

Background: Apelin and apelin receptor (APJ)-induced Gαi activation evokes a protective response in cardiac hypertrophy, whereas G-protein-independent signaling induces hypertrophy.
Results: Serine 348 is identified as one phosphorylation site in APJ, which is crucial for APJ interactions with GRK2/5, β-arrestin1/2, and its internalization.
Conclusion: Mutation at serine 348 modulates G-independent signaling of APJ.
Significance: Understanding APJ signaling and regulation could provide novel clues for the development of functionally selective APJ ligands.
Keywords: 7-Helix Receptor, Bioluminescence Resonance Energy Transfer (BRET), G Protein-coupled Receptor (GPCR), Protein Phosphorylation, Signal Transduction, Apelin Receptor (APJ), Biased Signal Pathway
Abstract
Phosphorylation plays vital roles in the regulation of G protein-coupled receptor (GPCR) functions. The apelin and apelin receptor (APJ) system is involved in the regulation of cardiovascular function and central control of body homeostasis. Here, using tandem mass spectrometry, we first identified phosphorylated serine residues in the C terminus of APJ. To determine the role of phosphorylation sites in APJ-mediated G protein-dependent and -independent signaling and function, we induced a mutation in the C-terminal serine residues and examined their effects on the interaction between APJ with G protein or GRK/β-arrestin and their downstream signaling. Mutation of serine 348 led to an elimination of both GRK and β-arrestin recruitment to APJ induced by apelin-13. Moreover, APJ internalization and G protein-independent ERK signaling were also abolished by point mutation at serine 348. In contrast, this mutant at serine residues had no demonstrable impact on apelin-13-induced G protein activation and its intracellular signaling. These findings suggest that mutation of serine 348 resulted in inactive GRK/β-arrestin. However, there was no change in the active G protein thus, APJ conformation was biased. These results provide important information on the molecular interplay and impact of the APJ function, which may be extrapolated to design novel drugs for cardiac hypertrophy based on this biased signal pathway.
Introduction
G protein-coupled receptors (GPCRs),3 which constitute the largest superfamily of cell surface receptors involved in many cellular signaling and physiological responses, are activated by binding with peptides, lipids, small molecules, and hormones (1, 2). Consequently, GPCRs are extensively researched within the context of both potential and established drug targets (3).
Upon stimulation by agonists, GPCRs are activated and initiate intracellular signaling events. In addition to G protein-dependent signaling, the activated GPCRs may also interact with many intracellular proteins to expand their ability to transmit signals through G protein-independent signaling pathway, such as G protein-coupled receptor kinases (GRKs) and β-arrestins. In this process, the agonist-occupied receptor is phosphorylated by GRKs, subsequently recruiting cytoplasmic β-arrestins to the GRK-phosphorylated receptor (4). As intracellular adaptor and scaffolding proteins, β-arrestins play important roles in GPCRs desensitization, internalization, and intracellular trafficking and could activate signaling cascades independently of G protein activation (5, 6). For example, β-arrestin2 binds Raf-1 and ERK1/2 directly and MEK-1 indirectly, leading to a β-arrestin-dependent ERK activation (7, 8).
Apelin receptor (APJ) is a member of the family A of GPCRs with a range of physiological functions including the regulation of cardiovascular function and fluid homeostasis (9, 10). APJ remained an orphan GPCR until 1998 when the peptide apelin was isolated from bovine stomach extracts as its endogenous ligand (11). Subsequently, numerous researchers have showed that apelin has an important regulatory role in various physiological processes. Among them, apelin-13 exhibits much stronger activity than other active isoforms in regulating the cardiovascular function (11). The apelin/APJ system is currently envisaged to be an important therapeutic target for the treatment of heart failure, hypertension, and obesity-related diseases (12, 13).
The mechanisms of APJ signal transduction are still under active investigation. New research has found that APJ has a dual role in cardiac hypertrophy (14). Apelin activates the signals of APJ through the Gαi pathway and elicits a cardiac protective response. Although sustained overload activates APJ to induce cardiac hypertrophy via a G protein-independent fashion. Previous data suggested that the C-terminal portion of APJ are required for internalization and mediates receptor internalization through palmitoylation and phosphorylation (15). But it is still unclear how APJ mediates its G protein-dependent and -independent signaling pathways. It has been demonstrated that mutagenesis of key serine/threonine residues at receptor phosphorylation result in diminished β-arrestin binding after agonist stimulation in receptors such as the M2 muscarinic receptor, the AT1A angiotensin receptor, and the V2 vasopressin receptor (16–18). There are several putative phosphorylated amino acids at the C terminus of APJ, which has not been investigated.
In the work presented here, we first identified a key C-terminal amino acid residue required for GRKs/β-arrestins recruitment to activated APJ after agonist binding. We found that the key residue for APJ regulation is serine 348. Furthermore, we assessed the role of serine 348 in APJ internalization and G protein-independent ERK1/2 activation. The results suggest that serine 348 is required for GRKs and β-arrestin-mediated biased G protein independent signaling of APJ. Taken together, these results are helpful for screening novel and existing drugs of the cardiovascular system based on this biased signaling pathway.
EXPERIMENTAL PROCEDURES
Materials
Human apelin-13 and 125I-apelin-13 were purchased from Phoenix Pharmaceuticals (Burlingame, CA). The amino acid sequence of apelin-13 is Gln-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe, as previously described (19). Lipofectamine 2000 was obtained from Invitrogen. Forskolin, PTX, and anti-FLAG antibody were obtained from Sigma. Anti-HA-agarose were obtained from Pierce Chemical Co. Anti-phospho-ERK1/2 antibody, anti-ERK1/2 antibody, anti-HA antibody, anti-β-arrestin1 antibody, anti-β-arrestin2 antibody, and anti-β-actin antibody were purchased from Cell Signaling Technology.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293) cells were maintained at 37 °C and 5% CO2 in Complete Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (FCS). Transient transfections were carried out 24 h after seeding using Lipofectamine 2000 following the manufacturer's instructions.
Protein Preparation and Mass Spectrometry
For mass spectrometry experiments, HEK293 cells stably expressing WT APJ receptor were treated with 100 nm apelin-13 at 37 °C for 10 min. The cells were washed and lysed for 30 min on ice with RIPA buffer containing protease and phosphatase inhibitors. After centrifugation, the supernatant was reduced with 10 mm dithiothreitol (DTT) at 56 °C for 1 h and subsequently alkylated with 55 mm iodoacetamide at room temperature for 45 min in the dark. The sample was resolved by SDS-PAGE on 10% gels and stained with colloidal Coomassie Blue. Subsequently, the protein was digested with trypsin overnight at 37 °C, and then the fractions were passed through a single TiO2 Spin Tip for phosphopeptide selective enrichment. Phosphopeptides were then purified and washed as described (20, 21). Peptide composition was determined by LC-MS/MS mass spectrometry.
LC-MS/MS measurements were carried out by a Shimadzu HPLC system (Columbia, MD) coupled to an AB SCIEX Triple TOF 5600 mass spectrometer (SCIEX, Foster City, CA). The two buffers used for the reverse phase chromatography were buffer A (0.1% (v/v) formic acid in water) and buffer B (0.1% (v/v) formic acid in acetonitrile). The HPLC gradient consists of holding 0% buffer B for 3 min, followed by increasing to 5% buffer B in 5 min, to 35% buffer B in 35 min, then to 60% buffer B in 5 min. In the following 2 min, the gradient was increased to 80% for 2 min. Initial chromatographic conditions were restored in 1 min and maintained for 10 min. A flow rate of 300 nl/min was used to elute the peptides into the ionization source of the mass spectrometer.
The acquired MS/MS spectra were processed with the Mascot search engine (Matrix Science, London, UK) for database correlation analysis. The search parameters through Mascot were set for phosphorylation emphasis and to search for biological modifications. Trypsin was set as the enzyme, and the IPI human database was searched. Each filtered MS/MS spectra exhibiting possible phosphorylated peptide was manually checked and validated.
Plasmid Constructs and Mutagenesis
The pcDNA3.1(+)-APJ and pcDNA3.1(+)-HA tagged-APJ plasmids were purchased from Missouri S&T cDNA Resource Center (Rolla, MO). Rluc-β-arrestin1 and Rluc-β-arrestin2 were generated by PCR amplification with plasmids containing β-arrestin1/β-arrestin2 ORF (kindly provided by Prof. Karin Eidne (QEII Medical Centre, Nedlands, Australia)) as templates, the coding sequences of human β-arrestin without their stop codons and subcloned into pRluc-N1 (PerkinElmer Life Sciences Inc.). On the other hand, the cDNA encoding the hAPJ without its stop codon was subcloned into pEGFP-N1 (Clontech, Mountain View, CA). Plasmids GRK2-GFP2 and GRK5-GFP2 were kindly provided by Prof. Christian E. Elling, 7TM Pharma A/S, 2970 Horsholm, Denmark. FLAG-tagged β-arrestin1/β-arrestin2 was kindly provided by Prof. Vsevolod V. Gurevich (Department of Pharmacology, Vanderbilt University).
For construction of the Gαi2-Rluc and Gαq-Rluc fusion proteins, the coding sequence of humanized Rluc was PCR amplified and inserted in the coding sequence of each Gα subunit. Depending on the Gα subunit, the Rluc was inserted between either residues 91 and 92 of Gαi2, or residues 97 and 98 of Gαq.
A series of APJ mutants were generated by overlap extension PCR using high fidelity Pfu polymerase and mutagenic primers as described previously (22). The mutagenic APJ cDNA was cut sequentially with EcoRI and HindIII and then ligated back into the original pcDNA3.1(+). All mutational cDNAs were confirmed by sequence analysis of both strands. All constructs were verified by sequencing.
Cell Surface Expression Assay
HEK293 cells were transiently transfected with the same amount of pcDNA3.1(+) containing HA-tagged wild-type APJ or HA-tagged APJ-S335A, APJ-S345A, and APJ-S348A. Twenty-four hours after transfection, cells were fixed in 4% paraformaldehyde for 15 min at room temperature, washed, and incubated in blocking solution (3% BSA) for 1 h. Subsequently, cells were incubated with 1:1000 primary rabbit polyclonal anti-HA antibody overnight at 4 °C. After washing three times with PBS, the cells were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) at 1:1000 dilutions for 1 h at room temperature. After extensive washing, the immunoreactivity was detected by the addition of TMB Plus substrate (Santa Cruz Biotechnology), and the reaction was stopped with 0.2 m H2SO4. The absorbance at 450 nm was measured on a microplate reader (Bio-Rad). For each experiment, mock conditions corresponding to the transfection of vector without receptor were included. The expression levels of mutational receptors were calculated as a percentage of WT APJ expression using the formula: [(ODmutant − ODmock)/(ODwt − ODmock)] × 100%. Receptor internalization was measured with 100 nm apelin-13 treatment in 60 min at 37 °C by the above cell surface ELISA procedure. The percentages of mutational receptor internalization were defined as described previously (23) using the formula: [(ODbasal − ODmock) − (ODstimulated − ODmock)]/(ODbasal − ODmock) × 100%.
Radioligand Binding Assay
HEK293 cells were transiently transfected with the same amount of WT APJ and mutational APJs. 48 h after transfection, a washed cell membrane preparation was prepared as described previously (24). The interactions of 125I-apelin-13 with WT APJ or mutational APJ receptors were measured using radioligand binding displacement binding assays according to a previous report (9).
Confocal Microscopy
HEK293 cells were plated on poly-d-lysine-coated glass coverslips in 6-well plates, grown to 60% confluence, and transiently co-transfected with constant amounts of plasmids encoding for HA-APJ and EGFP-β-arrestins. Twenty-four hours post-transfection, medium was changed to serum-free DMEM, and the cells were incubated with 100 nm apelin-13 at different time intervals. Then, the cells were fixed in 4% paraformaldehyde for 15 min, washed with PBS, and incubated with 3% BSA in PBS/Triton X-100 (0.1%) for 1 h at room temperature. For the staining, anti-HA antibody was incubated as the first antibody overnight at 4 °C. After washing the cells with PBS, cells were incubated with IgG TRITC-conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. Following a wash step, the cells were mounted on glass slides with VECTASHIELD medium containing DAPI (Vector Laboratories Inc., Peterborough, UK). Images were observed with a ×63 oil immersion objective in a Leica model DMRE laser scanning confocal microscope (Leica, Milton Keynes, UK).
Dose-response and Real-time Kinetic BRET Assays
HEK293 cells were transiently transfected with Rluc-tagged and various EGFP (or GFP2)-tagged constructs. Twenty-four hours after transfection, cells were then harvested in HEPES-buffered phenol red-free complete medium containing 5% FCS and seeded in poly-d-lysine-coated 96-well white microplates (Corning 3600). All BRET measurements were performed according to the donor and acceptor pairs used (Table 1) by the Mithras LB940 plate reader (Berthold Technologies, Bad Wildbad, Germany) and MicroWin 2000 software as described previously.
TABLE 1.
Summary of substrate and filter setting used in BRET assays
| Method | Donor | Substrate | Donor emission | Acceptor | Acceptor emission |
|---|---|---|---|---|---|
| nm | nm | ||||
| BRET1 | Rluc | Coelenterazine h | 460 | EGFP | 535 |
| BRET2 | Rluc | Deep Blue C | 400 | GFP2 | 515 |
| eBRET | Rluc | EnduRenTM | 460 | EGFP | 535 |
Co-immunoprecipitation and Immunoblotting
Immunoprecipitations were performed as described previously (25). HEK293 cells were transiently transfected with HA-APJ (or HA-APJ348) and FLAG-β-arrestin1 (or β-arrestin2) or the vector control. 36 h after transfection, cells were serum-starved overnight and stimulated with 100 nm apelin-13 for 15 min. For co-immunoprecipitations, cells were lysed and the supernatant fractions were collected and then incubated with anti-HA-agarose beads overnight at 4 °C with end-over-end rotation. The beads were washed three times with TBST and precipitates were eluted with SDS sample buffer containing β-mercaptoethanol. The supernatants were then analyzed with SDS-PAGE and immunoblotting for anti-FLAG antibody immunoreactivity.
cAMP Assay
HEK293 cells were transiently transfected with the WT APJ and various mutational APJs (APJ-S335A, APJ-S345A, APJ-S348A) as described above. After 24 h, the cells were incubated with 3-isobutyl-1-methylxanthine (0.5 mm) and MgCl2 (10 mm) for 20 min and then stimulated for 10 min with forskolin (10 μm) in either the absence or presence of various concentrations of apelin-13 (0.1–10,000 nm) at 37 °C. Subsequently, cells were washed twice with ice-cold PBS and suspended in acetate buffer. Intracellular cAMP level was measured using the absorbance-based cAMP ELISA kits (Cell Biolabs, Inc.) according to the manufacturer's protocol.
Intracellular Calcium Assay
HEK293 cells expressing WT APJ or mutants were plated at 5 × 104 cells per well in a 96-well poly-d-lysine-coated black plate (Corning), respectively. In addition, un-transfected HEK293 cells were used as negative controls. Calcium fluorescences were detected using Fluo-4 NW Calcium Assay Kits (Invitrogen) according to the manufacturer's instructions. Before the detection, the cells were incubated at 37 °C for 30 min in the dye loading solution dissolved in assay buffer. Following incubation, the plates were washed twice with assay buffer, equilibrated at room temperature for an additional 30 min. Subsequently, all cells stimulated with 100 nm apelin-13 and calcium fluorescences were immediately detected with the Mithras LB940 plate reader (Berthold Technologies, Bad Wildbad, Germany). Basal readings were obtained for 5 s prior to agonist addition and subtracted from post-injection readings to obtain a ligand-induced Ca2+ response. Calcium fluorescence ratio was represented using the formula: ligand-induced Ca2+ readings/basal readings. Data were imported into GraphPad Prism 5 software for statistical analysis and graphing.
Activation of ERK1/2 Assay
HEK293 cells expressing WT APJ or APJ-S348A in six-well plates were grown to 70–80% confluences. After overnight serum starvation, the cells were treated with apelin-13. Subsequently, cells were washed, harvested, and lysed in RIPA lysis buffer. Ten micrograms of cell extracts were separated by 10% SDS-PAGE and proteins were transferred to polyvinylidene fluoride (PVDF) membranes. The phosphorylation status of ERK1/2 was detected by immunoblotting with the antibody against phospho-ERK1/2. As a control for loading, the same membranes were stripped of antibody and re-probed with anti-total ERK1/2 antibody. The band intensities were measured by densitometry analysis and the change in ERK phosphorylation in both samples was calculated as the phospho-ERK/ERK ratio (see Ref. 19 for details).
For β-arrestin1/2 shRNA experiments, HEK-293 cells stably expressing APJ or APJ-S348A were transiently transfected with either β-arrestin1 shRNA (Sigma, Clone ID NM_004041.3–828s21c1) or β-arrestin2 shRNA (Sigma, Clone ID NM_004313.3–309s21c1) and shRNA Negative Control Med GC (Sigma, pLKO.1-puro) according to the manufacturer's instructions using Lipofectamine 2000 reagent. Forty-eight hours later, cells were stimulated with apelin-13 and lysed for ERK1/2 assays.
Statistical Analysis
All data are shown as the mean ± S.E. Data were presented and analyzed using Prism 5.0 graphing software (GraphPad). Sigmoidal curves were fitted to the dose-response data using non-linear regression. Statistical analysis was performed using one-way analysis of variance followed by Tukey's multiple comparison post-test.
RESULTS
Identification of Phosphorylation Sites on APJ by Mass Spectrometry
Previous studies have shown that C-terminal serines of APJ are required for mediating receptor desensitization and internalization (15). However, the key residues involved in APJ regulation have not been defined. Putative APJ phosphorylation sites were obtained from NetPhos 2.0 prediction software (26) with APJ C-terminal serine 335, 345, and 348 mutants being chosen as above. Among them, sequence analysis revealed that serine 345 and 348 are highly conserved C-terminal phosphorylation sites between human, mouse, rat, and other creatures (Fig. 1A).
FIGURE 1.

Mutations at serines 335, 345, and 348 of APJ do not alter the receptor expression and location at cell membranes. A, C-terminal amino acid sequence alignment of APJ human, mouse, rat, rhesus monkey, chimpanzee, dog, chicken, and zebrafish residues. Putative APJ phosphorylation sites were obtained from NetPhos 2.0. Mutations from serine to alanine are marked in yellow. Gray shading represents transmembrane helix 7. B, cell surface expression of the WT and mutational APJs quantified by ELISA. The results were expressed as percentage of cell surface expression levels of the WT APJ after correction of the nonspecific expression in cells transfected with the empty vector (n = 4; one-way analysis of variance; ns, no significant difference p > 0.05). C, BRET data indicating proximity between the cell surface marker Venus-Kras and Rluc-tagged WT APJ or mutational receptors (n = 4; one-way analysis of variance; ns, no significant difference, p > 0.05). D, internalization of WT and mutational APJs after 100 nm apelin-13 stimulation in HEK293 cells measured by ELISA (n = 4; one-way analysis of variance; ***, p < 0.001 versus WT APJ group).
To identify precise phosphorylation sites, we treated the stably transfected HEK293 cells with 100 nm apelin-13 for 10 min. Cell lysis and protein extraction was resolved by SDS-PAGE and stained (Fig. 2A). Following reduction and alkylation as described under ”Experimental Procedures,“ we used mass spectrometry to better define the specific sites phosphorylated in APJ under apelin-13 stimulation. We conducted a mass spectrometric analysis of the phosphoacceptor sites on APJ and two serine phosphor-acceptor sites (Ser-345 and Ser-348) in the C-terminal tail of APJ are shown in Fig. 2B.
FIGURE 2.

Identification of phosphorylation sites on APJ by mass spectrometry. Coomassie Blue-stained SDS-PAGE gel with proteins extracted from HEK293 cells expressing WT APJ were treated with apelin-13 (100 nm) for 10 min (A). Molecular mass markers are indicated on the left. Whole cell protein was extracted and subjected to tryptic digestion, and the peptides were analyzed by mass spectrometry. Shown is a schematic indicating the phospho-acceptor sites in C-terminal tail of APJ. Also shown are typical LC-MS/MS traces that identify serines 345 and 348 as phosphorylation sites. The phosphorylated residue is highlighted in red (B).
Site-directed Mutagenesis and Expression of WT and Mutational APJs
To determine which phosphorylation site identified was most likely to be the most important for the function of APJ, we generated mammalian APJ expression plasmids by site-directed mutagenesis to construct C-terminal receptor serine to alanine mutants. After a series of mutants were constructed, we first investigated the cell surface expression level of WT APJ, APJ-S335A, APJ-S345A, and APJ-S348A in HEK293 cells. Both ELISA and BRET results showed that all three point mutants had little effect on the receptor cell surface expression compared with the WT APJ (Fig. 1, B and C). This data suggested that APJ mutants were correctly synthesized and trafficked to the plasma membrane.
Binding Properties of WT and Mutational APJs
To characterize the binding properties of APJ, WT and mutational APJs were transiently transfected into HEK293 cells. Competitive displacement studies of 125I-apelin-13 by apelin-13 (cold), showed no differences in IC50 values of the WT APJ and mutational APJs (Table 2). Also, the Bmax values for binding 125I-apelin-13 to the membrane preparation did not differ (Table 2). The results further verified that the mutagenesis of APJ C-terminal serine residues had little influence on agonist binding. At least, serines 335, 345, and 348 were not key residues for apelin binding. Upon agonist binding, the mutants were likely to be activated and transmit downstream signaling, which can be used for subsequent experiments.
TABLE 2.
Binding characteristics of WT and mutational APJs
Binding characteristics of 125I-apelin-13 to the membrane preparation from HEK-293 cells transfected with the same amount of WT APJ and mutational APJs. Results are mean ± S.E. of four experiments.
| Receptor | IC50 | Bmax |
|---|---|---|
| nm | fmol/mg protein | |
| WT APJ | 0.68 ± 0.08 | 8.26 ± 0.17 |
| APJ-S335A | 0.75 ± 0.04 | 7.92 ± 0.34 |
| APJ-S345A | 0.67 ± 0.05 | 8.43 ± 0.15 |
| APJ-S348A | 0.70 ± 0.09 | 8.37 ± 0.35 |
Functional Validation of APJ Mutants by Apelin-13-induced Inhibition of cAMP and Elevation of Ca2+
The effect of apelin-13 on cAMP signaling in HEK293 cells was previously studied (19, 22). Consistent with previous research, the forskolin-induced intracellular cAMP production were reduced significantly by apelin-13 in a dose-dependent manner in all groups (Fig. 3A). There is no significant difference between WT and all mutational APJs. Our data suggest that site-directed mutagenesis of APJ C-terminal serines 335, 345, or 348 were not required for G protein coupling to adenylate cyclase inhibition.
FIGURE 3.
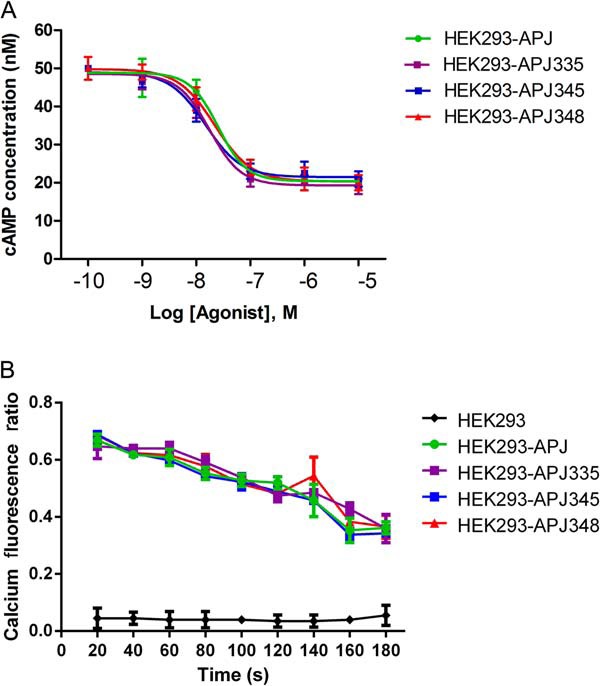
Apelin-13-induced cAMP and intracellular calcium accumulations are not affected by ser mutations. A, dose-response curve of cAMP accumulation for WT and mutational APJs. HEK293 cells expressing WT APJ or mutants were incubated for 20 min in 3-isobutyl-1-methylxanthine with various concentrations of apelin-13 (0.1–10,000 nm). Intracellular cAMP level was measured using the absorbance-based cAMP ELISA kits. Data represent four independent experiments. B, the variation of intracellular calcium fluorescence in 180 s. HEK293 cells expressing WT APJ or mutants were stimulated with apelin-13 (100 nm). The variation of intracellular calcium concentration was recorded by Fluo-4NW Calcium Assay Kits. Data represent four independent experiments.
We next determined whether mutational APJs could also impact apelin-13-induced elevation of intracellular calcium. The data showed that WT APJ and three mutants resulted in a significant increase of intracellular calcium (Fig. 3B). Taken together, the results suggest mutants are functional proteins and have not changed their role in activating the G protein-dependent signaling pathway.
Serine 348 Mutation of APJ Is Required for Apelin-13-induced Receptor Internalization
Upon ligand binding, most GPCRs are internalized. To evaluate whether serines 335, 345, or 348 play an important role in apelin-13-mediated APJ internalization, we compared the mutants engineered at these sites to the internalization kinetics of WT APJ by ELISA-based assay (Fig. 1D). In HEK293 cells expressing WT APJ, apelin-13 (100 nm) caused a robust time-dependent receptor internalization peaking at 30 min following addition of the agonist. Mutation of C-terminal serine 335 and 345 had no effect on apelin-13-induced receptor internalization, compared with the WT APJ. In contrast, mutation of serine 348 significantly blocked apelin-13-induced receptor internalization. After increasing the agonist concentration and duration time, apelin-13 (1–10,000 nm) failed to induce APJ-S348A internalization at all the time points in HEK293 cells (data not shown). Thus, serine 348 plays a key role in the internalization of APJ.
These results were also confirmed by confocal microscopy. To image cell surface expression and receptor internalization, HEK293 cells were transiently co-transfected HA-tagged WT or mutational APJ and EGFP-tagged β-arrestin1 or β-arrestin2. Fig. 4, A and B, shows that prior to agonist stimulation (at 0 min), the distribution of APJ (shown in red) was clearly localized at the cell surface, whereas β-arrestin1-EGFP or β-arrestin2-EGFP fluorescence protein was only evenly distributed throughout the cytosol (shown in green). Upon stimulation with the appropriate agonist apelin-13 (100 nm) for 15 min, a significant internalization was observed in HEK293 cells expressing WT APJ. Receptor internalization peaked at 30 min following agonist treatment, with the appearance of clusters showing moderate intensity yellow fluorescence representing co-localization of HA-APJ (in red) and β-arrestin1-EGFP or β-arrestin2-EGFP (in green). APJ was then redistributed back to the cell surface at 60 min. APJ-S335A and APJ-S345A showed a similar cell surface expression and internalization process compared with the WT APJ (data not shown). In contrast, although receptor expression was verified, apelin-13 failed to induce a significant shift in localization of APJ-S348A and no co-localization was observed with β-arrestin1 or β-arrestin2 after stimulation in 60 min (Fig. 4, C and D). Combined with the results obtained by the cell surface expression assay, these data together suggest that serine 348 is a crucial phosphor-regulation site of APJ, and mutation of serine 348 to alanine in APJ is sufficient to block apelin-13-induced APJ internalization.
FIGURE 4.

APJ-S348A mutation impairs agonist-induced receptor internalization in HEK293 cells. HEK293 cells were co-transfected with β-arrestin-EGFP and the HA-tagged APJ construct at 1:1 ratio over the ensuing time period with apelin-13 (100 nm) for 0, 15, 30, and 60 min. The redistribution and co-localization of HA-APJ (red) and β-arrestin1-EGFP (A) or β-arrestin2-EGFP (B) after agonist stimulation were shown. The redistribution and co-localization of the HA-APJ-S348A (red) and β-arrestin1-EGFP (C) or β-arrestin2-EGFP (D) after agonist stimulation were shown. The nuclei were stained with DAPI. Yellow indicates the co-localization of β-arrestin and receptor. The results shown are representative images of at least three independent experiments. Vertical arrows show plasma membrane, slanted arrows show endosomes. Scale bars, 10 μm.
Serine 348 Mutation of APJ Impairs Its Interaction with GRK2/5 and β-Arrestin1/2
For most GPCRs, agonist stimulation leads to GRK-mediated receptor phosphorylation followed by binding of β-arrestins to the phosphorylated receptor. In recent years, a number of studies have now utilized BRET to monitor the interactions between GPCRs and GRKs or β-arrestins (27–30). High-throughput screening of GPCR-specific drugs was also performed by a BRET assay (31). Therefore, the interaction between APJ and GRK2/5 or β-arrestins were also analyzed by BRET assays.
Agonist dose-response curves of GRK2/5 recruitment to APJ at 10 min post-stimulation were detected using BRET2. The data showed that apelin-13 caused a robust dose-dependent increase in BRET signal for WT APJ, APJ-S335A, and APJ-S345A, indicative of GRK2/5 being recruited to activated receptors (Fig. 5, A and B). The EC50 values were as follows: 6.58 ± 0.58 nm (WT APJ), 6.20 ± 1.56 nm (APJ-S335A), 5.69 ± 1.42 nm (APJ-S345A) with GRK2; 9.45 ± 2.58 nm (WT APJ), 8.20 ± 1.85 nm (APJ-S335A), and 7.34 ± 1.17 nm (APJ-S345A) with GRK5. The potency of GRK2 (or GRK5) recruitment to APJ-S335A or APJ-S345A was not reduced sufficiently to reach statistical significance compared with WT APJ. In contrast, the APJ-S348A mutant was unable to recruit GRK2/5 at any dose tested. Similarly, agonist dose-response curves of β-arrestin1/2 recruitment to WT APJ at 15 min were also detected using BRET1. The data showed that apelin-13 caused a robust dose-dependent increase in BRET signal for WT APJ, APJ-S335A, and APJ-S345A, indicative of β-arrestin1/2 being recruited to activated receptors (Fig. 5, C and D). The EC50 values were as follows: 5.69 ± 0.71 nm (WT APJ), 5.06 ± 1.05 nm (APJ-S335A), and 5.55 ± 0.94 nm (APJ-S345A) with β-arrestin1; 8.29 ± 1.25 nm (WT APJ), 7.25 ± 0.73 nm (APJ-S335A), and 7.55 ± 1.03 nm (APJ-S345A) with β-arrestin2. The potency of β-arrestin1/2 recruitment to APJ-S335A or APJ-S345A was not reduced sufficiently to reach statistical significance compared with WT APJ. In contrast, the APJ-S348A mutant was unable to recruit β-arrestin1/2 at any dose tested. This is consistent with GRK2/5 being recruited to the activated receptor in a ligand-dependent manner.
FIGURE 5.

APJ-S348A mutation impairs its interaction with GRKs and β-arrestins by real-time BRET assays. A–D, dose-response curve of the recruitment of GRKs or β-arrestins to the WT and mutational APJs measured by BRET. HEK293 cell transiently transfected Rluc-tagged receptors and GRK2-GFP2 (A), Rluc-tagged receptors and GRK5-GFP2 (B), EGFP-tagged receptors and Rluc-β-arrestin1 (C), or EGFP-tagged receptors and Rluc-β-arrestin2 (D) were treated with increasing doses of apelin-13. The BRET signals between receptor and GRKs/β-arrestins were recorded at 10 (for BRET2) or 15 min (for BRET1) after agonist stimulation. The results represent four independent experiments. E–H, kinetic-response curve of the recruitment of GRKs or β-arrestins to the WT and mutational APJs measured by BRET. Real-time analysis of the interaction between WT APJ (or mutational APJs) and GRK2 (E), WT APJ (or mutational APJs) and GRK5 (F), WT APJ (or mutational APJs) and β-arrestin1 (G), or WT APJ (or mutational APJs) and β-arrestin2 (H) using BRET in living cells. Transiently co-transfected HEK293 cells were incubated with DeepBlue C (for BRET2) or EnduRenTM (for eBRET) then the BRET signal was recorded after apelin-13 stimulation. The results represent four independent experiments.
We subsequently examined the effects of the mutational C-terminal serine residue on interaction between APJ with GRK2/5 or β-arrestin1/2, using real-time BRET assays. In cells expressing Rluc-APJ and GRK2-GFP2 or GRK5-GFP2 there was a rapid increase of the BRET2 signal after apelin-13 treatment, which peaked at 10 min (Fig. 5, E and F), and is similar to APJ with mutations at serine 335 and 345. However, the APJ-S348A mutant seriously decreased the BRET2 signal, suggesting this serine 348 to alanine mutation blocks the interaction between APJ and GRK2/5. Similarly, we also observed a vanished BRET signal caused by mutation at serine 348 of APJ in HEK293 cells expressing Rluc-β-arrestins and EGFP-tagged APJ-S348A after apelin-13 stimulation, indicating that serine 348 is also crucial for the binding β-arrestins to APJ (Fig. 5, G and H).
The interaction between APJ and β-arrestins has also been confirmed by a cellular co-immunoprecipitation assay. The results indicated that co-immunoprecipitations could be detected in cells transfected with both FLAG-β-arrestin1 (Fig. 6A) or β-arrestin2 (Fig. 6B) and HA-APJ plasmids with apelin-13 treatment, but not in cells transfected with either vector alone. The co-immunoprecipitations could not be detected in the cells expressing both FLAG-β-arrestin1 (Fig. 6A) or β-arrestin2 (Fig. 6B) and HA-APJ348. Taken together, these data suggest that mutation of serine 348 to alanine in APJ greatly prevents recruitment with GRK2/5 and β-arrestin1/2, in which APJ-S348A may fail to induce the β-arrestin-dependent signaling pathway.
FIGURE 6.

APJ-S348A mutation impairs its interaction with β-arrestins by CO-IP assays. A, HEK293 cells were transfected with expression vectors of both HA-APJ and FLAG-β-arrestin1 fusion proteins (co-transfected 1) or with either vector alone. The same cells were transfected with HA-APJ348 and FLAG-β-arrestin1 fusion proteins (co-transfected 2) or with either vector alone. Samples containing either HA-APJ or FLAG-β-arrestin1 were mixed (Mix 1) and samples containing either HA-APJ348 or FLAG-β-arrestin1 were also mixed (Mix 2). B, HEK293 cells were transfected with expression vectors of both HA-APJ and FLAG-β-arrestin2 fusion proteins (co-transfected 1) or with either vector alone. The same cells were transfected with HA-APJ348 and FLAG-β-arrestin2 fusion proteins (co-transfected 2) or with either vector alone. Samples containing either HA-APJ or FLAG-β-arrestin2 were mixed (Mix 1) and samples containing either HA-APJ348 or FLAG-β-arrestin2 were also mixed (Mix 2). Confirmation of the expression of appropriate constructs was obtained by immunoblotting cell lysates with either anti-FLAG or anti-HA antibodies (lower panels). The cell lysates were subsequently immunoprecipitated (IP) with anti-HA-agarose beads and followed by immunoblotted (IB) with anti-FLAG or anti-HA antibodies (upper panels).
Serine 348 Mutation of APJ Impairs Its Desensitization and Resensitization Characteristics
To establish the role of the APJ Ser-348 residue in desensitization and resensitization of apelin-13 signaling, we incubated HEK293 cells expressing WT APJ or APJ-S348A with apelin-13 (100 nm) for various time intervals, washed the cells, and then measured the effect of apelin-13 on cAMP levels. In HEK293 cells expressing WT APJ, pretreatment with apelin-13 for 3 h resulted in a sustained desensitization of apelin-13-induced decreases in the cAMP level. However, HEK293 cells expressing APJ-S348A could not induce obvious desensitization of apelin-13-induced decreases in the cAMP concentration (Fig. 7A). This result is consistent with other data that relates to its impaired interaction with β-arrestins.
FIGURE 7.

APJ-S348A mutation impairs its desensitization and resensitization characteristics. A, time course of APJ (or APJ-S348A) desensitization in HEK293 cells. Apelin-13 pretreated cells (100 nm for various time intervals) were stimulated with 100 nm apelin-13 for 10 min and the subsequent cAMP response was measured. B, time course of APJ (or APJ-S348A) resensitization in HEK293 cells. Apelin-13-pretreated cells (100 nm for 3 h to induce maximal receptor desensitization) were briefly washed, and allowed to recover for various time intervals before a second 100 nm apelin-13 stimulus for 10 min and measurement of the subsequent cAMP accumulation by ELISA. Data correspond to the mean ± S.E. from four independent experiments. APJ-S348A group compared with WT APJ group, ***, p < 0.01.
In a different set of experiments, the recovery kinetics of APJ response were also observed. After pretreatment of HEK293 cells expressing WT APJ with 100 nm apelin-13 for 3 h (to induce maximal receptor desensitization), the cells were washed to remove agonist and allowed to rest for various intervals before addition of 100 nm apelin-13 for 10 min. A 70% recovery of apelin-13 responsiveness was apparent after a 30-min resting period, and full recovery was achieved with cells cultured for 2 h before addition of apelin-13 (Fig. 7B). At all time intervals, basal and vehicle-stimulated cAMP levels were not different between treated and untreated cells (data not shown).
Serine 348 Mutation of APJ Does Not Alter Its Interaction with G Protein
Previous studies revealed that apelin activates APJ signals through the Gαi/q pathway (15, 32–34). To determine the function of the C-terminal serine to alanine substitutions of APJ, we examined their effects on the interaction between receptor and G protein by real-time BRET assays. The results showed that addition of apelin-13 to cells co-expressing EGFP-tagged WT APJ (or mutants) and Rluc-tagged Gαq resulted in a rapid and significant increased ligand-induced BRET signal indicative of Gαq being recruited to the activated receptor of both WT APJ and mutants. The BRET signal peaked at 5 min after treatment and decreased rapidly thereafter (Fig. 8A). Similar treatment of cells co-expressing APJ (or mutants) and Gαi2 also induced an increase in BRET signal with a similar kinetic profile indicative of Gαi2 being recruited to the activated receptors (Fig. 8B). Overall, BRET data clearly show that the three mutants and the Gαq (or Gαi2) interaction are similar to WT APJ. Taken together, these results suggest that APJ mutagenesis of the phosphorylation site cannot impact the binding of G protein and further activate intracellular downstream signaling via G protein-dependent pathway.
FIGURE 8.
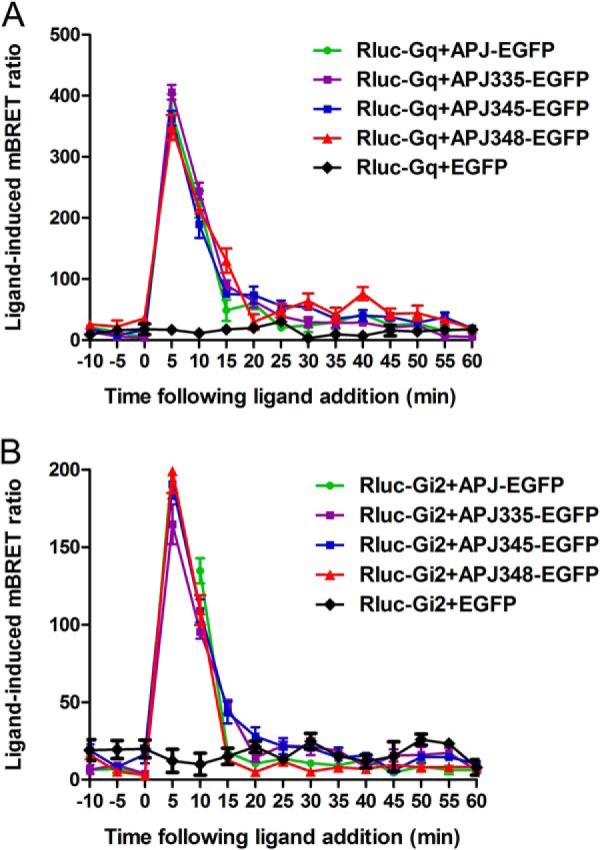
APJ-S348A mutation does not alter its interaction with G protein. Real-time analysis of the interaction between APJ (or APJ-S335A, APJ-S345A, APJ-S348A) and Gq (A) or Gαi2 (B) using eBRET in living cells. HEK293 cells were transiently transfected with EGFP-tagged various receptors and Rluc-Gq (or Rluc-Gαi2) then preincubated for 2 h with EnduRen and detected as above. The results are mean ± S.E. of at least four independent experiments.
Serine 348 Mutation of APJ Decreased G Protein-independent ERK Activation
Previously, we reported apelin-13-induced ERK1/2 activation via the wild-type receptor in stable transfected HEK293-APJ cells (19, 22). To observe whether the mutational APJ receptor affects ERK1/2 signaling, both fast and slow ERK1/2 activation, we examined time- and dose-dependent effects on ERK1/2 signaling activation, respectively. The results showed that a significantly increased ERK1/2 phosphorylation by apelin-13 appeared after 2 min treatment in WT APJ, with the maximal level at 5 min, and then phospho-ERK1/2 levels were kept high until 60 min (Fig. 9A, upper). In contrast, in mutational APJ-S348A, ERK1/2 phosphorylation was consistent with WT APJ 5 min after agonist stimulation. Subsequently, the phospho-ERK1/2 level decreased significantly to the near basal level from 15 min, which is GRKs/β-arrestins dependent (Fig. 9A, middle).
FIGURE 9.

The effects of Ser-348 mutation on ERK1/2 phosphorylation. A, time-dependent ERK1/2 phosphorylation in HEK293 cells expressing APJ (upper) or APJ-S348A (middle) after 100 nm apelin-13 stimulation. The time-dependent curves of the two groups were quantified and are shown (lower). B and C, dose-dependent ERK1/2 phosphorylation in HEK293 cells expressing APJ (upper) or APJ-S348A (middle) after apelin-13 stimulation for 15 (B) or 5 min (C). The dose-dependent effects of the two groups were quantified (lower). D, effect of PTX on apelin-13-induced ERK1/2 phosphorylation. HEK293 cells expressing APJ (upper) or APJ-S348A (middle) were pretreated with PTX (200 ng/ml) for 12 h, the cells were stimulated with apelin-13 (100 nm) for 5 min. The effects of PTX were quantified (lower). The density of the bands corresponding to 44 and 42 kDa was quantified with an imaging densitometer, normalized to total ERK. The data correspond to the mean ± S.E. from four independent experiments. APJ-S348A group compared with WT APJ group, ***, p < 0.01.
As for dose-dependent ERK phosphorylation in mutational APJ-S348A, we also found that apelin-13 (from 0.1 to 1000 nm) could not induce ERK1/2 activation as indicated for 15 min (Fig. 9B, middle). These data are distinct from those of WT APJ (Fig. 9B, upper), which shows dose-dependent trends. However, when duration of agonist was reduced, we observed dose-dependent ERK activation in both groups at 5 min (Fig. 9C), which has no statistical difference between HEK293 cells expressing WT APJ and APJ-S348A.
Next, we observed the same effect of pertussis toxin (PTX) on apelin-13-induced ERK1/2 phosphorylation in both groups. Pretreatment of HEK293-APJ or APJ-S348A cells with PTX for 12 h, which selectively deactivated Gαi/o-protein to prevent its interaction with the receptor, mostly abrogated the apelin-13-induced ERK1/2 activation (Fig. 9D), indicating that apelin-13-activated ERK1/2 by a G protein-dependent activation pathway was not affected by mutation at Ser-348.
We hypothesized that the ERK1/2 phosphorylation at later time points is mostly independent of G protein activity and regulated by β-arrestins. To test the potential role of β-arrestins, we analyzed the time course of apelin-13-induced ERK1/2 phosphorylation after depleting the cellular levels of β-arrestin1 or β-arrestin2 by transfecting shRNA specifically directed against each isoform. In the presence of a non-targeted control shRNA, apelin-13-induced ERK1/2 phosphorylation in cells expressing APJ is identical to that observed without any shRNA transfection (compare Figs. 9A and 10A). However, the ERK1/2 phosphorylation level was reduced significantly by both β-arrestin1 and β-arrestin2 shRNA individually on WT APJ beyond 15 min (Fig. 10A). The data are confirmed later and slow ERK1/2 activity are β-arrestin dependent. As for the role of β-arrestin shRNA on mutational APJ-S348A, we found that apelin-13-induced ERK1/2 signaling did not differ. These were at a very low level and have no statistically significant difference (Fig. 10B).
FIGURE 10.

Effects of β-arrestin shRNA on APJ stimulated ERK1/2 phosphorylation. HEK293 cells stably expressing APJ (A) or APJ-S348A (B) were transfected with the indicated shRNA. Serum-starved cells were treated with 100 nm apelin-13 and cell lysates were analyzed for ERK1/2 phosphorylation and β-arrestin (C). The densities of the bands corresponding to 44 and 42 kDa were quantified with an imaging densitometer, normalized to total ERK. The data correspond to the mean ± S.E. from four independent experiments. ***, p < 0.01 compared with the control treatment group.
DISCUSSION
The apelin receptor APJ is a member of GPCRs that play an important role in the regulation of cardiovascular function and central control of body fluid homeostasis (9, 10). In view of the extensive function of the apelin/APJ receptor in mammalian physiology and pathophysiology, APJ is rapidly emerging as an important therapeutic target for the treatment of heart failure, hypertension, and obesity-related diseases (12, 13).
Classically, GPCRs undergo a conformational change that regulates heterotrimeric G protein coupling and activation upon ligand binding, which promotes the generation of diffusible second messengers such as cAMP, calcium, or phosphoinositides. Aside from heterotrimeric G proteins, two protein families also specifically interact with the majority of GPCRs to regulate intracellular signaling pathways: GRKs and β-arrestins, which are involved in the control of the desensitization, internalization, and recycling of GPCRs (35). Previous studies found that APJ presented its rapid dissociation from β-arrestins following apelin-13 treatment and activation followed a typical rapid recycling pathway (36). In our studies, BRET assays revealed the kinetics of the recruitment of β-arrestins and GRK2/5 to activated APJ receptor after agonist apelin-13 stimulation.
A specific serine/threonine cluster within the C terminus of most GPCRs has been identified as the key sites that undergo GRK phosphorylation and β-arrestins association (37). To complicate matters, different ligands can favor specific phosphorylation events that raise the possibility of ligand-specific phosphorylation (38). The C terminus of the APJ receptor also possesses multiple serine and threonine residues, which are potential sites of phosphorylation by GRK. However, the key phosphorylation sites that have critical functions remain unknown. To identify key residues that modulate the interaction with GRK and β-arrestins, we have applied LC-MS/MS analytical methods to define the phosphorylated sites of APJ upon stimulation with apelin-13. Subsequently, we utilized site-directed mutagenesis to introduce serine to alanine mutations at the identified phosphorylation sites (serine 335, 345 and 348) and found that mutation at S348A abolishes the association of APJ with GRK2, GRK5, β-arrestin1 or β-arrestin2, and GRK/β-arrestin-dependent internalization after agonist treatment. These results are consistent with previous studies that the internalization of GPCRs is an important process for the regulation of GPCR signaling involving GRK and β-arrestin bindings (39). In general, many activated GPCRs are phosphorylated by GRKs, followed by β-arrestin recruitment and β-arrestin-mediated internalization via clathrin-coated pits (40). In this course, plenty of researches revealed a major role of the C terminus in efficient β-arrestin recruitment and receptor internalization, such as angiotesin II type 1 receptor (41), human neuropeptide Y2 receptor (42), thyrotropin-releasing hormone receptor (43), and bradykinin B2 receptor (44).
The extracellular signal-regulated kinase (ERK) is the best characterized signaling mechanism that can be stimulated by β-arrestins. β-Arrestins can scaffold various members of ERK in complexes with activated GPCRs and promote ERK1/2 activation along (5). The angiotensin type 1A receptor (AT1AR) and protease-activated receptor-2 have been shown to activate ERK in this manner (7, 8). Similarly, suppression of β-arrestin2 using specific small interfering RNA (siRNA) also eliminated the AT1AR-induced ERK activation (45). In addition, some GPCRs have been shown to activate ERK1/2 signaling cascades both in G protein-dependent and G protein-independent (β-arrestins dependent) manners (45). It is also in agreement with the fact that the mechanism of ERK1/2 activation by G protein- and β-arrestin-dependent manners show distinct activation kinetics: G protein-dependent activation is rapid and β-arrestin-dependent activation is slower (45). In a G protein-dependent process, both diacylglycerol accumulation and PKC activation play important roles to trigger downstream signaling within 5 min of stimulation (46), in contrast, G protein-independent (β-arrestins dependent) ERK1/2 activation mainly occurred after 15 min (7, 8). In our results, APJ-S348A failed to recruit β-arrestins. In contrast, it had no demonstrable impact on apelin-induced G protein activation and G protein-dependent downstream signaling. To further determine whether the mutation of APJ at Ser-348 affects activation of ERK1/2 dependence on either G protein or β-arrestins pathways, we observed the kinetic pattern that characterizes ERK1/2 activation by these two pathways. We utilized mutational APJ-S348A (which is incapable of activating β-arrestins) and pertussis toxin (an inhibitor of Gαi), to isolate the two pathways in HEK293 cells. We found that ERK1/2 phosphorylation peaked at 5 min after agonist stimulation and PTX treatment significantly decreased ERK1/2 phosphorylation. These data confirmed that apelin-13 activates ERK1/2 via a G protein-dependent activation pathway. Furthermore, the phosphor-ERK1/2 level after 15 min of APJ with mutation at Ser-348 was significantly lower compared with that of WT APJ. As for the dose-dependent response, apelin-13 (from 0.1 to ∼1000 nm) could induce ERK1/2 signaling activation in a dose-dependent manner in HEK293 cells expressing WT APJ. Moreover, the mutation at Ser-348 abolishes most slow ERK1/2 activation (15 min) at all concentrations, but does not affect fast ERK1/2 activation (5 min). Further studies utilizing shRNA directed against various β-arrestin later confirmed that ERK1/2 activity is β-arrestin dependent. These data suggested that β-arrestin-regulated G protein-independent ERK signaling was affected by point mutation at serine 348, whereas mutational APJ-S348A still triggers the activation of ERK1/2 via the G protein-dependent activation pathway.
The existence of biased ligands has enriched the complex signaling networks of GPCRs. The complexity of their actions provides both challenges and opportunities for drug screening and development. In the case of a biased ligand, binding of a biased ligand to an unbiased receptor results in a biased response such as altering the balance between G protein-dependent and β-arrestin-dependent signal transduction and eliciting differentiated pharmacological effects in vivo (47). For example, the selective β-arrestin-biased ligand of AT1AR [Sar1,d-Ala8]angiotesin II (TRV120027), reduced mean arterial pressure, increased cardiac performance, and preserved cardiac stroke volume in rats, whereas classical angiotensin receptor antagonists decreased overall cardiac performance (48). Furthermore, a G protein-biased ligand TRV130 at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine (49).
Similarly, in the case of a biased receptor, binding of an unbiased ligand to the biased receptor also results in a biased response. Biased receptors that have been generated by mutating key residues involved in G protein coupling or GRK/β-arrestin binding. For example, the AT1A receptor mutant AT1AR (DRY/AAY), in which residues of the highly conserved DRY motif have been mutated to AAY fails to activate classical heterotrimeric G protein signaling but leads to recruitment of β-arrestin2 and activation of ERK1/2 (50). Alanine substitution of AT1R caused a marked reduction of both inositol phosphate signaling and receptor internalization (51). In addition, the β2-adrenergic receptor mutant β2AR (TYY) was incapable of G protein activation, which contains mutations of three residues crucial for G protein coupling (52).
In our studies, we directly detected the ligand-induced phosphorylation sites of APJ and identified their roles by mutagenesis. In view of our mutagenesis results that serine 348 to alanine substitution of APJ abolished its capability to interact with GRK/β-arrestin, but had no change on G protein activation, we now have enough reasons to believe that Ser-348 is the key amino acid important for APJ phosphorylation and trafficking. Meanwhile, APJ-S348A was also incapable of β-arrestin-dependent receptor internalization and ERK1/2 activation. Taken together, APJ-S348A shows the characteristics of a biased receptor, which when attenuated was incapable of β-arrestin coupling. Recently, the dual role of APJ on cardiac hypertrophy has been revealed (14). Apelin-induced Gαi activation has been demonstrated as a benefiting effect on cardiac contractility (53–55), and a vasodilator activity that protects against angiotensin-II-induced cardiovascular fibrosis and atheroma (56, 57). In contrast, stretch causes hypertrophy through diminishing G protein activation, whereas augmenting β-arrestins recruitment. The APJ-mediated GRK/β-arrestin signals induced by stretch signals increase cardiomyocyte cell size and play a crucial role in causing hypertrophy in a G protein-independent fashion (Fig. 11). It has been suggested that identification of biased ligands or receptor, which make G protein-dependent signals of APJ override its G protein-independent signals, could be a productive therapeutic strategy. Taken together with our results, the mutation at Ser-348 of APJ significantly jeopardized the GRK/β-arrestin signals, whereas having little effect on its coupled G protein activation. These findings highlight Ser-348 of APJ as an important target to develop novel treatment without eliciting the negative side effects by GRKs/β-arrestins-dependent signaling.
FIGURE 11.
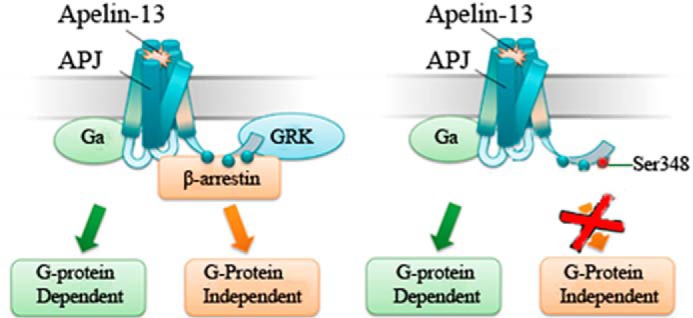
Proposed model on G protein- dependent or β-arrestin-dependent signal transduction of WT and mutant APJ. Apelin-13 binds APJ and triggers intracellular signaling events, including G protein-dependent and G protein-independent signaling pathways (left). Agonist fails to elicit β-arrestin recruitment and terminates β-arrestin-dependent signaling after Ser-348 of APJ was substituted with Ala (right).
In conclusion, the most striking finding of the present study is the identification of a key phosphorylation site in the C terminus of APJ for its binds to GRKs/β-arrestins, which make APJ with mutation at Ser-348 a biased receptor. Pharmacologically, this emphasizes that the key phosphorylation residue of APJ Ser-348 offers the potential for screening the G protein- or β-arrestin-dependent drugs based on the concept of biased signaling and developing therapeutic medicines for cardiovascular and metabolic diseases.
Acknowledgments
We thank Dr. Bee K. Tan, University of Warwick and Dr. Zhongbo Chen, King's College London, for help in preparing the manuscript. We also acknowledge MSc. Jiamiao Hu, University of Warwick, for assistance in drafting this manuscript and preparation of mass spectrometry assay.
This work was supported by National Nature Science Foundation of China Grants 30971081, 31271243, and 81070961, and the Taishan Scholar Construction Special Fund.
- GPCR
- G protein-coupled receptors
- GRK
- G protein-coupled receptor kinase
- APJ
- apelin receptor
- BRET
- bioluminescence resonance energy transfer
- TRITC
- tetramethylrhodamine isothiocyanate
- PTX
- pertussis toxin
- AT1AR
- angiotensin type 1A receptor
- OD
- optical density
- Rluc
- Renilla reniformis luciferase.
REFERENCES
- 1. Bockaert J., Pin J. P. (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 3. Ma P., Zemmel R. (2002) Value of novelty? Nat. Rev. Drug Discov. 1, 571–572 [DOI] [PubMed] [Google Scholar]
- 4. Kohout T. A., Lefkowitz R. J. (2003) Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 63, 9–18 [DOI] [PubMed] [Google Scholar]
- 5. Luttrell L. M., Lefkowitz R. J. (2002) The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 6. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 7. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katugampola S. D., Maguire J. J., Matthewson S. R., Davenport A. P. (2001) [125I]-(Pyr(1))Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br. J. Pharmacol. 132, 1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reaux A., De Mota N., Skultetyova I., Lenkei Z., El Messari S., Gallatz K., Corvol P., Palkovits M., Llorens-Cortès C. (2001) Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J. Neurochem. 77, 1085–1096 [DOI] [PubMed] [Google Scholar]
- 11. Tatemoto K., Hosoya M., Habata Y., Fujii R., Kakegawa T., Zou M. X., Kawamata Y., Fukusumi S., Hinuma S., Kitada C., Kurokawa T., Onda H., Fujino M. (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 251, 471–476 [DOI] [PubMed] [Google Scholar]
- 12. Giddings A. S. R., Thomas J., Tajuba J., Bortoff K., Maitra R. (2010) Development of a functional HTS assay for the APJ receptor. Int. J. High Throughput Screening 1, 39–47 [Google Scholar]
- 13. Castan-Laurell I., Dray C., Attané C., Duparc T., Knauf C., Valet P. (2011) Apelin, diabetes, and obesity. Endocrine 40, 1–9 [DOI] [PubMed] [Google Scholar]
- 14. Scimia M. C., Hurtado C., Ray S., Metzler S., Wei K., Wang J., Woods C. E., Purcell N. H., Catalucci D., Akasaka T., Bueno O. F., Vlasuk G. P., Kaliman P., Bodmer R., Smith L. H., Ashley E., Mercola M., Brown J. H., Ruiz-Lozano P. (2012) APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masri B., Morin N., Pedebernade L., Knibiehler B., Audigier Y. (2006) The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J. Biol. Chem. 281, 18317–18326 [DOI] [PubMed] [Google Scholar]
- 16. Pals-Rylaarsdam R., Gurevich V. V., Lee K. B., Ptasienski J. A., Benovic J. L., Hosey M. M. (1997) Internalization of the m2 muscarinic acetylcholine receptor: arrestin-independent and -dependent pathways. J. Biol. Chem. 272, 23682–23689 [DOI] [PubMed] [Google Scholar]
- 17. Qian H., Pipolo L., Thomas W. G. (2001) Association of β-arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol. Endocrinol. 15, 1706–1719 [DOI] [PubMed] [Google Scholar]
- 18. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., Caron M. G. (1999) Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274, 32248–32257 [DOI] [PubMed] [Google Scholar]
- 19. Bai B., Tang J., Liu H., Chen J., Li Y., Song W. (2008) Apelin-13 induces ERK1/2 but not p38 MAPK activation through coupling of the human apelin receptor to the Gi2 pathway. Acta Biochim. Biophys. Sin. 40, 311–318 [DOI] [PubMed] [Google Scholar]
- 20. Blethrow J. D., Tang C., Deng C., Krutchinsky A. N. (2007) Modular mass spectrometric tool for analysis of composition and phosphorylation of protein complexes. PloS One 2, e358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choi B. K., Cho Y. M., Bae S. H., Zoubaulis C. C., Paik Y. K. (2003) Single-step perfusion chromatography with a throughput potential for enhanced peptide detection by matrix-assisted laser desorption/ionization-mass spectrometry. Proteomics 3, 1955–1961 [DOI] [PubMed] [Google Scholar]
- 22. Li Y., Chen J., Bai B., Du H., Liu Y., Liu H. (2012) Heterodimerization of human apelin and κ opioid receptors: roles in signal transduction. Cell Signal. 24, 991–1001 [DOI] [PubMed] [Google Scholar]
- 23. Hawtin S. R. (2005) Charged residues of the conserved DRY triplet of the vasopressin V1a receptor provide molecular determinants for cell surface delivery and internalization. Mol. Pharmacol. 68, 1172–1182 [DOI] [PubMed] [Google Scholar]
- 24. Chen J., Randeva H. S. (2004) Genomic organization of mouse orexin receptors: characterization of two novel tissue-specific splice variants. Mol. Endocrinol. 18, 2790–2804 [DOI] [PubMed] [Google Scholar]
- 25. Wang C., Pan Y., Zhang R., Bai B., Chen J., Randeva H. S. (2014) Heterodimerization of mouse orexin type 2 receptor variants and the effects on signal transduction. Biochim. Biophys. Acta 1843, 652–663 [DOI] [PubMed] [Google Scholar]
- 26. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 27. See H. B., Seeber R. M., Kocan M., Eidne K. A., Pfleger K. D. (2011) Application of G protein-coupled receptor-heteromer identification technology to monitor β-arrestin recruitment to G protein-coupled receptor heteromers. Assay Drug Dev. Technol. 9, 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hasbi A., Devost D., Laporte S. A., Zingg H. H. (2004) Real-time detection of interactions between the human oxytocin receptor and G protein-coupled receptor kinase-2. Mol. Endocrinol. 18, 1277–1286 [DOI] [PubMed] [Google Scholar]
- 29. Jorgensen R., Holliday N. D., Hansen J. L., Vrecl M., Heding A., Schwartz T. W., Elling C. E. (2008) Characterization of G-protein coupled receptor kinase interaction with the neurokinin-1 receptor using bioluminescence resonance energy transfer. Mol. Pharmacol. 73, 349–358 [DOI] [PubMed] [Google Scholar]
- 30. Namkung Y., Dipace C., Javitch J. A., Sibley D. R. (2009) G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem. 284, 15038–15051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kocan M., Pfleger K. D. (2009) Detection of GPCR/β-arrestin interactions in live cells using bioluminescence resonance energy transfer technology. Methods Mol Biol 552, 305–317 [DOI] [PubMed] [Google Scholar]
- 32. Habata Y., Fujii R., Hosoya M., Fukusumi S., Kawamata Y., Hinuma S., Kitada C., Nishizawa N., Murosaki S., Kurokawa T., Onda H., Tatemoto K., Fujino M. (1999) Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim. Biophys. Acta 1452, 25–35 [DOI] [PubMed] [Google Scholar]
- 33. Masri B., Lahlou H., Mazarguil H., Knibiehler B., Audigier Y. (2002) Apelin(65–77) activates extracellular signal-regulated kinases via a PTX-sensitive G protein. Biochem. Biophys. Res. Commun. 290, 539–545 [DOI] [PubMed] [Google Scholar]
- 34. Yue P., Jin H., Xu S., Aillaud M., Deng A. C., Azuma J., Kundu R. K., Reaven G. M., Quertermous T., Tsao P. S. (2011) Apelin decreases lipolysis via Gq, Gi, and AMPK-dependent mechanisms. Endocrinology 152, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lefkowitz R. J., Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 36. Lee D. K., Ferguson S. S., George S. R., O'Dowd B. F. (2010) The fate of the internalized apelin receptor is determined by different isoforms of apelin mediating differential interaction with β-arrestin. Biochem. Biophys. Res. Commun. 395, 185–189 [DOI] [PubMed] [Google Scholar]
- 37. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., Caron M. G. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J. Biol. Chem. 276, 19452–19460 [DOI] [PubMed] [Google Scholar]
- 38. Butcher A. J., Prihandoko R., Kong K. C., McWilliams P., Edwards J. M., Bottrill A., Mistry S., Tobin A. B. (2011) Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286, 11506–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tobin A. B., Butcher A. J., Kong K. C. (2008) Location, location, location: site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gurevich E. V., Gurevich V. V. (2006) Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kule C. E., Karoor V., Day J. N., Thomas W. G., Baker K. M., Dinh D., Acker K. A., Booz G. W. (2004) Agonist-dependent internalization of the angiotensin II type one receptor (AT1): role of C-terminus phosphorylation in recruitment of β-arrestins. Regul. Pept. 120, 141–148 [DOI] [PubMed] [Google Scholar]
- 42. Walther C., Nagel S., Gimenez L. E., Mörl K., Gurevich V. V., Beck-Sickinger A. G. (2010) Ligand-induced internalization and recycling of the human neuropeptide Y2 receptor is regulated by its carboxyl-terminal tail. J. Biol. Chem. 285, 41578–41590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones B. W., Song G. J., Greuber E. K., Hinkle P. M. (2007) Phosphorylation of the endogenous thyrotropin-releasing hormone receptor in pituitary GH3 cells and pituitary tissue revealed by phosphosite-specific antibodies. J. Biol. Chem. 282, 12893–12906 [DOI] [PubMed] [Google Scholar]
- 44. Zimmerman B., Simaan M., Akoume M. Y., Houri N., Chevallier S., Séguéla P., Laporte S. A. (2011) Role of ssarrestins in bradykinin B2 receptor-mediated signalling. Cell Signal. 23, 648–659 [DOI] [PubMed] [Google Scholar]
- 45. Ahn S., Shenoy S. K., Wei H., Lefkowitz R. J. (2004) Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525 [DOI] [PubMed] [Google Scholar]
- 46. Luttrell L. M., Gesty-Palmer D. (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 62, 305–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Violin J. D., DeWire S. M., Yamashita D., Rominger D. H., Nguyen L., Schiller K., Whalen E. J., Gowen M., Lark M. W. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 [DOI] [PubMed] [Google Scholar]
- 49. DeWire S. M., Yamashita D. S., Rominger D. H., Liu G., Cowan C. L., Graczyk T. M., Chen X. T., Pitis P. M., Gotchev D., Yuan C., Koblish M., Lark M. W., Violin J. D. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344, 708–717 [DOI] [PubMed] [Google Scholar]
- 50. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gáborik Z., Jagadeesh G., Zhang M., Spät A., Catt K. J., Hunyady L. (2003) The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology 144, 2220–2228 [DOI] [PubMed] [Google Scholar]
- 52. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2-adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 53. Szokodi I., Tavi P., Földes G., Voutilainen-Myllylä S., Ilves M., Tokola H., Pikkarainen S., Piuhola J., Rysä J., Tóth M., Ruskoaho H. (2002) Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 91, 434–440 [DOI] [PubMed] [Google Scholar]
- 54. Ashley E. A., Powers J., Chen M., Kundu R., Finsterbach T., Caffarelli A., Deng A., Eichhorn J., Mahajan R., Agrawal R., Greve J., Robbins R., Patterson A. J., Bernstein D., Quertermous T. (2005) The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 65, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jia Y. X., Pan C. S., Zhang J., Geng B., Zhao J., Gerns H., Yang J., Chang J. K., Tang C. S., Qi Y. F. (2006) Apelin protects myocardial injury induced by isoproterenol in rats. Regul. Pept. 133, 147–154 [DOI] [PubMed] [Google Scholar]
- 56. Chun H. J., Ali Z. A., Kojima Y., Kundu R. K., Sheikh A. Y., Agrawal R., Zheng L., Leeper N. J., Pearl N. E., Patterson A. J., Anderson J. P., Tsao P. S., Lenardo M. J., Ashley E. A., Quertermous T. (2008) Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J. Clin. Invest. 118, 3343–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Siddiquee K., Hampton J., Khan S., Zadory D., Gleaves L., Vaughan D. E., Smith L. H. (2011) Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J. Hypertens. 29, 724–731 [DOI] [PMC free article] [PubMed] [Google Scholar]