp248 was proposed to serve this function in S. typhimurium FlgJC although its replacement did not greatly reduce catalytic activity; only a semiquantitative zymogram assay was used to assess enzyme activity (16). Although this residue is also invariant among the FlgJs of the Enterobacteriaceae, comprising motif VII, it would appear to be positioned to the side of the active site cleft and too far from any other acidic residues (Fig. 2, B and C). With the understanding of the crystal structure of the Sphingomonas sp. FlgJC, Glu185 was identified to function as the catalytic acid/base residue (20, 21). The homologous Glu184 of S. typhimurium FlgJC is positioned directly opposite Glu223 on the other side of the putative active site cleft in the predicted structure of the enzyme at a distance of 11.4 Å away (Fig. 2, B and C).
To confirm their participation in the catalytic mechanism of S. typhimurium FlgJC, both Glu184 and Glu223 were replaced with glutamine by site-directed mutagenesis using pACFH5 as the template. These two forms of FlgJC were purified to apparent homogeneity using the same protocol used for the wild-type form of the enyzme (data not shown) and CD spectroscopy was used to confirm that their secondary structures were not altered with the amino acid replacement. The resulting spectra were found to be indistinguishable from that of the wild-type enzyme (data not shown).
The specific activity of each of the FlgJ forms was determined using the turbidometric assay with 0.4 mg ml−1 M. luteus whole cells as substrate at pH 4 (Fig. 4B). The activity of (Glu223 → Gln)FlgJC was barely detectable and its calculated specific activity was 0.14% of that of the wild-type enzyme (Table 5). (Glu184 → Gln)FlgJC was also weakly active but it did retain 8.8% wild-type activity under the conditions employed.
TABLE 5.
Specific activities of FlgJC and its engineered variants
| Enzyme | Specific activity |
|
|---|---|---|
| ΔOD595 min−1mg protein−1 | % | |
| Wild-type FlgJC | 3.29 × 10−1 | 100 |
| (Glu184 → Gln) | 2.90 × 10−2 | 8.82 |
| (Glu223 → Gln) | 4.51 × 10−4 | 0.14 |
DISCUSSION
This study presents the first biochemical analysis of the enzymatic activity of FlgJ, an important protein involved in the flagellar biosynthetic pathway of many Gram-negative bacterial pathogens. Contrary to what has been proposed/discussed in the past, the enzyme is neither a muramidase nor an LT but rather it functions as an endo-acting β-N-acetylglucosaminidase. Moreover, two acidic residues appear to be essential for this catalytic activity.
Despite being identified 15 years ago (16), the nature of the lytic activity of FlgJ had not been determined, likely because of the significant technical limitations associated with studying PG-degrading enzymes. The heterogeneity of both the substrate and resulting lytic products in terms of stem peptide composition and extent of cross-linking, as well as any modification to the glycan chains, contribute to the complexity in the analysis of the PG-related material. With the lack of a soluble, defined and commercially available substrate, researchers based their assessment and interpretations on zymogram and/or turbidometric assays coupled with amino acid sequence alignments and structural homologies.
Our initial attempts at identifying reaction products released from FlgJC involved RP-HPLC coupled with ESI-MS/MS. This has been used successfully in the past to characterize the activity of exo-acting lytic enzymes that release a limited number of soluble dissacharide-based muropeptides, which proved to be relatively easy to identify (e.g. Ref. 42). However, a low abundance of these products was observed in the soluble fraction of FlgJC reactions suggesting endo-type activity where glycan reaction products would remain attached, either directly or through cross-linking, to the PG sacculi substrate. To overcome this limitation, we developed a novel assay that permitted the direct labeling of hydrolytic reaction products with the stable 18O isotope of [18O]H2O coupled with MS and compositional analyses. A major advantage of this assay is that it allows for (i) precise determination of reaction products generated by hydrolytic enzymes in both the soluble and insoluble fractions where the latter require subsequent enzymatic or acid digestion prior to analysis, and (ii) simultaneous discrimination between hydrolytic and non-hydrolytic reactions.
In the present study, the observed increase in production of glucosaminitol over time (after NaBH4 reduction of reaction products) coupled with the introduction of 18O into only GlcNAc residues facilitated the identification of FlgJ as being hydrolytic and, specifically, a β-N-acetylglucosaminidase. Furthermore, that the majority of the labeled GlcNAc was found in the insoluble fraction of reactions indicated FlgJ is endo-acting. This latter finding was not unexpected given the function of FlgJ to create pores within the existing PG sacculus for flagella assembly. S. typhimurium possesses 6–8 peritrichous flagella (43) as well as the ability to become hyperflagellated during periods of stress (44). Thus, flagellar assembly would require the activity of an endo-acting lytic to create these pores along the PG sacculus at varying positions distinct from regions of continuing growth or division where other autolysins (e.g. LTs) function (11–13, 15).
This study introduces the first characterized β-N-acetylglucosaminidase found in Gram-negative bacteria. Based on structural (primary, secondary, and tertiary) information, FlgJC from Sphingomonas sp. strain A1 had been classified as a member of the glycoside hydrolase family GH73 (20) of the CAZy database. This family is comprised largely of β-N-acetylglucosaminidases from Gram-positive bacteria. Hence, perhaps it should have been predicted earlier, if not expected, that FlgJ functions as a β-N-acetylglucosaminidase. Presumably, the lack of prior identification of this activity in Gram-negative bacteria together with knowledge of its structure possessing a lysozyme-fold led to is incorrect assignment by some as a muramidase. Identification of enzyme specificity became complicated with observations that FlgJ shares sequence identity with family 1E LTs, enzymes that also possess the lysozyme-fold and represent the major class of autolysin with glycolytic activity in Gram-negative bacteria (15, 41). Given this, the misassignment of FlgJ as an LT is understandable.
Another commonality shared between many β-N-acetylglucosaminidases and LTs is their putative substrate-assisted mechanisms of action. Typical glycolytic enzymes catalyze either single- or double-displacement reactions involving the direct participation of two catalytic residues (usually Glu and/or Asp), which are appropriately spaced across the active site. However, the family of GH18, GH20, GH84, and likely, GH85 β-N-acetylglucosaminidases have been shown to invoke the anchimeric assistance of the N-acetyl group of their GlcNAc-based substrate in their mechanism of action (45–47). In this mechanism, the carbonyl oxygen of the acetyl group of substrate substitutes for a nucleophilic catalytic residue to generate an oxazolinium ion intermediate that stabilizes the oxocarbenium ion transition state formed by acid catalysis during glycolytic cleavage (Fig. 9). Assistance in rendering the carbonyl oxygen nucleophilic is provided by a second acidic amino acid residue positioned appropriately to deprotonate the N-acetamido nitrogen, but distant from the catalytic acid/base residue. A similar mechanism of action has been proposed for the LTs of families GH23, GH102, GH103, and GH104 (Ref. 48, and references therein). With the GH73 enzymes, the situation is much less clear.
FIGURE 9.

Proposed mechanisms of inverting and retaining β-N-acetylglucosaminidases. A, with the single-displacement mechanism, a catalytic acid protonates the oxygen of the β-1,4 glycosidic linkage facilitating the departure of the first product, whereas a catalytic base abstracts a proton from water allowing it to directly attack the anomeric C-1 carbon of the GlcNAc residue with inversion of configuration to the α-GlcNAc product. B, in the anchimeric double-displacement mechanism, acid catalysis proceeds as described above, whereas the carbonyl oxygen of the substrate acetamido group acts as a nucleophile, with assistance from the catalytic base residue through deprotonation of the amide N, leading to the formation of the oxazolinium ion intermediate. The catalytic acid then acts as a base to abstract a proton from the nucleophilic water, which attacks the C-1 of the GlcNAc residue to hydrolyze the oxazolinium ion leading to retention of anomeric configuration.
The structures of three GH73 enzymes are now known, those of L. monocytogenes Auto (PDB 3F17), S. pneumoniae LytB (PDB 4Q2W), and FlgJ from Sphingomonas sp. (PDB 2ZYC). All share an α/β-hydrolase-fold with a deep cleft that accommodates their glycan substrates and a conserved active site. With each, the identities of the catalytic acid/base residues is clear and they (21, 49, 50), together with the homolog of Staphylococcus warneri Atl (51), have been proven experimentally. These findings were confirmed in the current study with the significant decrease in catalytic activity found with S. typhimurium (Glu184 → Gln)FlgJC. The residual activity observed may reflect the relatively low pH at which the enzyme assay was performed that may have facilitated acid catalysis.
The identification of a catalytic nucleophile/base residue, however, has been inconclusive with varying results obtained from different mutational studies. These studies have supported either a single displacement, inverting mechanism of action (21, 52) or one involving substrate assistance (49, 51). Our replacement of S. typhimurium FlgJC Glu223 with Gln led to an almost complete loss of activity (0.14% residual) strongly suggesting the direct participation of this residue as the base catalyst in an inverting mechanism of action. However, the residue is predicted to be positioned 11.4 Å from the catalytic acid Glu184 (Fig. 2), which represents an outer limit for the distance between catalytic residues in an inverting enzyme (53) (Fig. 9). Interestingly, Glu223 is located on a flexible loop that forms the active site cleft and this loop has been proposed to undergo a conformational change upon binding substrate (21), an implication that has been made for other GH73 N-acetylglucosaminidases such as Auto (52). Thus, it remains to be established whether this loop does indeed close to permit an inverting mechanistic pathway, or instead positions Glu233 allowing it to assist the deprotonation of the amide of an N-acetyl group in an anchimeric mode of action resulting in the retention of anomeric configuration in the lytic product.
Knowledge of the stereochemistry of the initial reaction products would help to identify whether the enzyme catalyzes an inverting or a retaining mechanism. Typically, this is assessed by conducting substrate hydrolyzes in an NMR for the immediate analysis of initial products but this experiment would prove technically challenging, if not impossible, for FlgJ given its endo activity on the totally insoluble PG substrate. However, this investigation could also be assisted by inhibition studies with GlcNAc-thiazoline, a mechanism-based inhibitor of N-acetylglucosaminidases that use substrate-assisted catalysis (CAZy database) (24).
In a recent study, Roure et al. (22) demonstated that endogenous LTs are required for full motility of both Helicobacter pylori and S. typhimurium. However, these lytic enzymes were not required for insertion or assembly of the flagella complex within cells walls (mutants deficient in the genes encoding the specific LTs still possessed flagella) but rather for the proper functioning of the flagellar motor protein MotB. The LTs were postulated to provide 1,6-anhydromuramoyl residues through which MotB could bind covalently to secure the motor complex within the PG sacculus. It was assumed that pores within the PG network of the sacculus would be large enough to accommodate the flagellum complex without any specific remodeling. Our observations, together with earlier findings on the requirement of FlgJ, suggest that the N-acetylglucosaminidase activity associated with this protein is required to generate the appropriate pores for flagella insertion that are then modified by an LT to provide binding sites for the motor complex. Why two distinct lytic enzymes would be required for this purpose rather than the use of an LT alone to create both the pore and appropriate binding sites for the flagellum remains unknown. It is also curious that these Gram-negative cells utilize an N-acetylglucosaminidase for this lysis and not a muramidase. It is possible that the N-acetylglucosaminidase domain of FlgJ represents an evolutionary link to flagellated Gram-positive bacteria given that N-acetylglucosaminidases represent their major autolysins (13) and they are indeed required for flagella insertion in these bacteria (Ref. 22 and references therein). Regardless, the low pH activity optimum of FlgJC of 4.0 is consistent with its autolytic role that would require the localization of the enzyme to the periplasm at the cytoplasmic membrane-PG interface. It would be expected that the pH of this microenvironment would be low due to the maintenance of a proton-motive force, the source of energy that will be used by the flagellar motor for motility.
Motility of pathogenic bacteria is a major virulence factor as it allows them to reach and colonize their specific environmental niches. Thus, targeting the assembly of motility appendages essential for bacterial survival may aid in the fight against bacterial infections and diseases. Knowledge of the specific type of lytic activity associated with flagella assembly is essential if the enyzmes responsible are to be targeted for inhibitor development. Moreover, our development of a novel assay based on the incorporation of 18O into hydrolytic products will greatly facilitate the characterization of other PG lytic enzymes that continue to be discovered as the metabolism of this important bacterial cell wall polymer is further explored.
Supplementary Material
Acknowledgments
We thank Dyanne Brewer and Armen Charchoglyan of the Mass Spectrometry Facility (Advanced Analysis Centre, University of Guelph) and Josh Kaplansky for expert technical assistance and both Dave Sychantha and Chris Vandenende for helpful discussions.
This work was supported by Canadian Institutes of Health Research Team Grant TGC114045 (to A. J. C.) and an Ontario Graduate Scholarship (to P. J. M.) from the Province of Ontario.

This article contains supplemental Fig. S1.
- PG
- peptidoglycan
- MurNAc
- N-acetylmuramic acid
- GlcNAc
- N-acetylglucosamine
- LT
- lytic transglycosylase
- Mlt
- membrane-bound lytic transglycosylase
- Slt
- soluble lytic transglycosylase
- Ni2+-NTA
- Ni2+-nitrilotriacetic acid
- HPAEC-PAD
- high pH anion-exchange chromatography-pulsed amperometric detection
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- PDB
- Protein Data Bank.
REFERENCES
- 1. Moens S., Vanderleyden J. (1996) Functions of bacterial flagella. Crit. Rev. Microbiol. 22, 67–100 [DOI] [PubMed] [Google Scholar]
- 2. Berger C. N., Shaw R. K., Brown D. J., Mather H., Clare S., Dougan G., Pallen M. J., Frankel G. (2009) Interaction of Salmonella enterica with basil and other salad leaves. ISME J. 3, 261–265 [DOI] [PubMed] [Google Scholar]
- 3. Eaves-pyles T., Murthy K., Liaudet L., Virág L., Ross G., Soriano F. G., Salzman A. L. (2001) Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: IκBα degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 166, 1248–1260 [DOI] [PubMed] [Google Scholar]
- 4. Apel D., Surette M. G. (2008) Bringing order to a complex molecular machine: the assembly of the bacterial flagella. Biochim. Biophys. Acta 1778, 1851–1858 [DOI] [PubMed] [Google Scholar]
- 5. Young G. M., Schmiel D. H., Miller V. L. (1999) A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. U.S.A. 96, 6456–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nambu T., Inagaki Y., Kutsukake K. (2006) Plasticity of the domain structure in FlgJ, a bacterial protein involved in flagellar rod formation. Genes Genet. Syst. 81, 381–389 [DOI] [PubMed] [Google Scholar]
- 7. Kubori T., Shimamoto N., Yamaguchi S., Namba K., Aizawa S. (1992) Morphological pathway of flagellar assembly in Salmonella typhimurium. J. Mol. Biol. 226, 433–446 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki T., Iino T., Horiguchi T., Yamaguchi S. (1978) Incomplete flagellar structures in non-flagellate mutants of Salmonella typhimurium. J. Bacteriol. 133, 904–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H., Yonekura K., Murata K., Hirai T., Oosawa K., Namba K. (1998) A structural feature in the central channel of the bacterial flagellar FliF ring complex is implicated in type III protein export. J. Struct. Biol. 124, 104–114 [DOI] [PubMed] [Google Scholar]
- 10. Demchick P., Koch A. L. (1996) The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 178, 768–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vollmer W., Bertsche U. (2008) Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 1778, 1714–1734 [DOI] [PubMed] [Google Scholar]
- 12. van Heijenoort J. (2011) Peptidoglycan hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev. 75, 636–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vollmer W., Joris B., Charlier P., Foster S. (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286 [DOI] [PubMed] [Google Scholar]
- 14. Höltje J.-V., Mirelman D., Sharon N., Schwarz U. (1975) Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 124, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scheurwater E., Reid C. W., Clarke A. J. (2008) Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 40, 586–591 [DOI] [PubMed] [Google Scholar]
- 16. Nambu T., Minamino T., Macnab R. M., Kutsukake K. (1999) Peptidoglycan-hydrolyzing activity of the FlgJ protein, essential for flagellar rod formation in Salmonella typhimurium. J. Bacteriol. 181, 1555–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dijkstra A. J., Keck W. (1996) Peptidoglycan as a barrier to transenvelope transport. J. Bacteriol. 178, 5555–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das M., Chopra A. K., Wood T., Peterson J. W. (1998) Cloning, sequencing and expression of the flagellin core protein and other genes encoding structural proteins of the Vibrio cholerae flagellum. FEMS Microbiol. Lett. 165, 239–246 [DOI] [PubMed] [Google Scholar]
- 19. Hirano T., Minamino T., Macnab R. M. (2001) The role in flagellar rod assembly of the N-terminal domain of Salmonella FlgJ, a flagellum-specific muramidase. J. Mol. Biol. 312, 359–369 [DOI] [PubMed] [Google Scholar]
- 20. Hashimoto W., Ochiai A., Momma K., Itoh T., Mikami B., Maruyama Y., Murata K. (2009) Crystal structure of the glycosidase family 73 peptidoglycan hydrolase FlgJ. Biochem. Biophys. Res. Commun. 381, 16–21 [DOI] [PubMed] [Google Scholar]
- 21. Maruyama Y., Ochiai A., Itoh T., Mikami B., Hashimoto W., Murata K. (2010) Mutational studies of the peptidoglycan hydrolase FlgJ of Sphingomonas sp. strain A1. J. Basic Microbiol. 50, 311–317 [DOI] [PubMed] [Google Scholar]
- 22. Roure S., Bonis M., Chaput C., Ecobichon C., Mattox A., Barrière C., Geldmacher N., Guadagnini S., Schmitt C., Prévost M. C., Labigne A., Backert S., Ferrero R. L., Boneca I. G. (2012) Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol. Microbiol. 86, 845–856 [DOI] [PubMed] [Google Scholar]
- 23. de la Mora J., Ballado T., González-Pedrajo B., Camarena L., Dreyfus G. (2007) The flagellar muramidase from the photosynthetic bacterium Rhodobacter sphaeroides. J. Bacteriol. 189, 7998–8004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mark B. L., Vocadlo D. J., Knapp S., Triggs-Raine B. L., Withers S. G., James M. N. (2001) Crystallographic evidence for substrate-assisted catalysis in a bacterial beta-hexosaminidase. J. Biol. Chem. 276, 10330–10337 [DOI] [PubMed] [Google Scholar]
- 25. de la Mora J., Osorio-Valeriano M., González-Pedrajo B., Ballado T., Camarena L., Dreyfus G. (2012) The C terminus of the flagellar muramidase SltF modulates the interaction with FlgJ in Rhodobacter sphaeroides. J. Bacteriol. 194, 4513–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Viollier P. H., Shapiro L. (2003) A lytic transglycosylase homologue, PleA, is required for the assembly of pili and the flagellum at the Caulobacter crescentus cell pole. Mol. Microbiol. 49, 331–345 [DOI] [PubMed] [Google Scholar]
- 27. Clarke A. J. (1993) Compositional analysis of peptidoglycan by high performance anion-exchange chromatography. Anal. Biochem. 212, 344–350 [DOI] [PubMed] [Google Scholar]
- 28. Hash J. H. (1967) Measurement of bacteriolytic enzymes. J. Bacteriol. 93, 1201–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blackburn N. T., Clarke A. J. (2000) Assay for lytic transglycosylases: a family of peptidoglycan lyases. Anal. Biochem. 284, 388–393 [DOI] [PubMed] [Google Scholar]
- 30. Lood R., Raz A., Molina H., Euler C. W., Fischetti V. A. (2014) A highly active and negatively charged Streptococcus pyogenes lysine with a rare d-alanyl-l-alanine endopeptidase activity protects mice against streptococcal bacteremia. Antimicrob. Agents Chemother. 58, 3073–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NJ [Google Scholar]
- 32. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bennett-Lovsey R. M., Herbert A. D., Sternberg M. J., Kelley L. A. (2008) Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70, 611–625 [DOI] [PubMed] [Google Scholar]
- 34. Kelley L. A., MacCallum R. M., Sternberg M. J. (2000) Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299, 499–520 [DOI] [PubMed] [Google Scholar]
- 35. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 36. Moynihan P. J., Clarke A. J. (2010) O-Acetylation of peptidoglycan in Gram-negative bacteria: identification and characterization of peptidoglycan O-acetyltransferase in Neisseria gonorrhoeae. J. Biol. Chem. 285, 13264–13273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pfeffer J. M., Weadge J. T., Clarke A. J. (2013) Mechanism of action of Neisseria gonorrhoeae O-acetylpeptidoglycan esterase, an SGNH serine esterase. J. Biol. Chem. 288, 2605–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sreerama N., Venyaminov S. Y., Woody R. W. (1999) Estimation of the number of helical and strand segments in proteins using CD spectroscopy. Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sreerama N., Venyaminov S. Y., Woody R. W. (2000) Estimation of protein secondary structure from CD spectra: Inclusion of denatured proteins with native protein in the analysis. Anal. Biochem. 287, 243–251 [DOI] [PubMed] [Google Scholar]
- 40. van Stokkum I. H., Spoelder H. J., Bloemendal M., van Grondelle R., Groen F. C. (1990) Estimation of protein secondary structure and error analysis from CD spectra. Anal. Biochem. 191, 110–118 [DOI] [PubMed] [Google Scholar]
- 41. Blackburn N. T., Clarke A. J. (2001) Identification of four families of peptidoglycan lytic transglycosylases. J. Mol. Evol. 52, 78–84 [DOI] [PubMed] [Google Scholar]
- 42. Blackburn N. T., Clarke A. J. (2002) Characterization of soluble and membrane-bound family 3 lytic transglycosylases from Pseudomonas aeruginosa. Biochemistry 41, 1001–1013 [DOI] [PubMed] [Google Scholar]
- 43. Fujii M., Shibata S., Aizawa S-I. (2008) Polar, peritrichous, and lateral flagella belong to three distinguishable flagellar families. J. Mol. Biol. 379, 273–283 [DOI] [PubMed] [Google Scholar]
- 44. Harshey R. M., Matsuyama T. (1994) Dimorphic transistion in Escherichia coli and Salmonella typhimurium: surface-induced differentiation into hyperflagellate swarmer cells. Proc. Natl. Acad. Sci. U.S.A. 91, 8631–8635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Terwisscha van Scheltinga A. C., Armand S., Kalk K. H., Isogai A., Henrissat B., Dijkstra B. W. (1995) Stereochemistry of chitin hydrolysis by a plant chitinase/lysozyme and x-ray structure of a complex with allosamidin: evidence for substrate assisted catalysis. Biochemistry 34, 15619–15623 [DOI] [PubMed] [Google Scholar]
- 46. Tews I., Perrakis A., Oppenheim A., Dauter Z., Wilson K. S., Vorgias C. E. (1996) Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nat. Struct. Biol. 3, 638–648 [DOI] [PubMed] [Google Scholar]
- 47. Drouillard S., Armand S., Davies G. J., Vorgias C. E., Henrissat B. (1997) Serratia marcescens chitobiase is a retaining glycosidase utilizing substrate acetamido group participation. Biochem. J. 328, 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reid C. W., Blackburn N. T., Legaree B. A., Auzanneau F.-I., Clarke A. J. (2004) Inhibition of membrane-bound lytic transglysosylase by NAG-thiazoline. FEBS Lett. 574, 73–79 [DOI] [PubMed] [Google Scholar]
- 49. Inagaki N., Iguchi A., Yokoyama T., Yokoi K. J., Ono Y., Yamakawa A., Taketo A., Kodaira K. (2009) Molecular properties of the glucosaminidase AcmA from Lactococcus lactis MG1363: mutational and biochemical analyses. Gene 447, 61–71 [DOI] [PubMed] [Google Scholar]
- 50. Bai X.-H., Chen H.-J., Jiang Y. L., Wen Z., Huang Y., Cheng W., Li Q., Lei Q., Zhang J.-R., Chen Y., Zhou C. Z. (2014) Structure of pneumococcal peptidoglycan hydrolase LytB reveals insights into the bacterial cell wall remodeling and pathogenesis. J. Biol. Chem. 289, 23403–23416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yokoi K. J., Sugahara K., Iguchi A., Nishitani G., Ikeda M., Shimada T., Inagaki N., Yamakawa A., Taketo A., Kodaira K. (2008) Molecular properties of the putative autolysin Atl(WM) encoded by Staphylococcus warneri M: mutational and biochemical analyses of the amidase and glucosaminidase domains. Gene 416, 66–76 [DOI] [PubMed] [Google Scholar]
- 52. Bublitz M., Polle L., Holland C., Heinz D. W., Nimtz M., Schubert W. D. (2009) Structural basis for autoinhibition and activation of Auto, a virulence-associated peptidoglycan hydrolase of Listeria monocytogenes. Mol. Microbiol. 71, 1509–1522 [DOI] [PubMed] [Google Scholar]
- 53. McCarter J. D., Withers S. G. (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 4, 885–892 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.