

Background: The RASSF1A tumor suppressor regulates apoptosis and the cell cycle.
Results: Loss of microtubule association does not affect the proapoptotic function of RASSF1A but abolishes its ability to modulate the cell cycle and suppress transformation.
Conclusion: Regulation of the cell cycle supersedes the proapoptotic effects of RASSF1A for suppression of the transformed phenotype.
Significance: This study provides further and unexpected insights into RASSF1A function.
Keywords: Apoptosis, Cell Cycle, Microtubule-associated Protein (MAP), Oncogene, Ras Protein, Ras, Tumor Suppressor
Abstract
The Ras association domain family protein 1A (RASSF1A) is arguably one of the most frequently inactivated tumor suppressors in human cancer. RASSF1A modulates apoptosis via the Hippo and Bax pathways but also modulates the cell cycle. In part, cell cycle regulation appears to be dependent upon the ability of RASSF1A to complex with microtubules and regulate their dynamics. Which property of RASSF1A, apoptosis induction or microtubule regulation, is responsible for its tumor suppressor function is not known. We have identified a short conserved motif that is essential for the binding of RASSF family proteins with microtubule-associated proteins. By making a single point mutation in the motif, we were able to generate a RASSF1A variant that retains wild-type apoptotic properties but completely loses the ability to bind microtubule-associated proteins and complex with microtubules. Comparison of this mutant to wild-type RASSF1A showed that, despite retaining its proapoptotic properties, the mutant was completely unable to induce cell cycle arrest or suppress the tumorigenic phenotype. Therefore, it appears that the cell cycle/microtubule effects of RASSF1A are key to its tumor suppressor function rather than its apoptotic effects.
Introduction
RASSF1A is a member of the RASSF2 family of tumor suppressors. There are six members of the classic RASSF family, which share ∼30–50% amino acid homology. All family members exhibit tumor suppressor properties and contain a Ras association domain that allows them to interact with the Ras oncoprotein (1, 2). RASSF proteins have no intrinsic enzymatic activity but, instead, are thought to act as scaffolding proteins. RASSF1A is the best characterized member and is inactivated by epigenetic silencing at high frequency in a broad range of tumors.
RASSF1A forms a complex with microtubules (3–6) and enhances their polymerization (5, 7). This property allows RASSF1A to modulate the cell cycle (1, 2, 8, 9). RASSF1A coimmunoprecipitates with α-, β-, and γ-tubulins (3–5, 10), but exactly how RASSF1A interacts with tubulin remains unclear. RASSF1A has been shown to bind directly to several microtubule-associated proteins (MAPs), such as MAP1B, MAP4, and C19ORF5 (MAP1S) (3, 11, 12). Because these proteins bind tubulin directly, the interaction of RASSF1A with tubulin could be indirect via MAPs.
In addition to microtubules and the cell cycle, RASSF1A also controls at least two apoptotic pathways, Hippo and Bax (13–16). RASSF1A activates the Hippo pathway by directly binding the kinases MST1 and MST2 (17, 18). It activates Bax by directly binding the Bax activator MOAP-1 (13, 14). Therefore, RASSF1A may both promote apoptosis and restrict the cell cycle, but which property is most responsible for its tumor suppressor properties is not known.
The precise domain of RASSF1A that is involved in its interaction with microtubules has not been clearly defined. Previously, deletion mutagenesis has been used to identify residues 120–185 as essential for the interaction with microtubules (5). A subsequent report suggested that association of RASSF1A with microtubules requires two regions within the protein, a five-amino stretch within the ATM phosphorylation site spanning residues 131–135 and a region in the C-terminal SARAH domain spanning residues 300–305 (4). Both of these deletion mutants suffered defects in their tumor-suppressing activity. However, the large structural disruption likely caused by the deletions makes it difficult to interpret their effects on the biology of the protein.
We have now identified a single point mutant of RASSF1A that completely loses the ability to associate with microtubules. This loss correlates with a loss of binding with all three MAPs known to bind RASSF1A directly. The mutant loses the ability to induce cell cycle arrest but retains the same apoptotic activity as the wild-type protein. Examination of the tumor-suppressive properties of the mutant showed that it was completely defective for the ability to suppress the soft agar growth of tumor cells. We identify the true minimal microtubule association domain of RASSF1A and show that it is the same as the minimum domain required to bind MAPs. Moreover, we show that it is the association with MAPs and the ability to regulate the cell cycle that is essential for the tumor suppressor properties of RASSF1A, not its apoptotic effects.
EXPERIMENTAL PROCEDURES
Tissue Culture and Cell Lines
HEK-293T, COS-7, and NCI-H1299 cells were obtained from the ATCC and cultured in DMEM and RPMI 1640 medium (Corning, Manassas, VA), respectively, supplemented with 10% FBS (Valley Biologicals, VA) and 1% penicillin/streptomycin (Corning). Transient transfections were performed with 1 μg of each plasmid DNA using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. NCI-H1299 stable transfectants were generated by transfecting cells with 2 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen) and selecting in 400 μg/ml G418. Stable cells were used as an early passage pooled population.
Plasmids and DNA
The pcDNA-HA, FLAG and GFP-RASSF1A, pZIP-HA-RASSF1A, and pCGN-K-Ras12V expression constructs have been described previously (3, 5, 19, 20). KATE-tagged RASSF1A was generated by subcloning a BamHI/EcoRI fragment from pcDNA3-HA-RASSF1A into pmKate2C (Evrogen, Moscow, Russia). The NORE1A expression construct was obtained from Origene (Rockville, MD), and HA- and FLAG-tagged NORE1A expression constructs were generated by inserting a BglII/EcoRI fragment into the BamHI/EcoRI sites of pcDNA3.1-HA or FLAG. The FLAG-C19ORF5, FLAG-Mst2, GFP-MAP1A, Myc-Mst1, and GFP-MAP4 expression constructs were provided by Drs. Farida Latif (21, 22), C. Chien (23), and J. Chernoff (24), respectively. GFP-tubulin was obtained from Clontech Laboratories (Mountain View, CA). The FAL mutants of RASSF1A and NORE1A were generated using the QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA) and PCR, respectively. For RASSF1A, the Leu residue at position 244 was mutated to a Ser, and for NORE1A, the FAL sequence (amino acids 317–319) was mutated to three glycine residues.
Immunoprecipitation and Western Blot Analysis
Total cell lysates were prepared by lysing the cells in modified radioimmune precipitation assay buffer (150 mm NaCl, 50 mm Tris (pH 7.5), and 1% Nonidet P-40) supplemented with 100 μg/ml leupeptin, 100 μg/ml aprotinin, and 1 mm sodium orthovanadate. Immunoprecipitations were performed using HA-, FLAG- (Sigma), or GFP-conjugated Sepharose beads (Allele Biotechnology, San Diego, CA). HA and FLAG antibodies were obtained from Sigma, anti-GFP and TFIIH antibodies from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and anti-p38 antibodies from Cell Signaling Technology (Danvers, MA). HRP-conjugated Trueblot secondary antibodies were purchased from eBioscience (San Diego, CA), and Western blot analyses were developed using a Pierce ECL detection system (Thermo Scientific, Rockford, IL).
Immunofluorescence
Fluorescence microscopy was performed on cells grown in glass bottom microwell dishes (MatTek Corp., Ashland, MA), and images were captured with an Olympus 1 × 50-FLA inverted fluorescent microscope (Optical Elements Corp., Dulles, VA) with an attached Spot Junior digital camera.
Cell Cycle Analysis
Cells grown in 60 mm dishes were analyzed by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences) essentially as described previously (25). The percentages of cells in G1 and G2 phases of the cell cycle were estimated using CellQuest software (BD Biosciences).
Subcellular Fractionation
Nuclear and cytoplasmic fractions were prepared using the NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific, Rockford, IL) according to the instructions of the manufacturer.
Statistical Analysis
Data are reported as mean ± S.D. The statistical significance of differences between the mean values was assessed by Student's t test. Data were considered significant at p < 0.05.
RESULTS
RASSF Family Members All Contain a Conserved FAL Motif That Is Not Required for Dimerization or Ras Binding
The six members of the classical RASSF family (RASSF1–6) show structural homology, but there are few areas where the primary structure is completely conserved in all members (1, 2). One of the few such regions is a three-amino acid FAL motif within the Ras association domain that is common to all of them (Fig. 1A). RASSF proteins can bind to the Ras oncoprotein (19, 26–28) and can homo- and heterodimerize with one another (26). The crystal structure of NORE1A (RASSF5) bound to Ras has been solved (29), and although the crystal structure of RASSF1A bound to Ras has not yet been elucidated, the residues in RASSF1A required for its interface with Ras have been determined (29), and the conserved FAL motif resides in both regions in RASSF1A and NORE1A required for their interactions with Ras (29). Therefore, we hypothesized that this conserved motif could either be essential for Ras binding or represent the core of a universal dimerization domain. To test these questions, we disrupted this domain in RASSF1A by substituting a serine residue for the leucine residue in the FAL sequence (RASSF1A-FAL) and measured the relative ability of this point mutant to homodimerize and heterodimerize with wild-type RASSF1A and NORE1A, respectively (Fig. 1, B and C) and to bind Ras (Fig. 1D). Although a very modest reduction in homodimerization was observed, disruption of the conserved FAL motif failed to block Ras binding or heterodimerization. Overall, the results suggest that this motif is not essential for dimerization of the RASSF family members or their interaction with Ras.
FIGURE 1.

A, sequence alignment encompassing a portion of the RA domain of RASSF family members with the conserved FAL motif in the boxed area. B and C, HEK-293T cells were cotransfected with expression constructs for HA-tagged RASSF1A wild-type or the FAL mutant and wild-type FLAG-tagged RASSF1A (B) or NORE1A (C), and equal amounts of protein were immunoprecipitated (IP) for FLAG. The immunoprecipitate was fractionated on SDS gels and immunoblotted (IB) with anti-HA and anti-FLAG antibodies. D, HEK-293T cells were cotransfected with GFP-tagged expression constructs for wild-type RASSF1A or the FAL mutant and HA-tagged K-Ras, and equal amounts of protein were immunoprecipitated for GFP. The immunoprecipitate was fractionated on SDS gels and immunoblotted with anti-GFP and anti-HA antibodies.
The Conserved FAL Motif in RASSF1A Is Required for Interaction with Microtubules
While examining the point mutant protein for the ability to dimerize by fluorescence microscopy, we noticed a dramatic change in its subcellular localization compared with the wild-type RASSF1A protein. Wild-type RASSF1A localized to the microtubules, as expected (3–5). However, RASSF1A-FAL localized predominantly to the nucleus, with faint, diffuse staining in the cytoplasm (Fig. 2A). Cotransfection of RASSF1A-FAL with wild-type RASSF1A relocalized it to the microtubules, along with the wild-type RASSF1A (Fig. 2B), and it showed strong colocalization with NORE1A in the nucleus (Fig. 2B). The relocalization of the RASSF1A-FAL mutant to microtubules in the presence of wild-type RASSF1A is probably due to its retained ability to homodimerize (Fig. 1B). In addition to its nuclear localization, the FAL mutant also appeared in cytoplasmic speckles (Fig. 2C) that colocalized with pDsRed2-Mito, a red-tagged fusion protein specific for mitochondria (Clontech), indicating that RASSF1A-FAL could also localize to mitochondria. This was not observed for wild-type RASSF1A. Therefore, the conserved FAL appears to be essential for the interaction with microtubules. To confirm that the RASSF1A-FAL mutant did not interact with tubulin, we cotransfected RFP-tagged RASSF1A-FAL and GFP-tagged tubulin into COS-7 cells and observed the subcellular localization of the proteins using fluorescence microscopy. Wild-type RASSF1A showed strong colocalization with tubulin in the cells, whereas the RASSF1A-FAL mutant failed to colocalize with tubulin (Fig. 2D). In addition, tubulin formed a fine meshwork structure within the cytoplasm when it was cotransfected with the empty vector, but in the presence of RASSF1A, the fine network of fibers became much thicker, indicative of polymerized, stabilized tubulin (Fig. 2D). The RASSF1A-FAL mutant did not cause thickening of the tubulin fibers (Fig. 2D), suggesting that it is defective for tubulin polymerization.
FIGURE 2.
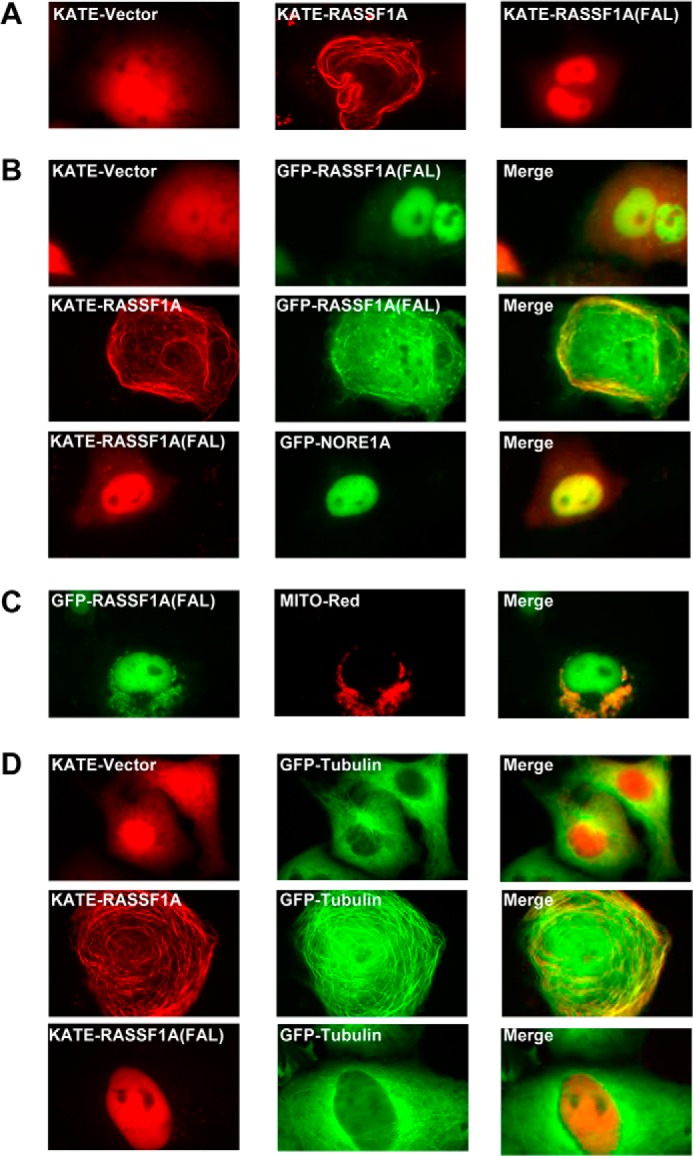
A–D, COS-7 cells were transfected with KATE-tagged wild-type RASSF1A or the FAL mutant (A); cotransfected with GFP- or KATE-tagged RASSF1A(FAL) and KATE-tagged wild-type RASSF1A or empty vector and GFP-tagged NORE1A (B); cotransfected with GFP-tagged RASSF1A(FAL) and MITO-Red (C); and cotransfected with expression constructs for GFP-tagged tubulin and KATE-tagged wild-type RASSF1A, RASSF1A(FAL), or empty vector (D). 24 h later, images were captured using a fluorescence microscope (magnification, ×100).
RASSF1A-FAL Fails to Interact with MAPs
RASSF1A has been reported to directly bind several MAPs, such as MAP1B and C19ORF5 (3, 11, 21). These MAPS are known to play a role in the regulation of tubulin dynamics. It seems reasonable to suppose that the interaction of RASSF1A with tubulin is via MAPs, but this has never been confirmed. Because the FAL mutant does not associate with microtubules, we determined its ability to interact with various MAPs, and we found that it does not interact with C19ORF5 (Fig. 3A), MAP1A (Fig. 3B), or MAP4 (Fig. 3C). This is not simply due to the apparent lack of cytoplasmic localization of the FAL mutant because fractionation experiments on the HEK-293T cells transfected with either wild-type RASSF1A or the FAL mutant show no significant difference in subcellular localization of the two proteins (Fig. 3D). This is in apparent contrast with the fluorescence data. Other RASSF family members have been reported to associate with MAPs, for example NORE1A (30). Therefore, the conserved FAL motif may be critical for RASSF family members to interact with MAPs. To test this, we completely disrupted the FAL motif in NORE1A by substituting the FAL sequence in NORE1A (Fig. 1A) with three glycine residues (NORE1A-FAL). We then used this mutant in coimmunoprecipitation experiments with the MAP C19ORF5. Similar to RASSF1A, wild-type NORE1A interacted with C19ORF5, but the NORE1A-FAL mutant did not (Fig. 4A). Mutation of the conserved FAL motif within NORE1A did not interfere with the ability of NORE1A to homo- or heterodimerize with RASSF1A or RASSF2 (Fig. 4, B and C).
FIGURE 3.

A–C, HEK-293T cells were cotransfected with HA-tagged RASSF1A wild-type or the FAL mutant and FLAG-tagged C19ORF5 (A), GFP-tagged MAP1A (B), or GFP-tagged MAP4 (C), and equal amounts of protein were immunoprecipitated (IP) with anti-HA (A) or anti-GFP (B and C) antibodies. The immunoprecipitates were fractionated on SDS gels and Western blotted with anti-FLAG, anti-HA, and anti-GFP antibodies. Levels of MAP1A were not measured by Western blot analysis because the size of MAP1A precluded it from entering the gel. However, the intensity of GFP was the same for each transfection, as determined by fluorescence microscopy. IB, immunoblot. D, HEK-293T cells were transfected with HA-tagged wild-type RASSF1A or RASSF1A(FAL). Cells were lysed, and cytoplasmic (Cyt) and nuclear (Nuc) fractions were prepared and analyzed by Western blotting using an anti-HA antibody. p38 and TFIIH were used as markers for the cytoplasmic and nuclear fractions, respectively.
FIGURE 4.
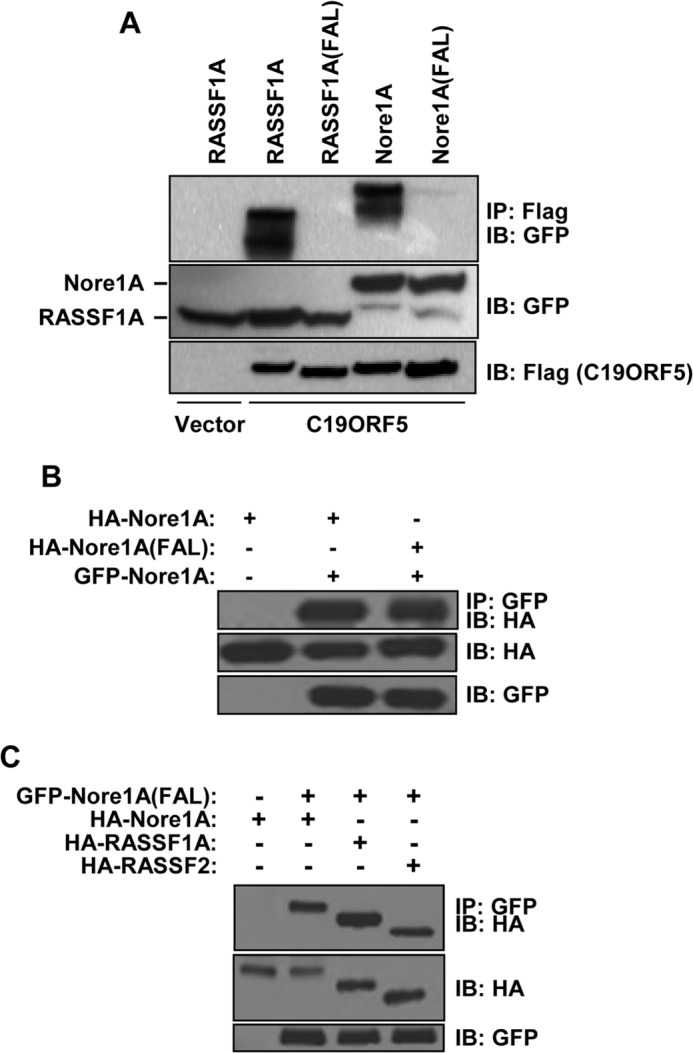
A–C, HEK-293T cells were cotransfected with expression constructs for GFP-tagged wild-type RASSF1A, RASSF1A(FAL), wild-type NORE1A or NORE1A(FAL), and FLAG-tagged C19ORF5 (A); HA-tagged NORE1A wild-type or the NORE1A FAL mutant and GFP-tagged NORE1A wild-type (B); and GFP-tagged NORE1A(FAL) and HA-tagged NORE1A, RASSF1A, or RASSF2 (C). 24 h later, equal amounts of protein were immunoprecipitated (IP) with anti-HA (A) and anti-GFP (B and C) antibodies. The immunoprecipitates were fractionated on SDS gels and immunoblotted (IB) with antibodies to FLAG, HA, and GFP.
Inactivation of the FAL Motif Blocks the Ability of RASSF1A to Suppress Growth and Restrict the Cell Cycle
RASSF1A acts, in part, to restrict the cell cycle. To determine whether the ability to bind MAPs and modulate microtubule dynamics is essential for tumor suppression, we stably expressed either wild-type RASSF1A or the FAL mutant in H1299 cells that were null for RASSF1A (31). Western blot analysis was used to confirm equivalent levels of expression of the RASSF1A proteins (Fig. 5A, inset). We then characterized the cell lines for biological phenotype. Cell proliferation analysis showed that wild-type RASSF1A significantly reduced the growth of the H1299 cells, whereas the cells expressing the FAL mutant grew at the same rate as the vector control cells (Fig. 5A). To determine whether the inability of RASSF1A-FAL to inhibit cell growth was due to cell cycle effects, we transfected HEK-293T cells with expression constructs for either wild-type RASSF1A or the FAL mutant and a vector control and measured the cell cycle profile of the cells 24 h later. Although wild-type RASSF1A induced a G2 cell cycle arrest, the FAL mutant failed to do so (Fig. 5B).
FIGURE 5.

A, NCI-H1299 lung cancer cells were stably transfected with vectors expressing HA-tagged wild-type or the FAL mutant of RASSF1A. Inset, expression of the exogenous protein was confirmed by Western blotting with an anti-HA antibody. 2 × 104 cells/well were plated in 6-well plates, and cell growth was determined by counting the number of cells at the indicated times. Data are plotted as the mean ± S.D. of two independent experiments performed in duplicate. *, p < 0.05 compared with vector control and FAL-transfected cells. B, HEK-293 cells were transfected with wild-type RASSF1A, RASSF1A(FAL), or empty vector control, and, 24 h later, cell cycle progression was measured using FACS analysis. Data are mean ± S.D. of three independent experiments. *, p < 0.05 compared with vector-transfected cells.
The FAL Mutant of RASSF1A Retains Wild-type Apoptotic Properties
In addition to binding MAPs and regulating microtubules, RASSF1A also promotes apoptosis. RASSF1A can stimulate the proapoptotic Hippo pathway by binding the MST kinases (17). It also binds the Bax-activating protein MOAP-1 to activate Bax-mediated apoptosis (13, 14). To determine whether the RASSF1A-FAL point mutant was defective for apoptosis, we performed coimmunoprecipitation experiments with HA-tagged RASSF1A or the FAL mutant and Myc-tagged Mst1 or FLAG-tagged Mst2 in HEK-293T cells and found that the FAL mutant interacted with both Mst1 and Mst2 to the same degree as wild-type RASSF1A (Fig. 6A). To test whether this interaction had any effect on the Hippo pathway, we measured the levels of phosphorylated YAP, a key downstream effector of the Hippo signaling pathway, in H1299 cells stably expressing either wild-type RASSF1A or the FAL mutant (Fig. 5A) and found elevated levels of phosphorylated YAP in both the wild-type and FAL-expressing cells compared with the vector control cells (Fig. 6B). We next performed apoptosis assays and found no significant difference between the mutant and the wild-type protein (Fig. 6, C and D). In fact, the FAL mutant showed slightly higher activation of caspase 3/7 than the wild-type protein in certain apoptosis assays (Fig. 6D). These results show that microtubule association of RASSF1A is not required for it to mediate its apoptotic function. Therefore, the FAL mutant allows us to separate these two aspects of RASSF1A biology. RASSF1A is a negative effector of K-Ras (1), and because the FAL mutant was still able to interact with K-Ras (Fig. 1D), we hypothesized that the FAL mutant would not affect K-Ras signaling. To test this, we used activation of Elk-1, a transcription factor that is activated by K-Ras (32), as a readout of Ras activity and, somewhat surprisingly, found that, although wild-type RASSF1A significantly inhibited K-Ras-induced activation of Elk-1, the FAL point mutant was defective for inhibiting K-Ras signaling (Fig. 6E). Both K-Ras and Elk-1 have been found to be associated with microtubules (33–35). Therefore, the inability of the FAL mutant to inhibit K-Ras-induced activation of Elk-1 while still maintaining the ability to interact with K-Ras may be due to its inability to bind to microtubules. Different pools of Ras have been identified at various subcellular locations, and the different subcellular localization of Ras influences the nature of Ras signaling (36). Therefore, it is possible that the interaction of the FAL mutant and K-Ras is occurring at additional subcellular structures.
FIGURE 6.

A, HEK-293T cells were cotransfected with HA-tagged RASSF1A wild-type or the FAL mutant and Myc-tagged Mst1 (left panel) or FLAG-tagged Mst2 (right panel). 24 h later, equal amounts of protein were immunoprecipitated (IP) with an anti-HA antibody. The immunoprecipitates were fractionated on SDS gels and Western blotted with anti-HA, anti-Myc, and anti-FLAG antibodies. IB, immunoblot. B, endogenous levels of YAP and phospho-YAP in H1299 cells stably expressing wild-type RASSF1A and the FAL mutant were measured by Western blot analysis using YAP- and phospho-YAP-specific antibodies. C, COS-7 cells were transfected with KATE-tagged wild-type RASSF1A, RASSF1A(FAL) or empty vector, and pCaspase 3 sensor, which produces a fluorescent protein that is sensitive to caspase cleavage. Cells positive for caspase activation were scored. The results are an average of four separate assays. *, p < 0.05 compared with vector-transfected cells. D, HEK-293 cells were transfected with HA-tagged RASSF1A wild-type or the FAL mutant and, 24 h later, analyzed for caspase activity using the Caspase-Glo® 3/7 reagent. Data are mean ± S.D. of triplicate experiments and expressed as relative light units (RLU). *, p < 0.05 compared with vector control cells. E, HEK-293 cells were cotransfected with expression constructs for HA-tagged RASSF1A wild-type or RASSF1A(FAL), Gal-Elk-1, and KATE-tagged K-Ras(12)V together with a 5xGal luciferase reporter construct. 24 h later, cells were lysed, and luciferase activity was measured (left panel). Data are mean ± S.D. of duplicate experiments. *, p < 0.05 compared with cells transfected with K-Ras alone. To ensure equivalent levels of protein expression, equal amounts of the lysates were fractionated on SDS gels and analyzed by Western blotting using anti-HA and anti-KATE antibodies (right panel). β-Actin was used as a loading control. F, H1299 cells stably expressing either wild-type RASSF1A, the FAL mutant, or vector control were plated in 6-well plates in growth medium supplemented with 0.5% serum, and cell growth was determined by counting the number of cells 1 day and 4 days after plating. Data are mean ± S.D. of duplicate experiments. *, p < 0.05 compared with vector control cells; **, p < 0.05 compared with wild-type RASSF1A-expressing cells. G, H1299 cells were plated in soft agar and scored for growth 14 days later. Data are mean ± S.D. of two experiments performed in duplicate. *, p < 0.05 compared with vector control and FAL-expressing cells.
Mutation of the FAL Motif Abrogates the Tumor Suppressor Activity of RASSF1A
RASSF1A can inhibit the cell cycle and induce apoptosis. Which property is primarily responsible for its tumor suppressor activity remains unclear. The experiments described above show that the RASSF1A-FAL mutant is totally defective for the ability to bind MAPs and modulate the cell cycle but is wild-type for the ability to induce apoptosis. When we examined the ability of the stably transfected NCI-H1299 cells to grow in low serum and soft agar, we found that the RASSF1A-FAL mutant had lost the ability to suppress agar growth and was able to induce a significant increase in cell growth after serum starvation, in contrast to the wild-type protein, which suppressed the growth (Fig. 6, F and G).
DISCUSSION
RASSF1A is one of the most frequently inactivated tumor suppressors identified. It is inactivated at high frequency by epigenetic silencing in a broad range of tumors. It is also inactivated by point mutations in ∼15% of human tumors (1, 8). Some tumor types, for example clear cell renal carcinomas, exhibit defects in RASSF1A at almost 100% (1). Exactly how RASSF1A loss facilitates transformation remains unclear. In part, this is because it has multiple biological effects, the best characterized of which are regulation of microtubule dynamics and the induction of apoptosis.
The interaction of RASSF1A with microtubules allows it to modulate microtubule polymerization (3, 7, 37), cell cycle progression, and mitotic spindle dynamics (5, 10). Exactly how RASSF1A associates with and regulates microtubules is unclear. We have reported previously that deletion of amino acids 120–185 abolishes the ability of RASSF1A to interact with microtubules (5), and deletion of two short amino acid stretches (amino acids 131–135 and 300–305) also results in loss of RASSF1A microtubule binding (4). In addition, deletions within a highly basic region of RASSF1A (amino acids 165–258) and of the Ras association (RA) domain (11) also results in loss of microtubule binding (10). These previous attempts to define the microtubule association domain of RASSF1A gave results that are somewhat contradictory and confusing. However, all utilized gross deletions that may impact protein folding. We now report that disruption of a conserved FAL motif (Fig. 1A, amino acids 242–244) by a single amino acid substitution abolishes the ability of RASSF1A to interact with microtubules. Because this conserved motif is located within the larger, highly basic domain of RASSF1A, a domain previously reported to be required for microtubule association (10), our data suggest that at least the minimal region required for RASSF1A to associate with microtubules is this FAL sequence.
Whether RASSF1A binds directly to tubulin or whether it is tethered to the microtubules via its interactions with MAPs (3, 4, 11) is not known. Our data demonstrating the inability of the FAL microtubule binding-defective mutant of RASSF1A to interact with multiple different MAPs (Fig. 3) and microtubules supports a model in which the interaction between RASSF1A and microtubules is indirect via binding to MAPs. This would mean that the FAL motif is conserved between RASSF family members to allow them all to associate with MAPs. This model is confirmed by the observation that mutating the FAL motif in the RASSF family member NORE1A has a similar effect on its interaction with MAPs and implies that all other RASSF family members may interact with MAPs via their FAL motifs.
In addition to binding MAPs and modulating tubulin dynamics, RASSF1A also promotes apoptosis by at least two pathways. It interacts with MOAP-1 (13, 14), an activator of Bax, as well as with the Mst kinases (17) to stimulate the proapoptotic Hippo pathway. To date, however, it is uncertain whether RASSF1A-mediated tumor suppression requires apoptosis, tubulin modulation/cell cycle control, or both. In this study, by using a single point mutant that specifically abrogates the MAP binding functions of RASSF1A while leaving the apoptotic pathways intact, we have shown that it is the MAP/microtubule effects of RASSF1A that are the key to its ability to suppress growth and transformation rather than the apoptotic effects. These results are in contrast to previous findings that microtubule binding was required for RASSF1A-mediated apoptosis in response to TNF-α (4). This discrepancy may be explained by the use of more disruptive deletion mutations rather than the single point mutant used in this study.
The inability of RASSF1A to interact with microtubules resulted in atypical localization of the protein. We found that it was localized predominantly in the nucleus as well as in a speckled pattern in the cytoplasm that stained positive for a mitochondrion-specific marker. Similar patterns of expression have been observed with other microtubule binding-defective mutants of RASSF1A (3–5, 38). Wild-type RASSF1A has also been found in the nucleus (39). Microtubule association of RASSF1A may be so predominant that it masks these other patterns of subcellular distribution of steady-state levels of RASSF1A. Therefore, RASSF1A may have additional mitochondrial and nuclear functions that are as yet undetermined. However, because RASSF1A has been implicated in binding the DNA repair protein XPA (40), the nuclear location may indicate a potential role in DNA repair. The RASSF1A binding MAP designated C19ORF5 also interacts with the protein LRPPRC, a mitochondrion-associated, leucine-rich protein implicated in autophagy (41). Therefore, RASSF1A may potentially link microtubules to mitochondria to modulate autophagic cell death, and, therefore, the RASSF1A-FAL mutant may prove to be a useful tool for revealing the role of RASSF1A in mitochondria.
In summary, we have identified the minimal, shared MAP binding motif in RASSF proteins. Disruption of this site separates the ability of the protein to induce apoptosis from its ability to induce G2 arrest. The FAL mutant allowed us to demonstrate that it is the non-apoptotic functions of RASSF1A that appear to be the most important for its tumor suppressor phenotype.
This work was supported, in whole or in part, by National Institutes of Health Grants 1P20 RR18733 and R01 CA133171-01A2 (to G. J. C.).
- RASSF
- Ras association (RalGDS/AF-6) domain family
- MAP
- microtubule-associated protein.
REFERENCES
- 1. Donninger H., Vos M. D., Clark G. J. (2007) The RASSF1A tumor suppressor. J. Cell Sci. 120, 3163–3172 [DOI] [PubMed] [Google Scholar]
- 2. van der Weyden L., Adams D. J. (2007) The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 1776, 58–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dallol A., Agathanggelou A., Fenton S. L., Ahmed-Choudhury J., Hesson L., Vos M. D., Clark G. J., Downward J., Maher E. R., Latif F. (2004) RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res. 64, 4112–4116 [DOI] [PubMed] [Google Scholar]
- 4. El-Kalla M., Onyskiw C., Baksh S. (2010) Functional importance of RASSF1A microtubule localization and polymorphisms. Oncogene 29, 5729–5740 [DOI] [PubMed] [Google Scholar]
- 5. Vos M. D., Martinez A., Elam C., Dallol A., Taylor B. J., Latif F., Clark G. J. (2004) A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res. 64, 4244–4250 [DOI] [PubMed] [Google Scholar]
- 6. Song M. S., Song S. J., Ayad N. G., Chang J. S., Lee J. H., Hong H. K., Lee H., Choi N., Kim J., Kim H., Kim J. W., Choi E. J., Kirschner M. W., Lim D. S. (2004) The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 6, 129–137 [DOI] [PubMed] [Google Scholar]
- 7. Liu L., Tommasi S., Lee D. H., Dammann R., Pfeifer G. P. (2003) Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene 22, 8125–8136 [DOI] [PubMed] [Google Scholar]
- 8. Agathanggelou A., Cooper W. N., Latif F. (2005) Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 65, 3497–3508 [DOI] [PubMed] [Google Scholar]
- 9. Dammann R., Schagdarsurengin U., Seidel C., Strunnikova M., Rastetter M., Baier K., Pfeifer G. P. (2005) The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol. Histopathol. 20, 645–663 [DOI] [PubMed] [Google Scholar]
- 10. Rong R., Jin W., Zhang J., Sheikh M. S., Huang Y. (2004) Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene 23, 8216–8230 [DOI] [PubMed] [Google Scholar]
- 11. Liu L., Vo A., McKeehan W. L. (2005) Specificity of the methylation-suppressed A isoform of candidate tumor suppressor RASSF1 for microtubule hyperstabilization is determined by cell death inducer C19ORF5. Cancer Res. 65, 1830–1838 [DOI] [PubMed] [Google Scholar]
- 12. Donninger H., Barnoud T., Nelson N., Kassler S., Clark J., Cummins T. D., Powell D. W., Nyante S., Millikan R. C., Clark G. J. (2011) RASSF1A and the rs2073498 Cancer Associated SNP. Front Oncol. 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baksh S., Tommasi S., Fenton S., Yu V. C., Martins L. M., Pfeifer G. P., Latif F., Downward J., Neel B. G. (2005) The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol. Cell 18, 637–650 [DOI] [PubMed] [Google Scholar]
- 14. Vos M. D., Dallol A., Eckfeld K., Allen N. P., Donninger H., Hesson L. B., Calvisi D., Latif F., Clark G. J. (2006) The RASSF1A tumor suppressor activates Bax via MOAP-1. J. Biol. Chem. 281, 4557–4563 [DOI] [PubMed] [Google Scholar]
- 15. Avruch J., Praskova M., Ortiz-Vega S., Liu M., Zhang X. F. (2006) Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol. 407, 290–310 [DOI] [PubMed] [Google Scholar]
- 16. Donninger H., Allen N., Henson A., Pogue J., Williams A., Gordon L., Kassler S., Dunwell T., Latif F., Clark G. J. (2011) Salvador protein is a tumor suppressor effector of RASSF1A with hippo pathway-independent functions. J. Biol. Chem. 286, 18483–18491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo C., Tommasi S., Liu L., Yee J. K., Dammann R., Pfeifer G. P. (2007) RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705 [DOI] [PubMed] [Google Scholar]
- 18. Matallanas D., Romano D., Yee K., Meissl K., Kucerova L., Piazzolla D., Baccarini M., Vass J. K., Kolch W., O'Neill E. (2007) RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vos M. D., Ellis C. A., Bell A., Birrer M. J., Clark G. J. (2000) Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J. Biol. Chem. 275, 35669–35672 [DOI] [PubMed] [Google Scholar]
- 20. Fiordalisi J. J., Johnson R. L., 2nd, Ulkü A. S., Der C. J., Cox A. D. (2001) Mammalian expression vectors for Ras family proteins: generation and use of expression constructs to analyze Ras family function. Methods Enzymol. 332, 3–36 [DOI] [PubMed] [Google Scholar]
- 21. Dallol A., Cooper W. N., Al-Mulla F., Agathanggelou A., Maher E. R., Latif F. (2007) Depletion of the Ras association domain family 1, isoform A-associated novel microtubule-associated protein, C19ORF5/MAP1S, causes mitotic abnormalities. Cancer Res. 67, 492–500 [DOI] [PubMed] [Google Scholar]
- 22. Cooper W. N., Hesson L. B., Matallanas D., Dallol A., von Kriegsheim A., Ward R., Kolch W., Latif F. (2009) RASSF2 associates with and stabilizes the proapoptotic kinase MST2. Oncogene 28, 2988–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chien C. L., Lu K. S., Lin Y. S., Hsieh C. J., Hirokawa N. (2005) The functional cooperation of MAP1A heavy chain and light chain 2 in the binding of microtubules. Exp. Cell Res. 308, 446–458 [DOI] [PubMed] [Google Scholar]
- 24. Creasy C. L., Chernoff J. (1995) Cloning and characterization of a human protein kinase with homology to Ste20. J. Biol. Chem. 270, 21695–21700 [DOI] [PubMed] [Google Scholar]
- 25. Donninger H., Binder A., Bohm L., Parker M. I. (2008) Differential effects of novel tumour-derived p53 mutations on the transformation of NIH-3T3 cells. Biol. Chem. 389, 57–67 [DOI] [PubMed] [Google Scholar]
- 26. Ortiz-Vega S., Khokhlatchev A., Nedwidek M., Zhang X. F., Dammann R., Pfeifer G. P., Avruch J. (2002) The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene 21, 1381–1390 [DOI] [PubMed] [Google Scholar]
- 27. Clark J., Freeman J., Donninger H. (2012) Loss of RASSF2 enhances tumorigencity of lung cancer cells and confers resistance to chemotherapy. Mol. Biol. Int. 2012, 705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matallanas D., Romano D., Al-Mulla F., O'Neill E., Al-Ali W., Crespo P., Doyle B., Nixon C., Sansom O., Drosten M., Barbacid M., Kolch W. (2011) Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 44, 893–906 [DOI] [PubMed] [Google Scholar]
- 29. Stieglitz B., Bee C., Schwarz D., Yildiz O., Moshnikova A., Khokhlatchev A., Herrmann C. (2008) Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J. 27, 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moshnikova A., Kuznetsov S., Khokhlatchev A. V. (2008) Interaction of the growth and tumour suppressor NORE1A with microtubules is not required for its growth-suppressive function. BMC Res. Notes 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burbee D. G., Forgacs E., Zöchbauer-Müller S., Shivakumar L., Fong K., Gao B., Randle D., Kondo M., Virmani A., Bader S., Sekido Y., Latif F., Milchgrub S., Toyooka S., Gazdar A. F., Lerman M. I., Zabarovsky E., White M., Minna J. D. (2001) Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 93, 691–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malumbres M., Pellicer A. (1998) RAS pathways to cell cycle control and cell transformation. Front. Biosci. 3, d887–d912 [DOI] [PubMed] [Google Scholar]
- 33. Chen Z., Otto J. C., Bergo M. O., Young S. G., Casey P. J. (2000) The C-terminal polylysine region and methylation of K-Ras are critical for the interaction between K-Ras and microtubules. J. Biol. Chem. 275, 41251–41257 [DOI] [PubMed] [Google Scholar]
- 34. Demir O., Korulu S., Yildiz A., Karabay A., Kurnaz I. A. (2009) Elk-1 interacts with neuronal microtubules and relocalizes to the nucleus upon phosphorylation. Mol. Cell Neurosci. 40, 111–119 [DOI] [PubMed] [Google Scholar]
- 35. Thissen J. A., Gross J. M., Subramanian K., Meyer T., Casey P. J. (1997) Prenylation-dependent association of Ki-Ras with microtubules: evidence for a role in subcellular trafficking. J. Biol. Chem. 272, 30362–30370 [DOI] [PubMed] [Google Scholar]
- 36. Casar B., Arozarena I., Sanz-Moreno V., Pinto A., Agudo-Ibáñez L., Marais R., Lewis R. E., Berciano M. T., Crespo P. (2009) Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol. Cell. Biol. 29, 1338–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnette C., Efimova N., Zhu X., Clark G. J., Kaverina I. (2014) Microtubule segment stabilization by RASSF1A is required for proper microtubule dynamics and Golgi integrity Mol. Biol. Cell 25, 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rong R., Jiang L. Y., Sheikh M. S., Huang Y. (2007) Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 26, 7700–7708 [DOI] [PubMed] [Google Scholar]
- 39. Thaler S., Hähnel P. S., Schad A., Dammann R., Schuler M. (2009) RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 69, 1748–1757 [DOI] [PubMed] [Google Scholar]
- 40. Dammann R., Li C., Yoon J. H., Chin P. L., Bates S., Pfeifer G. P. (2000) Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 25, 315–319 [DOI] [PubMed] [Google Scholar]
- 41. Liu L., Vo A., Liu G., McKeehan W. L. (2005) Putative tumor suppressor RASSF1 interactive protein and cell death inducer C19ORF5 is a DNA binding protein. Biochem. Biophys. Res. Commun. 332, 670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]