

Background: RIPK4 and IRF6 are important for epidermal development. However, whether they function together to regulate keratinocyte differentiation has not been addressed.
Results: RIPK4 directly activates IRF6, resulting in expression of the transcriptional regulators GRHL3 and OVOL1.
Conclusion: RIPK4 and IRF6 promote keratinocyte differentiation by functioning as a signaling axis.
Significance: This study reveals how mutations in RIPK4 may cause epidermal disorders.
Keywords: Differentiation, Keratinocyte, Protein Kinase C (PKC), Protein Phosphorylation, Transcription Factor, Grainyhead-like 3, Interferon Regulatory Factor 6, OVO-like 1, Receptor-interacting Protein Kinase 4, Popliteal Pterygium
Abstract
Receptor-interacting protein kinase 4 (RIPK4) and interferon regulatory factor 6 (IRF6) are critical regulators of keratinocyte differentiation, and their mutation causes the related developmental epidermal disorders Bartsocas-Papas syndrome and popliteal pterygium syndrome, respectively. However, the signaling pathways in which RIPK4 and IRF6 operate to regulate keratinocyte differentiation are poorly defined. Here we identify and mechanistically define a direct functional relationship between RIPK4 and IRF6. Gene promoter reporter and in vitro kinase assays, coimmunoprecipitation experiments, and confocal microscopy demonstrated that RIPK4 directly regulates IRF6 trans-activator activity and nuclear translocation. Gene knockdown and overexpression studies indicated that the RIPK4-IRF6 signaling axis controls the expression of key transcriptional regulators of keratinocyte differentiation, including Grainyhead-like 3 and OVO-like 1. Additionally, we demonstrate that the p.Ile121Asn missense mutation in RIPK4, which has been identified recently in Bartsocas-Papas syndrome, inhibits its kinase activity, thereby preventing RIPK4-mediated IRF6 activation and nuclear translocation. We show, through mutagenesis-based experiments, that Ser-413 and Ser-424 in IRF6 are important for its activation by RIPK4. RIPK4 is also important for the regulation of IRF6 expression by the protein kinase C pathway. Therefore, our findings not only provide important mechanistic insights into the regulation of keratinocyte differentiation by RIPK4 and IRF6, but they also suggest one mechanism by which mutations in RIPK4 may cause epidermal disorders (e.g. Bartsocas-Papas syndrome), namely by the impaired activation of IRF6 by RIPK4.
Introduction
The epithelial cells that cover the external surfaces of the body (e.g. epidermal and oral keratinocytes) serve a number of important functions. One such function is to provide a barrier defense against mechanical trauma, chemicals, and infection (1). The formation of this barrier during embryonic development relies upon a tightly regulated balance between keratinocyte proliferation and differentiation. Proliferation is restricted to the basal layer, where epidermal stem cells periodically lose the capacity to proliferate and initiate a program of terminal keratinocyte differentiation and migrate toward the surface (2). The development of various ectodermal structures (e.g. lips and mouth, digits, and external genitalia) also relies upon these processes being tightly regulated (2). Keratinocyte proliferation and differentiation must also be tightly regulated post-development to maintain barrier integrity and to prevent pathological conditions (e.g. squamous cell carcinoma) (3).
Interferon regulatory factor 6 (IRF6) is a critical transcriptional regulator of keratinocyte differentiation (4–6). Irf6-deficient mice exhibit a range of epidermal defects. For instance, their epidermis is characterized by a greatly expanded spinous layer and absence of the granular and cornified layers, resulting in defective epidermal barrier function (4). Irf6-deficient mice also have epidermal adhesions at several sites, including in the oral cavity. Although the signaling pathways in which IRF6 operates are unclear, its transcriptional activation of the transcription factors Grainyhead-like 3 (GRHL3) and OVO-like 1 (OVOL1) is important for keratinocyte differentiation (7, 8).
Receptor interacting protein kinase 4 (RIPK4) is also a critical regulator of keratinocyte differentiation (9, 10). The external orifices of Ripk4-deficient mice, including the mouth, are fused. The epidermis is greatly expanded and dysregulated, and the mice die at birth, most likely from suffocation (9, 10). Prior studies have indicated that RIPK4 functions in several signaling pathways, including the PKC, NF-κB, and Wnt/β-catenin pathways (10–16).
Mutations in IRF6 cause popliteal pterygium syndrome (17), a developmental epidermal disorder characterized by orofacial clefting, skin webbing, syndactyly, and genital deformities (18). Mutations in RIPK4 have been identified recently in Bartsocas-Papas syndrome (BPS)3 (19, 20). BPS is a more severe form of popliteal pterygium and causes death early in life (21, 22). RIPK4 and IRF6 mutations have also been identified in squamous cell carcinoma (23).
How RIPK4 and IRF6 regulate keratinocyte differentiation is not well understood. However, given the phenotypic similarities between Ripk4- and Irf6-deficient mice, along with the similarities in the developmental defects in individuals with mutations in RIPK4 and IRF6, we sought to establish whether a direct functional relationship exists between these two proteins. We demonstrate here that not only do RIPK4 and IRF6 function in the same PKC-regulated signaling pathway to promote keratinocyte differentiation, but RIPK4 also directly activates IRF6. We also provide insights into how a specific missense mutation in RIPK4 may cause BPS.
EXPERIMENTAL PROCEDURES
Reagents
Cell culture medium and supplements, FCS, SuperScript III reverse transcriptase, random primers, dNTPs, TaqMan Universal Master Mix II, Lipofectamine RNAiMAX, Lipofectamine 2000, Silencer Select RIPK4 siRNA and control non-targeting siRNA, precast 10% NuPAGE gels, mouse anti-V5 antibodies, Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 594-conjugated goat anti-mouse IgG antibodies, and ProLong Gold Antifade reagent (containing DAPI) were from Invitrogen. [γ-32P]ATP (3000 Ci/mmol) was from PerkinElmer Life Sciences. FuGENE6, Passive Lysis Buffer, and the Dual-Glo luciferase assay system were from Promega. The QuikChange II site-directed mutagenesis kit was from Agilent Technologies, and the mutagenic primers were synthesized by GeneWorks. cOmplete protease inhibitors were from Roche. The ON-TARGETplus IRF6 siRNA and non-targeting control siRNA were from Millennium Science. The anti-IRF6 and anti-PCNA antibodies were from Cell Signaling Technology. Anti-β-tubulin and HRP-conjugated anti-FLAG (M2) antibodies and anti-FLAG-agarose were from Sigma-Aldrich. Anti-HSP90 antibody was from BD Biosciences, and anti-ERK2 antibody was from Santa Cruz Biotechnology.
Expression Vectors and Mutagenesis
The IRF6 expression vectors pEF-HA-IRF6 (expresses an N-terminal HA-tagged version of human IRF6), pEF-HA-IRF6 S413A (Ser-413 replaced by alanine), pEF-HA-IRF6 S424A (Ser-424 replaced by alanine), pEF-HA-IRF6 S413A/S424A (Ser-413 and Ser-424 replaced by alanine), pEF-HA-IRF6 S413E (Ser-413 replaced by glutamic acid), pEF-HA-IRF6 S424E (Ser-424 replaced by glutamic acid), pEF-HA-IRF6 S413E/S424E (Ser-413 and Ser-424 replaced by glutamic acid), and pEF-V5-IRF6 (expresses an N-terminal V5-tagged version of IRF6) have been described previously (24). Expression vectors encoding FLAG-tagged versions of wild-type and kinase-dead (K51R) mouse Ripk4 were provided by Dr. Shiv Pillai (Harvard Medical School) (12). The Ripk4 expression vector pFLAG-Ripk4 I121N (Ile-121 replaced by asparagine) was created using the primer pair 5′-GGG ACC TGC GCT TTC GCA ACG TGC ACG AG-3′ and 5′-CTC GTG CAC GTT GCG AAA GCG CAG GTC CC-3′ and a QuikChange II site-directed mutagenesis kit. The GST expression vector pGEX-IRF6 CTD (expresses the C-terminal domain of IRF6 fused to GST) was created by PCR using the primer pair 5′-ACG GAA TTC GCT CGG ATG ATC TAC GAG ATG-3′ and 5′-CAG AAG CTT TTA CTG GGG AGG CAG GGC AG-3′. The PCR product was digested with EcoRI and HindIII and cloned into pGEX2TH, and then the sequence was verified.
Bacterial Expression and Purification of GST Proteins
Transformed BL21(DE3) Escherichia coli were grown at 37 °C in 2YT broth (1.6% tryptone, 1% yeast extract, 0.5% NaCl, pH 7.0) with constant shaking. When the culture A600 reached ∼0.6, protein expression was induced with isopropyl 1-thio-β-d-galactopyranoside (0.4 mm), and the culture was incubated at 25 °C for 2–3 h with constant shaking. The bacteria were lysed by sonication in ice-cold PBS containing 1% Triton X-100, DNase I (30 μg/ml) and protease inhibitors. The lysates were clarified by centrifugation (27,000 × g for 40 min at 4 °C), and the GST proteins (GST-IRF6 CTD and GST) were purified on an FPLC system using GSTrap FF columns (GE Healthcare) and concentrated using Amicon Ultra-4 centrifugal filters (Millipore).
Cell Culture
The human oral keratinocyte cell line OKF6/TERT-2 (25) was cultured in keratinocyte serum-free medium supplemented with 25 μg/ml bovine pituitary extract, 2 ng/ml EGF, 0.4 mm CaCl2, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-1. Primary human epidermal keratinocytes (Lonza) were cultured in KGM-GoldTM medium according to the protocol of the supplier. HEK293T cells were cultured in DMEM supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-1. Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
Cell Lysis and Western Blotting
Cells were washed twice with ice-cold PBS and then lysed (20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 10% glycerol, 10 mm NaF, 10 mm β-glycerol phosphate, and cOmplete protease inhibitors) on ice for 60 min. The lysates were clarified by centrifugation (13,000 × g for 10 min at 4 °C), and protein concentrations were measured using a protein assay kit (Bio-Rad). Lysates were subjected to electrophoresis on 10% NuPAGE gels, followed by Western blotting according to standard protocols. Immunoreactive bands were visualized using ECL reagents (Millipore) and a LAS-3000 Imager (Fujifilm) or by exposure to x-ray film (Fujifilm). Films were scanned using a GS-800 calibrated imaging densitometer (Bio-Rad).
Immunoprecipitation Assays
V5-IRF6 was immunoprecipitated by incubating 1 mg of cell lysate with 1 μg of anti-V5 antibody and 20 μl of protein G-Sepharose for 4 h at 4 °C with constant mixing. FLAG-Ripk4 was immunoprecipitated by incubating 1 mg of cell lysate with 10 μl of anti-FLAG-agarose for 4 h at 4 °C with constant mixing. In both cases, the beads were then washed four times with lysis buffer.
In Vitro Kinase Assays
Anti-FLAG (Ripk4) immunoprecipitates were incubated for 30 min at 30 °C in kinase assay buffer (20 mm Hepes (pH 7.4), 25 mm MgCl2, 3 mm MnCl2, 10 mm β-glycerol phosphate, 10 μm ATP, and 10 μCi of [γ-32P]ATP) containing 2 μg of GST-IRF6 CTD or GST. Reactions were terminated by the addition of SDS-PAGE sample buffer and heating at 95 °C for 5 min. Aliquots of the reactions were then subjected to SDS-PAGE. The gels were stained with colloidal Coomassie G-250 and dried, and 32P incorporation was detected using a Typhoon Trio PhosphorImager (GE Healthcare).
Gene Promoter Reporter Assays
HEK293T cells were seeded in 12-well tissue culture plates at a density of 3 × 105 cells/well and transfected (in duplicate) the next day using FuGENE6 transfection reagent. The total amount of plasmid in each transfection was kept constant using empty vector. The cells were lysed 24 h post-transfection with passive lysis buffer and assayed for firefly and Renilla luciferase activity using the Dual-Glo luciferase assay system. Renilla luciferase activity was used to normalize transfection efficiencies. The IFNβ gene promoter reporter plasmid was provided by Dr. Ashley Mansell (Monash Institute of Medical Research). The NF-κB-dependent E-selectin (ELAM-1) gene promoter reporter plasmid was as described previously (26). The control Renilla reporter plasmid, pRL-TK, was from Promega.
Immunofluorescent Staining and Confocal Microscopy
OKF6/TERT-2 cells and HEK293T cells (seeded on glass coverslips) were fixed with 4% paraformaldehyde, solubilized with 0.1% Triton X-100, and blocked in 5% goat serum. The cells were then incubated overnight (at 4 °C) with a rabbit anti-IRF6 antibody, a mouse anti-FLAG antibody, or a mouse anti-β-tubulin antibody for 16 h at 4 °C. Following three washes with PBS, the cells were incubated with an Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody or an Alexa Fluor 594-conjugated goat anti-mouse IgG antibody for 60 min at room temperature. The cells were washed three times with PBS and finally mounted on glass microscope slides using ProLong Gold Antifade reagent containing DAPI. Mounted coverslips were allowed to cure in the dark for 24 h. Images were acquired on an Olympus FV1000 scanning confocal microscope. No anti-IRF6 or anti-FLAG staining was apparent in HEK293T cells transfected with empty vector only. The effects of phorbol 12-myristate 13-acetate (PMA) on IRF6 subcellular localization were analyzed using ImageJ software. Three random fields of cells were analyzed for each treatment condition. Binary images were obtained by thresholding to define the nuclear region of interest, which was subsequently used for measuring IRF6 nuclear fluorescence. Thereafter, IRF6 cytoplasmic fluorescence was determined by subtracting the nuclear fluorescence from the total cell fluorescence. IRF6 nuclear localization was calculated by dividing the nuclear fluorescence by the cytoplasmic fluorescence and is presented as a ratio of nuclear to cytoplasmic IRF6.
Real-time PCR
Total RNA was purified using an RNAeasy mini kit (Qiagen). RNA (1 μg) was reverse-transcribed into cDNA using random primers and SuperScript III reverse transcriptase. Real-time PCR was performed (in triplicate) with an Applied Biosystems Prism 7900HT sequence detection system and TaqMan assays (Invitrogen) for the following genes: GRHL3 (Hs00297962_m1), IRF6 (Hs00196213_m1), IVL (Hs00902520_m1), KRT13 (Hs00999762_m1), MAD1 (Hs00965581_m1), OVOL1 (Hs00190060_m1), RIPK4 (Hs01062501_m1), and Ripk4 (Mm00458366_m1). Messenger RNA levels relative to those of the endogenous control gene, HPRT, were calculated using the ΔΔCt method (27).
Gene Silencing and Overexpression
A reverse transfection protocol was used for the transfection of OKF6/TERT-2 cells and primary human epidermal keratinocytes. For gene silencing, siRNAs were diluted to 120 nm with 100 μl of Opti-MEM I reduced serum medium (Invitrogen) and then mixed with 100 μl of Opti-MEM medium containing 1 μl of Lipofectamine RNAiMAX transfection reagent, followed by incubation at room temperature for 20 min. For gene overexpression, the expression plasmid (1 μg of total plasmid) was diluted in 100 μl of Opti-MEM medium and then mixed with 100 μl of Opti-MEM medium containing 1 μl of Lipofectamine 2000 transfection reagent, followed by incubation at room temperature for 20 min. For both gene silencing and overexpression, keratinocytes (2–5 × 105 cells in 1 ml of antibiotic-free growth medium) were seeded into 12-well plates, and the transfection mixture was added. For gene knockdown experiments, the medium was replaced after 16 h, and the cells were treated with PMA 48 or 72 h post-transfection. For gene overexpression experiments, the cells were treated with PMA 24 h post-transfection without medium change.
Statistical Analysis
Data combined from three or more independent experiments are given as mean ± S.E. Statistical analyses were performed using GraphPad Prism software version 6.01 (GraphPad Software, La Jolla, CA). Differences between two groups were evaluated using Student's t test. For multiple comparisons, statistical analysis was performed using a one-way analysis of variance and Sidak's or Dunnett's test as a post hoc test. p <0.05 was considered to be statistically significant.
RESULTS
Regulation of IRF6 by RIPK4
To establish whether a functional relationship exists between RIPK4 and IRF6, we first investigated whether the trans-activator activity of IRF6 is regulated by RIPK4. IRF6 trans-activator activity, which was measured using an IFNβ gene promoter reporter plasmid (24), was strongly induced, in a concentration-dependent manner, by wild-type RIPK4 (Fig. 1A) but not by the kinase-dead RIPK4 mutant RIPK4 K51R (Fig. 1B). Consistent with earlier findings (13, 15), RIPK4 also activated NF-κB in a concentration- (Fig. 1C) and kinase-dependent (Fig. 1D) manner.
FIGURE 1.
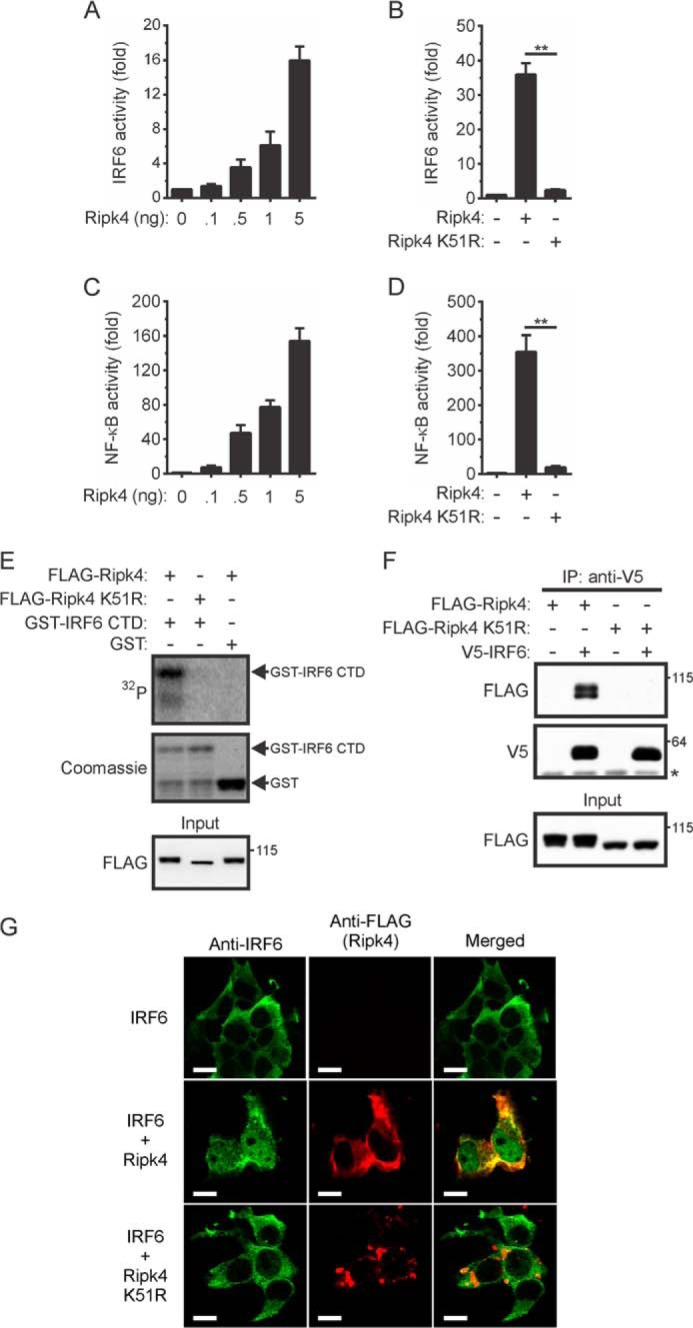
Activation of IRF6 by RIPK4. A–D, HEK293T cells were transfected with an IFNβ (A and B) or ELAM-1 (C and D) gene promoter reporter plasmid together with expression vectors encoding IRF6 (A and B) and the indicated Ripk4 proteins (A–D). Gene reporter activity was measured 24 h post-transfection. IRF6 (A and B) and NF-κB (C and D) activity are shown as fold increases. The data are combined from three independent experiments. **, p < 0.01. E, HEK293T cells transiently expressing FLAG-Ripk4 and FLAG-Ripk4 K51R were lysed 24 h post-transfection. The Ripk4 proteins were immunoprecipitated from the lysates using anti-FLAG antibodies, and their ability to phosphorylate GST-IRF6 CTD was measured. The lysates (input) were subjected to Western blotting with an anti-FLAG antibody. Data are representative of two independent experiments. F, HEK293T cells transiently expressing the indicated proteins were lysed 24 h post-transfection. IRF6 was immunoprecipitated (IP) from the lysates using anti-V5 antibodies, followed by Western blotting with anti-FLAG and anti-V5 antibodies. The asterisk indicates the position of the anti-V5 antibody heavy chain. The lysates (input) were subjected to Western blotting with an anti-FLAG antibody. The positions of molecular weight standards (in kilodaltons) are indicated. The data are representative of three independent experiments. G, HEK293T cells transiently expressing V5-IRF6 alone or together with either FLAG-Ripk4 or FLAG-Ripk4 K51R were stained with anti-IRF6 (green) and anti-FLAG (red) antibodies. Scale bars = 10 μm. Data are representative of four independent experiments.
The phosphorylation of regulatory serine residues in the C-terminal domain (CTD) of IRFs represents a critical step in their inducible activation (28). To determine whether RIPK4 can directly phosphorylate the IRF6 CTD, in vitro kinase assays were performed using a GST-IRF6 CTD fusion protein as substrate. RIPK4 phosphorylated the GST-IRF6 CTD protein but not the GST control (Fig. 1E). Because of the instability of the GST-IRF6 CTD fusion protein, a degradation product was also evident in the Coomassie-stained gel (Fig. 1E). The ability of RIPK4 to interact with IRF6 was also investigated by performing coimmunoprecipitation experiments. IRF6 interacted, either directly or as part of a complex, with wild-type RIPK4. However, no interaction with the RIPK4 K51R mutant was detected (Fig. 1F, top panel). Because of autophosphorylation (12), a doublet was detected when wild-type, but not kinase-dead, RIPK4 was expressed in HEK293T cells (Fig. 1F, bottom panel).
The ability of RIPK4 to regulate IRF6 nuclear translocation was also tested. IRF6 was detected in the cytoplasm of transfected cells. However coexpression of wild-type RIPK4, but not RIPK4 K51R, resulted in the partial nuclear translocation of IRF6 (Fig. 1G). RIPK4 localized exclusively to the cytoplasm, where it exhibited a degree of colocalization with the cytoplasmic pool of IRF6 (Fig. 1G). Collectively, the results in Fig. 1 indicate that RIPK4 can directly activate IRF6 and induce its nuclear translocation.
Characterization of PKC-regulated Oral Keratinocyte Differentiation
PKC is an important regulator of keratinocyte differentiation (29–33), and previous studies have indicated that RIPK4 functions in the PKC pathway (10–14). PMA is a potent PKC activator and a strong inducer of keratinocyte terminal differentiation (34). Therefore, we assessed whether PMA treatment of OKF6/TERT-2 cells (hereafter referred to as OKF6 cells), which have the characteristics of primary human oral keratinocytes (25), would provide a suitable model for oral keratinocyte differentiation. PMA treatment induced marked morphological changes (Fig. 2A) and strongly suppressed cell proliferation (Fig. 2B). This correlated with decreased levels of the proliferation marker proliferating cell nuclear antigen (PCNA) (Fig. 2, C and D). Differentiation was further assessed by measuring the expression levels of the early and late terminal differentiation markers involucrin (IVL) and keratin 13 (KRT13), respectively (35, 36). Both genes were strongly up-regulated by PMA treatment (Fig. 2, E and F). GRHL3, OVOL1, and mitotic arrest-deficient 1 (MAD1), which themselves are important transcriptional regulators of keratinocyte differentiation (37–42), were also up-regulated in PMA-treated cells (Fig. 2, G–I). OVOL1 expression was most rapidly up-regulated (Fig. 2H), whereas the induction of MAD1 gene expression was slowest (Fig. 2I). Collectively, these data demonstrate that PMA treatment of OKF6 cells provides a good model for investigating the regulation of keratinocyte differentiation by RIPK4 and IRF6.
FIGURE 2.
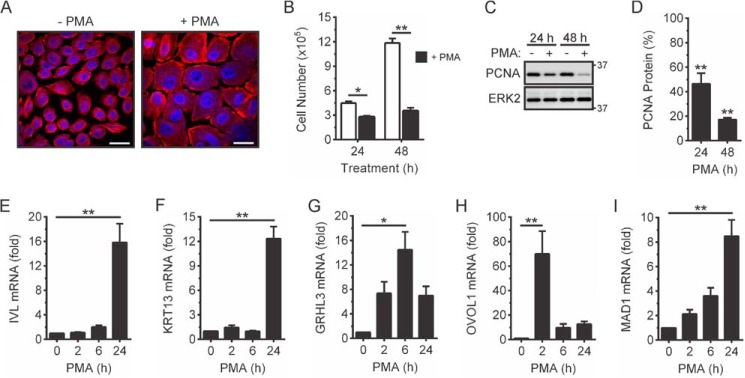
PMA-inducible OKF6 cell differentiation. A–D, OKF6 cells were cultured in the presence of 100 ng/ml PMA or 0.1% DMSO vehicle for up to 48 h. A, the cells were fixed 48 h post-treatment and stained with an anti-β-tubulin antibody (red), and nuclei were stained with DAPI (blue). Scale bars = 20 μm. B, the viable cell number was enumerated by cell counting. *, p < 0.05; **, p < 0.01. C and D, cell lysates were subjected to Western blotting with anti-PCNA and anti-ERK2 (loading control) antibodies (C). The positions of molecular weight standards (in kilodaltons) are indicated. D, PCNA protein levels in DMSO-treated cells in C were given an arbitrary value of 100%. **, p < 0.01. E–I, OKF6 cells were treated with 100 ng/ml PMA or 0.1% DMSO for the indicated times. IVL (E), KRT13 (F), GRHL3 (G), OVOL1 (H), and MAD1 (I) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05; **, p < 0.01. All graphical data are combined from three independent experiments.
IRF6 Is Important for PKC-regulated Keratinocyte Differentiation
To establish whether IRF6 mediates PKC-regulated keratinocyte differentiation, we first investigated the ability of PMA to induce IRF6 nuclear translocation. The staining of OKF6 cells with anti-IRF6 antibodies revealed that IRF6 was present in both the cytoplasm and nucleus of a substantial proportion of cells, whereas, in other cells, it localized exclusively to the cytoplasm (Fig. 3A). PMA did not appear to cause an overall increase in IRF6 nuclear localization (Fig. 3, A and B). However, a significant increase in IRF6 nuclear translocation was observed when the cells were pretreated with the nuclear export inhibitor leptomycin B (LMB) (Fig. 3, A and B). Therefore, PKC activation induces the transient nuclear translocation of IRF6. Gene promoter reporter assays demonstrated that PKC activation also induces IRF6 activity (Fig. 3C).
FIGURE 3.
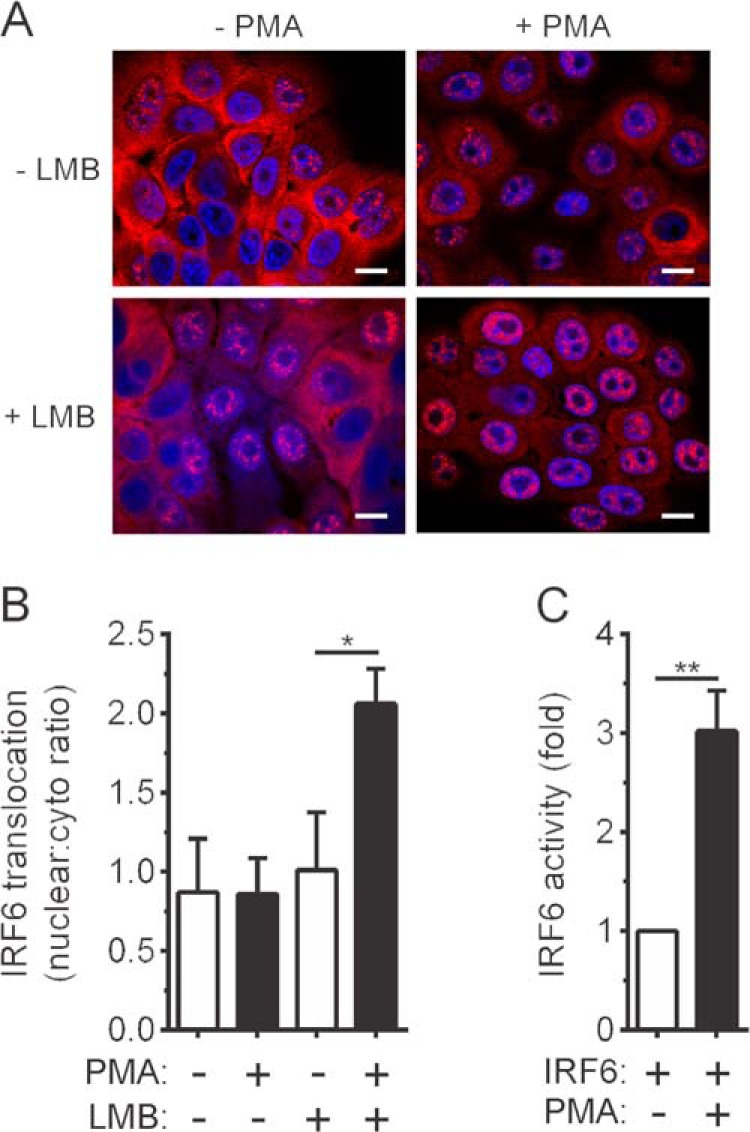
PMA-inducible IRF6 nuclear translocation and activation. A and B, OKF6 cells were pretreated with 20 ng/ml leptomycin B (LMB) for 60 min or left untreated. They were then treated with 100 ng/ml PMA or 0.1% DMSO vehicle for 30 min. A, the cells were stained with an anti-IRF6 antibody (red), and nuclei were stained with DAPI (blue). Scale bars = 10 μm. B, quantified data for IRF6 nuclear translocation in A is presented as the ratio of nuclear to cytosolic IRF6. *, p < 0.05. C, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with an IRF6 expression vector. The cells were treated 24 h post-transfection with PMA or DMSO for 6 h, and gene reporter activity was measured. IRF6 activity is shown as a fold increase. **, p < 0.01. The data in B and C are combined from three and four independent experiments, respectively.
We next investigated the importance of IRF6 for PKC-regulated keratinocyte differentiation. As shown in Fig. 4, the IRF6-targeting siRNA, which substantially reduced IRF6 mRNA (Fig. 4A) and protein levels (data not shown), significantly inhibited the up-regulation of the keratinocyte differentiation markers IVL and KRT13 in PMA-treated OKF6 cells (Fig. 4, B and C). It also reduced the inhibitory effects of PMA on cell proliferation, as measured by cell number (Fig. 4D) and PCNA levels (Fig. 4, E and F). Likewise, IRF6 knockdown inhibited the up-regulation of GRHL3 and OVOL1 expression (Fig. 4, G and H), a finding that is also consistent with these transcriptional regulators of keratinocyte differentiation being IRF6 target genes (7, 8). The up-regulation of MAD1 expression was also inhibited by IRF6 knockdown (Fig. 4I). The kinetics of PMA-induced GRHL3, OVOL1, and MAD1 expression were unaffected (data not shown). In addition to OKF6 cells (oral keratinocytes), the PMA-inducible up-regulation of GRHL3, OVOL1, and IVL expression in epidermal keratinocytes was also IRF6-dependent (Fig. 4, J–L). These data indicate that IRF6 functions in the PKC pathway to regulate keratinocyte differentiation.
FIGURE 4.

Effects of IRF6 knockdown on PMA-inducible keratinocyte differentiation. A–L, OKF6 cells (A–I)and (J–L) human epidermal keratinocytes were transfected with IRF6-targeting (black columns) or control non-targeting (white columns) siRNA. A–I, 48 h post-transfection (A), IRF6 mRNA levels were measured by real-time PCR. IRF6 mRNA levels in cells transfected with the control non-targeting siRNA were given an arbitrary value of 100%. **, p < 0.01. B and C, OKF6 cells were treated with 100 ng/ml PMA or 0.1% DMSO vehicle for 24 h. IVL (B) and KRT13 (C) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05. D–F, OKF6 cells were treated with 100 ng/ml PMA or 0.1% DMSO for 48 h and (D) viable cell number then enumerated by cell counting. *, p < 0.05. E, cells were lysed and subjected to Western blotting with anti-PCNA and anti-ERK2 (loading control) antibodies. F, PCNA protein levels in cells transfected with the control non-targeting siRNA and subsequently treated with DMSO as in E were given an arbitrary value of 100%. *, p < 0.05. G–I, OKF6 cells were treated with 100 ng/ml PMA or 0.1% DMSO for 2 h (OVOL1), 6 h (GRHL3), and 24 h (MAD1). GRHL3 (G), OVOL1 (H), and MAD1 (I) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05. J–L, 72 h post-transfection, epidermal keratinocytes were treated with 100 ng/ml PMA or 0.1% DMSO for 6 h (GRHL3 and OVOL1) and 24 h (IVL). GRHL3 (J), OVOL1 (K), and IVL (L) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05; **, p < 0.01. All graphical data are combined from at least three independent experiments.
RIPK4 Is Important for the Induction of IRF6-dependent Differentiation Genes by PKC
Given the ability of RIPK4 to activate IRF6 (Fig. 1), we investigated the importance of RIPK4 for the PMA-inducible expression of the above IRF6-dependent differentiation genes. RIPK4 knockdown (Fig. 5A) inhibited the up-regulation of the IRF6 target genes GRHL3 and OVOL1 in PMA-treated OKF6 cells (Fig. 5, B and C). It also inhibited the up-regulation of MAD1 expression (Fig. 5D). Consistent with its importance for keratinocyte differentiation (9, 10), RIPK4 knockdown concomitantly inhibited the up-regulation of IVL and KRT13 expression in PMA-treated OKF6 cells (Fig. 5, E and F). PMA-inducible GRHL3, OVOL1, and IVL expression in epidermal keratinocytes were likewise inhibited by RIPK4 knockdown (Fig. 5, G–I).
FIGURE 5.

Effects of RIPK4 knockdown and overexpression on PMA-inducible keratinocyte differentiation. A–I, OKF6 cells (A–F) and human epidermal keratinocytes (G–I) were transfected with RIPK4-targeting (black columns) or control non-targeting (white columns) siRNA. A–F, 48 h post-transfection (A) RIPK4 mRNA levels were measured by real-time PCR. RIPK4 mRNA levels in cells transfected with the control non-targeting siRNA were given an arbitrary value of 100%. **, p < 0.01. B–F, OKF6 cells were treated with 100 ng/ml PMA or 0.1% DMSO vehicle for 2 h (OVOL1), 6 h (GRHL3), and 24 h (MAD1, IVL, and KRT13). GRHL3 (B), OVOL1 (C), MAD1 (D), IVL (E), and KRT13 (F) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05; **, p < 0.01. G–I, 72 h post-transfection, the epidermal keratinocytes were treated with 100 ng/ml PMA or 0.1% DMSO vehicle for 6 h (GRHL3 and OVOL1) and 24 h (IVL). GRHL3 (G), OVOL1 (H), and IVL (I) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05. J–M, OKF6 cells were transfected with a FLAG-Ripk4 expression plasmid (black columns) or empty plasmid (white columns). 24 h post-transfection (J), Ripk4 mRNA levels were measured by real-time PCR and are shown relative to those of HPRT. ND, not detected. Cell lysates were subjected to Western blotting with anti-FLAG (Ripk4) and anti-HSP90 (loading control) antibodies. K–M, the cells were treated with 100 ng/ml PMA or 0.1% DMSO for 2 h (OVOL1), 6 h (GRHL3), and 24 h (IVL). GRHL3 (K), OVOL1 (L), and IVL (M) mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. **, p < 0.01. The data are combined from three independent experiments.
As a complementary approach, the effects of RIPK4 overexpression were also investigated. The anti-RIPK4 antibody failed to detect either transfected or endogenous RIPK4. Therefore, the level of overexpression could not be determined. However, the ectopic expression of RIPK4 in OKF6 cells (Fig. 5J) significantly enhanced the up-regulation of GRHL3, OVOL1, and IVL gene expression by PMA (Fig. 5, K–M). GRHL3, OVOL1, and IVL basal expression were not affected by RIPK4 overexpression (Fig. 5, K–M). Taken together, this indicates that RIPK4 functions upstream of IRF6 in the PKC pathway to promote keratinocyte differentiation. However, we cannot exclude the possibility that RIPK4 may promote keratinocyte differentiation by also activating additional transcriptional regulators (e.g. NF-κB).
A BPS-associated Mutation Inhibits RIPK4-mediated IRF6 Activation
Several mutations in RIPK4 were identified recently in BPS (19, 20), including a missense mutation (p.Ile121Asn) in the kinase domain (20). When expressed in HEK293T cells, a single RIPK4 I121N band was detected by Western blotting (Fig. 6A). This contrasted with wild-type RIPK4 but was similar to the kinase-dead RIPK4 mutant RIPK4 K51R, therefore suggesting that RIPK4 I121N is catalytically inactive. This was confirmed by performing in vitro kinase assays on RIPK4 I121N that had been immunoprecipitated from transfected cells. RIPK4 I121N could not autophosphorylate (Fig. 6B) and, hence, could not phosphorylate the GST-IRF6 CTD fusion protein (Fig. 6C). Gene promoter reporter assays demonstrated that the RIPK4 I121N mutant cannot activate IRF6 (Fig. 6D) or NF-κB (data not shown). RIPK4 I121N was also unable to form a complex with coexpressed IRF6 (Fig. 6E) or induce IRF6 nuclear translocation (Fig. 6F). The p.Ile121Asn mutation, therefore, prevents the activation of IRF6 (and NF-κB) by RIPK4.
FIGURE 6.
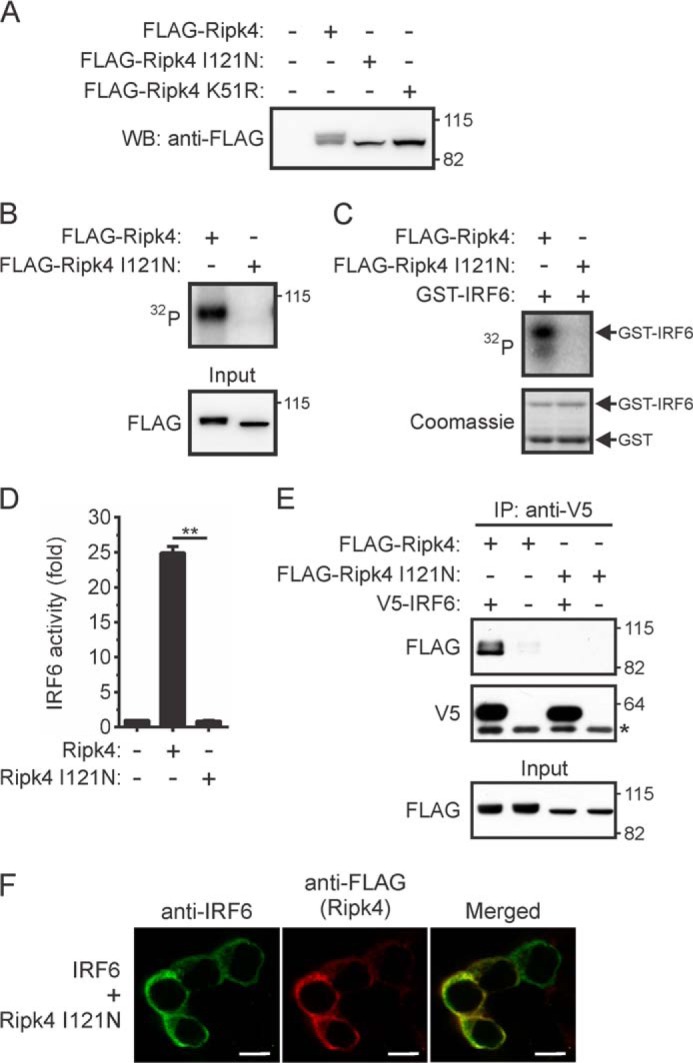
Effects of a BPS-associated mutation on the activation of IRF6 by RIPK4. A–C, HEK293T cells transiently expressing FLAG-Ripk4, FLAG-Ripk4 I121N, or FLAG-Ripk4 K51R were lysed 24 h post-transfection. A, the lysates were subjected to Western blotting (WB) with an anti-FLAG antibody. The positions of molecular weight standards (in kilodaltons) are indicated. B and C, FLAG-Ripk4 and FLAG-Ripk4 I121N were immunoprecipitated from the lysates using anti-FLAG antibodies and their abilities to autophosphorylate (B) and phosphorylate GST-IRF6 CTD (C) were measured. The data are representative of two independent experiments. D, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with expression vectors encoding IRF6 and either Ripk4 or Ripk4 I121N. Gene reporter activity was measured 24 h post-transfection, and IRF6 activity is shown as the fold increase. **, p < 0.01. The data are combined from three independent experiments. E, HEK293T cells transiently expressing the indicated proteins were lysed 24 h post-transfection. IRF6 was immunoprecipitated (IP) from the lysates using anti-V5 antibodies, followed by Western blotting with anti-FLAG and anti-V5 antibodies. The asterisk indicates the position of the anti-V5 antibody heavy chain. The lysates (input) were subjected to Western blotting with an anti-FLAG antibody. The data are representative of three independent experiments. F, HEK293T cells transiently coexpressing IRF6 and Ripk4 I121N were stained with antibodies against IRF6 (green) and anti-FLAG (red) antibodies. Scale bars = 10 μm. The data are representative of four independent experiments.
Ser-413 and Ser-424 in IRF6 Are Important for Its RIPK4-mediated Activation
As mentioned, phosphorylation represents a critical step in IRF activation. We recently identified Ser-413 and Ser-424 (Fig. 7A) as regulatory phosphorylation sites in IRF6 (24). Therefore, the importance of these residues for the activation of IRF6 by RIPK4 was investigated. The mutation of Ser-413 to alanine (i.e. IRF6 S413A) significantly reduced the activation of IRF6 by RIPK4 (Fig. 7B). RIPK4-mediated IRF6 activation was severely inhibited by the mutation of Ser-424 to alanine (i.e. IRF6 S424A), whereas the IRF6 S413A/S424A mutant was inactive in this assay (Fig. 7B). Therefore, Ser-413 and Ser-424 are both critical for the activation of IRF6 by RIPK4. This conclusion is further supported by the findings that combined mutation of Ser-413 and Ser-424 to the phosphomimetic glutamic acid resulted in the IRF6 mutant (i.e. IRF6 S413E/S424E) exhibiting trans-activator activity comparable with that of wild-type IRF6 following its coexpression with RIPK4 and that the IRF6 S413E/S424E trans-activator function was not further potentiated by RIPK4 coexpression (Fig. 7C). RIPK4 did, however, enhance the activities of the IRF6 S413E and IRF6 S424E mutants (Fig. 7C).
FIGURE 7.

Identification of RIPK4-dependent regulatory phosphorylation sites in IRF6. A, schematic of IRF6 in which the positions of Ser-413 and Ser-424 are indicated. DBD, DNA-binding domain; IAD, IRF association domain. B and C, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with expression vectors encoding the indicated proteins. Gene reporter activity was measured 24 h post-transfection, and IRF6 activity is shown as the fold increase. *, p < 0.05; **, p < 0.01. The data are combined from three independent experiments.
RIPK4 Is Also Important for PKC-regulated IRF6 Expression
It has been reported recently that IRF6 can regulate its own transcription (8). In view of the data above, we investigated whether RIPK4 can regulate IRF6 expression. Activation of the PKC pathway by PMA resulted in robust up-regulation of IRF6 expression in OKF6 cells (Fig. 8A) as well as epidermal keratinocytes (Fig. 8B). Notably, RIPK4 knockdown inhibited the up-regulation of IRF6 expression (Fig. 8, A and B). Conversely, RIPK4 overexpression potentiated IRF6 expression in PMA-treated cells (Fig. 8C). IRF6 basal expression was not significantly affected by RIPK4 knockdown (Fig. 8, A and B), or by RIPK4 overexpression (Fig. 8C). Similarly, RIPK4 expression was also unaffected by IRF6 knockdown (data not shown). Collectively, these data indicate that RIPK4 also plays a role in the regulation of IRF6 gene expression by the PKC pathway.
FIGURE 8.

Regulation of PMA-inducible IRF6 expression by RIPK4. A and B, OKF6 cells (A) and human epidermal keratinocytes (B) were transfected with RIPK4-targeting (black columns) or control non-targeting (white columns) siRNA. 48 (A) or 72 h (B) post-transfection, the cells were treated with 100 ng/ml PMA or 0.1% DMSO vehicle for the indicated times. C, OKF6 cells were transfected with a FLAG-Ripk4 expression plasmid (black columns) or empty plasmid (white columns). 24 h post-transfection, the cells were treated with 100 ng/ml PMA or 0.1% DMSO for the indicated times. A–C, IRF6 mRNA levels were measured by real-time PCR and are shown as the fold increase relative to DMSO-treated cells. *, p < 0.05; **, p < 0.01. The data are combined from three independent experiments.
DISCUSSION
RIPK4 and IRF6 are key regulators of keratinocyte differentiation (4–6, 9, 10), and their mutation causes the related congenital syndromes BPS and popliteal pterygium syndrome, respectively (17, 19, 20). These syndromes are characterized by various epidermal abnormalities, including orofacial clefting, skin webbing, syndactyly, and genital deformities. The data presented here suggest that not only do RIPK4 and IRF6 function in the same signaling pathway to regulate keratinocyte differentiation, but RIPK4 also directly regulates IRF6. Our findings may therefore help to explain how mutations in RIPK4 cause epidermal disorders such as BPS.
The molecular pathways in which RIPK4 and IRF6 operate to regulate keratinocyte differentiation are not well understood, although prior studies indicate that RIPK4 functions in the PKC pathway (10–14). PKC activation promotes keratinocyte differentiation by inducing the expression of various differentiation-associated genes, including IVL (29–33). We have shown here that RIPK4 and IRF6 are important for the induction of keratinocyte differentiation by the PKC pathway. Significantly, IRF6 nuclear translocation and trans-activator function were induced downstream of PKC signaling. Similarly, transfection studies demonstrated the regulation of IRF6 nuclear translocation and trans-activator function by RIPK4. Therefore, RIPK4 and IRF6 likely mediate keratinocyte differentiation, at least in part, by operating as a signaling axis downstream of PKC activation. RIPK4 has also been shown recently to regulate Wnt signaling by enhancing the stability of the LEF-1/TCF transcriptional coactivator β-catenin (16). Therefore, the RIPK4-IRF6 axis may also be a feature of other signaling pathways (e.g. Wnt pathway) that contribute to the regulation of epidermal development.
The inhibitory effects of RIPK4 and IRF6 knockdown on keratinocyte differentiation coincided with reduced GRHL3, OVOL1, and MAD1 expression. GRHL3 regulates keratinocyte differentiation and epidermal development through its interaction with the transcription factor LMO4 and by recruiting the Trithorax complex to the promoters of differentiation-associated genes (e.g. transglutaminase 1) (37–39). OVOL1 promotes differentiation by repressing c-Myc transcription (40), whereas MAD1 antagonizes c-Myc transcriptional activity (43). Interestingly, the strength and duration of c-Myc activity appear to influence the balance between keratinocyte proliferation and differentiation (44). Given that GRHL3 and OVOL1 are direct IRF6 target genes (7, 8), RIPK4 likely promotes keratinocyte differentiation, at least in part, by inducing their IRF6-mediated transcription. Whether IRF6 also directly controls MAD1 transcription is not known. However, its delayed induction in response to PMA suggests that IRF6 indirectly regulates MAD1 expression.
PKCδ, which has been shown previously to form a complex with RIPK4 (11), is an important regulator of keratinocyte differentiation (45–47). For example, PKCδ promotes IVL gene expression by up-regulating the expression of the transcription factor Kruppel-like factor 4 (KLF4) (47). Significantly, KLF4 has been found recently to be an IRF6 target gene (7, 8). Therefore, PKCδ may regulate KLF4 and, hence, IVL transcription via RIPK4-mediated IRF6 activation. This would position PKCδ so that it could also control the expression of other transcriptional regulators of keratinocyte differentiation, including GRHL3 and OVOL1.
Our findings may also provide a molecular basis for how mutations in RIPK4 cause BPS (19, 20). As shown here, the p.Ile121Asn mutation inhibits the kinase activity of RIPK4, thereby preventing it from phosphorylating and activating IRF6. BPS is a more severe developmental disorder than those where IRF6 is mutated (e.g. popliteal pterygium syndrome) (17), which is consistent with the positioning of RIPK4 upstream of IRF6. Prior studies have demonstrated an important role for NF-κB in the regulation of keratinocyte differentiation (48–50). In agreement with Kalay et al. (20), we found that the p.Ile121Asn mutation also abolishes RIPK4-mediated NF-κB activation. The mutation also compromises Wnt signaling (16). Therefore, the severe manifestation of the p.Ile121Asn mutation is likely to not only be due to impaired signaling by the RIPK4-IRF6 axis but also to be a consequence of defective signaling by additional developmental pathways, such as the NF-κB and Wnt pathways.
We recently identified Ser-413 and Ser-424 as regulatory phosphorylation sites in IRF6 (24). Our data here suggest that RIPK4 likely activates the IRF6 trans-activator function by phosphorylating both Ser-413 and Ser-424. A sequential, two-step phosphorylation model has been proposed for IRF activation (51). In this model, phosphorylation of residues in “site 2” (e.g. Ser-396 in IRF3) alleviates autoinhibition and facilitates the phosphorylation of residues in “site 1” (e.g. Ser-386 in IRF3), which is required for IRF3 activation and dimerization (51). Ser-413 and Ser-424 correspond, respectively, to Ser-386 and Ser-396 in IRF3. On the basis of the activation model for IRF3 and our mutagenesis data for IRF6, Ser-424 likely functions as a gatekeeper phosphorylation site controlling the subsequent phosphorylation of Ser-413, which is required for full IRF6 activation by RIPK4.
Significantly, Ser-424 is mutated in popliteal pterygium syndrome (52) as well as squamous cell carcinoma (23). The latter study also identified several mutations in RIPK4. The p.Asp205Val mutation, for example, causes replacement of an invariant aspartic acid residue in the kinase domain of RIPK4 (53), which might be expected to compromise RIPK4 activity, and hence IRF6 activation. IRF6 expression levels have also been reported to inversely correlate with tumor invasiveness in squamous cell carcinoma (8, 54). Therefore, impaired signaling by the RIPK4-IRF6 axis or loss of IRF6 per se may be important in squamous cell carcinoma.
RIPK4 is also shown here to be important for the regulation of IRF6 expression by the PKC pathway. IRF6 gene expression has been shown previously to be regulated by the binding of the p63 transcription factor to an enhancer element located 9.7 kb upstream of the IRF6 transcription start site (55, 56). However, IRF6-binding sites in the IRF6 gene have been identified, leading to the suggestion that IRF6 can also mediate its own expression (8). Its activation of IRF6 may therefore allow RIPK4 to further promote keratinocyte differentiation by also up-regulating IRF6 expression.
In conclusion, we have identified and mechanistically defined a previously unrecognized regulatory relationship between RIPK4 and IRF6 in the control of keratinocyte differentiation. This could help to explain how mutations in RIPK4 cause BPS. These findings may also be relevant to the development of squamous cell carcinoma.
This work was supported by National Health and Medical Research Council Project Grant 628769 and the Oral Health CRC.
- BPS
- Bartsocas-Papas syndrome
- PMA
- phorbol 12-myristate 13-acetate
- CTD
- C-terminal domain
- PCNA
- proliferating cell nuclear antigen
- IVL
- involucrin
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Madison K. C. (2003) Barrier function of the skin: “la raison d'etre” of the epidermis. J. Invest. Dermatol. 121, 231–241 [DOI] [PubMed] [Google Scholar]
- 2. Blanpain C., Fuchs E. (2009) Epidermal homeostasis: a balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 10, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ratushny V., Gober M. D., Hick R., Ridky T. W., Seykora J. T. (2012) From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest. 122, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ingraham C. R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K. J., Malik M. I., Dunnwald M., Goudy S. L., Lovett M., Murray J. C., Schutte B. C. (2006) Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richardson R. J., Dixon J., Malhotra S., Hardman M. J., Knowles L., Boot-Handford R. P., Shore P., Whitmarsh A., Dixon M. J. (2006) Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 38, 1329–1334 [DOI] [PubMed] [Google Scholar]
- 6. Biggs L. C., Rhea L., Schutte B. C., Dunnwald M. (2012) Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J. Invest. Dermatol. 132, 50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de la Garza G., Schleiffarth J. R., Dunnwald M., Mankad A., Weirather J. L., Bonde G., Butcher S., Mansour T. A., Kousa Y. A., Fukazawa C. F., Houston D. W., Manak J. R., Schutte B. C., Wagner D. S., Cornell R. A. (2013) Interferon regulatory factor 6 promotes differentiation of the periderm by activating expression of Grainyhead-like 3. J. Invest. Dermatol. 133, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Botti E., Spallone G., Moretti F., Marinari B., Pinetti V., Galanti S., De Meo P. D., De Nicola F., Ganci F., Castrignanò T., Pesole G., Chimenti S., Guerrini L., Fanciulli M., Blandino G., Karin M., Costanzo A. (2011) Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl. Acad. Sci. U.S.A. 108, 13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holland P., Willis C., Kanaly S., Glaccum M., Warren A., Charrier K., Murison J., Derry J., Virca G., Bird T., Peschon J. (2002) RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr. Biol. 12, 1424–1428 [DOI] [PubMed] [Google Scholar]
- 10. Rountree R. B., Willis C. R., Dinh H., Blumberg H., Bailey K., Dean C., Jr., Peschon J. J., Holland P. M. (2010) RIP4 regulates epidermal differentiation and cutaneous inflammation. J. Invest. Dermatol. 130, 102–112 [DOI] [PubMed] [Google Scholar]
- 11. Bahr C., Rohwer A., Stempka L., Rincke G., Marks F., Gschwendt M. (2000) DIK, a novel protein kinase that interacts with protein kinase Cδ. Cloning, characterization, and gene analysis. J. Biol. Chem. 275, 36350–36357 [DOI] [PubMed] [Google Scholar]
- 12. Chen L., Haider K., Ponda M., Cariappa A., Rowitch D., Pillai S. (2001) Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem. 276, 21737–21744 [DOI] [PubMed] [Google Scholar]
- 13. Muto A., Ruland J., McAllister-Lucas L. M., Lucas P. C., Yamaoka S., Chen F. F., Lin A., Mak T. W., Núñez G., Inohara N. (2002) Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-κ B activation induced by phorbol ester. J. Biol. Chem. 277, 31871–31876 [DOI] [PubMed] [Google Scholar]
- 14. Moran S. T., Haider K., Ow Y., Milton P., Chen L., Pillai S. (2003) Protein kinase C-associated kinase can activate NFκB in both a kinase-dependent and a kinase-independent manner. J. Biol. Chem. 278, 21526–21533 [DOI] [PubMed] [Google Scholar]
- 15. Meylan E., Martinon F., Thome M., Gschwendt M., Tschopp J. (2002) RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-κ B and is processed during apoptosis. EMBO Rep. 3, 1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang X., McGann J. C., Liu B. Y., Hannoush R. N., Lill J. R., Pham V., Newton K., Kakunda M., Liu J., Yu C., Hymowitz S. G., Hongo J. A., Wynshaw-Boris A., Polakis P., Harland R. M., Dixit V. M. (2013) Phosphorylation of Dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science 339, 1441–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kondo S., Schutte B. C., Richardson R. J., Bjork B. C., Knight A. S., Watanabe Y., Howard E., de Lima R. L., Daack-Hirsch S., Sander A., McDonald-McGinn D. M., Zackai E. H., Lammer E. J., Aylsworth A. S., Ardinger H. H., Lidral A. C., Pober B. R., Moreno L., Arcos-Burgos M., Valencia C., Houdayer C., Bahuau M., Moretti-Ferreira D., Richieri-Costa A., Dixon M. J., Murray J. C. (2002) Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorlin R. J., Sedano H. O., Cervenka J. (1968) Popliteal pterygium syndrome: a syndrome comprising cleft lip-palate, popliteal and intercrural pterygia, digital and genital anomalies. Pediatrics 41, 503–509 [PubMed] [Google Scholar]
- 19. Mitchell K., O'Sullivan J., Missero C., Blair E., Richardson R., Anderson B., Antonini D., Murray J. C., Shanske A. L., Schutte B. C., Romano R. A., Sinha S., Bhaskar S. S., Black G. C., Dixon J., Dixon M. J. (2012) Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am. J. Hum. Genet. 90, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalay E., Sezgin O., Chellappa V., Mutlu M., Morsy H., Kayserili H., Kreiger E., Cansu A., Toraman B., Abdalla E. M., Aslan Y., Pillai S., Akarsu N. A. (2012) Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am. J. Hum. Genet. 90, 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bartsocas C. S., Papas C. V. (1972) Popliteal pterygium syndrome: evidence for a severe autosomal recessive form. J. Med. Genet. 9, 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hennekam R. C., Huber J., Variend D. (1994) Bartsocas-Papas syndrome with internal anomalies: evidence for a more generalized epithelial defect or new syndrome? Am. J. Med. Genet. 53, 102–107 [DOI] [PubMed] [Google Scholar]
- 23. Stransky N., Egloff A. M., Tward A. D., Kostic A. D., Cibulskis K., Sivachenko A., Kryukov G. V., Lawrence M. S., Sougnez C., McKenna A., Shefler E., Ramos A. H., Stojanov P., Carter S. L., Voet D., Cortés M. L., Auclair D., Berger M. F., Saksena G., Guiducci C., Onofrio R. C., Parkin M., Romkes M., Weissfeld J. L., Seethala R. R., Wang L., Rangel-Escareño C., Fernandez-Lopez J. C., Hidalgo-Miranda A., Melendez-Zajgla J., Winckler W., Ardlie K., Gabriel S. B., Meyerson M., Lander E. S., Getz G., Golub T. R., Garraway L. A., Grandis J. R. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwa M. Q., Nguyen T., Huynh J., Ramnath D., De Nardo D., Lam P. Y., Reynolds E. C., Hamilton J. A., Sweet M. J., Scholz G. M. (2014) Interferon regulatory factor 6 differentially regulates Toll-like receptor 2-dependent chemokine gene expression in epithelial cells. J. Biol. Chem. 289, 19758–19768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dickson M. A., Hahn W. C., Ino Y., Ronfard V., Wu J. Y., Weinberg R. A., Louis D. N., Li F. P., Rheinwald J. G. (2000) Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 20, 1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schindler U., Baichwal V. R. (1994) Three NF-κ B binding sites in the human E-selectin gene required for maximal tumor necrosis factor α-induced expression. Mol. Cell Biol. 14, 5820–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tamura T., Yanai H., Savitsky D., Taniguchi T. (2008) The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 [DOI] [PubMed] [Google Scholar]
- 29. Denning M. F. (2004) Epidermal keratinocytes: regulation of multiple cell phenotypes by multiple protein kinase C isoforms. Int. J. Biochem. Cell Biol. 36, 1141–1146 [DOI] [PubMed] [Google Scholar]
- 30. Matsui M. S., Chew S. L., DeLeo V. A. (1992) Protein kinase C in normal human epidermal keratinocytes during proliferation and calcium-induced differentiation. J. Invest. Dermatol. 99, 565–571 [DOI] [PubMed] [Google Scholar]
- 31. Długosz A. A., Yuspa S. H. (1994) Protein kinase C regulates keratinocyte transglutaminase (TGK) gene expression in cultured primary mouse epidermal keratinocytes induced to terminally differentiate by calcium. J. Invest. Dermatol. 102, 409–414 [DOI] [PubMed] [Google Scholar]
- 32. Eckert R. L., Crish J. F., Efimova T., Dashti S. R., Deucher A., Bone F., Adhikary G., Huang G., Gopalakrishnan R., Balasubramanian S. (2004) Regulation of involucrin gene expression. J. Invest. Dermatol. 123, 13–22 [DOI] [PubMed] [Google Scholar]
- 33. Chew Y. C., Adhikary G., Wilson G. M., Reece E. A., Eckert R. L. (2011) Protein kinase C (PKC) δ suppresses keratinocyte proliferation by increasing p21(Cip1) level by a KLF4 transcription factor-dependent mechanism. J. Biol. Chem. 286, 28772–28782 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Watt F. M. (1989) Terminal differentiation of epidermal keratinocytes. Curr. Opin. Cell Biol. 1, 1107–1115 [DOI] [PubMed] [Google Scholar]
- 35. Watt F. M. (1983) Involucrin and other markers of keratinocyte terminal differentiation. J. Invest. Dermatol. 81, 100s-103s [DOI] [PubMed] [Google Scholar]
- 36. Fuchs E. (1988) Keratins as biochemical markers of epithelial differentiation. Trends Genet. 4, 277–281 [DOI] [PubMed] [Google Scholar]
- 37. Ting S. B., Caddy J., Hislop N., Wilanowski T., Auden A., Zhao L. L., Ellis S., Kaur P., Uchida Y., Holleran W. M., Elias P. M., Cunningham J. M., Jane S. M. (2005) A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science 308, 411–413 [DOI] [PubMed] [Google Scholar]
- 38. Yu Z., Lin K. K., Bhandari A., Spencer J. A., Xu X., Wang N., Lu Z., Gill G. N., Roop D. R., Wertz P., Andersen B. (2006) The Grainyhead-like epithelial transactivator Get-1/Grhl3 regulates epidermal terminal differentiation and interacts functionally with LMO4. Dev. Biol. 299, 122–136 [DOI] [PubMed] [Google Scholar]
- 39. Hopkin A. S., Gordon W., Klein R. H., Espitia F., Daily K., Zeller M., Baldi P., Andersen B. (2012) GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet. 8, e1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nair M., Teng A., Bilanchone V., Agrawal A., Li B., Dai X. (2006) Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J. Cell Biol. 173, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teng A., Nair M., Wells J., Segre J. A., Dai X. (2007) Strain-dependent perinatal lethality of Ovol1-deficient mice and identification of Ovol2 as a downstream target of Ovol1 in skin epidermis. Biochim. Biophys. Acta 1772, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Werner S., Beer H. D., Mauch C., Lüscher B., Werner S. (2001) The Mad1 transcription factor is a novel target of activin and TGF-β action in keratinocytes: possible role of Mad1 in wound repair and psoriasis. Oncogene 20, 7494–7504 [DOI] [PubMed] [Google Scholar]
- 43. Lüscher B. (2012) MAD1 and its life as a MYC antagonist: an update. Eur J. Cell Biol. 91, 506–514 [DOI] [PubMed] [Google Scholar]
- 44. Watt F. M., Frye M., Benitah S. A. (2008) MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat. Rev. Cancer 8, 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deucher A., Efimova T., Eckert R. L. (2002) Calcium-dependent involucrin expression is inversely regulated by protein kinase C (PKC)α and PKCδ. J. Biol. Chem. 277, 17032–17040 [DOI] [PubMed] [Google Scholar]
- 46. Adhikary G., Chew Y. C., Reece E. A., Eckert R. L. (2010) PKC-δ and -η, MEKK-1, MEK-6, MEK-3, and p38-δ are essential mediators of the response of normal human epidermal keratinocytes to differentiating agents. J. Invest. Dermatol. 130, 2017–2030 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 47. Chew Y. C., Adhikary G., Xu W., Wilson G. M., Eckert R. L. (2013) Protein kinase C δ increases Kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J. Biol. Chem. 288, 17759–17768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bushdid P. B., Brantley D. M., Yull F. E., Blaeuer G. L., Hoffman L. H., Niswander L., Kerr L. D. (1998) Inhibition of NF-κB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature 392, 615–618 [DOI] [PubMed] [Google Scholar]
- 49. Kanegae Y., Tavares A. T., Izpisúa Belmonte J. C., Verma I. M. (1998) Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Nature 392, 611–614 [DOI] [PubMed] [Google Scholar]
- 50. Gugasyan R., Voss A., Varigos G., Thomas T., Grumont R. J., Kaur P., Grigoriadis G., Gerondakis S. (2004) The transcription factors c-rel and RelA control epidermal development and homeostasis in embryonic and adult skin via distinct mechanisms. Mol. Cell Biol. 24, 5733–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Panne D., McWhirter S. M., Maniatis T., Harrison S. C. (2007) Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282, 22816–22822 [DOI] [PubMed] [Google Scholar]
- 52. Matsuzawa N., Kondo S., Shimozato K., Nagao T., Nakano M., Tsuda M., Hirano A., Niikawa N., Yoshiura K. (2010) Two missense mutations of the IRF6 gene in two Japanese families with popliteal pterygium syndrome. Am. J. Med. Genet. A 152A, 2262–2267 [DOI] [PubMed] [Google Scholar]
- 53. Meylan E., Tschopp J. (2005) The RIP kinases: crucial integrators of cellular stress. Trends Biochem. Sci. 30, 151–159 [DOI] [PubMed] [Google Scholar]
- 54. Restivo G., Nguyen B. C., Dziunycz P., Ristorcelli E., Ryan R. J., Özuysal Ö. Y., Di Piazza M., Radtke F., Dixon M. J., Hofbauer G. F., Lefort K., Dotto G. P. (2011) IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 30, 4571–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thomason H. A., Zhou H., Kouwenhoven E. N., Dotto G. P., Restivo G., Nguyen B. C., Little H., Dixon M. J., van Bokhoven H., Dixon J. (2010) Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120, 1561–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rahimov F., Marazita M. L., Visel A., Cooper M. E., Hitchler M. J., Rubini M., Domann F. E., Govil M., Christensen K., Bille C., Melbye M., Jugessur A., Lie R. T., Wilcox A. J., Fitzpatrick D. R., Green E. D., Mossey P. A., Little J., Steegers-Theunissen R. P., Pennacchio L. A., Schutte B. C., Murray J. C. (2008) Disruption of an AP-2α binding site in an IRF6 enhancer is associated with cleft lip. Nat. Genet. 40, 1341–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]