

Background: NccX is the membrane-anchored periplasmic metal sensor of the NccYXH transmembrane signal transduction complex.
Results: Detergent-induced redistribution of the hydrophobic interactions led to a non-native x-ray structure.
Conclusion: Molecular dynamics simulations along with in vivo assays reconciled the structural data with a physiological model of NccX dimer.
Significance: Complementary investigations are needed to rationalize three-dimensional structures obtained in non-native conditions.
Keywords: Bacterial Signal Transduction, Computational Biology, Membrane Protein, Metal Ion-Protein Interaction, Molecular Dynamics, Transmembrane Domain, X-ray Crystallography, NccX, Cupriavidus metallidurans, Dodecylphosphocholine
Abstract
The x-ray structure of NccX, a type II transmembrane metal sensor, from Cupriavidus metallidurans 31A has been determined at a resolution of 3.12 Å. This was achieved after solubilization by dodecylphosphocholine and purification in the presence of the detergent. NccX crystal structure did not match the model based on the extensively characterized periplasmic domain of its closest homologue CnrX. Instead, the periplasmic domains of NccX appeared collapsed against the hydrophobic transmembrane segments, leading to an aberrant topology incompatible with membrane insertion. This was explained by a detergent-induced redistribution of the hydrophobic interactions among the transmembrane helices and a pair of hydrophobic patches keeping the periplasmic domains together in the native dimer. Molecular dynamics simulations performed with the full-length protein or with the transmembrane segments were used along with in vivo homodimerization assays (TOXCAT) to evaluate the determinants of the interactions between NccX protomers. Taken as a whole, computational and experimental results are in agreement with the structural model of CnrX where a cradle-shaped periplasmic metal sensor domain is anchored into the inner membrane by two N-terminal helices. In addition, they show that the main determinant of NccX dimerization is the periplasmic soluble domain and that the interaction between transmembrane segments is highly dynamic. The present work introduces a new crystal structure for a transmembrane protein and, in line with previous studies, substantiates the use of complementary theoretical and in vivo investigations to rationalize a three-dimensional structure obtained in non-native conditions.
Introduction
Cupriavidus metallidurans is a Gram-negative bacillus best known for its outstanding ability to grow in the millimolar concentration range of many toxic heavy metals (1–4). To respond to environmental stress due to excess of these cations, C. metallidurans must sense extracellular changes and pass this information to the cytoplasm where transcription factors will be specifically activated to set up the appropriate efflux systems. The surplus of metal ions in the periplasm is commonly pumped back to the outside by redundant resistance, nodulation, and cell division heavy metal efflux pumps (3). For instance, pumping out the surplus of cobalt and nickel cations essentially involves the resistance, nodulation, and cell division efflux pumps CnrCBA in C. metallidurans CH34 or NccCBA in C. metallidurans 31A (3, 5). The transcription of the genes cnrCBA or nccCBA encoding the efflux pumps is under the control of the products of three additional genes, cnrYXH or nccYXH, also organized in operons (3, 6, 7). Although the Cnr and Ncc systems parallel each other in many respects, Cnr from C. metallidurans CH34 has emerged as the experimental model for their characterization (6–10). Metal sensing relies on CnrX, a dimeric type II transmembrane (TM)3 protein that displays a 13-kDa-large C-terminal metal sensor domain in the periplasm (9–12). Upon binding of nickel or cobalt to the sensor domain of CnrX, CnrY-mediated inhibition of the extracytoplasmic function σ factor CnrH (13) is relieved, and CnrH enables the RNA polymerase to transcribe cnrCBA resistance genes (6, 7).
To characterize the structural basis of metal sensing by CnrX, we have previously produced a soluble form of the periplasmic domain spanning residues 31–148. This soluble metal sensor domain is referred to as CnrXs in the following. The high resolution structures of CnrXs under the nickel-, cobalt-, and zinc-bound forms as well as in the apo form (10, 12) have shown that CnrXs is a cradle-shaped dimer that contains one physiologically relevant metal-binding site per monomer. They also pointed out the crucial role of the only methionine (Met123) (10, 12) in metal selectivity and affinity. Met123 is a key player of the allosteric switch corresponding to the first step of signal transduction (10, 12). Our understanding of subsequent signal propagation is limited by the lack of information on the TM domain of the metal sensor and on its relation with the bitopic protein CnrY that couples metal binding to CnrX in the periplasm to CnrH release in the cytoplasm. However, it has not been possible to purify CnrX as yet despite very good overproduction (10). In contrast with CnrX, we have successfully solubilized and purified NccX (10). NccX displays 76% overall sequence identity with CnrX with 80% identity between the soluble domains and 63% sequence identity between the TM anchors (see Fig. 1). A detailed analysis of the spectroscopic data obtained with the cobalt-bound forms of CnrXs or NccX led to the conclusion that CnrXs is a relevant model of the NccX metal sensor domain (10, 11). We have thus used NccX for a structural investigation of the full-length sensor protein of the CnrYXH/NccYXH complex. In the native state, CnrX/NccX was hypothesized to be flexibly anchored to the inner membrane with the two TM segments running perpendicular with respect to the periplasmic domain (see Fig. 2A). Here, we present the x-ray structure at a resolution of 3.12 Å of NccX purified and crystallized in the presence of foscholine-12 (dodecylphosphocholine (DPC) in the following). The structure of NccX differs markedly from the prediction. The structural data were reconciled with a physiological model of NccX dimer using molecular dynamics (MD) simulations and in vivo dimerization (TOXCAT) assays (14) as complementary approaches. It appeared that the detergent not only solubilized the membrane bilayer but also destabilized the hydrophobic dimer interface, thus leading to the redistribution of the hydrophobic interactions that maintained the native structure of NccX. The two main conclusions of this work are that first we present one of the rare complete structures of a bitopic, type II membrane protein and second we illustrate the need to use complementary approaches to extract meaningful biological information from a potentially artifactual crystal structure.
FIGURE 1.

CnrX and NccX sequence alignment and topology prediction generated by the OCTOPUS program. The differences are highlighted in gray. The sequence of NccX-TMsim is underlined and includes the transmembrane segment shown in bold. The letter code is as follows: i, inside (cytoplasm); M, membrane; o, outside (periplasm).
FIGURE 2.

X-ray structure of DPC-solubilized full-length NccX dimer. A, putative model of full-length NccX inserted in the inner membrane adapted from the model of CnrX (16). The two TM segments are supposed to anchor the periplasmic sensor domain. In the CnrXs construct that starts with Ser31, Gly39 was the first N-terminal (N-ter) residue to be structurally defined. B, x-ray structure of NccX determined in this study (Protein Data Bank code 4clv). Protomer A is colored sandy brown, and protomer B is colored steel blue. The side chains of some amino acids stressed in the text are shown as sticks. These amino acids are identified by their one-letter code, numbering in the sequence, and a lowercase letter corresponding to the protomer they belong to. The elements of secondary structure mentioned in the text are annotated (e.g. H2-H3 hairpin) and the location of the zinc ions is shown. The red portion in each H1 helix corresponds to the GXXXG motif. The orange portion corresponds to a stretch of about 10 residues (amino acids 31–40) that were present in the sequence of CnrXs (A) but not structurally defined (16, 19). This stretch includes Pro37 depicted as sticks and highlighted in yellow. Ten zinc ions were located in the asymmetric unit (gray spheres). Most of them have an incomplete coordination sphere at this resolution. The four zinc ions in the dotted square are those of the metal-binding sites detailed in C and D. The ligands of two zinc ions involved in interprotomer interactions are developed as sticks: these are H1 residues Glu36 and His32 contributing one oxygen and one nitrogen, respectively, and Glu106 from H3 of the opposite protomer that contributes one oxygen. C, superimposition of NccX and ZnA-CnrXs protomers. NccX is colored sandy brown, and ZnA-CnrXs is red. The portion of H1 in NccX colored yellow corresponds to the kink upstream at Pro37 that makes a break in the helix as mentioned in B. This kink marks the putative junction between the sensor domain and the TM segment (see “Discussion”). Note that the C-terminal (C-ter) helix H4 of NccX sticks out of the helix bundle. D, close-up view of the superimposition of the metal-binding sites (color code for the zinc ions as for proteins in C). Phe66 is not a metal ligand, but it interacts by π-π stacking with His119, thus providing an interaction between H2 and H3.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Full-length NccX
These steps were already described (10). Briefly, NccX was overproduced in Escherichia coli cells, and the membrane fraction was recovered by centrifugation. For solubilization of NccX, the membrane preparation was 10-fold diluted in a buffer composed of 50 mm Hepes, pH 7.5, 500 mm NaCl, 10 mm EDTA, 0.1% (w/v) (critical micellar concentration, 2) DPC (Anagrade from Anatrace) prior to Dounce homogenization and passage through a microfluidizer (M110-P, Microfluidics). After an incubation of 3 h on ice, the solubilized material was purified in two chromatographic steps consisting of a gel filtration on Superdex-200 (GE Healthcare) followed by an anion exchange on a Q Hi-Trap column (GE Healthcare). The fractions considered electrophoretically pure on SDS-PAGE were pooled, and NccX was concentrated at 13 mg/ml in the following buffer: 50 mm Hepes, pH 7.5, 100 mm NaCl, 0.1% (w/v) DPC. After concentration, the protein was aliquoted and stored at −80 °C for further use. Protein concentrations were determined using the Bradford protein assay with bovine serum albumin as standard (15).
Crystallization, Data Collection, Structure Determination, and Refinement
Crystals were obtained by mixing 1 μl of NccX at 13 mg/ml with 1 μl of reservoir solution consisting of 14% PEG 4000, 0.1 m sodium acetate, pH 4.5, 0.1 m zinc acetate. Crystals grew after 2 months at 20 °C. Prior to data collection, crystals were cryocooled using Paratone-N as cryoprotectant.
Diffraction data were collected to a resolution of 3.12 Å (Table 1) on the Proxima-1 beamline at SOLEIL (Saint Aubin, France) taking advantage of the Pilatus-6M detector. The beam energy was set to the zinc K-absorption edge. Data integration and scaling were done using the XDS package (16). The structure of NccX was determined by the single wavelength anomalous diffraction method. The SHELXD program (17) was used to locate the zinc substructure. Initial phases were obtained with PHASER (18) and improved with PARROT (19) by density modification and by taking advantage of the 2-fold non-crystallographic symmetry.
TABLE 1.
Data statistics of the NccX structure determination
FOM, figure of merit; r.m.s.d., root mean square deviation.
| Data collection | |
| Beamline | SOLEIL (Proxima-1) |
| Space group | P43212 |
| Cell (Å) | a = 53.8; c = 300.0 |
| λ (Å) | 1.28202 |
| ϕtotal (Δϕ) (°) | 360 (0.2) |
| Resolution (Å)a | 40-3.12 (3.20-3.12) |
| Total reflections | 203,269 (14,974) |
| Unique reflections | 14,994 (1,114) |
| Completeness | 100.0 (100.0) |
| Rmeas (%) | 6.6 (476.7) |
| I/σ(I) | 18.3 (0.62) |
| CC (1/2)b | 0.999 (0.721) |
| Wilson B (Å2) | 132.1 |
| Phasing | |
| Anom. corr. (%)c | 74 (3.8) |
| FOM | 0.36 |
| Refinement statistics | |
| Non-hydrogen protein atoms | 2,290 |
| Resolution (Å)a | 25-3.12 (3.49-3.12) |
| Reflections (total) | 8,604 |
| Reflections (working set) | 8,174 |
| Reflections (free set, 5%) | 430 |
| Rwork/Rfree (%) | 25.1/26/4 (28.2/29.5) |
| B (Å2) | 164.5 |
| Model geometry | |
| r.m.s.d. | |
| Bonds (Å) | 0.009 |
| Angle (°) | 1.04 |
| Clash scored | 2.41 |
| No. of Ramachandran outlier (%)e | 2 (0.7) |
| Molprobity scored | 1.68 |
a Values in parentheses refer to the outer resolution shell.
b See Ref. 23.
c Mean correlation factor between two randomly chosen subsets of anomalous intensity differences. In parentheses, resolution in Å at which anomalous correlation drops below 30%.
d According to Ref. 24.
e Percentage with respect to the total number of residues in the NccX model is given in parentheses.
Using the phased translation function of the MOLREP program (20) and using one chain of the homologous structure CnrXs (Protein Data Bank code 2y3d) (10) as template, two molecular replacement solutions were obtained. From that initial model and using single wavelength anomalous diffraction experimental electron density maps improved by density modification, iterative cycles of manual model building with Coot and refinement with autoBUSTER-TNT (21, 22) were carried out. Temperature factors were refined with the translation-libration-screw approach with a single translation-libration-screw group for the non-crystallographic symmetry dimer. Zinc atom positions were cross-checked by an anomalous Fourier synthesis using the final model. Coordinates and structure factors for the structure of NccX have been deposited in the Protein Data Bank with accession number 4clv. Structure figures were generated with Chimera (58).
Model Building and Simulation Parameters for NccX
Two models of NccX were constructed. The first one is based on the structure of DPC-solubilized NccX (this work) and was referred to as NccXDPC. The x-ray structure of NccX was oriented with VMD (25) so that its principal axes are superimposed with the coordinate system. All missing hydrogens were built with CHARMM and the CHARMM19 force field (26, 27). The structure was energy-minimized down to a gradient of 0.1 kcal/mol/Å subject to harmonic restraints of 5 kcal/mol/Å on heavy atoms. Simulations from NccXDPC include residues Thr6–Leu143 for protomer A and Thr6–Lys144 for protomer B. The second model, called NccXPred, was conceived to represent the structure predicted for NccX/CnrX proteins, based on the characterization of the periplasmic domain of CnrX, and used for control experiments. We started from the x-ray structure of the soluble sensor domain of CnrX in the nickel-bound form (Protein Data Bank code 2y39) defined from Gly39–Gln148 (10). The CnrXs dimer was built using the symmetry operations given in the Protein Data Bank file. Thanks to the high sequence similarity between NccX and CnrX (see Fig. 1), we could build a model of the soluble domain of NccX by mapping the residues between the two proteins one by one and determining the missing coordinates for the soluble part of NccX (Thr39–Gln148) from internal coordinates with CHARMM. In Protein Data Bank code 2y39, the coordinates of the N-terminal residues of CnrX from Met1 to His38 are absent (see Fig. 2A). To obtain NccXPred, these N-terminal missing residues, including the membrane-anchoring sequence, were initially built from internal coordinates only. To have a valid structure for the amino acid residues tethering the TM segment to the soluble part of the protein, we fitted the backbone atom coordinates of Pro37–Leu41 in the structure of NccXDPC to those calculated for NccXPred. Then after applying the same coordinate transformation to all atoms of NccXDPC from Thr6 to Glu36, we inserted these coordinates in the initial structure of NccXPred. Missing residues Met1–Arg5 and all missing hydrogens were built with CHARMM so that simulations from NccXPred included the full-length sequence for both protomers, i.e. residues Met1–Gln148. The structure was energy-minimized as described for NccXDPC. Both all-atom systems, NccXDPC and NccXPred were coarse grained (CG) (i.e. each amino acid was simplified to a couple of interacting spheres) with a script developed for GROMACS (28) before running simulations.
The CG NccXDPC protein contained 296 particles for chain A and 299 particles for chain B, whereas the CG NccXPred protein contained 638 particles. The structures were embedded either in CG 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) or in CG DPC and solvated with CG water molecules. The protein bears a total charge of −13 so that 13 Na+ ions were added to the system to ensure electric neutrality. CG-MD simulations were performed using the GROMACS software package (version 4.5.4) (29, 30). The proteins, lipids, ions, and water molecules were described with the MARTINI force field version 2.1 (31, 32). The system was simulated with periodic boundary conditions using a time step of 20 fs. During the simulations, the pressure was maintained at 1 atm using a Berendsen barostat with a coupling constant of 2 ps and a compressibility of 3 × 10−4 bar−1, and the temperature was maintained at 323.15 K with a Berendsen thermostat using a coupling constant of 1 ps. The pressure coupling was either isotropic, leaving all dimensions of the simulation box free to vary, or semi-isotropic in which case only the z dimension of the box could vary.
During the coarse graining process, helical structures are maintained as restraints in the GROMACS MD runs so that varying this number of restraints may have an incidence on the structure finally reached. As the helix H1 in NccXDPC propagated through the flexible linker in between the TM and periplasmic domains, the secondary structure status of residues 32–44 in the model was relaxed to allow flexibility to this sequence. Conditions for the simulations are given in the legend of the corresponding figures.
All atom coordinates corresponding to the last frame of the simulations using NccXDPC in DPC or POPC were converted to united atom resolution using the tool “backward” (33) after adding map files for DPC. The back-mapping was performed using a wrapper that relaxes the resulting structure through energy minimization and short MD-based relaxation. The size of the united atom systems increased by a factor varying from 8 to 12 with respect to the CG systems, reaching 326,184 and 89,102 atoms for the simulations in DPC and POPC, respectively. Two-nanosecond MD simulations in the constant number of particles, volume, and temperature ensemble followed by 5–100-ns MD simulations at constant pressure and temperature were subsequently run with a time step of 2 fs.
Self-assembly Simulations of the NccX TM Segments
To study the self-association of NccX TM segments, these were coarse grained as described previously for the whole protein. The sequence spanning Thr6–Ser40, thus encompassing the predicted TM segment (Leu9–Tyr29) of NccX, was used for simulations and called NccX-TMsim (Fig. 1). The structure of NccX in DPC (Protein Data Bank code 4clv; this work) provided the initial atomistic structure of TMsim monomers. All protein, lipids, and solvent CG particles were described with the MARTINI force field. Monomers of CG NccX-TMsim were inserted parallel to each other in a preformed POPC bilayer containing 492 lipids and 6,000 water particles at a distance of 6 nm from each other. The x, y, and z dimensions of the bilayer were 13.6, 12.2, and 8.7 nm, respectively. The z axis represents the normal to the bilayer plane (x, y). Prior to the insertion, the NccX-TMsim monomers were aligned along the z axis and randomly rotated about the z axis. The ionic strength of the system was set with 100 mm NaCl, which in addition contributed to neutralize the charges of the NccX-TMsim monomers.
The system was energy-minimized for 10,000 steps using the steepest descent integrator. Then MD simulations were performed at T = 300 K with a coupling time of 1 ps using a v-rescale algorithm. The pressure was coupled using a semi-isotropic scheme at P = 1 bar with a coupling time of 5 ps and a compressibility of 3 × 10−4 bar−1. A time step of 40 fs was used during the production period. Varying the initial orientation of the two monomers relative to each other as well as initial velocity, two distinct simulations of 60 μs were performed in POPC, resulting in a total simulation time of 120 μs.
TOXCAT Assays
The homodimerization of wild-type and mutant NccX (full-length or truncated) was assessed using the TOXCAT assay (14). The expression vectors pccKAN, pccGpA-WT, and pccGpA-G83I and the malE E. coli strain MM39 were kindly provided by Donald Engelman (Yale University, New Haven, CT). As in any TOXCAT experiment, the expression of the constructs produced chimeras in which the peptide or protein of interest was fused N-terminally with the dimerization-dependent transcriptional activator domain of ToxR from Vibrio cholerae and with the E. coli maltose-binding protein (MBP) at its C terminus. We inserted the NccX-TMsim segment used for simulation experiments and the full-length sequence of NccX (NccX-FL) between ToxR and MBP. The G16I/G20I (GGII) double mutation in NccX-TMsim or NccX-FL was generated using the Quik-Change mutagenesis kit (Agilent). The sequence of all the constructs was confirmed by DNA sequencing. The resulting plasmids were transformed into E. coli MM39 for further use. Oligomerization of the inserted sequences turns on the transcription of the chloramphenicol acetyltransferase (CAT) reporter gene proportionally to the strength of the interaction between TM segments. Insertion efficiency and proper orientation were tested on the basis of the ability to grow on M9 minimal medium plates with 0.4% maltose as the only carbon source. Membrane localization of NccX-FL was checked by cell fractionation and subsequent detection of the construct in the membrane fraction by Western blot with either CnrXs antibodies (10) or MBP antibodies (anti-MBP monoclonal antibody, HRP-conjugated, New England Biolabs). Briefly, the MM39 E. coli cells bearing either the ToxR-NccX-FL-MBP or the ToxR-GpA-MBP constructs and used for the TOXCAT assays were lysed by sonic oscillation in 25 mm Tris-HCl, pH 8.0, 2 mm EDTA. The resulting lysates were centrifuged at 4,500 × g for 20 min (Thermo Scientific). The pellet consisting of cell debris, unbroken cells, and inclusion bodies was washed in sonication buffer containing 0.5% Triton X-100 and centrifuged again at 4,500 × g for 20 min to obtain the inclusion bodies. The supernatant of the first centrifugation was subjected to a centrifugation of 118,000 × g for 60 min (Beckmann) to pellet the membranes. The resulting supernatant was used for electrophoretic and Western blot analysis, whereas the pellet was suspended in sonication buffer and centrifuged again the same way. The new supernatant and the membrane fraction were used for further analysis. CAT activity was assayed spectrophotometrically at room temperature by measuring the rates of chloramphenicol acetylation in the presence of dithiobisnitrobenzoate and acetyl coenzyme A (34). The rate of CoA formation was monitored by recording the linear increase of the absorbance at 412 nm due to its reaction with dithiobisnitrobenzoate and thionitrobenzoate dianion release. The background rate of acetyl-CoA hydrolysis in the absence of chloramphenicol was recorded and subtracted before calculating the specific activities, expressed as ΔA412·min−1·mg−1 of protein from total extracts. Each experiment was performed at least in triplicate. The wild-type and mutant forms of the glycophorin A (GpA) TM domain, GpA-WT and GpA-G83I, were used as positive and negative controls in the TOXCAT assays, respectively. Normalization for the relative expression level of each construct was carried out using Western blotting with MBP antibodies and densitometry with ImageJ.
RESULTS
X-ray Structure of NccX
The x-ray structure of DPC-solubilized full-length NccX crystallized in the presence of an excess of zinc was determined at a resolution of 3.12 Å (Fig. 2B). It shows a dimer with an all-α-helix secondary structure like CnrXs (Fig. 2A) (9, 10, 12). Only a few residues are missing in the 148-residue-long NccX because each protomer spans residues Ser4–Arg145 (sequence shown in Fig. 1). Both NccX and CnrXs are characterized by a long hairpin with the NccX helices H2 and H3 coinciding with CnrXs helices H2′ and H3′ (Fig. 2C). However, whereas CnrXs protomer is best described as a hooked hairpin with the short helices H1′ and H4′ (N- and C-terminal helices, respectively) folded back on the H2′-H3′ hairpin, full-length NccX is a three-helix bundle with the short C-terminal helix H4 sticking out at an angle of about 50° to avoid a steric clash with the H3 helix of the opposite protomer (Fig. 2). NccX helix H1 (residues 3–48) is a long helix whose C-terminal end corresponds to CnrXs helix H1′ and that extends upstream to include the TM domain and the linker in between the TM and periplasmic domains. Noteworthily, a kink upstream at Pro37 physically delineates the C-terminal part of H1 that coincides with CnrXs H1′ (residues 37–48). The NccX protomers A and B actually differ slightly in the angle of this kink.
The NccX structure was stabilized by numerous zinc ions, consistent with the presence of 100 mm zinc acetate in the crystallization conditions of NccX. The anomalous diffusion properties of Zn(II) allowed precisely locating 10 zinc ions in the asymmetric unit. Two Zn(II) ions were bound by each NccX protomer with the same trigonal bipyramidal coordination geometry at the same metal-binding sites (Fig. 2, C and D) as those argued to be an artifact in protomer A of zinc-bound CnrXs (ZnA-CnrXs) (10). Accordingly, NccX protomers were best superimposable to ZnA-CnrXs (root mean square deviation of 0.93 Å for 78 Cα atom pairs), which is loaded with two Zn(II) ions (Fig. 2C), than to ZnB-CnrXs (root mean square deviation of 0.99 Å for 48 Cα atom pairs), which is loaded with a single Zn(II) ion. Extra Zn(II) ions were also located outside the metal-binding site (Fig. 2B) at the interface between two monomers either at the dimer interface or at crystal contacts. Two zinc ions of particular importance for the conformation of the NccX dimer link a nitrogen of His32 and Glu36 from H1 of one protomer with an oxygen of Glu106 from H3 of the opposite protomer. The coordination sphere of these extra Zn(II) ions is supposed to be completed with counteranions or ligands from the solvent (undefined at this resolution).
The TM domains cross each other near their Tyr29 residue at an angle of about 55° (Fig. 2B), which brings both N-terminal Ser3 residues ∼34 Å apart. Moreover, the putative GXXXG dimerization motifs in the middle of the TM segments are distant by about 18 Å, although they are supposed to interact in vivo. Each GXXXG motif makes contacts with the C-terminal tip of H2 helix of the same protomer on one side and with the TM domain of a symmetry-related dimer in the crystal on the other side (Fig. 3). In this way, two NccX dimers interlock by the fork formed by their TM domains, which act as hydrophobic clamps. This crystal contact is contributed by the N-terminal halves of the TM domains that run antiparallel to each other (from Phe7 to Leu18). Nevertheless, parallel docking of H1 helices is still visible in the C-terminal half of the TM (from Leu22 to Tyr29), a portion that is delimited in between the kink at Pro37, and a second kink between Val21 and Leu22. The packing of H1 helices at the C-terminal half of the TM is likely a remnant of the physiological arrangement as suggested by in silico and in vivo experiments (see below).
FIGURE 3.

Crystal contacts involving the TM domains of two symmetry-related dimers of NccX. A, the TM domain of dimeric NccX is composed of the N-terminal half of both H1 helices (blue and cyan or red and yellow). The two H1 helices of a TM domain join together at Tyr29, and segments upstream (toward the N terminus) form a fork with an angle of about 55°. This angle is the result of crystal contacts involving two TM domains that are interlocked and run antiparallel to each other (cyan and yellow or blue and red, respectively) (lower panel). B, ribbon representation of residues 4–36 of two interlocked dimers corresponding to a close-up view of the area shaded in blue in the lower panel of A. The N-terminal and C-terminal halves of each TM domain engage in different types of helix-helix interactions. The N-terminal (N-ter) halves of TM helices down to the GXXXG motif form antiparallel dimers with their counterparts from symmetry-related NccX proteins (cyan and yellow or blue and red). The C-terminal halves of TM helices (downstream the GXXXG motif) form parallel dimers within NccX dimers. The color code is as follows: purple, GXXXG motifs; pink, atom contacts between helices with parallel docking within the NccX dimer; brick red, atom contacts between helices with antiparallel docking between interlocked NccX dimers. Some residues making the interface boundaries are indicated.
Actually, the crystal structure of NccX that was determined reflects many artifactual constraints and represents a model that is incompatible with the insertion and dimerization of TM domains inside the membrane. In the following, we used in silico simulations to investigate the link between the NccX crystal structure and the significant body of data obtained with the CnrX periplasmic domain (9, 10, 12).
Structure-based Models of NccX
In a first series of in silico experiments, NccXDPC, the CG model derived from the x-ray structure of NccX, was mixed with single molecules of either DPC or POPC in random orientations and positions. Several experiments with similar outcomes were performed, and Fig. 4 illustrates a representative MD simulation with DPC (A–C) or with POPC (D–F). With DPC, the end of the simulation shows that the DPC molecules organized as micelles and that the structure of NccX remained in a collapsed state (Fig. 4B). This suggests that the crystalline structure of NccX reflects a conformation of the DPC-solubilized protein. An all-atom simulation was performed, starting from the output of the CG simulation. This improved the representation of the system, and the results yielded by the CG approach were confirmed. For instance, Fig. 4C shows the association of a micelle of DPC with the hydrophobic tips of the H1 helices of each NccX protomer. With POPC, the lipid molecules quickly formed a lipid bilayer between two layers of water, thus mimicking the inner membrane (Fig. 4E). After a few microseconds, NccX was found inserted in the lipid bilayer by the two long N-terminal helices predicted to anchor the protein in the biological membrane. The parts of NccX that correspond to the TM and periplasmic domains clearly tend to partition in the membrane bilayer and the solvent, respectively. However, the hairpin formed by the loop connecting H2 and H3, which participates in the hydrophobic dimer interface (see above and Fig. 8), still contacted the lipid bilayer after 12 μs. Here as well, the all-atom simulation (Fig. 4F) confirmed the pertinence of the CG approach.
FIGURE 4.
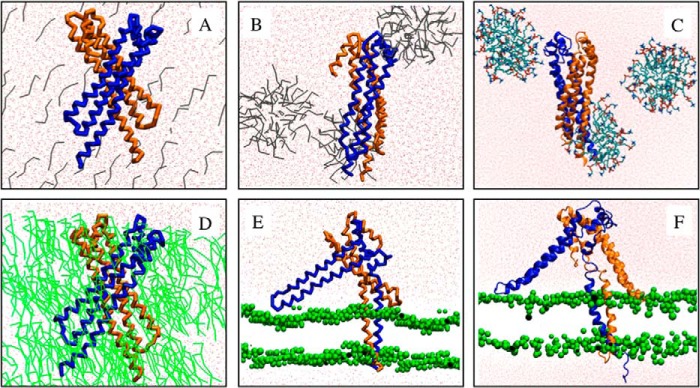
Simulations performed with NccXDPC. Upper panels, NccXDPC was mixed with DPC molecules in random position (A), and the result of a simulation of 5 μs is shown (B). Lower panels, NccXDPC was mixed with POPC molecules in random position (D), and the result of a simulation of 12 μs is shown (E). C and F correspond to 10- and 50-ns prolongations in a united atom representation of the CG simulations shown in B and E, respectively. The united atom simulation showed that the systems did not bring to light a quirk in the CG models. The two protomers of NccX are depicted in blue and brown. The water and DPC molecules are colored red and gray, respectively. POPC molecules are colored green and represented as sticks (D) or as beads (E and F). The Na+ and Cl− ions have been purposely omitted for clarity. The conditions of simulation are as follows. Upper panels, number of DPC, 116; number of Na+, 31; number of Cl−, 18; number of atoms (coarse grained), 27,900; number of atoms (all-atom), 326,184; initial box dimension, 13.0, 13.0, and 17.9 nm; final box dimension, 13.7, 13.7, and 17.9 nm. Lower panels, number of POPC, 167; number of Na+, 13; number of Cl−, 0; number of atoms (coarse grained), 7,108; number of atoms (all-atom), 89,102; initial box dimension, 9.5, 9.5, and 10.5 nm; final box dimension, 7.7, 7.7, and 13.6 nm.
FIGURE 8.
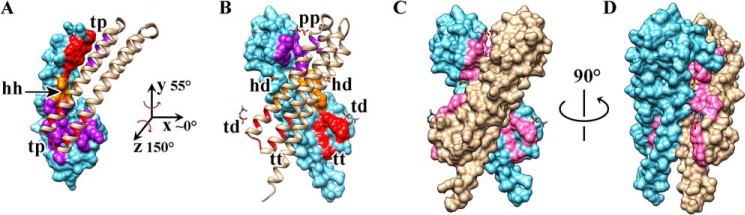
Redistribution of the residues contributing to the dimer interface of the NccX periplasmic domain upon DPC solubilization. A homology model of NccXs, the periplasmic domain of NccX, was built with Modeler (35) using the crystal structure of zinc-bound CnrXs as a template (Protein Data Bank code 2y3d). Chains A (tan) of the NccXs model and NccX crystal structure have been superimposed and are presented side by side as ribbons in A and in B, respectively. Chains B are shown as molecular surfaces (blue). The dimer interface of each protomer of NccXs was considered as a collection of three pieces: the tip of the H2-H3 hairpin (t; red), the body of helix H3 (h; orange), and the platform built by H3-H4 (p; purple). These pieces (t, h, and p) are colored on the molecular surface representation of protomer B of NccXs (A) or NccX (B). The phosphate moiety of some DPC molecules bound to the protein is depicted as orange sticks. Full polar heads of DPC are shown as orange (phosphate moiety) and blue (choline moiety) sticks. A, in NccXs, two protomers stick together via tip-to-platform contacts (tp) and helix-to-helix contacts (hh). B, in NccX, tip-to-platform contacts have been replaced by tip-to-DPC (td), tip-to-TM (tt), and platform-to-platform (pp) contacts. The helix-to-helix contacts have also been disrupted and replaced by helix-to-DPC (hd) contacts. As a result, the dimer interface in crystallized NccX has been completely remodeled as compared with that of NccXs with DPC and the TM N-terminal half sticking in (see text). Rotations about the y and z axes make NccXs chain B superimposable on NccX chain B. C, molecular surface representation of NccX with DPC moieties as sticks. The orientation of NccX is the same as in B. The residues colored in pink are those of the NccXs dimer interface. As a result, regions that were at least partially buried in NccXs and that are exposed to the solvent in NccX do appear in pink on a blue or tan background. D, the same representation as in C was rotated 90° as indicated. DPC moieties co-localized with regions in pink, thus emphasizing the role of DPC in the stabilization of the topology seen in the NccX crystal structure, which is at odds with the topology of the soluble periplasmic domain of NccX, as inferred by homology with CnrXs (10).
Membrane insertion of NccXDPC was examined throughout the simulation experiment. The crossing angles of H1 helices (Ω) showed a right-handed population with average negative values of about −26° (Fig. 5). The distribution of the tilt angles of the two TM helices with average values with respect to the bilayer normal of 22 ± 6° and 12 ± 7°, respectively (Fig. 5) indicates that the anchoring helices were in dynamic equilibrium while they remained inserted in the membrane over the full time of simulation.
FIGURE 5.
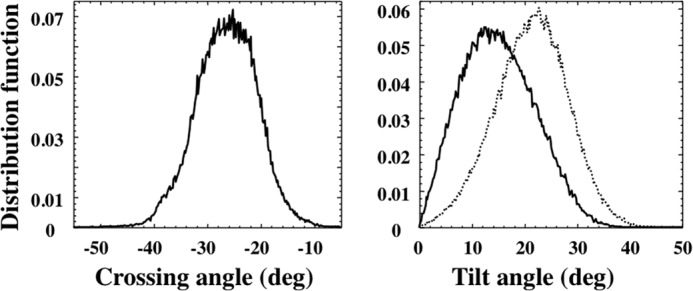
Distribution of crossing angles (left panel) and tilt angles (right panel) of the two protomers averaged over the time course (12 μs) of the simulation of NccXDPC in POPC shown in Fig. 4, D and E. deg, degrees.
As a control, a second series of simulations was performed with NccXPred, a plausible CG model for the native conformation of membrane-embedded NccX, i.e. with the TM helices running perpendicular with respect to the globular soluble domain (10) and packed with a right-handed negative crossing angle of about −35° as defined by Chothia et al. (36). NccXPred was either mixed with DPC in random orientation or inserted in a preformed POPC bilayer. Fig. 6 shows that the NccXPred model was not stable in DPC and that, similar to NccXDPC, it displayed a collapsed structure. In contrast, NccXPred remained inserted in the POPC membrane, keeping a similar global fold. These two series of simulations confirmed both the destabilization of the interprotomer hydrophobic interactions by DPC and the relevance of the model of TM insertion of the H1 helices.
FIGURE 6.

Simulations performed with NccXPred. Upper panels, NccXPred was mixed with DPC molecules in random position (A), and the result of a simulation of 5 μs is shown (B). Lower panels, NccXPred was mixed with POPC equilibrated as a membrane patch (C), and the result of a simulation of 14 μs is shown (D). The color code is the same as in Fig. 4. The Na+ and Cl− ions have been purposely omitted for clarity. The conditions of simulation are as follows. Upper panels, number of DPC, 117; number of Na+, 28; number of Cl−, 22; number of atoms, 27,899; initial box dimension, 13.0, 13.0, and 17.0 nm; final box dimension, 13.7, 13.7, and 17.9 nm. Lower panels, number of POPC, 267; number of Na+, 13; number of Cl−, 22; number of atoms, 10,125; initial box dimension, 10.5, 12.5, and 9.0 nm; final box dimension, 8.9, 10.6, and 12.3 nm.
In Silico and in Vivo Transmembrane Segment Interactions
To gain further insight into the possible interaction between TM segments, self-assembling simulations of the NccX TM segments embedded in POPC bilayer were performed using CG-MD. For these calculations, a 35-residue-long peptide called NccX-TMsim, encompassing the 21-residue-long TM segment and flanking residues, was used (Fig. 1). Two NccX-TMsim were inserted into a preformed POPC bilayer 6 nm away from each other. Fig. 7A shows that they self-assembled in less than 2 μs and that a long lasting helix dimer was spontaneously formed. Examination of the structure of this dimer during a typical 60-μs simulation revealed that the helices explored different conformational spaces (Fig. 7A). Although the main helix packing was left-handed (average value of the helix crossing angle Ω of about 10°), a right-handed population also existed with Ω of about −20°, which is in agreement with the right-handed crossing angle measured with NccXDPC in POPC. There is no clear period of alternation between both states. As NccX-TMsim contains the putative 16GXXXG20 interaction motif, the double mutant GGII was chosen to further analyze its role in dimerization by the TOXCAT assay in both NccX-TMsim and NccX-FL. The results of the TOXCAT assays are displayed in Fig. 7, B and C, and those of the corresponding controls are displayed in Fig. 7, D and E. The expression level of each construct assessed by Western blotting against MBP antibodies in whole-cell lysates (Fig. 7, B and C) was used to normalize the specific CAT activities.
FIGURE 7.

NccX transmembrane segment homodimerization revealed by CG-MD simulations and the TOXCAT assay. A, upper panel, time course of the interhelix distance over a typical 60-μs simulation. Lower panel, time course of the helix crossing angle (Ω) over the same simulation. Positive and negative crossing angles correspond to left-handed and right-handed packings, respectively. B, the ability of NccX TM helices (NccX-TMsim) to homodimerize was assessed with the TOXCAT reporter assay: GpA-WT and GpA-G83I were used as positive and negative controls, respectively. For each construct, CAT specific activity was normalized against GpA-WT, averaged over three independent experiments, and corrected for the production levels of the chimeric proteins ToxR-GpA-MBP and ToxR-NccX-TMsim-MBP that were assessed by Western blot with anti-MBP antibodies (lower panel). C, NccX-TMsim, taken as a reference, was compared with NccX-FL and with the double GGII mutants of either construct. Normalization of the CAT activities was that same as in B. D and E are controls of topology of the constructs used for the TOXCAT assays. D, MalE complementation assays confirmed the correct topology of all constructs but the full-length proteins. For the latter, the bulky metal sensor part of NccX likely hampered the ability of MBP to interact with the maltose transporter. E, the topology of both NccX-FL constructs was found to be correct as they were specifically detected in the membrane fraction. High CAT activity (C) demonstrated the correct orientation of the chimeric protein with ToxR in the cytoplasm. Total proteins were separated by SDS-PAGE and blotted onto a PVDF membrane. ToxR-NccX-FL-MBP was revealed by Western blot analysis with antibodies to CnrXs (upper panel) that cross-react with NccX (10) or with antibodies to MBP (lower panel). The ToxR-GpA-MBP construct was used as a negative control. A very faint band corresponding to ToxR-NccX-FL-MBP was visible in lanes L and S2, whereas this protein was highly detected in lane M corresponding to the membrane fraction. L, lysate obtained by sonic oscillation; IB, inclusion bodies; S1, supernatant of the first centrifugation used to pellet the membrane. The pellet was suspended in sonication buffer and centrifuged again. The new supernatant (S2) and the membrane fraction (M) were used for further analysis. deg, degrees. Error bars in B and C represent S.D.
In a first experiment, NccX-TMsim was compared with the widely used controls GpA-WT and GpA-G83I (14). Consistent with CG simulations, the dimerization of NccX-TMsim was detected in vivo by recording a CAT activity that represented only 18.6% of the CAT activity induced by the dimerization of GpA-WT (Fig. 7B). Then, NccX-TMsim was compared with NccX-FL with both constructs bearing the GGII double mutation or not (Fig. 7C). Interestingly, the CAT activity yielded by NccX-FL was 10-fold higher than that of NccX-TMsim taken as a reference. The GGII double mutation canceled the protein interaction in NccX-TMsim, whereas it had no significant effect in the full-length construct. These results demonstrate that the two N-terminal ends of dimeric full-length NccX are close to each other in agreement with the results of the simulations reported above (Fig. 4). They also indicate that although the GXXXG motif is important for NccX-TMsim interaction it has a minor impact for the dimerization of full-length NccX protomers that is mainly driven by the periplasmic domains.
Interpretation of the Detergent-induced Structural Rearrangements
The crystal structure of DPC-solubilized NccX differs markedly from the prediction shown in Fig. 2A. Despite the clear correspondence between the secondary structure elements of NccX and CnrXs, the cradle-shaped CnrXs dimer (Fig. 2A) is not reproduced even partially by the NccX dimer. Structural rearrangements probably occurred after solubilization by DPC. However, the 3.12-Å resolution was insufficient to accurately locate DPC molecules in the electron density. Still, the polar head of a detergent molecule was located near the side chains of Trp94 and Glu99 of each protomer, i.e. at the tip of the H2-H3 hairpin (Fig. 8). Furthermore, two phosphate moieties from DPC were observed close to the loop connecting H3 and H4, and two more moieties were located near Arg114 residues, i.e. near the middle of the H3 helices. Both sites belong to the CnrXs dimer interface (10), thus suggesting that DPC destabilized the interface of the native periplasmic domain. This was examined in more detail.
Compared with CnrXs, the dimer interface of NccX is noticeably different (Fig. 8). In CnrXs, H4′ is part of the helix bundle and contributes to the dimer interface by binding the partner protomer via the tip of its H2′-H3′ hairpin (10). This contact is no longer possible in NccX in which H4 sticks out of the helix bundle. Hence, the residues of the CnrXs dimer interface are pulled apart in NccX where the dimer interface buries 1,943 Å2, which is much less than 3,054 Å2 in CnrXs. A homology model of the periplasmic sensor domain of NccX, called NccXs (Fig. 8A), was built using the crystal structure of zinc-bound CnrXs as a template (the residues present in Protein Data Bank code 2y3d of CnrXs share 80% identity with the corresponding residues in NccX). In this model, the dimer interface consists of pairs of residues carried by both H3′ helices (Fig. 8A, hh contacts) or carried by H2′ and H4′ helices from either protomer (Fig. 8A, tp contacts) as in the CnrXs template (10). These interactions no longer existed in DPC-solubilized NccX as H2 and H1 of each protomer form an antiparallel coiled coil where H2 residues that were engaged in the NccXs dimer interface now interact with TM residues in H1 of the same protomer (Fig. 8B, tt contacts). A wider permutation of the residues in contact occurred as follows: the H2′(protomer A)-H4′ (protomer B) dimer interface in NccXs (Fig. 8A, tp contacts) was redistributed between the H1-H2 coiled coil dimerization motif in NccX and a new contact area where H4 residues face H3 residues that were previously part of the soluble NccXs dimer interface (Fig. 8B, pp contacts). Fig. 8 shows that DPC moieties co-localized with regions that were at least partially buried in NccXs and that are exposed to the solvent in NccX, thus emphasizing the role of DPC in the stabilization of the topology seen in the NccX crystal structure. Altogether, these data show that the residues contributing to the dimer interface of the native NccX periplasmic domain are redistributed upon DPC solubilization. One can conclude that the detergent in addition to solubilizing the membrane bilayer destabilized the hydrophobic interactions between subunits, thus allowing the two periplasmic domains to rearrange and collapse against the hydrophobic H1 helices.
DISCUSSION
Although the single pass membrane proteins represent almost half of all membrane proteins, few examples of structures containing an isolated TM helix already exist. This is the case for the rat and the human monoamine oxidase A (37, 38) whose membrane attachment not only involves a C-terminal TM helix but also hydrophobic patches at the surface of the protein. Such is also the case of the membrane-bound cytochrome c quinol dehydrogenase NrfH from Desulfovibrio vulgaris: NrfH is dimeric, and both TM helices pack together in a right-handed fashion (39). Most recently, the structure of a cytochrome P450 from yeast showed a single TM domain standing alone and playing an unforeseen role in soluble domain orientation outside the membrane, indicating that there are lessons to be learned by characterizing more single pass membrane proteins (40). To our knowledge, such is the scarce repertoire of structures available for bitopic membrane proteins. Specific difficulties in both purification and ordered crystallization can explain this fact. The structural characterization of full-length NccX, the membrane-anchored nickel, cobalt, and cadmium sensor protein of the NccYXH transmembrane signaling complex, also contributes to fill this void in the Protein Data Bank. NccX was efficiently extracted regardless of the detergent used (i.e. DPC, n-dodecyl β-d-maltopyranoside, n-octyl β-d-glucopyranoside, or tetraethylene glycol monooctyl ether (C8E4)), but only DPC allowed achieving both purification and crystallization. Although widely used in membrane protein investigation (41, 42), DPC has been seldom used for crystallization, although DPC micelles can be considered a membrane mimetic of choice for NMR studies because of the number of structures observed (41). Very recently, the effect of DPC on protein conformation was illustrated in two instances where solution structures obtained in DPC micelles and crystal structures obtained either in 3-lauramidopropyl-N,N-dimethylamine oxide micelles or lipid meso-phase were significantly different. In one example, the solution structure of E. coli diacylglycerol kinase showed a domain-swapped trimer, whereas the same enzyme and two variants crystallized in a lipid bilayer showed no sign of domain swapping: the implication is such that the architecture and the catalytic properties of the active sites are different in both structures (43, 44). In the other case, when the solution structure of the uncoupling protein UCP2 solubilized and purified in DPC was described (45), comparison with the high resolution x-ray structure of the bovine ATP/ADP mitochondrial carrier crystallized in 3-lauramidopropyl-N,N-dimethylamine oxide micelles (46) suggested that it carried biases affecting biological interpretation (47). This raises the question of the behavior of a membrane protein once extracted from its native membrane environment and of the techniques to be used to explore this behavior. Regarding UCP2, the physiological relevance of its solution structure was assessed with the help of theoretical calculations. Indeed, one major conclusion of this study was that combining functional and computational investigations is a promising general strategy that would be beneficial to analyze the structures of numerous membrane proteins (47). As a general guideline, alternative methods are required for structure validation, quality assessment, and enhancement (48). This is particularly true for the membrane proteins with an activity difficult to assess in vitro.
Such a strategy was adapted to the 3.12-Å resolution x-ray structure of NccX. Extensive work on the soluble domain alone made it obvious that the NccX crystal structure was at odds with the functional native form (9, 10, 12). We performed MD simulations and TOXCAT assays to rationalize the x-ray structure of DPC-solubilized NccX. Solubilization of the membrane bilayer and detergent destabilization of the main hydrophobic contacts of the dimer interface led to the crystallization of a protein in which the periplasmic domains of both protomers parted from each other and collapsed against the hydrophobic TM segments within H1 helices. That the detergent is responsible for the structural collapse of NccX can be inferred from the redistribution of hydrophobic interactions among the TM helix and a pair of hydrophobic patches within the periplasmic domain. Although the exposure of the TM helix followed membrane solubilization, it seems that DPC played an active role in dissociating the dimer of periplasmic domains because headgroups of DPC were found in close vicinity of the hydrophobic patches that kept them together. The sensitivity of the NccX structure to DPC solubilization might be a consequence of the intrinsic plasticity of this sensor, the conformation of which was proposed to be tuned from the metal-binding site via hydrophobic packing in the core of the periplasmic domain (10).
Additional constraints affected the TM segments. In the crystal, the TM segments of one dimer form a hydrophobic fork that interacts with the symmetry-related hydrophobic fork of another dimer to compose an interlocked tail-to-tail arrangement of dimers. This is a case of crystal packing with extreme consequences where the antiparallel docking of TM helices displaced the parallel, possibly native docking; i.e. an aberrant topology was produced that is incompatible with membrane insertion. Crystal packing was also supported by several zinc ions that were present in excess in the crystallization conditions. Interestingly, NccX protomers best superimposed with chain A of zinc-bound CnrXs, i.e. the chain that contained two zinc ions (10). This shows that each NccX protomer considered individually retained the characteristic fold of the protomers of CnrXs extended by most of the residues of the long N-terminal membrane-anchoring helix. From its C terminus to the N terminus, helix H1 contains several distinctive features. His46 and His42 at the C-terminal end are strictly conserved residues in the CnrX protein family and are essential residues of the metal-binding site (9, 10, 12). The residue His38 was found to chelate a Zn(II) ion in both NccX and protomer A of CnrXs crystallized in the presence of an excess of Zn(II). This residue is not strictly conserved (10), and the phenotype of CnrX is poorly affected by the H38R substitution (8). His38 belongs to a stretch of about 10 residues (residues 31–40) that was not resolved in the crystal structure of CnrXs and was proposed to make a flexuous junction between CnrXs and the TM segment, thus tethering the sensor domain to the membrane. The flexibility of this stretch is illustrated by its poor definition in the electron density maps of CnrXs and by secondary structure predictions suggesting a random coil in the full-length protein. That NccX or CnrX contains a proline or a glycine, respectively, at position 37 further argues for this hypothesis as these residues are well known helix breakers. We can thus speculate that the kink observed at Pro37 is the result of the constraints that arose with membrane dislocation and helix repacking in the presence of detergent. When the membrane was solubilized by DPC, constraints imposed by hydrophobic surface exclusion, crystal packing, and zinc-mediated interactions yielded a topology that does not fit seamlessly the sequence of NccX.
Going up to the N terminus, the membrane-inserted sequence of 21 residues extending from residue 29 to 9 contains a GXXXG motif that often mediates interactions between TM helices (14, 49–54). In addition, this TM sequence includes 11 β-branched residues (Leu, Val, and Ile) and seven small residues (Ala, Gly, and Ser). The strength of the interaction mediated by the GXXXG motif is enhanced by nearby β-branched residues (51), and a short interhelical distance due to the presence of small residues favors the TM interactions (52). However, the repartition of such residues is crucial (52). For instance, based on the seminal work of Engelman and co-workers (55) on the dimerization of GpA, the complete motif of interactions, LIXXGVXXGVXXT, was identified with a minimal consensus motif for stability defined as LXXXGXXXGXXXT. This shows that the GXXXG motif requires stabilization on both sides (52, 55). Both NccX and CnrX display a very incomplete motif in the TM region with a corresponding sequence of STXXG16XXXG20VXXA. This is in agreement with the minor role of the GXXXG motif in TM segment interactions observed as far as the full-length protein was considered. Finally, the rest of the N-terminal sequence extending from residue 8 to 1 is predicted to be located in the cytoplasm (Fig. 1) in agreement with its content in several neutral or polar residues.
MD simulations are of great interest to study lateral association and oligomerization of TM helices (56, 57). CG simulations performed in the presence of DPC resulted in NccX conformations matching the collapsed structure of DPC-solubilized NccX regardless of the initial model, be it NccXDPC or NccXPred, suggesting that the crystalline structure stems from solubilization of the membrane bilayer as well as from destabilization of the interprotomer hydrophobic interactions. Reciprocally, CG simulations provided a nice picture of the transition from NccXDPC to NccXPred when the former was placed in a POPC environment. The partition between the hydrophobic helices in the POPC membrane and the sensor domain in the aqueous phase is fast and easily reproducible even if the coordinate root mean square deviation of the soluble part with respect to NccXPred is still high at the end of the longest simulations. Relaxing the constraints of GROMACS on the assignment of the secondary structures during CG dynamics was not sufficient to allow complete NccX refolding. Altogether, our results strongly suggest that the periplasmic and TM domains of NccX form independent folding units and that the soluble domain already characterized is representative of the periplasmic domain in full-length NccX/CnrX (10). The main determinant that brings the dimer together is the periplasmic domain: this keeps NccX TM segments close to each other and could potentialize their dimerization via the GXXXG motif as exemplified by the in silico and in vivo characterization of NccX-TMsim interactions. Moreover, the interaction of the TM segments has been proved to be highly dynamic in the simulations reported here. CnrX/NccX and CnrY/NccY can engage in both periplasmic and transmembrane interactions (8, 10). How transmembrane signaling is performed by CnrYXH/NccYXH complexes once CnrX/NccX has bound nickel or cobalt is not known. Whether the dynamic interaction of TM segments is related to the function of CnrX/NccX in signaling and how these TM segments may help propagate the signal to CnrH/NccH with the relay of CnrY/NccY are two questions that require further work.
Even though it is imperfect, the structure of full-length NccX extends the very limited catalogue of structures available for fully characterized single pass transmembrane proteins. Besides, the results presented here emphasize the benefits of implementing complementary approaches to reach physiologically relevant information from detergent-solubilized proteins.
Acknowledgments
We thank Eve de Rosny, Juliette Trepreau, and Adeline Veyret for help during the first steps of purification of NccX. We thank beamline Proxima-1 (Synchrotron SOLEIL, Gif Sur Yvette) for provision of beamtime and Pierre Legrand for data collection, structure determination, and refinement of the reported structure of NccX. We also thank Dr. P. Fuchs (Paris Diderot University) for providing useful Python scripts for trajectory analyses with Gromacs. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS, National Institutes of Health Grant P41-GM103311).
The atomic coordinates and structure factors (code 4clv) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TM
- transmembrane
- CAT
- chloramphenicol acetyltransferase
- DPC
- dodecylphosphocholine
- GpA
- glycophorin A
- MBP
- maltose-binding protein
- POPC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- MD
- molecular dynamics
- CG
- coarse grained
- FL
- full length
- GGII
- G16I/G20I
- CnrXs
- soluble metal sensor domain of CnrX
- NccXs
- periplasmic sensor domain of NccX.
REFERENCES
- 1. Mergeay M., Nies D., Schlegel H. G., Gerits J., Charles P., Van Gijsegem F. (1985) Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 162, 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monchy S., Benotmane M. A., Janssen P., Vallaeys T., Taghavi S., van der Lelie D., Mergeay M. (2007) Plasmids pMOL28 and pMOL30 of Cupriavidus metallidurans are specialized in the maximal viable response to heavy metals. J. Bacteriol. 189, 7417–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von Rozycki T., Nies D. H. (2009) Cupriavidus metallidurans evolution of a metal-resistant bacterium. Antonie Van Leeuwenhoek 96, 115–139 [DOI] [PubMed] [Google Scholar]
- 4. Janssen P. J., Van Houdt R., Moors H., Monsieurs P., Morin N., Michaux A., Benotmane M. A., Leys N., Vallaeys T., Lapidus A., Monchy S., Médigue C., Taghavi S., McCorkle S., Dunn J., van der Lelie D., Mergeay M. (2010) The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS One 5, e10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nies D. H. (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 27, 313–339 [DOI] [PubMed] [Google Scholar]
- 6. Grass G., Grosse C., Nies D. H. (2000) Regulation of the cnr cobalt and nickel resistance determinant from Ralstonia sp. strain CH34. J. Bacteriol. 182, 1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tibazarwa C., Wuertz S., Mergeay M., Wyns L., van Der Lelie D. (2000) Regulation of the cnr cobalt and nickel resistance determinant of Ralstonia eutropha (Alcaligenes eutrophus) CH34. J. Bacteriol. 182, 1399–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grass G., Fricke B., Nies D. H. (2005) Control of expression of a periplasmic nickel efflux pump by periplasmic nickel concentrations. Biometals 18, 437–448 [DOI] [PubMed] [Google Scholar]
- 9. Pompidor G., Maillard A. P., Girard E., Gambarelli S., Kahn R., Covès J. (2008) X-ray structure of the metal-sensor CnrX in both the apo- and copper-bound forms. FEBS Lett. 582, 3954–3958 [DOI] [PubMed] [Google Scholar]
- 10. Trepreau J., Girard E., Maillard A. P., de Rosny E., Petit-Haertlein I., Kahn R., Covès J. (2011) Structural basis for metal sensing by CnrX. J. Mol. Biol. 408, 766–779 [DOI] [PubMed] [Google Scholar]
- 11. Trepreau J., de Rosny E., Duboc C., Sarret G., Petit-Hartlein I., Maillard A. P., Imberty A., Proux O., Covès J. (2011) Spectroscopic characterization of the metal-binding sites in the periplasmic metal-sensor domain of CnrX from Cupriavidus metallidurans CH34. Biochemistry 50, 9036–9045 [DOI] [PubMed] [Google Scholar]
- 12. Trepreau J., Grosse C., Mouesca J.-M., Sarret G., Girard E., Petit-Haertlein I., Kuennemann S., Desbourdes C., de Rosny E., Maillard A. P., Nies D. H., Covès J. (2014) Metal sensing and signal transduction by CnrX from Cupriavidus metallidurans CH34: role of the only methionine assessed by a functional, spectroscopic, and theoretical study. Metallomics 6, 263–273 [DOI] [PubMed] [Google Scholar]
- 13. Maillard A. P., Girard E., Ziani W., Petit-Härtlein I., Kahn R., Covès J. (2014) The crystal structure of the anti-σ factor CnrY in complex with the σ factor CnrH shows a new structural class of anti-σ factors targeting extracytoplasmic function σ factors. J. Mol. Biol. 426, 2313–2327 [DOI] [PubMed] [Google Scholar]
- 14. Russ W. P., Engelman D. M. (1999) TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. U.S.A. 96, 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 16. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 18. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cowtan K. (2010) Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 21. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER, Version 2.11.15, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 23. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) Graphical tools for macromolecular crystallography in PHENIX. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 26. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neria E., Fisher S., Karplus M. (1996) Simulation of activation free energies in molecular systems. J. Chem. Phys. 105, 1902–1921 [Google Scholar]
- 28. de Jong D. H., Singh G., Bennett W. F., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., Marrink S. J. (2013) Improved parameters for the MARTINI coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 [DOI] [PubMed] [Google Scholar]
- 29. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 30. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 31. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H. (2007) The MARTINI force field: coarse-grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 [DOI] [PubMed] [Google Scholar]
- 32. Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S. J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 [DOI] [PubMed] [Google Scholar]
- 33. Wassenaar T. A., Pluhackova K., Böckmann R. A., Marrink S. J., Tieleman D. P. (2014) Going backward: a flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 10, 676–690 [DOI] [PubMed] [Google Scholar]
- 34. Wei P., Liu X., Hu M.-H., Zuo L.-M., Kai M., Wang R., Luo S.-Z. (2011) The dimerization interface of the glycoprotein Ibβ transmembrane domain corresponds to polar residues within a leucine zipper motif. Protein Sci. 20, 1814–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 36. Chothia C., Levitt M., Richardson D. (1981) Helix to helix packing in proteins. J. Mol. Biol. 145, 215–250 [DOI] [PubMed] [Google Scholar]
- 37. Ma J., Yoshimura M., Yamashita E., Nakagawa A., Ito A., Tsukihara T. (2004) Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J. Mol. Biol. 338, 103–114 [DOI] [PubMed] [Google Scholar]
- 38. Son S. Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. (2008) Structure of human monoamine oxidase A at 2.2 Å resolution: the control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. U.S.A. 105, 5739–5744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodrigues M. L., Oliveira T. F., Pereira I. A., Archer M. (2006) X-ray structure of the membrane-bound cytochrome c quinol dehydrogenase NrfH reveals novel haem coordination. EMBO J. 25, 5951–5960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monk B. C., Tomasiak T. M., Keniya M. V., Huschmann F. U., Tyndall J. D., O'Connell J. D., 3rd, Cannon R. D., McDonald J. G., Rodriguez A., Finer-Moore J. S, Stroud R. M. (2014) Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. U.S.A. 111, 3865–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warschawski D. E., Arnold A. A., Beaugrand M., Gravel A., Chartrand É., Marcotte I. (2011) Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim. Biophys. Acta 1808, 1957–1974 [DOI] [PubMed] [Google Scholar]
- 42. Tulumello D. V., Deber C. M. (2012) Efficiency of detergents at maintaining membrane protein structures in their biologically relevant forms. Biochim. Biophys. Acta 1818, 1351–1358 [DOI] [PubMed] [Google Scholar]
- 43. Van Horn W. D., Kim H. J., Ellis C. D., Hadziselimovic A., Sulistijo E. S., Karra M. D., Tian C., Sönnichsen F. D., Sanders C. R. (2009) Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science 324, 1726–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li D., Lyons J. A., Pye V. E., Vogeley L., Aragão D., Kenyon C. P., Shah S. T., Doherty C., Aherne M., Caffrey M. (2013) Crystal structure of the integral membrane diacylglycerol kinase. Nature 497, 521–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berardi M. J., Shih W. M., Harrison S. C., Chou J. J. (2011) Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 476, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trézéguet V., Lauquin G. J., Brandolin G. (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44 [DOI] [PubMed] [Google Scholar]
- 47. Zoonens M., Comer J., Masscheleyn S., Pebay-Peyroula E., Chipot C., Miroux B., Dehez F. (2013) Dangerous liaisons between detergents and membrane proteins. The case of mitochondrial uncoupling protein 2. J. Am. Chem. Soc. 135, 15174–15182 [DOI] [PubMed] [Google Scholar]
- 48. Zhou H.-X., Cross T. A. (2013) Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 42, 361–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lemmon M. A., Flanagan J. M., Treutlein H. R., Zhang J., Engelman D. M. (1992) Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry 31, 12719–12725 [DOI] [PubMed] [Google Scholar]
- 50. Kleiger G., Grothe R., Mallick P., Eisenberg D. (2002) GXXXG and AXXXA: common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry 41, 5990–5997 [DOI] [PubMed] [Google Scholar]
- 51. Senes A., Gerstein M., Engelman D. M. (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol. 296, 921–936 [DOI] [PubMed] [Google Scholar]
- 52. Senes A., Engel D. E., DeGrado W. F. (2004) Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 14, 465–479 [DOI] [PubMed] [Google Scholar]
- 53. Moore D. T., Berger B. W., DeGrado W. F. (2008) Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure 16, 991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cymer F., Veerappan A., Schneider D. (2012) Transmembrane helix-helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim. Biophys. Acta 1818, 963–973 [DOI] [PubMed] [Google Scholar]
- 55. Melnyk R. A., Kim S., Curran A. R., Engelman D. M., Bowie J. U., Deber C. M. (2004) The affinity of GXXXG motifs in transmembrane helix-helix interactions is modulated by long-range communication. J. Biol. Chem. 279, 16591–16597 [DOI] [PubMed] [Google Scholar]
- 56. Psachoulia E., Marshall D. P., Sansom M. S. (2010) Molecular dynamics simulations of the dimerization of transmembrane α-helices. Acc. Chem. Res. 43, 388–396 [DOI] [PubMed] [Google Scholar]
- 57. Aci-Sèche S., Sawma P., Hubert P., Sturgis J. N., Bagnard D., Jacob L., Genest M., Garnier N. (2014) Transmembrane recognition of the semaphorin co-receptors neuropilin 1 and plexin A1: coarse-grained simulations. PLoS One 9, e97779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]