

Background: NMDA receptor plays roles central to various brain functions.
Results: Glycine, an amino acid widely presents in the brain, induces bidirectional modifications in NMDA receptor-mediated responses.
Conclusion: Trafficking of NMDA receptors at postsynaptic sites may underlie bidirectional modifications in NMDA receptor responses.
Significance: Revealing the molecular mechanism by which glycine dictates direction of persistent modifications in NMDA receptor function.
Keywords: Glycine Receptor, Ionotropic Glutamate Receptor, Membrane Trafficking, Membrane Transport, Synaptic Plasticity, LTD, LTP, NMDA Receptors, Glycine
Abstract
Glycine can persistently potentiate or depress AMPA responses through differential actions on two binding sites: NMDA and glycine receptors. Whether glycine can induce long-lasting modifications in NMDA responses, however, remains unknown. Here, we report that glycine induces long-term potentiation (LTP) or long-term depression (LTD) of NMDA responses (Gly-LTPNMDA or Gly-LTDNMDA) in a dose-dependent manner in hippocampal CA1 neurons. These modifications of NMDA responses depend on NMDAR activation. In addition, the induction of Gly-LTPNMDA requires binding of glycine with NMDARs, whereas Gly-LTDNMDA requires that glycine bind with both sites on NMDARs and GlyRs. Moreover, activity-dependent exocytosis and endocytosis of postsynaptic NMDARs underlie glycine-induced bidirectional modification of NMDA excitatory postsynaptic currents. Thus, we conclude that glycine at different levels induces bidirectional plasticity of NMDA responses through differentially regulating NMDA receptor trafficking. Our present findings reveal important functions of the two glycine binding sites in gating the direction of synaptic plasticity in NMDA responses.
Introduction
Glycine is a multifaceted bioactive molecule in the central nervous system (CNS) and exerts its effect through at least three targets (1, 2). First, glycine is a strychnine-insensitive co-agonist for classical NMDA receptors (NMDARs) and its binding with NMDARs GluN1 subunit is essential for NMDAR activation (3–5). Second, glycine is a primary inhibitory neurotransmitter in certain brain regions such as the spinal cord and brain stem and displays its inhibitory effect through activation of glycine receptors (GlyRs). Enormous evidence over the last decade has suggested that functional GlyRs are present in all regions of the hippocampus and play an important role in regulating cell excitability and synaptic plasticity (6, 7). These strychnine-sensitive GlyRs, if located postsynaptically, are mostly in extrasynaptic sites (8). Third, NMDAR GluN3A subunit has very high affinity for glycine with an apparent dissociation constant less than that of GluN1 (9).
Under most physiological conditions, the glycine concentration in cerebrospinal fluid is estimated to be in the low micromolar range (12). The affinity of glycine for NMDA receptors (EC50 = 0.5 μm) is substantially higher than that of GlyRs (EC50 = 0.67 mm; see Refs. 10 and 11) but lower than that of GluN3A. It seems GluN3A has the priority to bind to glycine. However, compared with the GluN1 and GluN2 subunits of NMDAR, the expression level of GluN3A is much lower in hippocampus (13, 14). Moreover, at CA1 hippocampal synapses GluN3A is only localized at extra- and perisynaptic sites but not postsynaptic sites of the plasma membrane (13, 15). Therefore, in the hippocampus the endogenous glycine released from the presynaptic site is more likely to exert its excitatory effect through postsynaptic NMDAR co-agonist binding (16, 17). Under some pathophysiological conditions, such as brain ischemia and epilepsy, glycine concentration in the synaptic cleft is elevated significantly (18, 19). This raises the possibility that excessive glycine accumulation in the synaptic cleft of the hippocampus can spill over to extrasynaptic sites and reach the dosage threshold of activating the functional GlyRs inhibitory effect. This possible bidirectional effect by glycine is supported by our recent findings that suggest that glycine dictates the direction of synaptic plasticity in a dose-dependent manner (6). Through its binding with NMDARs, glycine at relatively low levels (0.6 mm) in Mg2+-free perfusion medium induces long-term potentiation (LTP)3 of AMPA receptor-mediated excitatory postsynaptic currents (AMPA EPSCs) in CA1 neurons of hippocampal slices. In contrast, glycine at high levels (1.5 mm) activates both GlyRs and NMDARs and produces long-term depression (LTD).
Because NMDAR plays a central role in many brain functions including synaptic plasticity, learning, and memory (20, 21), up- or down-regulation of NMDAR number and/or function may contribute to the bidirectional modifications of AMPA EPSCs indirectly by glycine. Therefore, it is tempting to examine the role of glycine in eliciting persistent modifications of NMDAR-mediated synaptic responses. In the present study, we demonstrate that similar to its effect on AMPA responses, glycine at different levels induces bidirectional modifications in NMDA receptor-mediated synaptic responses in hippocampal CA1 neurons. These effects are mainly attributable to the modulatory effects of glycine on NMDARs and GlyRs. Moreover, activity-dependent exocytosis and endocytosis of postsynaptic NMDARs underlie glycine-induced bidirectional modifications of NMDA responses. Our findings reveal important functions of the two glycine binding sites in gating the direction of synaptic plasticity of NMDA receptor-mediated synaptic responses.
MATERIALS AND METHODS
Electrophysiological Recordings in Hippocampal Slices
Hippocampal slices were prepared from 2–3-week-old male Sprague-Dawley rats. After being anesthetized with ethyl ether and decapitated, the entire brain was removed and 400-μm coronal hippocampal brain slices were cut using a vibrating blade microtome in ice-cold oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mm) 126 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 1.25 KH2PO4, 26 NaHCO3, and 20 glucose (pH 7.4). Slices were stored in warm (34 °C) oxygenated ACSF and recovered for at least 1.5 h before being transferred to the recording chamber.
Whole-cell voltage-clamp recordings were made with patch pipettes containing (in mm): 132.5 Cs-gluconate, 17.5 CsCl, 2 MgCl2, 0.5 EGTA, 10 HEPES, 4 ATP, and 5 QX-314, with pH adjusted to 7.2 by CsOH. Hippocampal slices were continuously superfused in 34 °C ACSF bubbled with 95% O2, 5% CO2. Excitatory postsynaptic responses were evoked by stimulating Schaffer fibers through a constant current pulse delivered using a bipolar tungsten electrode. Synaptic responses were evoked at 0.05 Hz, except during glycine application. EPSCs were recorded at −65 mV. NMDAR-mediated EPSCs (NMDA EPSCs) were isolated with continuous low-Mg2+ (0.25 mm) ACSF perfusion containing bicuculline methiodide (BMI, 10 μm, Tocris) to block GABAA receptor-mediated inhibitory synaptic currents and NBQX (10 μm, Sigma) to block AMPAR-mediated excitatory synaptic currents (AMPA EPSCs). ACSF was always kept in 0.25 mm Mg2+ during the whole recordings. CA1 neurons were viewed under upright microscopy (ECLIPSE E600-FN, Nomasky, Nikon Corporation, Tokyo, Japan) and recorded using an Axopatch-200B amplifier (Molecular Devices, Palo Alto, CA). Changes in NMDA EPSCs amplitude were examined during the last 5 min of recording. Records were low-pass filtered at 2 kHz and acquired at 5–10 kHz. Recordings from each neuron lasted at least 60 to 80 min. The series resistance (Rs) in the recordings varied between 4 and 6 megaohm. For fear of re-sealing the ruptured membrane, changes in kinetics and amplitude of the EPSCs, caused by Rs, were always monitored during recording. Cells in which the Rs or capacitance deviated by >20% from initial values were excluded from the analysis. Cells with Rs >20 megaohm at any time during the recording were also excluded from the analysis. Data were collected with pClamp 9.2 software and analyzed using Clampfit 9.2 (Molecular Devices, Palo Alto, CA).
Immunofluorescence Labeling and Analysis
Fluorescent immunostaining was applied to determine the colocalization of GluN1 and PSD-95 in rat hippocampal slices. 400-μm slices were prepared and incubated for 10 min with glycine (0.6 mm or 1.5 mm). After a 15-min recovery time, slices were fixed in ice-cold 4% paraformaldehyde overnight, dehydrated in 30% sucrose for 24 h at 4 °C, and sectioned at 30–40 μm on a freezing microtome (Leica CM1900). Sections were made permeable in 0.3% Triton X-100 for 60 min (37 °C), then blocked with 10% cattle serum for 60 min. Sections were then incubated with goat anti-GluN1 antibody (1:200, Santa Cruz), mouse anti-PSD-95 antibody (1:100, Millipore) in PBS containing 10% cattle serum at 4 °C for 48 h. After thoroughly washing with PBS, sections were probed with FITC- and TRITC-conjugated secondary antibody (1:500, Jackson ImmunoResearch Laboratories) overnight at 4 °C. The sections were mounted with a mounting medium (Vectashield, Vector Laboratories) after rinsing with PBS.
A confocal imaging system (Olympus FV1000) with a ×60 oil immersion lens was used for image acquisition. A series of optical sections were collected at 0.5-μm steps and a resolution of 1024 × 1024 pixels. Each image was collected by averaging five scans. Because of variability in brightness, it was necessary to use different gain and contrast settings for different cells. To control for this, all measurements were expressed in terms of ratios. All measurements were performed using ImageJ (NIH) software. To quantify the staining, sections from 3 animals were used for quantitative analysis. Generally, 3–4 images of each slice were averaged to determine each value. A close-up view was obtained from one segment of an apical dendrite (about 50–150 μm away from the cell body layer). The data were analyzed with ANOVA LSD for statistical significance and expressed as mean ± S.E.
Subcellular Fractionation and Immunoblotting
Hippocampal slices were prepared as described in electrophysiological recordings. After a 1-h recovery, slices were incubated for 10 min in the presence of glycine (0.6 mm or 1.5 mm). After washing 3 times with fresh ACSF, slices were incubated for 10–15 min. Then slices were stored in liquid nitrogen immediately in cold 0.32 m sucrose containing 1 mm HEPES, 1 mm MgCl2, 1 mm NaHCO3, 20 mm sodium pyrophosphate, 20 mm β-phosphoglycerol, 0.2 mm dithiothreitol, 1 mm EDTA, 1 mm EGTA, 50 mm NaF, 1 mm Na3VO4, 1 mm p-nitrophenyl phosphate (pH 7.4), in the presence of protease inhibitors and phosphatase inhibitors: 1 mm phenylmethylsulfonyl fluoride (PMSF), 5 μg/ml of aprotinin, 5 μg/ml of leupeptin, 5 μg/ml of pepstatin A, and 16 μg/ml of benzamidine. The homogenate was centrifuged at 1,000 × g for 10 min. 200 μl were removed from the total protein fractions (H) and the remaining supernatant was centrifuged at 3,000 × g for 15 min to obtain the fraction of mitochondria and synaptosomes. The pellet was resuspended in 8 ml of hypotonic buffer with the presence of protease inhibitors and centrifuged at 100,000 × g for 1 h and then suspended again in 8 ml of buffer containing 75 mm KCl and 1% Triton X-100 and centrifuged at 100,000 × g for 1 h. The final pellet was homogenized 3 times in 20 mm HEPES. This fraction is regarded as a Triton-insoluble fraction. The Triton-insoluble fraction was used instead of the classical PSD (45).
Equal amounts of protein (20 μg) were separated by 10% SDS-PAGE and electrotransferred onto nitrocellulose membranes (0.45 mm; BioTrance NT, Ann Arbor, MI) for immunoblotting. Membranes were blocked with 3% (w/v) BSA (fraction V) in wash buffer (10 mm Tris, pH 7.4, 0.1% Tween 20 (w/v), and 100 mm NaCl) for 1 h at room temperature. Blotted proteins were probed with primary antibodies goat anti-GluN1 (1:800, Santa Cruz) and rabbit anti-tubulin (1:3000, CWBio). Signals were generated by enhanced chemiluminescent reagent (ECL, BioWorld), according to the manufacturer's protocol, and visualized by exposing with the Bio-Rad system. Quantification was performed using ImageJ. Results were expressed as fold versus control. Proteins were separated by SDS-PAGE using precast 7–10% gradient gels and blotted onto nitrocellulose filter (NC) membranes.
Peptide Studies
All drugs were bath applied in ACSF at 32 °C. SNAP-25 C-terminal peptides (the blocking peptide was Tat-MEKADANKTRI and the scrambled peptide was Tat-KANAKTDEIRM, 10 μm, ChinaPeptides Co., Ltd.) and the dynamin inhibitory peptide were used (Tat-QVPSRPNRAP and control peptide QVPSRPNRAP, 50 μm, ChinaPeptides Co., Ltd.).
Pharmacology
Channel blockers, including BMI (Tocris), NBQX (Sigma), dl-AP5 (Sigma), L689560 (Sigma), and strychnine (Sigma) and competitive antagonists TeTx (Sigma) and D15 (Tocris) were used.
Data Analysis
All populations are described as mean ± S.E. Data were analyzed by analysis of variance (ANOVA) or Student's t test. Within-group comparisons were performed using paired-sample t tests, and differences between groups were compared using independent-sample t test and ANOVA post hoc comparisons. A one-way ANOVA LSD test was used when equal variances were assumed. Differences were considered significant when p < 0.05 (*, p < 0.05; **, p < 0.01).
RESULTS
Glycine-induced Bidirectional Modification in NMDA EPSCs
To examine whether glycine induces changes in NMDAR-mediated synaptic responses, we performed whole-cell patch clamp recordings of evoked NMDAR-mediated excitatory postsynaptic currents (NMDA EPSCs) in CA1 pyramidal neurons in hippocampal slices. NMDA EPSCs were recorded at a −65 mV holding potential in a low-Mg2+ (0.25 mm) ACSF to relieve blockade of NMDARs by Mg2+. The ACSF perfusion medium contains BMI (10 μm) and NBQX (10 μm) to block GABAA and AMPA receptor-mediated synaptic currents, respectively (Fig. 1A). Following 10 min of stable baseline EPSCs recording, 0.6 mm glycine was applied for 10 min and then washed out immediately. Changes in EPSCs amplitude were examined during the last 10 min of recording. We found that 0.6 mm glycine significantly increased NMDA EPSCs and induced LTP of NMDA EPSCs (Gly-LTPNMDA, normalized amplitudes 1.31 ± 0.06, n = 5, p < 0.01, paired-samples t test; Fig. 1, B and F). This persistent modification in NMDAR-mediated synaptic responses was reproducible and consistent (i.e. observed in more than 80% of recorded cells), except when exogenous glycine reached 1.0 mm, in which no persistent change in NMDA EPSCs was observed (n = 6, p = 0.07). Further increasing the glycine concentration to 1.5 mm produced LTD of NMDA EPSCs (Gly-LTDNMDA, 0.62 ± 0.06, n = 6; p < 0.01, paired-samples t test). Notably, Gly-LTDNMDA was not due to the rundown of NMDA EPSCs caused by the deterioration of the recorded neurons (data not shown) or to an adverse effect on the recording of the neurons during glycine treatment, because glycine at this concentration does not display toxic effects on nerve cells (22–24). The above results suggest that exogenously applied glycine at different levels induces persistent bidirectional modifications in NMDAR-mediated synaptic responses.
FIGURE 1.

Glycine-induced bidirectional modifications in NMDA EPSCs. A, sample traces showing isolation and identification of evoked NMDAR-mediated currents using antagonists of AMPAR (NBQX, 10 μm), GABAAR (BMI, 10 μm), and NMDAR (dl-AP5, 50 μm). B, exogenous glycine applied at different concentrations displays different effects on evoked NMDA EPSCs. A 10-min treatment of 0.6 mm glycine caused significant potentiation of evoked NMDAR-mediated EPSCs when cells were held at −65 mV in a conventional whole cell patch configuration (n = 5). When the concentration of glycine was increased to 1.0 mm, no persistent change in NMDA EPSCs was observed (n = 6). In contrast, 1.5 mm glycine treatment induced LTD of NMDAR-mediated EPSCs (n = 6). Overlaid traces above the graph show changes in averaged NMDA EPSCs chosen at the times indicated. Unless stated otherwise, ACSF was always kept at a constant of 0.25 mm Mg2+ during the entire recording for this and the other figures. C and D, endogenous glycine at different concentrations displays different effects on evoked NMDA EPSCs. Blocking GlyT1 with specific GlyT1 antagonist NFPS (0.2 μm) in low-Mg2+ ACSF for 10 min, which increased endogenous glycine levels in the synaptic cleft, induced LTP of NMDA EPSCs (C; n = 8). In contrast, blocking GlyT1 with NFPS (2.0 mm) for 10 min produced LTD of NMDA EPSCs (D; n = 6). Extending the application time of NFPS (0.2 μm) from 10 to 30 min led to more endogenous glycine accumulation in the synaptic cleft and induced LTD of EPSCs (C; n = 5). Shortening the application time of NFPS (2.0 mm) from 10 to 4 min led to reduced endogenous glycine accumulation in the synaptic cleft and induced LTP of EPSCs (D; n = 6). E, acute NFPS treatment failed to affect NMDA EPSCs. Sample traces showing the absence of changes in evoked NMDA EPSCs upon brief puffing of NFPS at 0.20 (top panel) or 2.0 μm (top panel) concentrations. F, summary of data comparing persistent changes in evoked NMDA EPSCs induced by glycine or NFPS across a range of concentrations (glycine, 0.6, 1.0, and 1.5 mm, marked in black; NFPS, 0.2 and 2.0 μm, marked in red). *, p < 0.05; **, p < 0.01, compared with control, ANOVA LSD test.
We then investigated whether such changes in NMDA EPSCs could also be reproduced by endogenous glycine. To test this possibility, we inhibited endogenous glycine re-uptake with a selective blocker of type 1 glycine transporters (24, 25) to observe how the accumulation of extracellular glycine could affect NMDA EPSCs. A high-affinity glycine transporter type 1 (GlyT1) was reported in glial cells and glutamatergic neurons (26). N-[3-([1,1-Biphenyl]-4-yloxy)-3-(4-fluorophenyl)propyl]-N-methylglycine (NFPS) is a specific and potent GlyT1 blocker and displays dose-dependent GlyT1 inhibition (27). A 10-min application of a low concentration of NFPS (0.2 μm), which should cause a relatively low level of endogenous glycine accumulation in extracellular milieu, induced LTP of NMDA EPSCs (1.32 ± 0.06, n = 8, p < 0.01; Fig. 1, C and F). In contrast, high concentrations of NFPS (2.0 μm) in low Mg2+ perfusion medium elicited a persistent depression of NMDA EPSCs (0.83 ± 0.06, n = 6, p < 0.01, paired samples t test; Fig. 1, D and F). These results are consistent with the above data on exogenous glycine and provide further support to the notion that glycine at different levels induces bidirectional modifications in NMDA EPSCs.
To further confirm that the observed changes in NMDA EPSCs upon NFPS treatment are caused by accumulation of extracellular glycine at different levels, we modulated the duration of NFPS treatment. The assumption is that if the duration of NFPS treatment is extended to 0.2 μm, glycine accumulation will be higher, and, as a result, the direction of modification in NMDA EPSCs may change. To examine this assumption, we switch the 0.2 μm NFPS perfusing time from 10 to 30 min. We indeed observed Gly-LTDNMDA rather than Gly-LTPNMDA (0.89 ± 0.15, n = 5, p < 0.05; Fig. 1, C and E). In contrast, shortening the duration of 2.0 μm NFPS treatment from 10 to 4 min, which should lead to less extracellular glycine accumulation, induced Gly-LTPNMDA rather than Gly-LTDNMDA (1.47 ± 0.14, n = 6, p < 0.01; Fig. 1, D and E). These results re-emphasize that the amount of extracellular glycine is a critical factor that dictates the direction of glycine-induced synaptic plasticity in NMDA EPSCs.
Modification of NMDA EPSCs Depends on NMDAR Activation
To examine whether the induction of Gly-LTPNMDA and Gly-LTDNMDA requires the activation of NMDARs, we co-applied the competitive NMDAR antagonist dl-AP5 (50 μm) with glycine. Under this treatment, we failed to detect any obvious changes in NMDA EPSCs (0.6 mm Gly + AP5, 0.96 ± 0.13, n = 6, compared with baseline, p = 0.09, paired samples t test; 1.5 mm Gly + AP5, 1.02 ± 0.18, n = 6, compared with baseline, p = 0.45, paired samples t test; Fig. 2, A and C). In addition, we performed another parallel set of experiments in ACSF with normal Mg2+ concentration (1.0 mm) perfusion solution only when glycine was applied. Because of the Mg2+ block of NMDARs, this treatment could block the selective activation of synaptic NMDARs. Similar to the above results using AP5, no Gly-LTPNMDA or Gly-LTDNMDA were detected (0.6 mm Gly, 0.99 ± 0.09, n = 6, compared with baseline, p = 0.72, paired samples t test; 1.5 mm Gly, 0.98 ± 0.03, n = 6, compared with baseline, p = 0.07, paired samples t test; Fig. 2, B and C). These data strongly suggest that both LTPNMDA and LTDNMDA induced by glycine at different concentrations depend on NMDAR activation.
FIGURE 2.
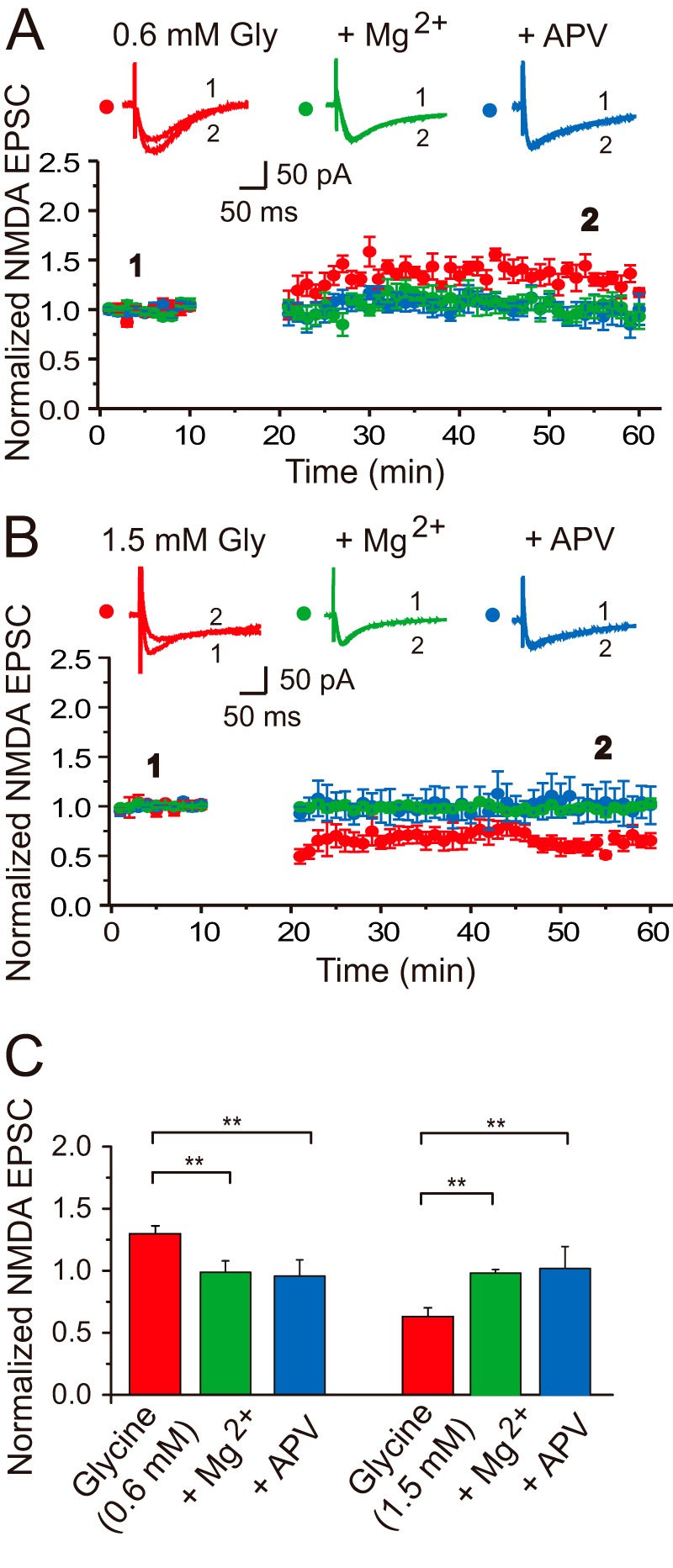
Glycine-induced bidirectional modifications in NMDA EPSCs require NMDAR activation. A, LTP of NMDA EPSCs depends on NMDAR activity. Gly-LTPNMDA was completely abolished by the competitive NMDAR antagonist dl-AP5 (50 μm, n = 6) or normal Mg2+ ACSF (n = 6) co-applied with glycine (0.6 mm). Sample traces above the graph show averaged NMDA EPSCs chosen at the times indicated. The plotting data for Gly-LTPNMDA was borrowed from Fig. 1B for comparison. B, LTD of NMDA EPSCs depends on NMDAR activity. Gly-LTDNMDA was completely abolished by the competitive NMDAR antagonist dl-AP5 (50 μm, n = 6) or normal Mg2+ ACSF (n = 6) co-applied with glycine (1.5 mm). The plotting data for Gly-LTDNMDA was borrowed from Fig. 1B for comparison. C, statistical plotting of data displaying the effects of antagonizing NMDAR functions by AP5 or Mg2+ on glycine-induced bidirectional modifications in NMDA EPSCs. **, p < 0.01, compared between indicated groups.
NMDAR Trafficking Underlies Modification in NMDA EPSCs
An important mechanism underlying persistent modification in neurotransmission is to change the receptor number at postsynaptic sites. To examine whether the glycine-induced changes in NMDA EPSCs are caused by alteration of postsynaptic NMDAR number, we performed immunofluorescence assays in hippocampal slices to determine the amount of synaptic NMDARs by quantifying colocalization of NMDARs with PSD-95, a postsynaptic marker. We detected a significant increase and decrease in postsynaptic expression of NMDARs shortly after 0.6 and 1.5 mm glycine treatments, respectively (normalized GluN1 intensity, 1.37 ± 0.03, n = 14; 0.69 ± 0.02, n = 14, compared with control, p < 0.01, one-way ANOVA LSD test; Fig. 3, A and B). This observation is further supported with Western blot data. Using a Western blot assay of the Triton-insoluble fraction of hippocampal tissue, which roughly represents the subcellular fraction at postsynaptic sites (28, 29), we found a dramatic elevation and decline of postsynaptic expression of NMDARs after 0.6 and 1.5 mm glycine treatments, respectively (1.24 ± 0.08, p < 0.01, n = 7, one-way ANOVA LSD test; 0.83 ± 0.04, p < 0.01, n = 7, one-way ANOVA LSD test; Fig. 3, E and F). Taken together, these results demonstrate that glycine at different levels induces bidirectional changes in NMDAR expression at postsynaptic sites.
FIGURE 3.

Glycine-induced bidirectional changes in postsynaptic localization of NMDARs. A, different effects of glycine on postsynaptic NMDAR localization: immunofluorescence evidence. 0.6 mm glycine treatment promotes postsynaptic localization of the GluN1 subunit, indicated by increased colocalization with PSD-95, whereas 1.5 mm glycine treatment decreased postsynaptic localization of GluN1 subunit (n = 14). Bar, 20 μm. Higher magnification of dendritic branches are in lower panels. Bar, 2 μm. B, statistical plots of data in A. C, low level of GluN3A expression at different developmental stages: Western blot assay. D, when 5-fold hippocampal tissues were harvested for the Western blot assay, developmental changes in GluN3A expression from P2 to adult were detected. E, different effects of glycine on postsynaptic NMDAR expression: Western blot evidence (n = 7). F, statistical plots of data in E. **, p < 0.01, compared with control, one-way ANOVA LSD test.
GluNR3A is another potential target of glycine. Therefore, glycine might also induce changes in GluN3A expression at postsynaptic sites, which in turn contributes to the overall change in NMDAR expression. To examine this possibility, we first determined the expression level of GluN3A using whole hippocampal tissue. We found that the GluN3A expression level is very low and hard to detect when using a regular amount of hippocampal tissues for the protein level assay (Fig. 3C; see Ref. 13). When we harvested 5-fold hippocampal tissues, however, we do detect GluN3A expression (Fig. 3D). Interestingly, we observed developmental changes in GluN3A expression from P2 to adult, which is consistent with previous findings (14, 30). Moreover, when we determined the GluN3A level in the Triton X-100-insoluble fraction of hippocampal tissue (P20), which roughly represents the subcellular fraction at postsynaptic sites, we did not detect any obvious changes in GluN3A expression (1.02 ± 0.02, p = 0.38, n = 7, one-way ANOVA LSD test; 1.01 ± 0.03, p = 0.75, n = 7, one-way ANOVA LSD test; Fig. 3, E and F). These results are also consistent with previous findings that GluN3 predominantly locates in extra- and perisynaptic sites of the hippocampus (15). Taking into consideration the fact that the other GluN3 subunit GluN3B is mainly expressed in motor neurons (31), these results suggest that GluN3 does not contribute to glycine-induced changes in NMDAR expression level.
How could these changes occur in postsynaptic expression of NMDARs following glycine treatment? It has been reported that receptor trafficking through exocytosis or endocytosis is a major route to adding or decreasing the receptor number at the postsynaptic surface (32). We determined next whether bidirectional trafficking of NMDARs contributes to glycine-induced bidirectional modification of NMDA EPSCs. To test this possibility, we loaded cells with the specific SNARE-dependent exocytosis inhibitor tetanus toxin (TeTx, 0.1 mm) by including it in the pipette solution during whole cell recording and examined whether Gly-LTPNMDA could be induced under this condition (33, 34). We found that TeTx completely blocked Gly-LTPNMDA induction (0.99 ± 0.06, n = 6; p > 0.05; Fig. 4, A and C). To further confirm our observation, we used a peptide corresponding to the C-terminal region of SNAP-25 to acutely disturb the association of endogenous SNAP-25 with its SNARE complex binding partners, syntaxin-1 and VAMP2. This interfering peptide (corresponding to residues 182–192 of SNAP-25) contains the nonphosphorylatable residue Ala in place of Ser at residue 187 flanked on either side by 5 amino acids (35). Loading interfering peptide (10 μm) in the recording pipette totally abolished Gly-LTPNMDA (Fig. 4, A and C). In contrast, when we used a scrambled peptide with the same residue composition in a randomized order that renders it incapable of blocking excytosis, glycine could still induce potentiation of NMDA currents (1.34 ± 0.08, n = 6, p < 0.01). These results indicate that SNARE-dependent NMDAR exocytosis is responsible for increased surface expression of NMDARs.
FIGURE 4.

NMDAR trafficking underlies glycine-induced modification in NMDA EPSCs. A, Gly-LTPNMDA was abolished by infusing SNARE-dependent exocytosis inhibitor TeTx (0.1 mm) or SNAP-25 C-terminal peptides (10 μm) in the pipette solution. Overlaid traces above the graph show changes in averaged NMDA EPSCs under various conditions chosen at the times indicated. The plotting data for Gly-LTPNMDA was borrowed from Fig. 1B for comparison. B, Gly-LTDNMDA was reversed to Gly-LTPNMDA by infusing dynamin-dependent endocytosis inhibitor D15 (0.1 mm) or dynamin inhibitory peptide (50 μm) in the pipette solution. The magnitude of resultant Gly-LTPNMDA is comparable with that of Gly-LTPNMDA induced by 0.6 mm glycine. The plotting data for Gly-LTDNMDA was borrowed from Fig. 1B for comparison. C, summary of data displaying changes in NMDA EPSCs following treatments of various exocytosis or endocytosis blockers. **, p < 0.01, compared between the indicated groups. D, inhibiting exocytosis reversed the enhancement in postsynaptic NMDAR expression induced by glycine at 0.6 mm (n = 5). As a control, scramble peptide failed to display a similar effect (n = 5). Pep., SNAP-25 peptide; Scr., scramble peptide. E, inhibiting endocytosis reversed the decrease in postsynaptic NMDAR expression induced by glycine at 1.5 mm (n = 5). The control cell impermeable membrane peptide failed to display a similar effect (n = 5). T, Tat-QVPSRPNRAP peptide; Q, QVPSRPNRAP peptide.
To examine the possible contribution of NMDAR endocytosis in the induction of Gly-LTDNMDA, we loaded cells with an inhibitor of dynamin-dependent endocytosis termed D15 during recordings. D15 is a cell membrane-impermeable peptide that possesses the PPPQVPSRRNRAPPG sequence and can specifically interfere with the binding of dynamin to amphiphysin (36–38). Unexpectedly, we found that D15 (2 mm) reversed the polarity of synaptic plasticity from Gly-LTDNMDA to Gly-LTPNMDA (1.35 ± 0.08, n = 6, p < 0.01; Fig. 4, B and C), with the resultant potentiation magnitude comparable with Gly-LTPNMDA induced by 0.6 mm glycine alone (p > 0.05). To further confirm this observation, we also made use of a dynamin inhibitory peptide, QVPSRPNRAP, which also competitively binds to the amphiphysin SH3 domain and prevents endocytosis when applied intracellularly (7, 39, 40). Tat-QVPSRPNRAP is a membrane-permeable form of this peptide. Tat protein (Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg), which was obtained originally from the cell membrane transduction domain of the human immunodeficiency virus type 1 (HIV-1), was fused to the constructed peptides and resulted in the fusion peptide Tat-QVPSRPNRAP. This manipulation allowed the constructed peptides to easily cross the membrane and exert their effects intracellularly (41). Consistent with the results using D15, briefly perfusing slices with both 1.5 mm glycine and interfering peptide (50 μm) resulted in Gly-LTPNMDA (1.34 ± 0.12, n = 7, p < 0.01; Fig. 4, B and C). As a negative control, we applied the non-membrane-permeable form of the peptide (without the TAT) to bath solution and found that it did not affect the internalization of NMDARs (0.69 ± 0.08, n = 7, p < 0.01). These results indicate that dynamin-dependent NMDAR endocytosis underlies Gly-LTDNMDA. The reversal of plasticity polarity may be caused by competition between the dichotomous effects (excitatory versus inhibitory or exocytosis versus endocytosis) mediated by the two glycine binding sites. When endocytosis or the suppressive effect was removed, the exocytosis or excitatory effect was uncovered. Taken together, our observation strongly suggests that bidirectional NMDAR trafficking, which results in an increase or decrease of the number of postsynaptic NMDARs, is responsible for glycine-induced bidirectional modifications of NMDA EPSCs.
If these interfering peptides do disturb intracellular NMDAR trafficking, then we should observe correlated changes in NMDAR expression at postsynaptic sites. To examine this possibility, we determined the postsynaptic NMDAR expression under various treatments. We found a significant elevation of NMDAR expression after 0.6 mm glycine treatment for 20 min (1.33 ± 0.11, n = 5, p < 0.05; Fig. 4D). Pretreatment of the receptor exocytosis inhibitor SNAP-25 peptide (10 μm) reversed this enhancement (1.04 ± 0.05, n = 5, p = 0.44). As a control, scramble peptide failed to display a similar effect. In contrast, 1.5 mm glycine induced a decrease in NMDAR expression (0.81 ± 0.04, n = 5, p < 0.01; Fig. 4E). Notably, this decrease was reversed to elevation in NMDAR expression upon pretreatment with the endocytosis inhibitor, 50 μm Tat-QVPSRPNRAP (1.10 ± 0.04, n = 5, p < 0.05). This result is consistent with our electrophysiological observations showing reversal from Gly-LTDNMDA to Gly-LTPNMDA (Fig. 4B).
The reversal of Gly-LTDNMDA to Gly-LTPNMDA by blocking endocytosis could be caused by a counter NMDAR exocytosis. If this assumption is true, then we can make the two following predictions: first, inhibiting NMDAR exocytosis upon 1.5 mm glycine application would induce Gly-LTDNMDA with greater depression magnitude; second, the Gly-LTPNMDA that was reversed from Gly-LTDNMDA upon endocytosis blockade would be abolished by further treatment with the exocytosis blocker. To test these predictions, we applied either TeTx or SNAP25 in the recording pipette solution upon 1.5 mm glycine application (0.70 ± 0.07, n = 6, compared with baseline, p < 0.01; 0.50 ± 0.12, n = 5, p < 0.01; 0.50 ± 0.10, n = 6, p < 0.01; Fig. 5, A and B). Changes in EPSCs amplitude were examined during the last 10 min of recording. We indeed detected Gly-LTDNMDA with a greater depression magnitude. In addition, we co-applied TeTx or SNAP25 peptide with D15 in the recording pipette solution to identify whether competing trafficking pathways were operating. We found that the reversed Gly-LTPNMDA upon D15 treatment alone was abolished and neither Gly-LTPNMDA nor Gly-LTDNMDA was induced by 1.5 mm glycine (0.94 ± 0.10, n = 6, compared with baseline, p > 0.05; Fig. 5, C and D), indicating that NMDAR exocytosis contributes to the reversed Gly-LTPNMDA. Moreover, D15 alone did not display any obvious effects on basal NMDA responses (1.01 ± 0.07, n = 6, p = 0.25; Fig. 5, E and F). These observations highly support our predictions and provide new evidence to the notion that NMDAR exocytosis and endocytosis underlie Gly-LTPNMDA and Gly-LTDNMDA, respectively, and these two receptor trafficking processes may compete with each other under glycine application.
FIGURE 5.

The reversal of Gly-LTDNMDA to Gly-LTPNMDA was abolished by further inhibiting exocytosis with either TeTx or SNAP-25. A, inhibiting NMDAR exocytosis with either TeTx (n = 5) or SNAP-25 (n = 6) interfering peptide upon 1.5 mm glycine application induced Gly-LTDNMDA with a greater depression magnitude. The plotting data for Gly-LTDNMDA was borrowed from Fig. 1B for comparison. B, statistical plots of data in A. Changes in EPSCs amplitude were examined during the last 10 min of recording. C, co-applied TeTx or SNAP25 peptide with D15 in the recording pipette solution abolished reversed Gly-LTPNMDA upon D15 treatment alone. Neither Gly-LTPNMDA nor Gly-LTDNMDA was induced by 1.5 mm glycine under these conditions, indicating that the reversal of Gly-LTDNMDA to Gly-LTPNMDA was caused by a counter NMDAR exocytosis uncovered after blocking endocytosis (n = 6). D, statistical plots of data in A. **, p < 0.01, compared with control, one-way ANOVA LSD test. E, D15 alone failed to display any obvious effects on basal NMDA response (n = 6). F, statistical plots of data in E, compared with control, one-way ANOVA LSD test.
Role of Glycine Binding Sites in Modification of NMDA EPSCs
In the hippocampus, glycine can exert its action NMDAR co-agonist binding site (site B) and extrasynaptic GlyR site (site A), which mediate excitatory and inhibitory actions, respectively. Because NMDARs have a substantially higher affinity for glycine than GlyRs (10, 11), the potentiation effect by glycine at a relatively low level is more likely via its action on NMDAR co-agonist binding site (site B). As the level of glycine increases, it may further activate GlyRs (site A) largely located at the extrasynaptic site and display an inhibitory effect in hippocampal neurons. We assumed that this dichotomous property of glycine may take a role in bidirectional modification of NMDA EPSCs. To examine this hypothesis, we used antagonists of the two glycine binding sites to determine whether and how the observed changes in NMDA EPSCs could be affected by these blockers. A specific glycine site B antagonist, L689560 (60 μm), was applied together with glycine in low-Mg2+ ACSF. As a result, Gly-LTPNMDA was completely abolished (0.89 ± 0.13, n = 6, compared with baseline, p > 0.05, paired samples t test; Fig. 6, A and C). However, in slices treated with the specific GlyR antagonist strychnine (1 μm), no significant change in the magnitude of Gly-LTPNMDA was detected (1.32 ± 0.10, n = 6, compared with baseline, p < 0.01, paired-samples t test; Fig. 6, A and C). Thus, the induction of Gly-LTPNMDA was dependent on glycine binding with NMDARs.
FIGURE 6.

Role of glycine binding sites in modifications of NMDA EPSCs induced by exogenous glycine application. A, Gly-LTPNMDA was abolished by blocking the binding between glycine and NMDAR with the specific antagonist L689560 (60 μm; n = 6), but was unaffected by GlyR antagonist strychnine (1 μm; n = 6). Overlaid traces above the graph show changes in averaged NMDA EPSCs under various conditions chosen at the times indicated. The plotting data for Gly-LTPNMDA was borrowed from Fig. 1B for comparison. B, Gly-LTDNMDA was abolished by blocking the binding between glycine and NMDAR with the specific antagonist L689650 (60 μm; n = 6) but was reversed to Gly-LTPNMDA upon treatment of strychnine (1 μm; n = 6). Notably, Gly-LTDNMDA was abolished but not reversed to Gly-LTPNMDA by co-application of L689560 (L689) with strychnine (L689 + strychnine). The plotting data for Gly-LTDNMDA was borrowed from Fig. 1B for comparison. C, summary of data displaying the effects of L689560 and strychnine on Gly-LTPNMDA and Gly-LTDNMDA. ** p < 0.01, compared between the indicated groups. D and E, the effects of antagonists for the two glycine binding sites on NMDAR expression at postsynaptic sites. Pretreatment of L689560 (60 μm) reversed the enhancement of postsynaptic NMDAR expression induced by 0.6 mm glycine (D; n = 4), whereas pretreatment of strychnine (1 μm) reversed the attenuation of postsynaptic NMDAR expression induced by 1.5 mm glycine (E; n = 5).
Because glycine at high levels may exert its effect on both the NMDAR co-agonist binding site (site B) and GlyRs (site A), we further investigated the mechanisms underlying Gly-LTDNMDA and determined how Gly-LTDNMDA was affected by the antagonists of the two glycine binding sites. Interestingly, we found that Gly-LTDNMDA was totally abolished by L689560 (60 μm) but was reversed to Gly-LTPNMDA upon treatment of strychnine (1 μm; 1.02 ± 0.11, n = 6, paired samples t test, p > 0.05; 1.19 ± 0.12, n = 6, compared with baseline, p < 0.01; Fig. 6, B and C), indicating that GlyRs was also involved in Gly-LTDNMDA induction. In addition, co-application of the two antagonists displayed similar blocking effects on Gly-LTDNMDA without further reversing Gly-LTDNMDA to Gly-LTPNMDA (Fig. 6, B and C). This was not unexpected, because L689560 blocks glycine binding at NMDARs, which was also required for Gly-LTPNMDA induction.
To further examine whether these antagonists for the two binding sites take their roles through changing the number of NMDAR at postsynaptic sites, we determined the postsynaptic NMDAR expression under treatments of these antagonists. We found pretreatment of L689560 (60 μm) reversed the enhancement of NMDAR expression induced by 0.6 mm glycine (1.17 ± 0.04, n = 4, p < 0.05; 0.91 ± 0.05, n = 4, p = 0.35; 1.16 ± 0.02, n = 4, p < 0.05; Fig. 6D), whereas pretreatment of strychnine (1 μm) reversed the attenuation of NMDAR expression induced by 1.5 mm glycine (0.79 ± 0.11, n = 5, p < 0.05; 1.02 ± 0.02, n = 5, p = 0.50; 1.13 ± 0.02, n = 5, p < 0.01; Fig. 6E).
To confirm the role of these two binding sites in glycine-induced modifications of NMDA EPSCs, we next examined how the antagonists for these binding sites affect Gly-LTPNMDA and Gly-LTDNMDA induced by endogenous glycine accumulation following GlyT1 blockade. LTP elicited by 10 min perfusion of NFPS (0.2 μm) was abolished by L689560 (60 μm; 0.96 ± 0.17, n = 5, compared with control, p < 0.01; Fig. 7, A and C), whereas Gly-LTDNMDA elicited by 10 min perfusion of NFPS (2.0 μm) was reversed to Gly-LTPNMDA by strychnine (1 μm; 1.64 ± 0.19, n = 5, p < 0.01; Fig. 7, B and C). Combined with the above results, these findings provide strong evidence that glycine induces bidirectional modifications of NMDA response by separately targeting the two glycine binding sites. Taking into consideration the accompanying alteration of postsynaptic NMDAR expression upon treatment of these antagonists, we propose glycine-induced changes in NMDAR trafficking and subsequent changes in postsynaptic NMDAR number may contribute to the modification of NMDA responses.
FIGURE 7.
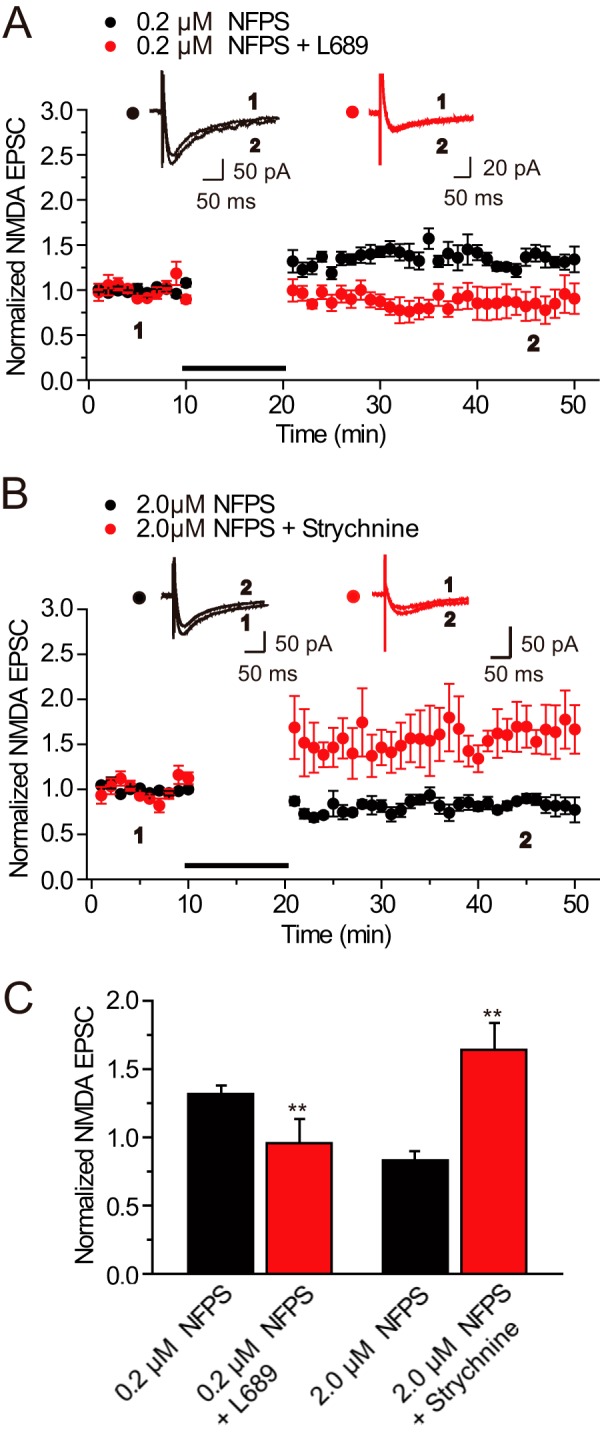
Role of glycine binding sites in modifications of NMDA EPSCs induced by endogenous glycine accumulation. A, Gly-LTPNMDA induced by endogenous glycine accumulation following 10 min perfusion of NFPS (0.2 μm) was totally abolished by L689560 (60 μm, n = 5). B, Gly-LTDNMDA induced by endogenous glycine accumulation following a 10-min perfusion of NFPS (2.0 μm) was reversed to Gly-LTPNMDA upon treatment of strychnine (1 μm, n = 5). The plotting data for Gly-LTDNMDA was borrowed from Fig. 1B for comparison. C, statistical plots of data in A and B.
DISCUSSION
NMDARs mainly serve as the molecular trigger for synaptic changes. Activity-dependent changes in NMDAR-mediated synaptic strength are of great importance because they involve potentiation or depression of the molecular trigger itself (42). In the present study, we demonstrate that glycine, an important amino acid released by both glial cells and glutamatergic neurons, elicits persistent bidirectional modifications in NMDAR-mediated synaptic responses at different concentrations. Further investigation elucidated the mechanisms by which glycine exerts its role in the synaptic plasticity of NMDAR-mediated synaptic responses.
Glycine has been reported to potentiate the NMDA response through an allosteric activation of NMDARs in cultured mouse brain neurons (4). This effect is observed when glycine was co-applied with NMDA. It is still unclear whether this potentiation effect is acute or longer in duration. Our present findings that glycine at a relative low level can induce long-lasting facilitation of NMDA EPSCs is the first demonstration of persistent modification of NMDA EPSCs by glycine. These findings, however, are in clear contrast to a previous study (43). In that study, glycine at a similar dosage induced LTP of AMPA receptor-mediated synaptic responses but failed to display significant effects on miniature NMDAR-mediated synaptic responses (mEPSCsNMDA). This inconsistency could be caused by difficulty in detecting changes in relatively smaller spontaneous EPSC amplitudes, as well as greater variability of NMDA versus AMPA responses (44). The small change (<30%) in mEPSCsNMDA is not easily detected. Compared with mEPSCsNMDA, evoked NMDA EPSCs recorded in neurons in slices are usually much larger with relatively smaller variability.
Glycine exerts its inhibitory effect mainly through the activation of GlyRs and the subsequent opening of GlyR-gated chloride channels. In the present study, we show that, in contrast to the induction of LTPNMDA by glycine at relatively low concentrations, glycine at high concentrations produces LTDNMDA. We further demonstrate that decreasing the NMDAR number through endocytosis underlies the LTDNMDA. Persistent suppression of NMDA EPSCs can be blocked by either L689560 or strychnine, suggesting GlyRs may also have a role in this effect. One study has revealed that stimulation of the glycine site can initiate signaling through the NMDAR complex and prime the receptors for clathrin-dependent endocytosis (7). Our present observation of glycine-induced NMDA EPSC suppression and NMDAR endocytosis supports this finding. Notably, the priming effects of glycine in that study were observed in the persistent presence of the GlyRs blocker strychnine, thus failing to monitor the possible involvement of GlyRs in the effect of glycine on the NMDAR-mediated function. Here we demonstrate that the activation of GlyR by glycine has additional contributions to the glycine-induced NMDAR endocytosis as well as suppression of NMDAR function. Thus, it would be expected that antagonizing glycine binding to both sites upon a high level glycine treatment may uncover the potentiation and excitatory effects on NMDAR. However, because glycine-induced potentiation in NMDAR currents require glycine binding at the NMDAR site, the resultant NMDA currents fail to display any obvious change. Moreover, it is still unclear how the effect of GlyR activation is transduced to NMDAR internalization.
Our present results strongly suggest that the concentration is a very important determinant for the effect of glycine on NMDAR-mediated neurotransmission. This may be largely attributable to differences in the affinity of glycine for the NMDAR co-agonist binding site (site B) and GlyRs (site A). NMDA receptors have a substantially higher affinity for glycine than GlyRs (10, 11). When the level of endogenous glycine is low, it typically prefers to act through the NMDAR co-agonist binding site. Under this condition, glycine induces potentiation of NMDAR function (Fig. 8A). When the level of glycine gets higher and crosses the “set point” of the NMDAR endocytosis mechanism, it may activate GlyRs (Fig. 8B). This increase of the glycine level could occur under some pathophysiological states, such as seizure and ischemia. As a result, the inhibitory effect may be stronger than the excitatory effect and the net effect mediated by glycine is a depression of the NMDA response.
FIGURE 8.
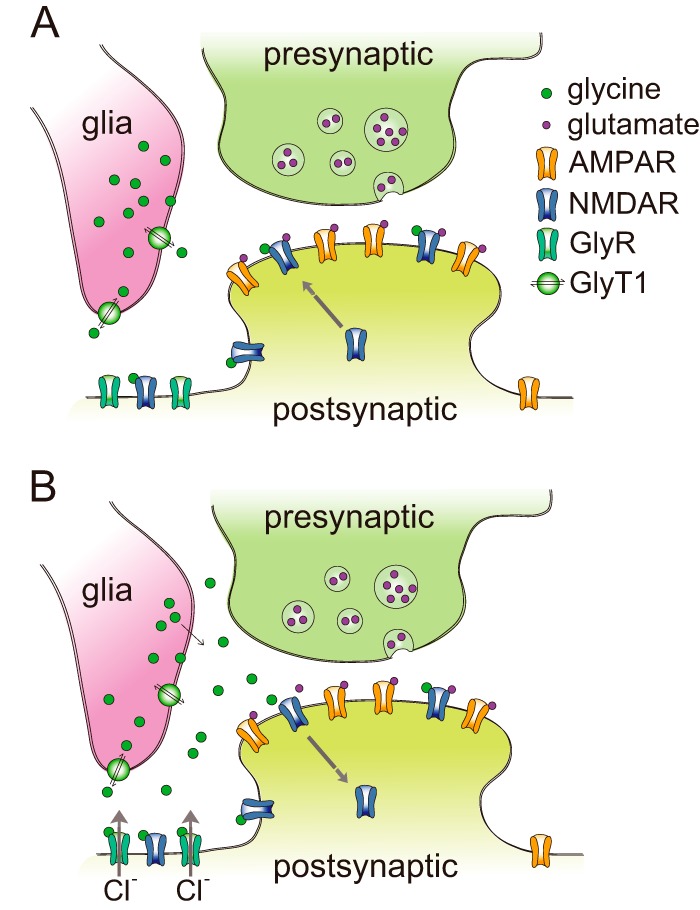
A model of dose-dependent bidirectional modifications of NMDA responses by glycine. A, under physiological conditions, the level of endogenous glycine is usually low. Glycine prefers to act through the NMDAR co-agonist binding site to induce NMDAR exocytosis and resultant potentiation of NMDAR function. B, under some pathophysiological states, the dysfunction of the glycine transporter causes accumulation of glycine in the synaptic cleft. Excessive glycine can then be recruited to two additional sites. Glycine spills over to neighboring extrasynaptic sites and activates GlyRs located there. As a result, glycine induces NMDAR endocytosis and depression of NMDAR response.
Our findings reveal that glycine is more than a co-agonist for NMDAR and may also bind to both GlyRs and NMDARs and induce NMDAR endocytosis. Because NMDARs are involved in a variety of physiological and pathological processes throughout the central nervous system, the bidirectional modifications of NMDA function by glycine we observed here may have a general implication in understanding the regulation of nervous system function in normal and disease states.
Acknowledgments
We thank Dr. Yong Li and Jingjing Wang for technical support.
This work was supported by National Natural Science Foundation of China (NSFC) Grant 31025011, Major State Basic Research Program of China Grant 2013CB733801, the Doctoral Fund of the Ministry of Education of China Grant 20103234110004, and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University (to W. L.).
- LTP
- long-term potentiation
- LTD
- long-term depression
- EPSC
- excitatory postsynaptic currents
- ACSF
- artificial cerebrospinal fluid
- BMI
- bicuculline methiodide
- NBQX
- 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- TRITC
- tetramethylrhodamine isothiocyanate
- ANOVA
- analysis of variance
- NFPS
- N-[3-([1,1-biphenyl]-4-yloxy)-3-(4-fluorophenyl)propyl]-N-methylglycine
- TeTx
- tetanus toxin
- GlyT1
- glycine transporter type 1.
REFERENCES
- 1. Keck T., White J. A. (2009) Glycinergic inhibition in the hippocampus. Rev. Neurosci. 20, 13–22 [DOI] [PubMed] [Google Scholar]
- 2. Xu T. L., Gong N. (2010) Glycine and glycine receptor signaling in hippocampal neurons: diversity, function and regulation. Prog. Neurobiol. 91, 349–361 [DOI] [PubMed] [Google Scholar]
- 3. Danysz W., Parsons C. G. (1998) Glycine and N-methyl-d-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol. Rev. 50, 597–664 [PubMed] [Google Scholar]
- 4. Johnson J. W., Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531 [DOI] [PubMed] [Google Scholar]
- 5. Kemp J. A., Leeson P. D. (1993) The glycine site of the NMDA receptor: five years on. Trends Pharmacol. Sci. 14, 20–25 [DOI] [PubMed] [Google Scholar]
- 6. Chen R. Q., Wang S. H., Yao W., Wang J. J., Ji F., Yan J. Z., Ren S. Q., Chen Z., Liu S. Y., Lu W. (2011) Role of glycine receptors in glycine-induced LTD in hippocampal CA1 pyramidal neurons. Neuropsychopharmacology 36, 1948–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nong Y., Huang Y. Q., Ju W., Kalia L. V., Ahmadian G., Wang Y. T., Salter M. W. (2003) Glycine binding primes NMDA receptor internalization. Nature 422, 302–307 [DOI] [PubMed] [Google Scholar]
- 8. Danglot L., Rostaing P., Triller A., Bessis A. (2004) Morphologically identified glycinergic synapses in the hippocampus. Mol. Cell. Neurosci. 27, 394–403 [DOI] [PubMed] [Google Scholar]
- 9. Yao Y., Mayer M. L. (2006) Characterization of a soluble ligand binding domain of the NMDA receptor regulatory subunit NR3A. J. Neurosci. 26, 4559–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chattipakorn S. C., McMahon L. L. (2002) Pharmacological characterization of glycine-gated chloride currents recorded in rat hippocampal slices. J. Neurophysiol. 87, 1515–1525 [DOI] [PubMed] [Google Scholar]
- 11. Vyklický L., Jr., Benveniste M., Mayer M. L. (1990) Modulation of N-methyl-d-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J. Physiol. 428, 313–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Westergren I., Nyström B., Hamberger A., Nordborg C., Johansson B. B. (1994) Concentrations of amino acids in extracellular fluid after opening of the blood-brain barrier by intracarotid infusion of protamine sulfate. J. Neurochem. 62, 159–165 [DOI] [PubMed] [Google Scholar]
- 13. Pérez-Otaño I., Luján R., Tavalin S. J., Plomann M., Modregger J., Liu X. B., Jones E. G., Heinemann S. F., Lo D. C., Ehlers M. D. (2006) Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat. Neurosci. 9, 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong H. K., Liu X. B., Matos M. F., Chan S. F., Pérez-Otaño I., Boysen M., Cui J., Nakanishi N., Trimmer J. S., Jones E. G., Lipton S. A., Sucher N. J. (2002) Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J. Comp. Neurol. 450, 303–317 [DOI] [PubMed] [Google Scholar]
- 15. Yuan T., Bellone C. (2013) Glutamatergic receptors at developing synapses: the role of GluN3A-containing NMDA receptors and GluA2-lacking AMPA receptors. Eur. J. Pharmacol. 719, 107–111 [DOI] [PubMed] [Google Scholar]
- 16. Li Y., Krupa B., Kang J. S., Bolshakov V. Y., Liu G. (2009) Glycine site of NMDA receptor serves as a spatiotemporal detector of synaptic activity patterns. J. Neurophysiol. 102, 578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Sacchi S., Pollegioni L., Basu A. C., Coyle J. T., Bolshakov V. Y. (2013) Identity of endogenous NMDAR glycine site agonist in amygdala is determined by synaptic activity level. Nat. Commun. 4, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Globus M. Y., Busto R., Martinez E., Valdés I., Dietrich W. D., Ginsberg M. D. (1991) Comparative effect of transient global ischemia on extracellular levels of glutamate, glycine, and γ-aminobutyric acid in vulnerable and nonvulnerable brain regions in the rat. J. Neurochem. 57, 470–478 [DOI] [PubMed] [Google Scholar]
- 19. Lasley S. M. (1991) Roles of neurotransmitter amino acids in seizure severity and experience in the genetically epilepsy-prone rat. Brain Res. 560, 63–70 [DOI] [PubMed] [Google Scholar]
- 20. Constantine-Paton M., Cline H. T. (1998) LTP and activity-dependent synaptogenesis: the more alike they are, the more different they become. Curr. Opin. Neurobiol. 8, 139–148 [DOI] [PubMed] [Google Scholar]
- 21. Zoghbi H. Y., Gage F. H., Choi D. W. (2000) Neurobiology of disease. Curr. Opin. Neurobiol. 10, 655–660 [DOI] [PubMed] [Google Scholar]
- 22. Barth A., Nguyen L. B., Barth L., Newell D. W. (2005) Glycine-induced neurotoxicity in organotypic hippocampal slice cultures. Exp. Brain Res. 161, 351–357 [DOI] [PubMed] [Google Scholar]
- 23. Wallis R. A., Panizzon K. L., Nolan J. P. (1995) Glycine-induced CA1 excitotoxicity in the rat hippocampal slice. Brain Res. 685, 115–125 [PubMed] [Google Scholar]
- 24. Zhang L. H., Gong N., Fei D., Xu L., Xu T. L. (2008) Glycine uptake regulates hippocampal network activity via glycine receptor-mediated tonic inhibition. Neuropsychopharmacology 33, 701–711 [DOI] [PubMed] [Google Scholar]
- 25. Martina M., Gorfinkel Y., Halman S., Lowe J. A., Periyalwar P., Schmidt C. J., Bergeron R. (2004) Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J. Physiol. 557, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cubelos B., Giménez C., Zafra F. (2005) Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb. Cortex 15, 448–459 [DOI] [PubMed] [Google Scholar]
- 27. Tsen G., Williams B., Allaire P., Zhou Y. D., Ikonomov O., Kondova I., Jacob M. H. (2000) Receptors with opposing functions are in postsynaptic microdomains under one presynaptic terminal. Nat. Neurosci. 3, 126–132 [DOI] [PubMed] [Google Scholar]
- 28. Caputi A., Gardoni F., Cimino M., Pastorino L., Cattabeni F., Di Luca M. (1999) CaMKII-dependent phosphorylation of NR2A and NR2B is decreased in animals characterized by hippocampal damage and impaired LTP. Eur. J. Neurosci. 11, 141–148 [DOI] [PubMed] [Google Scholar]
- 29. Gardoni F., Bellone C., Cattabeni F., Di Luca M. (2001) Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J. Biol. Chem. 276, 7609–7613 [DOI] [PubMed] [Google Scholar]
- 30. Henson M. A., Roberts A. C., Pérez-Otaño I., Philpot B. D. (2010) Influence of the NR3A subunit on NMDA receptor functions. Prog. Neurobiol. 91, 23–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chatterton J. E., Awobuluyi M., Premkumar L. S., Takahashi H., Talantova M., Shin Y., Cui J., Tu S., Sevarino K. A., Nakanishi N., Tong G., Lipton S. A., Zhang D. (2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415, 793–798 [DOI] [PubMed] [Google Scholar]
- 32. Mabb A. M., Ehlers M. D. (2010) Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell Dev. Biol. 26, 179–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kang N., Xu J., Xu Q., Nedergaard M., Kang J. (2005) Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 94, 4121–4130 [DOI] [PubMed] [Google Scholar]
- 34. Tojima T., Akiyama H., Itofusa R., Li Y., Katayama H., Miyawaki A., Kamiguchi H. (2007) Attractive axon guidance involves asymmetric membrane transport and exocytosis in the growth cone. Nat. Neurosci. 10, 58–66 [DOI] [PubMed] [Google Scholar]
- 35. Lau C. G., Takayasu Y., Rodenas-Ruano A., Paternain A. V., Lerma J., Bennett M. V., Zukin R. S. (2010) SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 30, 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lüscher C., Xia H., Beattie E. C., Carroll R. C., von Zastrow M., Malenka R. C., Nicoll R. A. (1999) Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24, 649–658 [DOI] [PubMed] [Google Scholar]
- 37. Morishita W., Marie H., Malenka R. C. (2005) Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat. Neurosci. 8, 1043–1050 [DOI] [PubMed] [Google Scholar]
- 38. Peng Y., Zhao J., Gu Q. H., Chen R. Q., Xu Z., Yan J. Z., Wang S. H., Liu S. Y., Chen Z., Lu W. (2010) Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus 20, 646–658 [DOI] [PubMed] [Google Scholar]
- 39. Kittler J. T., Delmas P., Jovanovic J. N., Brown D. A., Smart T. G., Moss S. J. (2000) Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J. Neurosci. 20, 7972–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lissin D. V., Gomperts S. N., Carroll R. C., Christine C. W., Kalman D., Kitamura M., Hardy S., Nicoll R. A., Malenka R. C., von Zastrow M. (1998) Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 95, 7097–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aarts M., Liu Y., Liu L., Besshoh S., Arundine M., Gurd J. W., Wang Y. T., Salter M. W., Tymianski M. (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298, 846–850 [DOI] [PubMed] [Google Scholar]
- 42. Lau C. G., Zukin R. S. (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 [DOI] [PubMed] [Google Scholar]
- 43. Lu W., Man H., Ju W., Trimble W. S., MacDonald J. F., Wang Y. T. (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254 [DOI] [PubMed] [Google Scholar]
- 44. Han L., Campanucci V. A., Cooke J., Salter M. W. (2013) Identification of a single amino acid in GluN1 that is critical for glycine-primed internalization of NMDA receptors. Mol. Brain 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yan J. Z., Xu Z., Ren S. Q., Hu B., Yao W., Wang S. H., Liu S. Y., Lu W. (2011) Protein kinase C promotes N-methyl-d-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. J. Biol. Chem. 286, 25187–25200 [DOI] [PMC free article] [PubMed] [Google Scholar]