

Background: Phosphorylation and sumoylation are post-translational modifications of the Parkinson disease protein α-synuclein.
Results: α-Synuclein inclusion clearance is impaired in yeast when sumoylation is inhibited; phosphorylation of α-synuclein can compensate SUMO impairment.
Conclusion: Sumoylation stimulates autophagy clearance of α-synuclein inclusions, whereas phosphorylation promotes autophagy and proteasome degradation.
Significance: A complex molecular post-translational cross-talk is required in yeast to clear toxic inclusions.
Keywords: α-Synuclein, Autophagy, Post-translational Modification, Proteasome, Yeast
Abstract
Parkinson disease is associated with the progressive loss of dopaminergic neurons from the substantia nigra. The pathological hallmark of the disease is the accumulation of intracytoplasmic inclusions known as Lewy bodies that consist mainly of post-translationally modified forms of α-synuclein. Whereas phosphorylation is one of the major modifications of α-synuclein in Lewy bodies, sumoylation has recently been described. The interplay between α-synuclein phosphorylation and sumoylation is poorly understood. Here, we examined the interplay between these modifications as well as their impact on cell growth and inclusion formation in yeast. We found that α-synuclein is sumoylated in vivo at the same sites in yeast as in human cells. Impaired sumoylation resulted in reduced yeast growth combined with an increased number of cells with inclusions, suggesting that this modification plays a protective role. In addition, inhibition of sumoylation prevented autophagy-mediated aggregate clearance. A defect in α-synuclein sumoylation could be suppressed by serine 129 phosphorylation by the human G protein-coupled receptor kinase 5 (GRK5) in yeast. Phosphorylation reduced foci formation, alleviated yeast growth inhibition, and partially rescued autophagic α-synuclein degradation along with the promotion of proteasomal degradation, resulting in aggregate clearance in the absence of a small ubiquitin-like modifier. These findings suggest a complex interplay between sumoylation and phosphorylation in α-synuclein aggregate clearance, which may open new horizons for the development of therapeutic strategies for Parkinson disease.
Introduction
Parkinson disease (PD)3 is the second most common neurodegenerative disorder after Alzheimer disease. Pathologically, it is characterized by loss of dopaminergic neurons in the substantia nigra pars compacta of the brain and the accumulation of cytoplasmic inclusions termed Lewy bodies (1, 2). Lewy bodies are composed of different proteins such as α-synuclein, ubiquitin, Synphilin-1, or cytoskeletal proteins (3). The small neuronal protein α-synuclein (αSyn) consists of 140 amino acids and represents the major component of Lewy bodies (4). In addition, mutations (5–7) and multiplications (8) of the SNCA gene, coding for αSyn, cause familial forms of PD, further supporting the involvement of αSyn in pathogenesis. However, the precise molecular mechanisms underlying αSyn toxicity are still unclear. Several studies reported that αSyn is subjected to various post-translational modifications that can alter αSyn inclusion formation and cytotoxicity (9). These include sumoylation, phosphorylation, ubiquitination (10–12), or nitration (13, 14).
It has been shown that sumoylation negatively regulates αSyn aggregation by promoting its solubility (15). Besides αSyn, there are additional examples of proteins involved in neurodegenerative diseases that are SUMO targets (16, 17). The predominant αSyn phosphorylation site (>90%) is serine 129 (Ser-129) in Lewy bodies (18, 19). Several kinases such as G protein-coupled receptor kinases or Polo-like kinases 1–3 and casein kinases 1 and 2 can phosphorylate αSyn on Ser-129 in human cells (18–24). Phosphorylation of αSyn by GRK5 plays a crucial role in the pathogenesis of PD (25). PLK2 is the most efficient Polo-like kinase phosphorylating αSyn on Ser-129 (26–28). The role of αSyn phosphorylation under physiological conditions and in inclusion formation and pathogenesis remains controversial. In Alzheimer disease, increased Tau phosphorylation can stimulate its sumoylation (29). There is also additional evidence indicating that the cross-talk between phosphorylation and sumoylation can affect substrates in different ways (30), suggesting this might also modulate αSyn function, distribution, and/or aggregation.
The molecular mechanisms involved in the clearance of αSyn aggregates is a central question for elucidating the αSyn-related toxicity. Soluble αSyn can be targeted to the 26 S proteasome for degradation (31–34) or can be degraded by the autophagy-lysosomal pathway (33–36). The budding yeast Saccharomyces cerevisiae has been extensively used as a powerful system to study the basic molecular mechanisms involved in αSyn-mediated cytotoxicity (37–40). We showed that aggregate clearance of αSyn depends mainly on the autophagy pathway (38). Here, we addressed the question of whether the cross-talk between specific post-translational modifications of αSyn modulates the processing of inclusions through degradation by autophagy or the proteasome. For the first time, we demonstrate an interplay between αSyn sumoylation and phosphorylation to control protein turnover. αSyn is sumoylated in yeast cells at the same site as in human cells and can be efficiently phosphorylated on Ser-129 by the heterologously expressed human G protein-coupled receptor kinase 5 (GRK5). Interestingly, we found that sumoylation exhibits a protective role against αSyn toxicity and inclusion formation, and likewise, phosphorylation alleviates αSyn-mediated toxicity in SUMO-deficient cells by partially rescuing autophagic aggregate clearance and promoting proteasome-mediated degradation of αSyn. Altogether, our findings support that a deeper understanding of the interplay between different post-translational modifications in αSyn might open novel opportunities for therapeutic intervention in PD and other synucleinopathies.
EXPERIMENTAL PROCEDURES
Yeast Strains, Plasmids, Transformation, and Growth Conditions
Plasmids and S. cerevisiae strains are listed in Tables 1 and 2. Wild-type (WT) αSyn encoding the cDNA sequence (referred to as SNCA) or the A30P mutant sequence was cloned into the integrative pRS306 and pRS304 vectors or into the yeast pRS426 overexpression vector (41) preceded by the GAL1 promoter and followed by CYC1 terminator. The K96R/K102R mutant constructs and the S129A mutant were generated by site-directed mutagenesis using Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies). Plasmids pME3945 and pME3597 were used as templates for generation of the desired amino acid substitutions. Human kinases GRK5 and PLK2 were cloned into the SmaI restriction site of pME2792 yeast vector proceeded by the GPD1 and GAL1 promoter, respectively. All constructs were analyzed by sequencing. For microscopy analysis, all αSyn variants were tagged at the C terminus with GFP via the KLID linker (38).
TABLE 1.
Yeast plasmids used in this study
| Plasmid | Description | Source |
|---|---|---|
| pME2795 | pRS426-GAL1-promoter, CYC1-terminator, URA3, 2 μm, pUC origin, AmpR | 38 |
| pME3764 | pME2795 with GAL1::SNCAA30P::GFP (KLID linker) | 38 |
| pME3759 | pME2795 with GFP | 38 |
| pME3760 | pME2795 with GAL1::SNCAWT | 38 |
| pME3945 | pRS306 with SNCAWT::GFP (KLID linker), CYC1-terminator, URA3, integrative, pUC origin, AmpR | 38 |
| pME3596 | pRS304 with GAL1-promoter, CYC1-terminator, TRP1, integrative, pUC origin, AmpR | This study |
| pME3597 | pME3596 with GAL1::SNCAWT | This study |
| pME3598 | pME3596 with GAL1::SNCAA30P | This study |
| pME4089 | pME3596 with GAL1:: SNCAK96R/K102R | This study |
| pME4090 | pME3596 with GAL1:: SNCAA30P/K96R/K102R | This study |
| pME4091 | pRS306 with GAL1:: SNCAK96R/K102R ::GFP (KLID linker), CYC1-terminator, URA3, integrative, pUC origin, AmpR | This study |
| pME2792 | pRS423 -GAL1-promoter, CYC1-terminator, HIS3, 2 μm, pUC origin, AmpR | This study |
| pME4092 | pME2792 with GAL1::PLK2 | This study |
| pME4093 | pME2792 with GPD::GRK5 | This study |
| pME4094 | pME3596 with GAL1:: SNCAS129A::GFP (KLID linker) | This study |
| pME4095 | pME2795 with GAL1::SNCAWT::HIS6 | This study |
| pME4097 | pME2795 with GAL1:: SNCAK96R/K102R::GFP (KLID linker) | This study |
| D1374 | YIplac211-ADH1::HIS6::SMT3 | 82 |
TABLE 2.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| W303-1A | MAT a; ura3-1; trp1-1; leu2–3_112; his3-11; ade2-1; can1-100 | EUROSCARF |
| BY4741 | Mat a; his3D1; leu2D0; met15D0; ura3D0 | EUROSCARF |
| Δatg1 | BY4741; Mat a; his3D1; leu2D0; met15D0; ura3D0; YGL180W::kanMX4 | EUROSCARF |
| Δatg7 | BY4741; Mat a; his3D1; leu2D0; met15D0; ura3D0; YHR171W::kanMX4 | EUROSCARF |
| smt3ts | S542: MAT α, smt3-331 | 48 |
| RH3601 | smt3ts containing two genomic copies GAL1::SNCAWT::GFP in URA3 locus | This study |
| RH3602 | W303 containing two genomic copies GAL1::SNCAK96R/K102R::GFP in URA3 locus | This study |
| ulp1ts | 82 | |
| RH3603 | ulp1ts containing YIplac211-ADH-His6-Smt3 in HIS3 locus | This study |
| RH3604 | RH3603 containing GAL1::SNCAWT integrated in TRP1 locus | This study |
| RH3605 | RH3603 containing GAL1::SNCAA30P integrated in TRP1 locus | This study |
| RH3606 | RH3603 containing GAL1::SNCAK96R/K102R integrated in TRP1 locus | This study |
| RH3607 | smt3ts containing 2 genomic copies GAL1::SNCAS129A::GFP in TRP1 locus | This study |
S. cerevisiae strains W303-1A, smt3ts, and ulp1ts were used for transformations performed by standard lithium acetate protocol (42). Transformations into the temperature-sensitive smt3ts and ulp1ts strains were performed at 25 °C. All strains were grown in synthetic complete medium (SC) (43) lacking the nutrient amino acid (uracil, histidine, or tryptophan) corresponding to the marker, and supplemented with 2% raffinose or 2% galactose. αSyn expression was induced by shifting yeast cells cultivated overnight in raffinose to galactose medium (A600 = 0.1).
Spotting Assay
For growth test on solid medium, yeast cells were pre-grown in minimal medium containing 2% raffinose lacking the corresponding marker to mid-log phase. Cells were normalized to equal densities, serially diluted 10-fold starting with an A600 of 0.1, and spotted on SC plates containing either 2% glucose or 2% galactose and lacking the corresponding marker. smt3ts mutant cells were incubated at permissive temperature (25 °C) and restrictive temperature (30 °C). After 3 days of incubation, the plates were photographed.
Fluorescence Microscopy and Quantifications
Wild-type (W303-1A) yeast cells harboring αSyn were grown in SC selective medium containing 2% raffinose at 30 °C and smt3ts mutant cells at 25 °C overnight and transferred to 2% galactose containing medium for induction of αSyn expression for 6 h. Smt3ts mutant cells were induced at 25 and 30 °C. Fluorescent images were obtained with Zeiss Observer Z1 microscope equipped with CSU-X1 A1 confocal scanner unit (YOKOGAWA), QuantEM: 512SC (Photometrics) digital camera, and SlideBook 5.0 software package (Intelligent Imaging Innovations). For quantification of aggregation, at least 300 cells were counted per strain and per experiment. The number of cells presenting inclusions was referred to the total number of cells counted. The values are the mean of at least three independent experiments.
Immunoblotting
Wild-type (W303-1A) yeast cells harboring αSyn were pre-grown at 30 °C in SC selective medium containing 2% raffinose. Cells were transferred to SC medium containing 2% galactose at A600 = 0.1 to induce the GAL1 promoter for 5 h. Smt3ts cells harboring αSyn were preincubated at 25 °C and later transferred to either 25 or 30 °C. Total protein extracts were prepared, and the protein concentrations were determined with a Bradford assay. 10 μg of each protein were subjected to 12% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were probed with mouse αSyn monoclonal antibody (AnaSpec, Fremont, CA), rabbit αSyn polyclonal antibody (Santa Cruz Biotechnology), or SUMO rabbit antibody (Rockland Immunochemicals Inc.). Rabbit cdc28 polyclonal antibody (Santa Cruz Biotechnology) or mouse monoclonal GAPDH antibody (Thermo Fisher Scientific) were used as loading controls. For detection of phosphorylated αSyn, mouse Ser-129 phospho-specific antibody (Wako Chemicals USA, Inc., Richmond, VA) was used.
Quantifications of Western Blots
Pixel density values for Western quantification were obtained from TIFF files generated from digitized x-ray films (Kodak) and analyzed with the ImageJ software (44). Before comparison, sample density values were normalized to the corresponding loading control. The adjusted density values were standardized to the control lane to get fold increase. The significance of differences was calculated using Student's t test or one-way ANOVA with Bonferroni's multiple comparison test. p value < 0.05 was considered to indicate a significant difference.
Ni2+-NTA Affinity Chromatography
Ulp1ts mutant cells carrying GAL1-SNCA integrations and His6-tagged Smt3 (His6-smt3) were pre-grown in 200 ml of SC medium containing 2% raffinose at 30 °C overnight. Total cells harvested by centrifugation were transferred to 2 liters of YEPD liquid medium containing 2% galactose for 12 h of induction. Cells were collected and lysed by 25 ml of 1.85 m NaOH containing 7.5% β-mercaptoethanol for 10 min on ice. Protein was precipitated in 25 ml of 50% trichloroacetic acid (TCA) and washed with 100% cold acetone. Proteins were suspended in 25 ml of buffer A (6 m guanidine HCl, 100 mm sodium phosphate, 10 mm Tris/HCl, pH 8.0) and rotated for 1 h at 25 °C. The supernatant was cleared by centrifugation; the pH was adjusted to 7.0 by 1 m Tris base and supplemented with imidazole to a final concentration of 20 mm. After equilibration of the His GraviTrap column (GE Healthcare) with 5 ml of buffer A containing 20 mm imidazole, proteins were applied to the column, and the flow-through fraction was collected for analysis. The column was washed with buffer A supplemented with 20 mm imidazole and then with buffer B (8 m urea, 100 mm sodium phosphate, 10 mm Tris, pH 6.3). Then the column was washed with buffer C (50 mm Tris, pH 8.0, 300 mm NaCl, 20 mm imidazole). Finally, the proteins were eluted four times with 1 ml of 200 mm imidazole resolved in buffer C. Protein concentration in the eluted fractions was determined with Bradford assay.
Promoter Shutoff Assays and Chemical Treatments
Yeast cells carrying αSyn were pre-grown in SC selective medium containing 2% raffinose overnight and then shifted to 2% galactose SC selective medium to induce the αSyn expression for 5 h. Then cells were shifted to SC medium supplemented with 2% glucose to shut off the promoter. At several time points after promoter shutoff, cells were visualized by fluorescence microscopy. For experiments with temperature-sensitive yeast strain smt3ts, preincubation was performed at 25 °C. Induction of αSyn expression and the promoter shutoff assay were performed at 25 and 30 °C. The reduction of the number of cells displaying αSyn inclusions was recorded and plotted on a graph. Drugs used in this study were carbobenzoxyl-leucinyl-leucinyl-leucinal (MG132) dissolved in dimethyl sulfoxide (DMSO) and phenylmethanesulfonyl fluoride (PMSF) in ethanol (EtOH). Drug treatment was conducted concomitantly with the shift to glucose-supplemented medium in promoter shutoff assays. PMSF was added to the cell suspension to a final concentration of 1 mm. An equal volume of ethanol was added to the cells as a control (45). MG132 treatment was performed as described previously (46). MG132 was applied to the cell suspension in a final concentration of 75 μm, and in parallel, an equal volume of DMSO was added to the cells as a control.
Immunoprecipitation
100 μg of protein purified by Ni2+-NTA was incubated with primary antibody (ubiquitin mouse monoclonal antibody, Millipore) at 4 °C for 2 h in Immunoprecipitation (IP) buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA) with freshly added 6 mm protease inhibitor mixture (Roche Applied Science), 2 mm DTT, 0.1% phosphatase inhibitor (Roche Applied Science) and then incubated with prewashed protein A-Sepharose beads in IP buffer overnight at 4 °C. Beads were washed three times with ice-cold IP buffer; the immunoprecipitated protein was dissolved from the beads by heating in 1× sample loading buffer at 95 °C for 10 min, and the samples were subjected to Western blot analyses using rabbit αSyn polyclonal antibody (Santa Cruz Biotechnology).
Southern Hybridization and Copy Number Determination
Several transformants were analyzed by Southern hybridization for verification of the integration of αSyn-GFP construct into the mutated genomic ura3-1 locus. Isolation of genomic DNA from S. cerevisiae was performed according to standard procedures. 10 μg of genomic DNA were subjected to restriction digestion with HindIII. The restriction fragments were resolved on a 1% agarose gel, transferred to a nitrocellulose membrane, cross-linked by UV irradiation for 5 min, and hybridized to a URA3 gene fragment probe. Copy numbers of the integrated vector were estimated according to the profile of the restriction fragments. One copy corresponded to 2.7 + 4.7 kb and two copies to 2.7 + 4.7 + 6.2 kb. For integration of αSyn-GFP into the mutated genomic trp1-1 locus, 10 μg of genomic DNA were subjected to restriction digestion with EcoRI. One copy of the integrated vector corresponded to 1.9 + 4.2-kb restriction digestion fragments and two copies to 1.9 + 4.2 + 4.6 kb.
RESULTS
αSyn Is Sumoylated in Yeast
First, we analyzed whether αSyn expressed in yeast cells is sumoylated. A temperature-sensitive strain of S. cerevisiae was used with a conditional defect in a gene for an isopeptidase for SUMO deconjugation in a temperature-sensitive manner (ulp1ts) (47). Genes for wild-type (WT) αSyn or the familial mutant A30P and the His6-tagged yeast SUMO protein Smt3 were integrated into the genome and co-expressed. Down-regulation of the gene for the ULP1 protease activity at the nonpermissive 30 °C resulted in an enrichment of SUMO-conjugated proteins (Fig. 1A). SUMO targets were isolated by Ni2+ affinity chromatography under denaturing conditions. The SUMO-modified protein with a molecular mass of ∼35 kDa can be separated from unmodified 17-kDa αSyn. Immunoblotting analysis with a monoclonal antibody against αSyn revealed significant sumoylation of both αSyn variants (Fig. 1B).
FIGURE 1.

αSyn is sumoylated in S. cerevisiae and impairment of sumoylation increases αSyn toxicity and foci formation. A, total protein extract of ulp1 temperature-sensitive yeast cells, defective in SUMO deconjugation, co-expressing integrated αSyn (driven by GAL1 promoter at TRP1 locus) and the His6-tagged yeast SUMO protein Smt3 (driven by ADH1 promoter at HIS3 locus). Enriched sumoylated proteins in the ulp1ts strain in comparison with the control W303 were detected by Western blot with Smt3 antibody (α-Smt3). EV, yeast cells, transformed with empty vector. B, nickel pulldown of His6-tagged yeast SUMO protein Smt3 (His6-smt3) in ulp1ts cells co-expressing αSyn. Sumoylated αSyn (αSyn and A30PSyn) was detected in the pulldown fractions with αSyn antibody (upper panel). Unmodified αSyn was detected in the flow-through. Yeast cells transformed with empty vector were used as a control. Western hybridization of the same blot with Smt3 antibody (lower panel) verified the Ni2+ pulldown (lower panel). C, Western hybridization with Smt3 antibody of total protein extract of smt3 temperature-sensitive yeast cells, co-expressing αSyn or empty vector (EV) at permissive (25 °C) or restrictive temperature (30 °C). D, spotting assay of conditional smt3ts mutant strain expressing αSyn-GFP or A30P-GFP at permissive (25 °C; +SUMO; Smt3 functional) or restrictive temperature (30 °C; −SUMO; Smt3 dysfunctional). GAL1-driven αSyn-GFP is expressed from two genomically integrated copies. GAL1-driven A30P-GFP is expressed from a 2-μm plasmid. GFP, expressed from the same promoter, is used as a control. Yeast cells were spotted in 10-fold dilutions on selection plates containing glucose (αSyn 'OFF') or galactose (αSyn 'ON'). E, fluorescence microscopy of smt3ts cells expressing αSyn-GFP or A30P-GFP at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). Scale bar, 1 μm. F, quantification of the percentage of cells displaying αSyn inclusions. W303 cells expressing αSyn-GFP or A30P-GFP at 25 or 30 °C were used in comparison with smt3ts cells expressing αSyn-GFP or A30P-GFP at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). At least 300 cells were counted per strain and experiment. Significance of differences was calculated with t test (***, p < 0.001, n = 3).
Next, we examined the effect of sumoylation on αSyn yeast cells defective in the SUMO-encoding gene (smt3ts) (48). The smt3-331 allele expresses a temperature-sensitive Smt3 mutant protein. The mutant Smt3 is dysfunctional at the restrictive temperature (30 °C), rendering the protein misfolded (49). The level of SUMO conjugates in the smt3ts strain was not changed at the nonpermissive temperature, presumably due to the accumulation of misfolded SUMO (Fig. 1C). This is supported by the earlier finding that the phenotype of smt3-331 can be suppressed by WSS1, which had been originally identified as a high copy number suppressor of the temperature-sensitive smt3-331 allele (49). The WSS1 protein acts as SUMO-dependent isopeptidase (50) and presumably detaches misfolded SUMO chains that are caused by the smt3-331 mutation. Consistently, misfolded SUMO chains are accumulated from SUMO conjugates at the nonpermissive temperature.
We previously showed that expression of WT αSyn from two copies is under the threshold for yeast growth inhibition (38). Thus, yeast smt3ts strains expressing WT αSyn from two genomically integrated gene copies were constructed, and the number of integrated copies was verified by Southern hybridization. Colony growth was compared in spotting assays between yeast cells with the GAL1 promoter-driven αSyn expression under inducing (galactose) or noninducing (glucose) conditions. All strains grew equally well at the permissive temperature (25 °C) when sumoylation was not impaired. Expression of WT αSyn resulted in growth inhibition in comparison with cells expressing GFP as a control, when sumoylation was down-regulated at the restrictive temperature (30 °C). Similar results were obtained for A30P, where high copy plasmid expression normally does not impair yeast growth, whereas defects in sumoylation resulted in a drastic growth inhibition (“+SUMO” versus “−SUMO” cells depleted of functional SUMO-conjugates in Fig. 1D). This suggests a protective role of the SUMO modification for αSyn-expressing yeast cells.
We then assessed whether αSyn-mediated cytotoxicity was related to the formation of αSyn inclusions. Inclusion formation was followed by live-cell imaging using GFP as a reporter. Quantification of the number of cells displaying fluorescent foci revealed significant increases in cells displaying αSyn foci in the absence of sumoylation (Fig. 1E). Control experiments with wild-type yeast (W303) excluded that the difference in the number of cells with inclusions is due to the temperature shift (Fig. 1F).
These results illustrate that inhibition of sumoylation in yeast has a strong growth impact on cells expressing αSyn. Growth impairment correlates with the increase of intracellular accumulation of WT αSyn or A30P αSyn fluorescent foci. This supports that sumoylation protects yeast growth by inhibiting inclusion formation of αSyn.
Protective Function of SUMO Requires Direct Modification of αSyn at Acceptor Sites That Are Conserved in Eukaryotic Cells
The protective function of SUMO could be due to a direct sumoylation of αSyn or due to an indirect effect through another SUMO target protein. Lys-96 and Lys-102 were mapped as major αSyn SUMO acceptor sites in higher cells (15, 29). Thus, we then analyzed whether the SUMO acceptor sites in αSyn were also conserved in yeast. Double lysine substitutions, K96R/K102R, of WT and A30P αSyn were generated. Ulp1ts yeast cells carrying His6-tagged Smt3 were transformed with the double lysine mutants, and SUMO conjugates were purified by Ni2+-NTA pulldown as above. Considerable amounts of probe were loaded on the gel to increase the detection of low signals. Immunodetection of αSyn revealed a significant reduction in sumoylation of the K96R/K102R variant and a complete sumoylation abolishment in the A30P variant carrying these substitutions (Fig. 2A). These results corroborate that Lys-96 and Lys-102 are conserved as major SUMO acceptor sites of αSyn in yeast.
FIGURE 2.

Lysine 96 and 102 are conserved as major sumoylation sites of αSyn in eukaryotes. A, lysine to arginine substitutions at positions 96 and 102 resulted in decreased αSyn sumoylation. αSyn and A30PSyn and the corresponding αSyn amino acid variants K96R/K102R were transformed into ulp1ts yeast cells expressing the yeast SUMO protein His6-Smt3. His6-tagged SUMO conjugates were pulled down by Ni2+-NTA. αSyn was detected by Western hybridization using αSyn antibody (upper panel). Western hybridization of the same blot with Smt3 antibody (α-Smt3) verified the Ni2+-NTA pulldown (lower panel). B, spotting assay of W303 yeast cells, carrying two copies of GAL1-driven αSyn-GFP and K96R/K102R-GFP. Yeast cells were spotted in 10-fold dilutions on selection plates containing glucose (αSyn 'OFF') or galactose (αSyn 'ON'). C, quantification of the percentage of cells displaying αSyn inclusions in W303 yeast background. Significance of differences was calculated with t test (***, p < 0.001, n = 3).
The major sumoylation sites of αSyn were replaced in cis to examine whether modification of αSyn as direct target of SUMO protects against growth inhibition of yeast. Strains expressing either WT αSyn or the K96R/K102R variant from two genomically integrated copies were constructed as above. Expression of the K96R/K102R variant resulted in growth inhibition in contrast to wild-type αSyn (Fig. 2B). Fluorescence microscopy studies revealed an increase in the percentage of cells with αSyn inclusions for the K96R/K102R mutant (Fig. 2C). These data support that direct sumoylation of αSyn at the conserved modification sites (Lys-96 and Lys-102) protects against cytotoxicity and reduces the formation of inclusions.
Phosphorylation of αSyn by Human Kinases GRK5 or PLK2 Varies When the Cellular SUMO Pool Is Lowered
αSyn is phosphorylated predominantly at the residue Ser-129 (18, 19). The effects of phosphorylation on αSyn-induced toxicity are complex with reports supporting negative as well as positive impacts on cells (23, 24, 51–54). Therefore, we next investigated the interplay between αSyn sumoylation and phosphorylation by examining how changes in sumoylation affect αSyn phosphorylation and whether this impacted αSyn toxicity. αSyn can be phosphorylated in yeast by endogenous kinases (55). Several kinase families are reported to phosphorylate αSyn at Ser-129 in higher eukaryotes (19–22, 27, 28, 56–58). Ser-129 phosphorylation correlates with GRK5 kinase activity (21, 25), and PLK2 was shown to be one of the main Polo-like kinases in mammalian cells that phosphorylates αSyn at Ser-129 (59).
We assessed the combined effects of sumoylation and αSyn Ser-129 phosphorylation (αSyn Ser(P)-129) by overexpressing the human kinases GRK5 or PLK2 from episomal 2-μm plasmids in the smt3ts cells expressing αSyn. Heterologous expression of kinases GRK5 or PLK2 resulted in increased phosphorylation of αSyn at Ser-129 in comparison with vector control cells (Fig. 3A). Quantification of αSyn Ser-129 phosphorylation levels in the presence or absence of functional SUMO revealed a significant increase of αSyn Ser-129 phosphorylation when either of the two kinases is expressed (Fig. 3B), whereas expression of GRK5 in the absence of SUMO had a less pronounced effect. Wild-type W303 yeast cells co-expressing K96R/K102R Syn-GFP together with GRK5 or PLK2 were hybridized with αSyn Ser(P)-129-specific antibody to examine the Ser-129 phosphorylation level of the sumoylation-deficient αSyn variant (Fig. 3C). Quantification of the Western blots revealed a significant increase of the phosphorylation level on Ser-129 of K96R/K102R Syn where the major sumoylation sites of αSyn were blocked. Both kinases, GRK5 or PLK2, increased phosphorylation similarly (Fig. 3D).
FIGURE 3.

Expression of the human kinases GRK5/PLK2 increases αSyn Ser-129 phosphorylation in yeast. A, Smt3ts mutant cells co-expressing αSyn and GRK5 or PLK2 at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). The phosphorylation level of αSyn on Ser-129 was detected by αSyn Ser-129 phosphorylation-specific antibody (αSyn pSer129) when expressed either alone (αSyn-GFP + empty vector (EV)) or in the presence of GRK5 or PLK2. Immunoblotting analysis of yeast cells expressing S129A-GFP with αSyn Ser(P)129 antibody (right panel) was used as a control for antibody specificity. B, quantification of αSyn Ser-129 phosphorylation level in the presence or absence of GRK5 and PLK2, respectively, at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). Densitometric analysis of the immunodetection of αSyn Ser(P)-129 was normalized to the total amount of αSyn and relative to αSyn + EV at permissive temperature (25 °C; +SUMO). Significance of differences was calculated with one-way ANOVA with Bonferroni's multiple comparison test (**, p < 0.01; ##, p < 0,05 versus empty vector, n = 4). C, Western hybridization of W303 yeast cells co-expressing K96R/K102R-GFP and GRK5 or PLK2. The phosphorylation level of sumoylation-deficient αSyn mutant on Ser-129 was visualized with αSyn Ser(P)-129. D, quantification of αSyn Ser-129 phosphorylation levels of sumoylation-deficient αSyn mutant in the presence or absence of GRK5 and PLK2. Densitometric analysis of the immunodetection of αSyn Ser(P)-129 was normalized to the total amount of αSyn. Significance of differences was calculated with one-way ANOVA (**, p < 0.01, n = 4).
Expression of GRK5 Alleviates αSyn-induced Cytotoxicity and Inclusion Formation in SUMO-deficient Strain
We investigated whether αSyn-mediated toxicity was altered when increased αSyn Ser-129 phosphorylation levels are combined with functional or dysfunctional SUMO. The effects of GRK5 or PLK2 were tested by spotting assays of smt3ts cells expressing αSyn (Fig. 4A). We found that increased GRK5 suppressed the growth defect associated with impaired sumoylation. Increased PLK2 levels resulted in a less pronounced improvement of growth in comparison with corresponding cells with GRK5 activity (Fig. 4A). The specificity of phosphorylation of GRK5 or PLK2 on Ser-129 was analyzed in greater detail by integrating two copies of an S129A mutant form of αSyn in the genome. S129E/S129D mutants were not included in the analysis because recent reports show that they fail to mimic the effect of αSyn phosphorylation (9, 60). In the presence of functional SUMO, co-expression of S129A with GRK5 had the same growth phenotype as that observed for cells co-expressing WT αSyn with GRK5. A slight growth retardation was observed by co-expression of S129A and PLK2 at the permissive temperature. In the absence of functional SUMO, neither kinase could rescue the growth defect of the mutant αSyn where the phosphorylation site was missing (Fig. 4A). These data indicate that the SUMO-dependent effect of GRK5 or PLK2 expression on yeast growth depends on the phosphorylation of αSyn on Ser-129. We then performed growth assays of cells expressing WT αSyn or the K96R/K102R variant to test whether the growth rescue by expression of GRK5 or PLK2 required direct sumoylation of αSyn. Co-expression of GRK5 and K96R/ K102R resulted in a striking recovery of growth (Fig. 4B). This suggests that GRK5 directly suppresses a sumoylation defect of αSyn. In contrast, expression of PLK2 did not significantly influence yeast growth. This suggests an indirect effect on αSyn toxicity caused by down-regulation of the sumoylation activity, which then allows a partial growth recovery by PLK2 expression.
FIGURE 4.

Increased αSyn Ser-129 phosphorylation level by GRK5/PLK2 expression alleviates the toxicity and reduces foci formation associated with impaired sumoylation in smt3ts cells. A, spotting assay of smt3ts cells co-expressing αSyn-GFP or S129A-GFP with GRK5 or PLK2 either at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). Yeast cells were spotted in 10-fold dilutions on selection plates containing glucose (αSyn 'OFF'; kinases 'OFF') or galactose (αSyn 'ON'; kinases 'ON'). B, spotting assay of W303 yeast cells, carrying two copies of GAL1-driven αSyn-GFP and K96R/K102R-GFP in the presence of GRK5 and PLK2 or empty vector. Yeast cells were spotted in 10-fold dilutions on selection plates containing glucose (αSyn 'OFF'; kinases 'OFF') or galactose (αSyn 'ON'; kinases 'ON'). C, fluorescence microscopy of smt3ts cells expressing αSyn in the presence or absence of GRK5 or PLK2 (left panel). Scale bar, 1 μm. Quantification of percentage of cells displaying αSyn inclusions in the presence or absence of GRK5/PLK2 (upright) and quantification of cells with foci formation expressing S129A-GFP with and without overexpression of GRK5/PLK2. Significance of differences was calculated with t test with respect to αSyn-GFP at the same temperature (**, p < 0.01, n = 3).
We investigated whether the growth recoveries of smt3ts cells expressing αSyn in the presence of GRK5 or PLK2 are associated with changes in inclusion formation. Phosphorylation of αSyn on Ser-129 by GRK5 or PLK2 in sumoylation-deficient cells correlated with decreased accumulation of αSyn foci (Fig. 4C). Quantification of the cells displaying αSyn inclusions revealed that both GRK5 and PLK2 promote a significant decrease in the percentage of cells bearing fluorescent foci. This effect was Ser-129-dependent, because co-expression of the S129A mutant with either kinase did not reveal decreased accumulation of αSyn foci in the absence of SUMO. These results suggest that increased levels of αSyn Ser-129 phosphorylation can suppress the αSyn-induced cytotoxicity in the SUMO-deficient mutant. PLK2 does not significantly influence yeast growth, although there seems to be a decrease in aggregate formation.
Phosphorylation Promotes Proteasome and Autophagy Degradation of αSyn, whereas Sumoylation Preferentially Stimulates Autophagy
We performed GAL1 promoter shutoff experiments and analyzed the impact of blocking these systems by drug treatments to compare the role of proteasome and autophagy-mediated degradation systems on the clearance of αSyn inclusions when sumoylation was inhibited. Expression of αSyn was induced for 4 h in galactose-containing medium, and the cells were then shifted to glucose-containing medium to repress the promoter. Cells were imaged 2 h after promoter shutoff, and the percentage of cells with inclusions was determined.
Shutoff studies were performed with the mutants Δatg1 and Δatg7, which render cells unable to perform autophagy. Atg1 is a serine/threonine kinase that acts in autophagy regulation and is essential for autophagy induction (61). Atg7 is an activator of Atg8 and is required for the formation of autophagic bodies (62). Deletion of ATG1 and ATG7 autophagy genes significantly reduced αSyn aggregate clearance 2 h after shutoff. In contrast, cells expressing K96R/K102R αSyn cleared inclusions in a similar manner to the isogenic wild-type strain (Fig. 5A).
FIGURE 5.
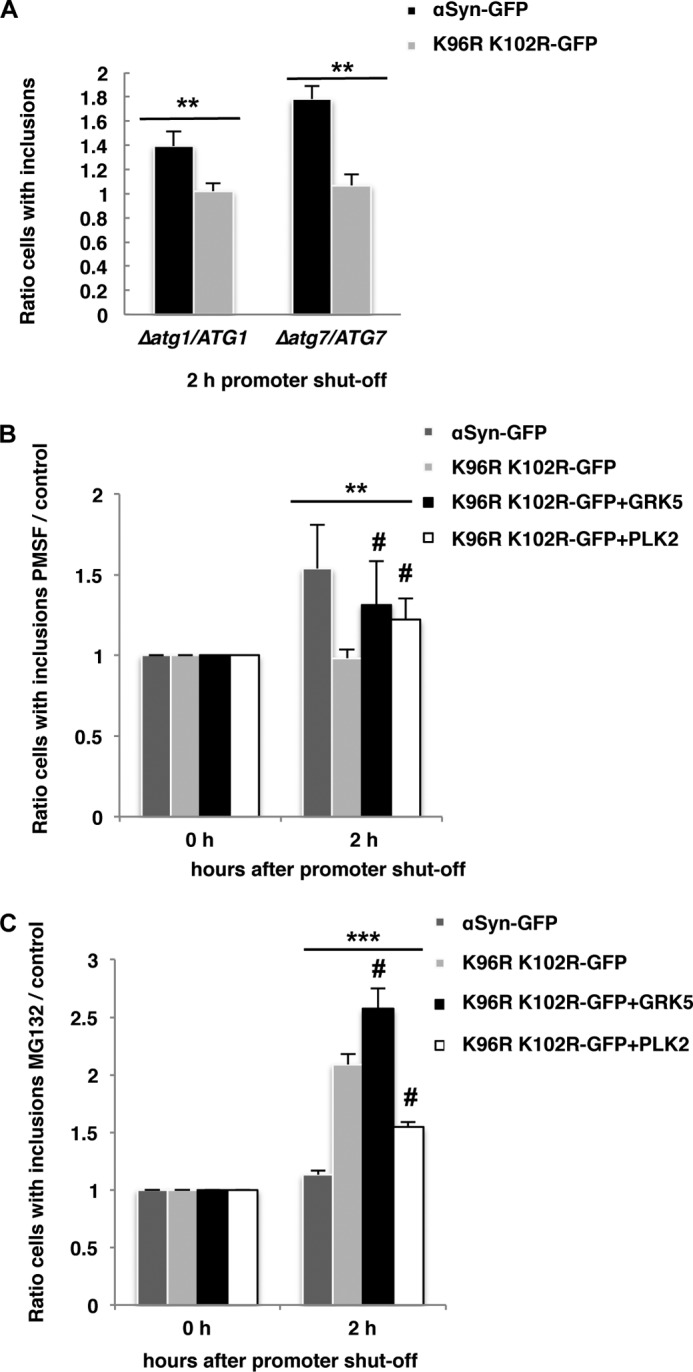
αSyn aggregate clearance upon promoter shutoff. A, inhibition of autophagy by deletion of ATG1 and ATG7. Expression of αSyn-GFP and K96R/K102R-GFP was induced for 4 h in galactose medium and then the cells were shifted to glucose medium. Quantification of the reduction of inclusions was done 2 h after the promoter shutoff and was presented as the ratio of aggregate clearance in the deletion strain to aggregate clearance in the isogenic wild-type strain. Significance of differences was calculated with t test (**, p < 0.01, n = 3). B, inhibition of the vacuolar degradation pathway by PMSF. Quantification was of cells expressing αSyn-GFP, K96R/K102R-GFP, and K96R/K102R-GFP and co-expressing GRK5 or PLK2, respectively. αSyn-GFP and K96R/K102R-GFP were expressed from two genomically integrated copies. After 4 h of induction of the protein expression in galactose medium, cells were shifted to glucose medium supplemented with 1 mm PMSF dissolved in ethanol (EtOH) or only EtOH as a control. Quantification of the reduction of inclusions was done 2 h after the promoter shutoff. Cells with inclusions were counted and presented as a ratio to the control (EtOH). C, inhibition of the proteasome with MG132. The protein expression was induced as above, and the cells were shifted to glucose medium supplemented with 75 μm MG132 and dissolved in DMSO or only DMSO as a control. Quantification of the reduction of inclusions was done 2 h after the promoter shutoff. Cells with inclusions were counted and presented as a ratio to the control (DMSO). Significance of differences was calculated with one-way ANOVA with Bonferroni's multiple comparison test (**, p < 0.01; ***, p < 0.001; n = 3; #, p < 0.05 versus K96R/K102R-GFP) (Bonferroni's multiple comparison test).
PMSF as an inhibitor of the autophagy/vacuolar pathway (38) was used in a second approach to study the contribution of autophagy/vacuole for aggregate clearance. PMSF inhibits the activity of numerous vacuolar serine proteases (63) without affecting proteasome function (64). PMSF affects autophagic body formation (65) and leads to accumulation of autophagosomes in the vacuole due to decreased degradation of the autophagic bodies (66). Inhibition of autophagic proteases with PMSF resulted in similar impairment in the clearance of inclusions as with the mutant Δatg1 strain (Fig. 5B). Cells expressing K96R/K102R αSyn cleared inclusions in a similar manner as control cells without drug (ethanol). This suggests that sumoylation supports the autophagy-dependent clearance of αSyn.
PMSF was also applied to assess the impact of GRK5 and PLK2 on the clearance of αSyn inclusions (Fig. 5B). Expression of GRK5 or PLK2 altered the inclusion clearance significantly and resulted in intermediate levels between WT and K96R/K102R, suggesting that expression of GRK5 or PLK2 can partially rescue the inclusion clearance through autophagy.
The impact of GRK5 or PLK2 expression and sumoylation on αSyn inclusion clearance by the proteasome was analyzed by applying the proteasome inhibitor MG132 (dissolved in DMSO) (67). Quantification of the results of promoter shutoff studies revealed equal inclusion clearance of wild-type αSyn in MG132-treated cells when compared with the control (DMSO) (Fig. 5C). In contrast, cells were unable to clear inclusions when αSyn sumoylation (K96R/K102R) and the proteasome (MG132) were blocked simultaneously. This corroborates that sumoylation-deficient αSyn is cleared by the proteasome. Expression of GRK5 in the sumoylation-deficient mutant promoted the proteasome-dependent clearing of inclusions significantly and, accordingly, MG132 treatment resulted in an increased percentage of cells with inclusions. Expression of PLK2 in the sumoylation-deficient mutant could only partially promote proteasomal degradation in comparison with wild-type αSyn, suggesting a minor impact on inclusion clearance by the proteasome in comparison with GRK5.
These findings indicate that sumoylated αSyn is primarily targeted to the autophagy pathway and nonsumoylated αSyn primarily to the proteasome. Inhibition of sumoylation results in inefficient autophagy-mediated aggregate clearance and directs the protein to the proteasome. Expression of the human kinase GRK5 promotes clearance of nonsumoylated αSyn to the autophagosome and the proteasome. PLK2 can efficiently phosphorylate nonsumoylated αSyn but shows only partial effects on aggregate clearance. This might be due to additional effects on other yet unidentified targets in yeast cells.
Ulp1 SUMO Isopeptidase Activity Increases αSyn Inclusion Formation
Decreased sumoylation of αSyn impairs inclusion clearance. Therefore, we analyzed whether increased sumoylation of αSyn affected the process of inclusion formation and clearance. Expression of αSyn-GFP or K96R/K102R αSyn in ulp1ts strain, deficient for SUMO de-conjugation, revealed a general decrease in inclusion formation in comparison with W303, suggesting that the loss of a general SUMO isopeptidase might have multiple effects on the cell (Figs. 1F and 6A). The growth of αSyn-GFP- and K96R/K102R αSyn-expressing cells in ulp1ts was inhibited similarly (Fig. 6B). The cells showed partial cytoplasmic GFP staining additionally to fluorescent foci (Fig. 6C), whereas expression of αSyn-GFP and K96R/K102R αSyn in the W303 background did not reveal any cytoplasmic GFP staining. Promoter shutoff experiments revealed a slower rate of inclusion clearance of the sumoylation-deficient K96R/K102R mutant in comparison with αSyn (Fig. 6D). The results suggest that an increase of the pool of sumoylated proteins by inhibition of SUMO de-conjugation can change the inclusion formation and localization of αSyn without changing its toxicity. One possible explanation might be that high pools of free SUMO necessary for sumoylation of αSyn are required to decrease its toxicity and impact on cell growth.
FIGURE 6.

αSyn aggregate clearance in ulp1ts cells. A, percentage of cells displaying αSyn-GFP inclusions after 6 h of induction of ulp1ts cells, expressing αSyn-GFP or K96R/K102R-GFP. B, spotting assay of ulp1ts cells expressing αSyn-GFP or K96R/K102R-GFP from 2 μm plasmid. Yeast cells were spotted in 10-fold dilutions on selection plates containing glucose (αSyn 'OFF') or galactose (αSyn 'ON'). C, fluorescence microscopy of ulp1ts cells expressing αSyn-GFP or K96R/K102R-GFP. After 6 h of induction of the protein expression in galactose medium, cells were shifted to glucose medium. Scale bar, 1 μm. D, quantification of the reduction of inclusions 2 h after the promoter shutoff. Cells with inclusions were counted and presented as a ratio to time point 0 h. Significance of differences was calculated with t test (**, p < 0.01, n = 3).
Phosphorylation Promotes Ubiquitination and Degradation of αSyn
Ubiquitination is the common post-translational modification for proteasome-dependent protein degradation and is usually primed by a kinase reaction. We showed that increased levels of αSyn phosphorylation on Ser-129 affect the clearance of inclusions by the proteasome. These results prompted us to analyze how sumoylation or phosphorylation influences ubiquitination of αSyn. Wild-type cells expressing His6-tagged αSyn were grown, and the protein was purified from cell extracts using Ni2+ affinity chromatography. Immunoprecipitation was performed with ubiquitin antibodies and revealed that αSyn monomers are mono-ubiquitinated in yeast (Fig. 7A), in agreement with findings in higher organisms (68–70). Phospho-antibodies showed that mono-ubiquitinated αSyn was simultaneously phosphorylated on Ser-129 (Fig. 7A).
FIGURE 7.

αSyn is ubiquitinated in yeast cells. A, αSyn-His6 protein was purified by Ni2+ pulldown and subjected to immunoprecipitation with ubiquitin antibody. The ubiquitinated and phosphorylated αSyn was detected by αSyn and αSyn Ser(P)-129-specific antibody, respectively. Empty vector (EV) was used as a control. B, Smt3ts cells expressing αSyn-His6 co-transformed with GRK5 or PLK2 and empty vector of the kinases (EV) as a control at permissive (25 °C; +SUMO) or restrictive temperature (30 °C; −SUMO). The purified αSyn protein from Ni2+ pulldown was subjected to ubiquitin immunoprecipitation (IP Ubi). As a control, the same experiments were performed without addition of ubiquitin antibody. The ubiquitinated αSyn was analyzed by Western hybridization with an antibody against αSyn. Western hybridization of the same blots after stripping with ubiquitin antibody (lower panels). A representative result is shown from three independent experiments.
We next assessed whether ubiquitination of αSyn was affected by sumoylation and whether phosphorylation altered αSyn ubiquitination. For this, His6-tagged αSyn was expressed from a 2-μm plasmid in smt3ts cells. The effect of GRK5 and PLK2 was investigated by co-expression of each kinase in the presence or absence of SUMO (Fig. 7B). Ni2+ affinity chromatography of His6-tagged αSyn was performed, followed by immunoprecipitation of the protein with an antibody against ubiquitin. Immunoblotting analysis revealed different patterns of ubiquitinated αSyn species ranging from 22 to 36 kDa (Fig. 7B). In the absence of GRK5 or PLK2, only a single molecular band at around 29 kDa was precipitated. The presence of either kinase resulted in multiple distinct bands, including a major band of 22 kDa. The additional smear pattern of modified αSyn to higher molecular weights was especially pronounced when sumoylation was down-regulated. This might be due to mono-ubiquitination (22 kDa), di-ubiquitination (29 kDa), or tri-ubiquitination, as described earlier (11). The expression of GRK5 resulted in larger effects on the ubiquitination of αSyn than those observed with PLK2, especially in the absence of SUMO. This was consistent with the stronger suppression of αSyn toxicity by GRK5 when SUMO was down-regulated, in comparison with PLK2.
GAL1 promoter shutoff assays were performed to determine the effect of sumoylation and increased Ser-129 phosphorylation by GRK5 and PLK2 on αSyn stability. As described above, the promoter was shut off after 4 h, and cells were collected at various time points. Immunoblotting analysis revealed a reduction in the level of αSyn with time. Phosphorylation of αSyn by GRK5 or PLK2 resulted in a slight decrease of the protein levels after 18 h in comparison with the control (Fig. 8A). We assessed the role of the proteasome and autophagy-mediated degradation systems on αSyn stability and analyzed the impact of blocking these systems by drug treatments. Inhibition of the proteasome with MG132 had slight impact on the stability of αSyn after 18 h, whereas inhibition of the vacuolar/autophagy pathway with PMSF resulted in increased protein stability (Fig. 8B). These results corroborate our previous findings (38). Western blot hybridization of MG132-treated cells with ubiquitin antibody confirmed the effectiveness of MG132 as a proteasome inhibitor (Fig. 9).
FIGURE 8.

Effect of sumoylation and increased αSyn Ser-129 phosphorylation by GRK5/PLK2 on αSyn protein stability. GAL1 promoter shutoff studies and drug treatments. A, Smt3ts yeast cells expressing αSyn with or without GRK5 or PLK2 at permissive temperature (25 °C) were induced for 4 h in galactose (αSyn on) and then transferred to glucose containing medium (αSyn off). Immunoblotting analysis was performed at the indicated time points after promoter shutoff with αSyn antibody and GAPDH antibody as loading control. B, W303 cells expressing αSyn, K96R/K102R (C), K96R/K102R + GRK5 (D), or K96R/K102R + PLK2 (E) were induced for 4 h in galactose (αSyn on) and then transferred to glucose containing medium (αSyn off). The glucose medium was supplemented with 75 μm MG132 or 1 mm PMSF. Immunoblotting analysis was performed at the indicated time points after promoter shutoff with αSyn antibody and GAPDH antibody as loading control. A representative result is shown from three independent experiments. Right panels, densitometric analysis of the immunodetection of αSyn-GFP relative to the GAPDH loading control. Significance of differences was calculated with one-way ANOVA with Bonferroni's multiple comparison test (***, p < 0.001; **, p < 0.01; *, p < 0.05; ##, p < 0.05 versus 0 h (Bonferroni's multiple comparison test)).
FIGURE 9.
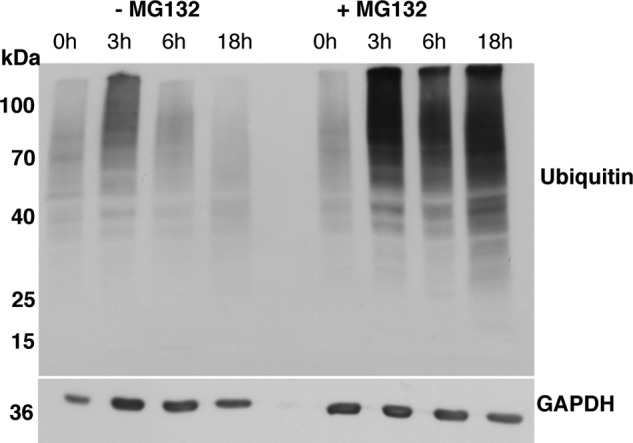
Inhibition of the proteasome with MG132. Promoter shutoff of yeast cells, expressing αSyn-GFP in the absence and presence of proteasome inhibitor MG132. Representative Western hybridization with ubiquitin antibody, showing accumulation of ubiquitinated species after treatment with MG132 in comparison with the control (no drug and 0 h of treatment).
We analyzed whether direct inhibition of αSyn sumoylation, by blocking the major sumoylation sites (K96R/K102R), affected the steady state of protein stability. GAL1 promoter shutoff experiments revealed that the SUMO-deficient K96R/K102R αSyn variant is a highly stable protein (Fig. 8C). We next tested whether increased αSyn phosphorylation by GRK5 and PLK2 could alter the protein stability of the SUMO-deficient K96R/K102R variant. Immunoblotting analysis after promoter shutoff revealed a significant reduction in the levels of αSyn. Phosphorylation of αSyn by GRK5 or PLK2 therefore destabilizes αSyn significantly when sumoylation is impaired (Fig. 8, D and E). Inhibition of the proteasome with MG132 and vacuolar/autophagy pathway with PMSF resulted in a significant increase in protein stability. The results support the data from aggregate clearance assays and suggest that GRK5 and PLK2 affect the protein stability of the SUMO-deficient mutant by directing the protein to the vacuole and proteasome for degradation.
We examined whether the effect depends directly on phosphorylation of Ser-129. GAL1 promoter shutoff assays were performed with S129A αSyn mutant in smt3ts strain at permissive (+SUMO) or restrictive temperature (−SUMO). S129A-αSyn mutant revealed a decrease in protein level after 18 h of promoter shutoff (Fig. 10). However, the mutant protein was much more stable than wild-type αSyn (Fig. 8A). Down-regulation of sumoylation at a restrictive temperature resulted in no significant decrease of the protein stability. The data suggest that phosphorylation of αSyn at Ser-129 decreases the protein stability, which is further affected by sumoylation. These data corroborate the results with the sumoylation-deficient K96R/K102R mutant. Expression of GRK5 or PLK2 did not affect the stability of S129A-αSyn. The results indicate that the effect of GRK5 and PLK2 expression on αSyn protein stability depends directly on Ser-129.
FIGURE 10.

Effect of sumoylation and GRK5/PLK2 expression on S129A-GFP protein stability. GAL1 promoter shutoff studies. A, Smt3ts yeast cells expressing S129A-GFP with or without GRK5 or PLK2 at permissive temperature (25 °C, +SUMO) and restrictive temperature (30 °C, −SUMO) were induced for 4 h in galactose (αSyn on) and then transferred to glucose-containing medium (αSyn off). Immunoblotting analysis was performed at the indicated time points after promoter shutoff with αSyn antibody and GAPDH antibody as loading control. A representative result is shown from three independent experiments. B, densitometric analysis of the immunodetection of S129A-GFP relative to the GAPDH loading control. Significance of differences was calculated with one-way ANOVA with Bonferroni's multiple comparison test (**, p < 0.01; ##, p < 0.05 versus 0 h (Bonferroni's multiple comparison test).
These results demonstrate that phosphorylation at Ser-129 promotes αSyn ubiquitination and decreases its stability. The data support a complex cross-talk between sumoylation- and phosphorylation-mediated ubiquitination of αSyn.
DISCUSSION
Here, we used S. cerevisiae as a model to investigate the molecular interplay between sumoylation and phosphorylation in the clearance of αSyn (summarized in Fig. 11). We uncovered a complex cross-talk between these post-translational modifications that impact ubiquitination and thereby influence the degradation of αSyn by autophagy or the 26 S proteasome. Ultimately, the differential processing of αSyn by these systems interferes with inclusion formation and cytotoxicity.
FIGURE 11.
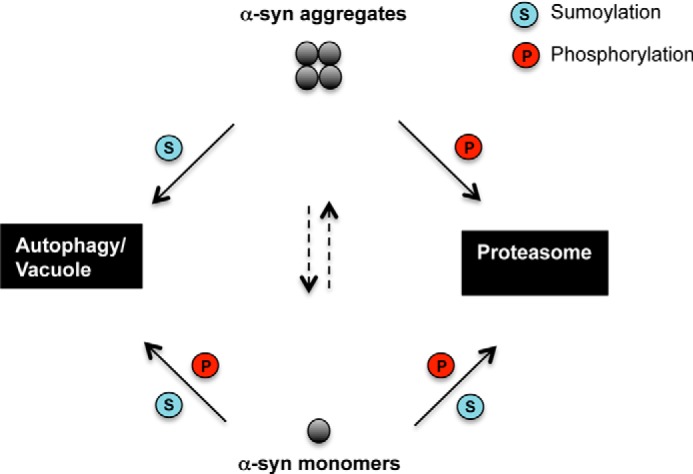
αSyn clearance and degradation in yeast. Proteasome and autophagy/vacuole as major degradation pathways are depicted. When synthesis of αSyn is switched off, wild-type yeast cells clear αSyn aggregates within hours and regain normal growth rates (38). In the presence of functional SUMO, the aggregates are primarily cleared by the autophagy/vacuolar pathway. When sumoylation is impaired, the aggregate clearance through autophagy/vacuolar pathway is prevented, and the proteasomal degradation is promoted. Increase of Ser-129 phosphorylation level by GRK5 or PLK2 rescues the autophagic aggregate clearance and additionally promotes the proteasomal degradation. For monomers, degradation of soluble αSyn monomers occurs through both pathways. Inhibition of αSyn sumoylation has a strong effect on monomer protein stability, significantly increasing the half-life of the protein and inhibiting the degradation through both pathways. Phosphorylation at Ser-129 by GRK5 or PLK2 decreases the protein stability and promotes degradation of soluble αSyn through proteasome and autophagy pathways.
αSyn undergoes numerous post-translational modifications such as phosphorylation, ubiquitination, nitration, acetylation, O-glycosylation, and sumoylation. αSyn was found to be a SUMO target in cultured cells and in a rat animal model of PD (15, 29), but the number of sumoylation studies of αSyn is very limited in comparison with those on other post-translational modification publications, limiting our understanding of the implications of sumoylation on αSyn biology. Here, we showed that both WT αSyn as well as the A30P mutant are sumoylated in vivo in yeast at Lys-96 and Lys-102, two sumoylation sites that are conserved in eukaryotes (15, 29). By decreasing the cellular SUMO pool, or by mutating the codons for the major SUMO sites, we determined that sumoylation protects yeast cells against αSyn-mediated cytotoxicity and inclusion formation. Previously, sumoylation was suggested to keep αSyn in solution and, therefore, decrease αSyn aggregation (15). Similarly, sumoylation was found to modulate the solubility of mutant huntingtin, Ataxin 7, androgen receptor, and STAT1, also reducing the toxicity of these proteins in other degenerative diseases (71–73). Consistently, impairment of sumoylation in yeast resulted in a significant increase in the number of cells displaying αSyn inclusions. This further supports the beneficial regulatory role of sumoylation in inhibiting αSyn inclusion formation in vivo.
Sumoylation and phosphorylation are both reversible dynamic processes, which can actively interfere with each other and modulate the molecular features of their substrates. The major αSyn phosphorylation site at Ser-129 is also used in yeast when human kinases are expressed (24). Here, we expressed and analyzed the effects of two of the most efficient human kinases in yeast, i.e. the G protein-coupled kinase GRK5 (21, 25) and the Polo-like kinase PLK2 (26, 28). Our study revealed a significant increase of αSyn phosphorylation at Ser-129 in yeast in the presence of human PLK2 or GRK5 kinases. PLK2 phosphorylated αSyn with similar efficiency in the presence or absence of functional SUMO. GRK5 phosphorylated preferentially αSyn in cells with an intact sumoylation machinery. The difference in the substrate specificity suggests that other mechanisms or phosphorylation of residues other than Ser-129 could facilitate αSyn clearance by overexpression of GRK5.
Several examples have been reported in the literature where phosphorylation depends on the sumoylation profile of target proteins (74, 75). Sumoylation can modulate the specific interaction with kinases or phosphatases by changing substrate surfaces and activity. In particular, sumoylation of protein-tyrosine phosphatase 1B has been shown to reduce catalytic activity and therefore change the phosphorylation status of substrates (76).
Accumulating evidence suggests that αSyn post-translational modifications modulate αSyn-mediated toxicity and aggregate formation (9, 15, 20, 23, 29, 68, 69, 77). However, there is still no consensus of the effects of different modifications on αSyn aggregation and toxicity (15, 23, 24, 54, 78). Although earlier studies did not observe the effects of αSyn phosphorylation at Ser-129 on αSyn-mediated toxicity and aggregation (54, 79), protective roles of αSyn Ser-129 phosphorylation were described in a strain-specific manner in yeast. Therefore, the specific genetic context was proposed to determine the sensitivity to changes in αSyn phosphorylation (80). This suggests a complex and subtle cross-talk between different modifications that can change features of the target protein, including inclusion formation, stability, and the affinity to the autophagic or the proteasome degradation pathways. Here, we focused on the interplay between αSyn sumoylation and Ser-129 phosphorylation. Increased αSyn Ser-129 phosphorylation induced by GRK5 can rescue yeast cells from αSyn-mediated cytotoxicity associated with sumoylation impairment. Alleviation of αSyn-mediated cytotoxicity in SUMO-deficient cells correlates with a decreased number of cells presenting αSyn intracellular inclusions. Expression of GRK5 induced a strong improvement on yeast growth when the sumoylation was impaired both in trans and in cis. We found that PLK2 might cause additional effects in yeast, in agreement with a recent study where we reported a specific role of PLK2 on αSyn inclusion formation and toxicity in yeast independent of the level of αSyn phosphorylation on Ser-129 (24).
The dynamic process of the αSyn aggregate formation depends on the equilibrium between synthesis and degradation, which determines the protein levels of αSyn. An important question is how αSyn degradation is distributed between the ubiquitin-proteasome system and the autophagy-lysosome/vacuole pathway (69). At low levels, αSyn seems to be preferentially degraded by the ubiquitin-proteasome system, whereas increased αSyn expression stimulates autophagy as the main degradation pathway (34). We previously found that autophagy represents the major pathway for aggregate clearance in yeast after the shutoff of further protein biosynthesis, allowing cells to recover from αSyn toxicity (38).
One of the major findings of this study is that sumoylation of αSyn promotes aggregate clearance by autophagy. αSyn clearance is impaired when sumoylation is inhibited either by reducing the cellular SUMO pool or by amino acid substitutions of the SUMO target sites of αSyn. Another major finding is that phosphorylation of αSyn by GRK5 can compensate for this effect. The protective role of PLK2, which can form a complex with αSyn and can also induce the autophagy pathway (23), seems to be more complicated and might include additional phosphorylation target proteins. The discrepancy between a clear PLK2 effect on inclusion formation and only a mild protective effect on yeast growth suggests that cellular survival does not only depend on inclusion clearance but requires additional protection pathways.
Sumoylation and phosphorylation are two post-translational modifications of αSyn, which protect against αSyn-induced toxicity. However, they represent distinct signals for the processing of αSyn by different degradation pathways. Whereas sumoylation primarily targets αSyn for autophagy, phosphorylation by kinases such as GRK5 has a dual effect because it partially rescues the autophagy pathway but also promotes increased ubiquitination and a reduced half-life of the protein. Phosphorylation is a well known priming reaction for ubiquitination (11, 81), and our data suggest that increased phosphorylation of αSyn presumably results in increased ubiquitination and proteasome-mediated degradation. Proteasome inhibitor studies further support that the phosphorylation-dependent degradation of αSyn is promoted by the proteasome. A dual modification that is interdependent allows a subtle fine-tuning as a molecular mechanism to selectively control αSyn turnover in response to sumoylation or phosphorylation input signals. Sumoylation might induce structural and conformational changes in αSyn and thus modulate the interaction with different kinases, which have various effects in the channeling to distinct degradation pathways.
Our study provides evidence, for the first time, that the degree of switching between autophagic and proteasomal degradation of αSyn is linked to a molecular cross-talk between sumoylation and phosphorylation. Sumoylation preferentially directs αSyn aggregates toward autophagy, and phosphorylation can shift the fate of αSyn to increased ubiquitination and proteasome degradation. Ultimately, a deeper understanding of this cross-talk will enable the design of effective strategies for directing αSyn for processing by the desired degradation machinery and may therefore constitute the basis for novel therapeutic strategies in PD and other synucleinopathies.
Acknowledgments
We thank Stefan Jentsch for providing the strain ulp1ts and plasmid D1374. We thank Maria Mayer for excellent technical assistance. We thank Rebekka Harting for carefully reading the manuscript.
This work was supported by the Cluster of Excellence and Deutsche Forschungsgemeinschaft Research Center Nanoscale Microscopy and Molecular Physiology of the Brain.
- PD
- Parkinson disease
- αSyn
- α-synuclein
- SUMO
- small ubiquitin-like modifier
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- ANOVA
- analysis of variance
- MG132
- carbobenzoxyl-leucinyl-leucinyl-leucinal.
REFERENCES
- 1. Obeso J. A., Rodriguez-Oroz M. C., Goetz C. G., Marin C., Kordower J. H., Rodriguez M., Hirsch E. C., Farrer M., Schapira A. H., Halliday G. (2010) Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661 [DOI] [PubMed] [Google Scholar]
- 2. Galvin J. E., Lee V. M., Trojanowski J. Q. (2001) Synucleinopathies: clinical and pathological implications. Arch. Neurol. 58, 186–190 [DOI] [PubMed] [Google Scholar]
- 3. Forno L. S., DeLanney L. E., Irwin I., Langston J. W. (1996) Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey. Adv. Neurol. 69, 217–228 [PubMed] [Google Scholar]
- 4. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 5. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 6. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 7. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 8. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- 9. Oueslati A., Fournier M., Lashuel H. A. (2010) Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145 [DOI] [PubMed] [Google Scholar]
- 10. Kuzuhara S., Mori H., Izumiyama N., Yoshimura M., Ihara Y. (1988) Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 75, 345–353 [DOI] [PubMed] [Google Scholar]
- 11. Hasegawa M., Fujiwara H., Nonaka T., Wakabayashi K., Takahashi H., Lee V. M., Trojanowski J. Q., Mann D., Iwatsubo T. (2002) Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076 [DOI] [PubMed] [Google Scholar]
- 12. Tofaris G. K., Razzaq A., Ghetti B., Lilley K. S., Spillantini M. G. (2003) Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411 [DOI] [PubMed] [Google Scholar]
- 13. Giasson B. I., Duda J. E., Murray I. V., Chen Q., Souza J. M., Hurtig H. I., Ischiropoulos H., Trojanowski J. Q., Lee V. M. (2000) Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290, 985–989 [DOI] [PubMed] [Google Scholar]
- 14. Hodara R., Norris E. H., Giasson B. I., Mishizen-Eberz A. J., Lynch D. R., Lee V. M., Ischiropoulos H. (2004) Functional consequences of α-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753 [DOI] [PubMed] [Google Scholar]
- 15. Krumova P., Meulmeester E., Garrido M., Tirard M., Hsiao H. H., Bossis G., Urlaub H., Zweckstetter M., Kügler S., Melchior F., Bähr M., Weishaupt J. H. (2011) Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krumova P., Weishaupt J. H. (2013) Sumoylation in neurodegenerative diseases. Cell. Mol. Life Sci. 70, 2123–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eckermann K. (2013) SUMO and Parkinson's disease. Neuromol. Med. 15, 737–759 [DOI] [PubMed] [Google Scholar]
- 18. Anderson J. P., Walker D. E., Goldstein J. M., de Laat R., Banducci K., Caccavello R. J., Barbour R., Huang J., Kling K., Lee M., Diep L., Keim P. S., Shen X., Chataway T., Schlossmacher M. G., Seubert P., Schenk D., Sinha S., Gai W. P., Chilcote T. J. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752 [DOI] [PubMed] [Google Scholar]
- 19. Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M. S., Shen J., Takio K., Iwatsubo T. (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164 [DOI] [PubMed] [Google Scholar]
- 20. Okochi M., Walter J., Koyama A., Nakajo S., Baba M., Iwatsubo T., Meijer L., Kahle P. J., Haass C. (2000) Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J. Biol. Chem. 275, 390–397 [DOI] [PubMed] [Google Scholar]
- 21. Pronin A. N., Morris A. J., Surguchov A., Benovic J. L. (2000) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522 [DOI] [PubMed] [Google Scholar]
- 22. Waxman E. A., Giasson B. I. (2008) Specificity and regulation of casein kinase-mediated phosphorylation of α-synuclein. J. Neuropathol. Exp. Neurol. 67, 402–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oueslati A., Schneider B. L., Aebischer P., Lashuel H. A. (2013) Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–E3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basso E., Antas P., Marijanovic Z., Gonçalves S., Tenreiro S., Outeiro T. F. (2013) PLK2 Modulates α-synuclein aggregation in yeast and mammalian cells. Mol. Neurobiol. 48, 854–862 [DOI] [PubMed] [Google Scholar]
- 25. Arawaka S., Wada M., Goto S., Karube H., Sakamoto M., Ren C. H., Koyama S., Nagasawa H., Kimura H., Kawanami T., Kurita K., Tajima K., Daimon M., Baba M., Kido T., Saino S., Goto K., Asao H., Kitanaka C., Takashita E., Hongo S., Nakamura T., Kayama T., Suzuki Y., Kobayashi K., Katagiri T., Kurokawa K., Kurimura M., Toyoshima I., Niizato K., Tsuchiya K., Iwatsubo T., Muramatsu M., Matsumine H., Kato T. (2006) The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J. Neurosci. 26, 9227–9238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salvi M., Trashi E., Marin O., Negro A., Sarno S., Pinna L. A. (2012) Superiority of PLK-2 as α-synuclein phosphorylating agent relies on unique specificity determinants. Biochem. Biophys. Res. Commun. 418, 156–160 [DOI] [PubMed] [Google Scholar]
- 27. Mbefo M. K., Paleologou K. E., Boucharaba A., Oueslati A., Schell H., Fournier M., Olschewski D., Yin G., Zweckstetter M., Masliah E., Kahle P. J., Hirling H., Lashuel H. A. (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inglis K. J., Chereau D., Brigham E. F., Chiou S. S., Schöbel S., Frigon N. L., Yu M., Caccavello R. J., Nelson S., Motter R., Wright S., Chian D., Santiago P., Soriano F., Ramos C., Powell K., Goldstein J. M., Babcock M., Yednock T., Bard F., Basi G. S., Sham H., Chilcote T. J., McConlogue L., Griswold-Prenner I., Anderson J. P. (2009) Polo-like kinase 2 (PLK2) phosphorylates α-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dorval V., Fraser P. E. (2006) Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J. Biol. Chem. 281, 9919–9924 [DOI] [PubMed] [Google Scholar]
- 30. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 31. Bennett M. C., Bishop J. F., Leng Y., Chock P. B., Chase T. N., Mouradian M. M. (1999) Degradation of α-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858 [DOI] [PubMed] [Google Scholar]
- 32. Tofaris G. K., Layfield R., Spillantini M. G. (2001) α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26 [DOI] [PubMed] [Google Scholar]
- 33. Webb J. L., Ravikumar B., Atkins J., Skepper J. N., Rubinsztein D. C. (2003) α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013 [DOI] [PubMed] [Google Scholar]
- 34. Ebrahimi-Fakhari D., Cantuti-Castelvetri I., Fan Z., Rockenstein E., Masliah E., Hyman B. T., McLean P. J., Unni V. K. (2011) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 31, 14508–14520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vogiatzi T., Xilouri M., Vekrellis K., Stefanis L. (2008) Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin Y., Klucken J., Patterson C., Hyman B. T., McLean P. J. (2005) The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates α-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 280, 23727–23734 [DOI] [PubMed] [Google Scholar]
- 37. Outeiro T. F., Lindquist S. (2003) Yeast cells provide insight into α-synuclein biology and pathobiology. Science 302, 1772–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petroi D., Popova B., Taheri-Talesh N., Irniger S., Shahpasandzadeh H., Zweckstetter M., Outeiro T. F., Braus G. H. (2012) Aggregate clearance of α-synuclein in Saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J. Biol. Chem. 287, 27567–27579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zabrocki P., Pellens K., Vanhelmont T., Vandebroek T., Griffioen G., Wera S., Van Leuven F., Winderickx J. (2005) Characterization of α-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 272, 1386–1400 [DOI] [PubMed] [Google Scholar]
- 40. Franssens V., Boelen E., Anandhakumar J., Vanhelmont T., Büttner S., Winderickx J. (2010) Yeast unfolds the road map toward α-synuclein-induced cell death. Cell Death Differ. 17, 746–753 [DOI] [PubMed] [Google Scholar]
- 41. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gietz D., St Jean A., Woods R. A., Schiestl R. H. (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guthrie C., Fink G. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol. 194, 1–863 [PubMed] [Google Scholar]
- 44. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Image processing with ImageJ. Biophotonics Int. 11, 36–42 [Google Scholar]
- 45. Lee D. H., Goldberg A. L. (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271, 27280–27284 [DOI] [PubMed] [Google Scholar]
- 46. Liu C., Apodaca J., Davis L. E., Rao H. (2007) Proteasome inhibition in wild-type yeast Saccharomyces cerevisiae cells. BioTechniques 42, 158–162 [DOI] [PubMed] [Google Scholar]
- 47. Soustelle C., Vernis L., Fréon K., Reynaud-Angelin A., Chanet R., Fabre F., Heude M. (2004) A new Saccharomyces cerevisiae strain with a mutant Smt3-deconjugating Ulp1 protein is affected in DNA replication and requires Srs2 and homologous recombination for its viability. Mol. Cell. Biol. 24, 5130–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dieckhoff P., Bolte M., Sancak Y., Braus G. H., Irniger S. (2004) Smt3/SUMO and Ubc9 are required for efficient APC/C-mediated proteolysis in budding yeast. Mol. Microbiol. 51, 1375–1387 [DOI] [PubMed] [Google Scholar]
- 49. Biggins S., Bhalla N., Chang A., Smith D. L., Murray A. W. (2001) Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 159, 453–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mullen J. R., Chen C. F., Brill S. J. (2010) Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol. Cell. Biol. 30, 3737–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen L., Feany M. B. (2005) α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663 [DOI] [PubMed] [Google Scholar]
- 52. Freichel C., Neumann M., Ballard T., Müller V., Woolley M., Ozmen L., Borroni E., Kretzschmar H. A., Haass C., Spooren W., Kahle P. J. (2007) Age-dependent cognitive decline and amygdala pathology in α-synuclein transgenic mice. Neurobiol. Aging 28, 1421–1435 [DOI] [PubMed] [Google Scholar]
- 53. Gorbatyuk O. S., Li S., Sullivan L. F., Chen W., Kondrikova G., Manfredsson F. P., Mandel R. J., Muzyczka N. (2008) The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 105, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Azeredo da Silveira S., Schneider B. L., Cifuentes-Diaz C., Sage D., Abbas-Terki T., Iwatsubo T., Unser M., Aebischer P. (2009) Phosphorylation does not prompt, nor prevent, the formation of α-synuclein toxic species in a rat model of Parkinson's disease. Hum. Mol. Genet. 18, 872–887 [DOI] [PubMed] [Google Scholar]
- 55. Wang S., Xu B., Liou L. C., Ren Q., Huang S., Luo Y., Zhang Z., Witt S. N. (2012) α-Synuclein disrupts stress signaling by inhibiting polo-like kinase Cdc5/Plk2. Proc. Natl. Acad. Sci. U.S.A. 109, 16119–16124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ellis C. E., Schwartzberg P. L., Grider T. L., Fink D. W., Nussbaum R. L. (2001) α-Synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem. 276, 3879–3884 [DOI] [PubMed] [Google Scholar]
- 57. Waxman E. A., Giasson B. I. (2011) Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J. Neurosci. Res. 89, 231–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakamoto M., Arawaka S., Hara S., Sato H., Cui C., Machiya Y., Koyama S., Wada M., Kawanami T., Kurita K., Kato T. (2009) Contribution of endogenous G-protein-coupled receptor kinases to Ser129 phosphorylation of α-synuclein in HEK293 cells. Biochem. Biophys. Res. Commun. 384, 378–382 [DOI] [PubMed] [Google Scholar]
- 59. Bergeron M., Motter R., Tanaka P., Fauss D., Babcock M., Chiou S. S., Nelson S., San Pablo F., Anderson J. P. (2014) In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 α-synuclein phosphorylation in mouse brain. Neuroscience 256, 72–82 [DOI] [PubMed] [Google Scholar]
- 60. Paleologou K. E., Schmid A. W., Rospigliosi C. C., Kim H. Y., Lamberto G. R., Fredenburg R. A., Lansbury P. T., Jr., Fernandez C. O., Eliezer D., Zweckstetter M., Lashuel H. A. (2008) Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J. Biol. Chem. 283, 16895–16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matsuura A., Tsukada M., Wada Y., Ohsumi Y. (1997) Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192, 245–250 [DOI] [PubMed] [Google Scholar]
- 62. Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. (1999) Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 10, 1367–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jones E. W. (2002) Vacuolar proteases and proteolytic artifacts in Saccharomyces cerevisiae. Methods Enzymol. 351, 127–150 [DOI] [PubMed] [Google Scholar]
- 64. Dubiel W., Ferrell K., Pratt G., Rechsteiner M. (1992) Subunit 4 of the 26 S protease is a member of a novel eukaryotic ATPase family. J. Biol. Chem. 267, 22699–22702 [PubMed] [Google Scholar]
- 65. Takeshige K., Baba M., Tsuboi S., Noda T., Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kametaka S., Matsuura A., Wada Y., Ohsumi Y. (1996) Structural and functional analyses of APG5, a gene involved in autophagy in yeast. Gene 178, 139–143 [DOI] [PubMed] [Google Scholar]
- 67. Lee D. H., Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 68. Nonaka T., Iwatsubo T., Hasegawa M. (2005) Ubiquitination of α-synuclein. Biochemistry 44, 361–368 [DOI] [PubMed] [Google Scholar]
- 69. Rott R., Szargel R., Haskin J., Shani V., Shainskaya A., Manov I., Liani E., Avraham E., Engelender S. (2008) Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328 [DOI] [PubMed] [Google Scholar]
- 70. Rott R., Szargel R., Haskin J., Bandopadhyay R., Lees A. J., Shani V., Engelender S. (2011) α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 18666–18671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mukherjee S., Thomas M., Dadgar N., Lieberman A. P., Iñiguez-Lluhí J. A. (2009) Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J. Biol. Chem. 284, 21296–21306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Janer A., Werner A., Takahashi-Fujigasaki J., Daret A., Fujigasaki H., Takada K., Duyckaerts C., Brice A., Dejean A., Sittler A. (2010) SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 19, 181–195 [DOI] [PubMed] [Google Scholar]
- 73. Droescher M., Begitt A., Marg A., Zacharias M., Vinkemeier U. (2011) Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J. Biol. Chem. 286, 18731–18746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hietakangas V., Anckar J., Blomster H. A., Fujimoto M., Palvimo J. J., Nakai A., Sistonen L. (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. U.S.A. 103, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yao Q., Li H., Liu B. Q., Huang X. Y., Guo L. (2011) SUMOylation-regulated protein phosphorylation, evidence from quantitative phosphoproteomics analyses. J. Biol. Chem. 286, 27342–27349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dadke S., Cotteret S., Yip S. C., Jaffer Z. M., Haj F., Ivanov A., Rauscher F., 3rd, Shuai K., Ng T., Neel B. G., Chernoff J. (2007) Regulation of protein tyrosine phosphatase 1B by sumoylation. Nat. Cell Biol. 9, 80–85 [DOI] [PubMed] [Google Scholar]
- 77. Reynolds M. R., Berry R. W., Binder L. I. (2007) Nitration in neurodegeneration: deciphering the “Hows” “nYs”. Biochemistry 46, 7325–7336 [DOI] [PubMed] [Google Scholar]
- 78. Smith W. W., Margolis R. L., Li X., Troncoso J. C., Lee M. K., Dawson V. L., Dawson T. M., Iwatsubo T., Ross C. A. (2005) α-Synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McFarland N. R., Fan Z., Xu K., Schwarzschild M. A., Feany M. B., Hyman B. T., McLean P. J. (2009) α-Synuclein Ser-129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sancenon V., Lee S. A., Patrick C., Griffith J., Paulino A., Outeiro T. F., Reggiori F., Masliah E., Muchowski P. J. (2012) Suppression of α-synuclein toxicity and vesicle trafficking defects by phosphorylation at Ser-129 in yeast depends on genetic context. Hum. Mol. Genet. 21, 2432–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 82. Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]