

Background: The mechanism by which Ral GTPases mediate malignant transformation is not well understood.
Results: Depletion of Ral leads to increases in ATM kinase p53, Ser-15 phosphorylation, p53 half-life, and p53-dependent expression of p21WAF.
Conclusion: Ral expression is required for maintaining low levels of p53.
Significance: Down-regulation of Ral reactivates p53 by enhancing its stability, and this contributes to suppression of malignant transformation.
Keywords: Apoptosis, Cancer Biology, p53, Ras Protein, Signal Transduction, K-Ras, RalA, RalB, Serine 15 Phosphorylation
Abstract
Ral GTPases are critical effectors of Ras, yet the molecular mechanism by which they induce malignant transformation is not well understood. In this study, we found the expression of K-Ras, RalB, and sometimes RalA, but not AKT1/2 and c-Raf, to be required for maintaining low levels of p53 in human cancer cells that harbor mutant K-Ras and wild-type p53. Down-regulation of K-Ras, RalB, and sometimes RalA increases p53 protein levels and results in a p53-dependent up-regulation of the expression of p21WAF. K-Ras, RalA, and RalB depletion increases p53 stability as demonstrated by ataxia telangiectasia-mutated kinase activation, increased Ser-15 phosphorylation, and a significant (up to 6-fold) increase in p53 half-life. Furthermore, depletion of K-Ras and RalB inhibits anchorage-independent growth and invasion and interferes with cell cycle progression in a p53-dependent manner. Depletion of RalA inhibits invasion in a p53-dependent manner. Thus, expression of K-Ras and RalB and possibly RalA proteins is critical for maintaining low levels of p53, and down-regulation of these GTPases reactivates p53 by significantly enhancing its stability, and this contributes to suppression of malignant transformation.
Introduction
Malignant transformation is a complex process that involves constitutive activation of proto-oncogenes and inactivation of tumor suppressor genes (1). The Ras family members K-, N-, and H-Ras are proto-oncogenes that are mutated and constitutively activated in one-third of all human cancers, with K-Ras mutations being the most prevalent (2). Mutant Ras proteins induce malignant transformation by persistently activating several downstream pathways (3). Among the most thoroughly studied downstream effectors of Ras proteins are those mediated by the Raf/MEK/ERK, the PI3K/Akt, and the RalGDS/Ral pathways. These three pathways have been shown to be required for Ras proteins to induce certain hallmarks of cancer such as uncontrolled proliferation, apoptosis evasion, angiogenesis, migration, and invasion (3). In addition to activating pathways that promote oncogenesis, mutant Ras proteins also antagonize tumor-suppressive pathways to transform cells.
The tumor suppressor p53 is the genome guardian that prevents malignant transformation by inducing cell cycle arrest, senescence, and apoptosis in response to stress signals such as oncogene activation (4, 5) and DNA damage (6, 7). p53 is a transcription factor that induces or represses the expression of many genes, including those involved in cell cycle progression and cell survival (8). Most human tumors contain nonfunctional p53, either because of p53 mutations or inactivation of p53-dependent pathways (4, 9). One mechanism by which tumors with wild-type p53 inactivate it is by overexpressing its negative regulator MDM2, an E3 ligase that induces p53 degradation (10). Another mechanism by which wild-type p53 is inactivated is by loss of the MDM2 antagonist ARF (11–13). Therefore, for oncogenes to transform cells with wild-type p53, they must trigger pathways that lead to the inactivation of p53. In the case of the Ras proteins, this is isoform-dependent (14–17). For example, expressing oncogenic H-Ras in primary human and rodent cells induces senescence and apoptosis through activation of p53 (17). Similarly, expressing mutant N-RasG12D in lymphoid tissue of transgenic mice leads to lymphocytes that are highly susceptible to senescence (14). Consistent with this, cells challenged with mutant H-Ras or mutant N-Ras protect themselves by inducing the expression of ARF (18–20), which antagonizes MDM2 function either by sequestering MDM2 in the nucleoli (13) or by directly inhibiting its ubiquitin ligase activity (11). This leads to increased p53 levels, which in turn leads to senescence and apoptosis (5). In contrast, primary mouse embryo fibroblasts that express oncogenic K-Ras fail to undergo senescence; instead, they proliferate as immortal cells (21). Consistent with these findings, recent studies have shown that overexpressing mutant K-Ras but not H- or N-Ras reduces p53 levels (16). One proposed mechanism by which mutant K-Ras reduces p53 levels may involve the activation of the E3 ligase SNAIL, which leads to ubiquitination of p53 and its proteasomal degradation (16).
Although not thoroughly investigated, some studies reported on the regulation of p53 by Raf and Akt, kinases known to mediate Ras malignant transformation in some cells. For example, in Ras-transformed cells, Raf promotes the degradation of p53 by inducing MDM2, and this leads to resistance to p53-dependent apoptosis following DNA damage (22). Furthermore, AKT phosphorylates MDM2 on Ser-186, which leads to ubiquitination and degradation of p53 (23). Whether Ral proteins, which are also known to mediate Ras malignant transformation, regulate p53 and whether this contributes to malignancy have not been investigated.
RalA and RalB GTPases are molecular switches that are on (active) when bound to GTP and off (inactive) when bound to GDP (24). RalGEFs such as RalGDS displace GDP for GTP to activate Ral proteins (25). Ral proteins can be activated by Ras as well as by other pathways that are independent of Ras (26, 27). The interest in Ral proteins has recently increased following the demonstration that in some cancers Ral pathways are more critical than Raf and AKT pathways in mediating Ras-driven malignant transformation (28). RalA and RalB share ∼82% sequence identity, yet they have been shown to have different contributions to malignant transformation processes, and this is cancer cell type-specific (29–33). For example, in pancreatic cancer cells, RalA promotes anchorage-independent growth in soft agar and tumor growth in vivo, whereas RalB promotes cell survival, invasion, and migration (30). In colon cancer cells, RalA has similar functions as in pancreatic cancer cells, but RalB antagonizes RalA-driven anchorage-independent growth (31). The reasons for these divergent effects are not known, but differences in localization (33) and post-translational modifications (28, 31, 32) could be contributing factors.
Although Ral proteins have been shown to induce many hallmarks of cancer, such as anchorage-dependent and -independent growth, migration, and invasion (34, 35), the molecular mechanism by which they accomplish this is not well understood. In addition, whether Ral proteins antagonize tumor suppressive pathways to transform cell is not known. For example, whether Ral proteins regulate the levels of the tumor suppressor p53 and whether this contributes to their ability to induce malignant transformation are not known. In this manuscript, we demonstrated that depletion of RalB and sometimes RalA proteins activates the ATM2 kinase, increases p53 Ser-15 phosphorylation, and leads to a significant increase in p53 half-life and stability. In addition, depletion of RalB and some times RalA proteins inhibits malignant transformation in a p53-dependent manner. Taken together, these results suggest that down-regulation of RalB and sometimes RalA leads to stabilization and reactivation of p53 to inhibit malignant transformation of cancer cells that harbor wild-type p53 and mutant K-Ras.
EXPERIMENTAL PROCEDURES
Cell Culture
The human tumor cell lines used for our study were grown in their respective media: H460, MCF7, OVCAR-5, and BxPC-3 in RPMI (Invitrogen); A549 in Kaighn's modification medium (F12K) (Sigma); Calu-1 in modified McCoy's 5a medium (Sigma); and Panc-1, HCT116, and HKH2 in Dulbecco's modified minimal essential medium (DMEM) (Invitrogen). These cell lines have not been authenticated. All cells were grown in their respective media containing 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified incubator at 5% CO2.
siRNAs and Antibodies
Nontargeting, K-Ras, RalA, RalB, p53, and p21, and c-Raf siRNAs were purchased from Dharmacon (Lafayette, CO), and AKT1, AKT2, and Atk3 siRNAs were purchased from Cell Signaling Technology (Danvers, MA). A second distinct set of siRNAs for K-Ras, RalA, and RalB was also purchased (Santa Cruz Biotechnology, Dallas, TX) and used only for Fig. 2. The following are the siRNA sequences used: from Dharmacon siRNA (All Smart Pools): nontargeting siRNA, UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, AUGAACGUGAAUUGCUCAA, and UGGUUUACAUGUCGACUAA; Kras siRNA, GCUCAGGACUUAGCAAGAA, GACAAAGUGUGUAAUUAUG, UAAGGACUCUGAAGAUGUA, and CGAAUAUGAUCCAACAAUA; RalA siRNA, GAGCUAAUGUUGACAAGGU, GAAAUUCGAGCGAGAAAGA, GCAGACAGCUAUCGGAAGA, and GGACUACGCUGCAAUUAGA; RalB siRNA, AGACAAGAAUGGCAAGAAA, UACCAAAGCUGACAGUUAU, GAAAUCAGAACAAAGAAGA, and GAAAGAUGUUGCUUACUAU; and from Santa Cruz Biotechnology: siRNA, nontargeting siRNA, UUCUCCGAACGUGUCACGU; Kras siRNA, GGAAGCAAGUAGUAAUUGAtt, CUAGAACAGUAGACACAAAtt, and GAACCUUUGAGCUUUCAUAtt; RalA siRNA, GACAGGUUUCUGUAGAAGAtt; RalB siRNA, GGUGAUCAUGGUUGGCAGCtt.
FIGURE 2.
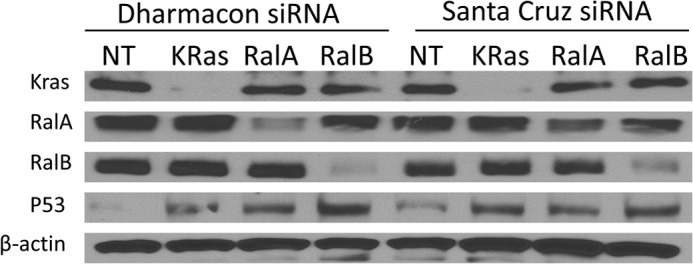
Depletion of RalA, RalB, or K-Ras increases p53 protein levels, confirmation with two distinct siRNA pools. H460 cells were transfected with the indicated siRNA for 48 h and processed for Western blotting analysis as described under “Experimental Procedures.” siRNAs were from Dharmacon (a smart pool of four distinct siRNAs for each gene), and siRNAs were from Santa Cruz Biotechnology (a smart pool of 3–5 distinct siRNAs for each gene). NT, nontargeting.
p21, c-Raf, p53 (DO-1), and AKT1/2 antibodies were also purchased from Santa Cruz Biotechnology. Cleaved poly(ADP-ribose) polymerase and phospho-p53 (Ser-15) antibodies were purchased from Cell Signaling. RalB and MDM2 antibodies were purchased from Millipore (Billerica, MA). β-Actin, vinculin, α-tubulin, and GAPDH antibodies were purchased from Sigma. K-Ras (OP-24) antibody was purchased from Calbiochem. RalA antibody was purchased from BD Biosciences. Peroxidase-conjugated rabbit anti-goat IgG, goat anti-mouse IgG, and mouse anti-rabbit IgG antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
siRNA Transfection
All cells used in this study were transfected with a similar siRNA transfection protocol using Lipofectamine RNAiMAX transfection reagent (Invitrogen). The cells were plated overnight to reach ∼40–50% confluence at the time of transfection. Opti-MEM-reduced serum medium (Invitrogen) and Lipofectamine RNAiMAX were mixed in one tube, and a mixture of Opti-MEM and siRNA was prepared in a separate tube, with the content of the two tubes mixed after 5 min of incubation at room temperature. Following this, the mixture was incubated for 20 min at room temperature and then added to the cells dropwise. After 24 h of transfection, appropriate media with 10% serum only were added until the desired time of transfection.
DNA Transfection
Cells were plated overnight to reach 70–80% confluence and transfected with DNA using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Briefly, Lipofectamine 2000 reagent (1.5 μl/μg of DNA) was diluted in Opti-MEM and allowed to equilibrate for 5 min at room temperature. DNA was diluted in Opti-MEM in a separate tube. The contents of the two tubes were mixed and incubated for 20 min to complex at room temperature and then added dropwise to cells that contained only 10% FBS-containing regular growth media. The cells were transfected for 6 h before the medium was changed to 10% FBS-only regular growth media.
Western Blotting
Cells were rinsed twice with ice-cold PBS and lysed on the plate in lysis buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 μg/ml leupeptin, and 1 mm PMSF. The lysate was collected by scraping and centrifuging at 13,000 × g for 13 min to remove the debris. The protein content was measured using the Bradford protein assay. Samples were resolved using SDS-PAGE and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% milk in Tris-buffered saline and Tween 20 (TBST) and then probed with the desired antibodies. Western Lightning ECL (PerkinElmer Life Sciences) was used for antibody detection.
Caspase Activation Assay
Caspase-3 activity was measured in cell lysates using a fluorogenic substrate. The assay measures the cleavage of amido-4-methylcoumarin groups from a caspase-3-specific substrate (Ac-DEVD-amido-4-methylcoumarin) (Biomol Research Labs, Inc., Plymouth Meeting, PA). Briefly, cells were lysed in a protease inhibitor-free lysis buffer (50 mm Tris, pH 8, 5 mm EDTA, 150 mm NaCl, and 0.5% IgePal). The lysate was collected by scraping. After clearing the lysate by centrifugation, protein was measured; 20 μg of protein and 20 μm caspase-3 substrate were added into 50 mm Tris, pH 7.5, reaction buffer in triplicate in a 96-well plate. The reactions were incubated for 1 h at 37 °C, and liberated amido-4-methylcoumarin groups were quantified by a fluorometer (WALLAC Victor 1420 Multilabel counter, PerkinElmer Life Sciences) with 355-nm excitation and 460-nm emission filters.
Soft Agar Colony Formation Assay
HCT116 and H460 cells were transfected with siRNA for 48 h; cells were trypsinized and counted, and then 2,000 cells/well were seeded in triplicate in 12-well culture plates in 0.3% top agar over a 0.6% bottom agar layer. Cultures were incubated for 2 weeks (H460) or 3 weeks (HCT116). Plates were scanned after overnight incubation with 2 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Calbiochem), in PBS. The colony numbers were visually determined and quantified.
Cell Invasion Assay
Analysis of cell invasion was performed using 24-well Matrigel invasion chambers (8 μm pore size; BD Biosciences) according to the manufacturer's instructions. Briefly, the inserts were rehydrated by adding warm serum-free medium for 2 h at 37 °C, 5% CO2 atmosphere. The medium was then removed from the inserts, and 750 μl of medium containing 20% FBS was added to each well of the plate, with inserts placed on top of the medium. Transfected A549, Panc-1, H460 (40,000 cells in 500 μl of serum-free media), and HPNE cells (20,000 cells) were added to the inserts. The cells were incubated for 48 h at 37 °C. Cell invasion was determined by staining the bottom of the insert with crystal violet (0.5% crystal violet in 25% methanol). The porous membrane was removed and placed on the slide. The number of cells that invaded were counted in three high power microscope (×40) fields and averaged as the mean number of invaded cells per field for each membrane.
Promoter-Reporter Transcriptional Assay
To determine the effects of K-Ras, RalA, or RalB depletion on p21 promoter activity, H460 or A549 cells (300,000 per well) were plated in 6-well plates overnight and then transfected with 3 μg of p21 and β-galactosidase promoter-reporter constructs using Lipofectamine 2000 as described above under “DNA Transfection.” Twelve hours after transfection, the cells were then transfected with different siRNAs using Lipofectamine RNAiMax or using Lipofectamine 2000 as described above. After 48 h of siRNA or DNA transfection, cells were rinsed with PBS and then lysed on plates using Reporter Lysis Buffer as described by the manufacturer (Promega BioScience, San Luis Obispo, CA). Cells were then freeze-thawed for efficient lysis. The lysate was centrifuged at 12,000 × g for 2 min to remove debris. The p21 promoter activity was detected by incubating the lysate with the assay substrate. Luciferase activity was detected using a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA), and β-galactosidase was read at 420-nm wavelength using a Bio-Tek plate reader. The p21 promoter activity was normalized to the respective β-galactosidase reading.
Determination of p53 Half-life
H460 or A549 cells were transfected with siRNA. Following 48 h of transfection, cells were treated with 50 μg/ml cycloheximide for 0, 0.5, 1, 2, or 4 h and then harvested and processed for Western blot analysis as indicated above.
Cell Cycle Analysis
A549 cells were transfected with different siRNAs for 48 h. After siRNA transfection was completed, cells were trypsinized and then centrifuged at 200 × g for 5 min to harvest the cells. The cells were rinsed with PBS twice and then resuspended in ethanol overnight. Following overnight fixation, cells were centrifuged. The supernatant was removed and rinsed with PBS. The cells were stained with propidium iodide stain containing 20 μg/ml propidium iodide, 0.1% (v/v) Triton in PBS, and 0.2 mg/ml DNase-free RNase A for 2 h at room temperature. Cell distribution on different cell cycle phase was determined using a flow cytometer.
ATM Kinase Assay
H460 cells were transfected with siRNA for nontargeting, K-Ras, RalA, and RalB for 48 h as described above, with cells then lysed in AKT lysis buffer (Cell Signaling Technology), and cell lysates were subjected to immunoprecipitation. Briefly, 50 ng of ATM antibody (Santa Cruz Biotechnology) was added to 500 μg of lysate that was previously pre-cleared with a protein A/G bead mix (EMD Millipore, Billerica, MA) and immunoprecipitated overnight at 4 °C. The immunocomplex was then captured with 100 μl of protein A/G bead slurry for 1 h at 4 °C. The ATM kinase assay was performed as described previously (36). Briefly, 50 μl of kinase buffer (25 mm Tris-HCl, pH 7.5, 5 mm β-glycerophosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, and 10 mm MgCl2) (Cell Signaling) was mixed with 0.1 mm ATP and 1 μg of GST-p53 (Cell Signaling) at 30 °C for 30 min. Reactions were stopped by the addition of SDS sample buffer followed by boiling. Reaction products were subjected to immunoblotting as described above.
RESULTS
Expression of K-Ras, RalA, or RalB, but Not AKT1/2 or c-Raf, Is Required for the Maintenance of Low Levels of p53 in Human Cancer Cells That Harbor Mutant K-Ras and Wild-type p53
About half of the human cancers lack the tumor-suppressive activity of p53 due to gene deletions or mutations (9, 37–40). The other half that expresses wild-type p53 harbors aberrant pathways that either inactivate p53 or suppress its expression levels (9, 37–40). Mutant K-Ras is known to down-regulate wild-type p53 levels, but little is known about the contributions of its downstream effectors. For example, Ral GTPases are critical mediators of K-Ras-driven malignant transformation, but whether they regulate wild-type p53 is not known. To address this important question, we first determined whether depletion of Ral GTPases affects the levels of p53 in A549 and H460 cell lines that harbor mutant K-Ras and wild-type p53. Because AKT and Raf are also critical to Ras malignant transformation, we also investigated the effects of their depletion on the levels of p53. To this end, A549 and H460 cells were transfected with either nontargeting siRNA or siRNAs to K-Ras, RalA, RalB, AKT1/2, or c-Raf, and the cells were processed for Western blotting as described under “Experimental Procedures.” Fig. 1A shows that in both A549 and H460 cells, depletion of K-Ras, but not AKT1/2 and c-Raf, resulted in increased p53 levels. Depletion of RalB in both cell lines also increased p53, and this increase was more pronounced (3.5-fold for both A549 and H460) than with K-Ras depletion (1.6- and 2.4-fold for H460 and A549, respectively). Furthermore, depletion of RalA increased the levels of p53 in H460 but had little effect in A549 (Fig. 1A). The increased p53 levels following depletion were confirmed by using two distinct pools of siRNA for each of K-Ras, RalA, and RalB genes (Fig. 2). To determine whether the increase in p53 levels is dependent on the mutation status of K-Ras and p53, we used other cell lines with mutant K-Ras and wild-type p53 (HCT116 colon cancer cells and OVCAR-5 ovarian cancer cells), wild-type K-Ras and wild-type p53 (MCF7 breast cancer cells and HCT116 cells), where the mutant K-Ras was deleted by homologous recombination (HKH2 cells), wild-type K-Ras, mutant p53 (BXPC3 pancreatic cancer cells), and mutant K-Ras and mutant p53 (Panc-1 pancreatic cancer cells). Fig. 1B shows that depletion of K-Ras, RalA, and RalB increased p53 levels in cells with mutant K-Ras and wild-type p53 (HCT116 and OVCAR-5) but not in cells with either wild-type K-Ras and/or mutant p53 (MCF7, BXPC3, and Panc-1). Furthermore, depletion of K-Ras, RalA, and RalB in the isogenic cell line HCT116 (HKH2), where the mutant K-Ras allele was deleted, had little effect on the levels of p53 (Fig. 1B). (In Fig. 1B, the effects of RalA and RalB siRNAs on the RalA and RalB levels varied; however, the percent deceases in the levels of the Ral proteins were similar between cell lines where there was an increase in p53 levels and those where there was no increase.) Our results suggest that, in human cancer cells that harbor mutant K-Ras and wild-type p53, the expression of K-Ras and RalB is required for the maintenance of low levels of p53. This was also the case for RalA except in A549 cells.
FIGURE 1.

Depletion of RalA and/or RalB increases p53 protein levels in cancer cells with wild-type p53 and mutant (mt) K-Ras. A, depletion of RalA and/or RalB but not AKT1/2 or c-Raf affects p53 protein levels in cells with mt K-Ras and WT p53. H460 and A549 cells were transfected with indicated siRNAs for 48 h and processed for Western blot analysis as described under “Experimental Procedures.” B, cancer cells that harbor mt K-Ras and WT p53 (HCT116 colon cancer cells and OVCAR-5 ovarian cancer cells), WT K-Ras and WT p53 (HKH2 and MCF7 breast cancer cells), WT K-Ras and mt p53 (BXPC3 pancreatic cancer cells), and mt K-Ras and mt p53 (Panc-1 pancreatic cancer cells) were transfected with the indicated siRNAs for 48 h and processed for Western blot analysis as described under “Experimental Procedures.” Data are representative of three (A549, H460, and HCT116) or two (OVCAR-5, Panc-1, HKH2, MCF7, and BXPC3) independent experiments. In H460 cells, densitometry quantification shows that depletion of K-Ras, RalA, and RalB increased p53 levels by 2.16 ± 0.33-, 2.23 ± 0.18-, and 3.36 ± 0.11-fold, respectively. These increases were statistically significant with p values of less than 0.05, 0.01, and 0.01, respectively. In A549 cells, densitometry quantification shows that depletion of K-Ras and RalB increased p53 levels by 1.89 ± 0.26- and 2.40 ± 0.55-fold, respectively. These increases were statistically significant, with p values of less than 0.05 and 0.05, respectively. Depletion of RalA did not affect the levels of p53 (1.02 ± 0.07-fold) in A549 cells. NT, nontargeting.
Depletion of RalA and RalB Leads to Up-regulation of p21 Expression in a p53-dependent Manner
p53 controls cell cycle progression by regulating at the transcriptional levels the expression of several genes that are involved in cell cycle progression such as p21 (41–44). Therefore, we next determined whether Ral GTPases are involved in the regulation of p21 and whether this is dependent on p53. Fig. 3A shows that, in A549 cells, depletion of K-Ras and RalB increased the protein levels of p53 and p21. Consistent with this, depletion of K-Ras and RalB also increased the p21 promoter transcriptional activity (Fig. 3B). In H460 cells, depletion of K-Ras, RalA, and RalB increased p21 expression at the protein (Fig. 3C) as well as the transcriptional (Fig. 3D) levels. We next determined whether this increase in p21 levels is dependent on the expression of p53. To this end, we took two approaches. In the first approach, because depleting K-Ras, RalA, and RalB increased p53 levels, we reasoned that co-depleting p53 should rescue the increase in p21 if the effects are p53-dependent. Therefore, we transfected A549 and H460 cells with K-Ras, RalA, or RalB siRNAs alone or in combination with p53 siRNA and evaluated the levels of p53 and p21 as described under “Experimental Procedures.” Fig. 3A shows that, in A549 cells, K-Ras and RalB siRNAs were unable to increase p21 protein levels in the absence of p53. In H460 cells, K-Ras, RalA, and RalB siRNAs were unable to increase p21 protein levels in the absence of p53 (Fig. 3C). Similar results were obtained with p21 promoter transcriptional activity for RalA and RalB but not for KRas (Fig. 3, B and D). Indeed, depletion of KRas resulted in an increase in the levels of p21 promoter transcriptional activity in a p53-independent manner, although the overall levels were lower in the absence of p53 (Figs. 3B and 2D). In the second approach, we used a cell line where K-Ras is mutated and both p53 alleles are deleted (human lung cancer Calu-1 cells), and we determined the effects of depleting K-Ras, RalA, and RalB. Fig. 3E shows that depletion of K-Ras, RalA, and RalB failed to increase p21 levels.
FIGURE 3.

Depletion of K-Ras, RalA, and RalB regulates p21 expression levels in a p53-dependent manner. A549 (A) and H460 (C) cells were transfected with nontargeting (NT), K-Ras, RalA, or RalB siRNAs alone or in combination with p53 siRNA for 48 h. Cells were harvested for Western blot analysis as described under “Experimental Procedures.” A549 (B) and H460 (D) cells were transfected with p21 promoter-reporter constructs followed by transfection with the indicated siRNAs. Cells were harvested and analyzed for p21 promoter activity as described under “Experimental Procedures.” E, Calu-1 cells were transfected with the indicated siRNAs and processed for Western blot analysis as described under “Experimental Procedures.” Extra lanes on p53 and p21 blots were from a control lysate (Panc-1 cells) used as a control for the Western blot analysis. Data are representative of three independent experiments except for Calu-1 cell experiments, which were done twice.
Depletion of RalA and RalB Activates ATM and Increases p53 Ser-15 Phosphorylation, p53 Stability and Half-life
Figs. 1 and 3 demonstrated that depletion of Ral GTPases increased the amount of functional p53 protein in human cancer cells with mutant K-Ras and wild-type p53. We next determined whether the increase in p53 levels was due to increased stability of the p53 protein. To this end, H460 cells were transfected with either nontargeting siRNA or siRNA to K-Ras, RalA, or RalB for 48 h followed by treatment with cycloheximide for 0, 0.5, 1, 2, or 4 h, with the cells then processed for Western blotting as described under “Experimental Procedures.” Figs. 4 and 5 show that, in H460 cells transfected with nontargeting siRNA, the protein levels of p53 decreased steadily after treatment with cycloheximide with a half-life between 30 and 60 min. In cells transfected with RalA, RalB, or K-Ras siRNA, p53 protein levels decreased after cycloheximide treatment but at a much slower rate, with half-lives greater than 4 h (RalA and RalB siRNAs) and greater than 2 h (K-Ras siRNA) (Fig. 4A and Fig. 5). Thus, in the absence of RalA and RalB, the p53 half-life was increased by greater than 6-fold, and in the absence of K-Ras it was increased by greater than 2-fold. Similar results were obtained in A549 cells where RalB depletion also increased the half-life of p53 from 30 min to 4 h (Fig. 6). Depletion of K-Ras, RalA, and RalB in H460 cells also increased the levels of MDM2, but in contrast to p53, it had little effect on its half-life (about 30 min with and without K-Ras, RalA, and RalB siRNAs). This suggests that depletion of K-Ras, RalA, or RalB stabilizes p53, which in turn induces the expression of MDM2, a known target for p53. Consistent with this, the ability of RalB siRNA to increase MDM2 levels was partially rescued by p53 siRNA, suggesting that RalB depletion up-regulates MDM2 levels in a p53-dependent manner.
FIGURE 4.

Depletion of K-Ras, RalA, and RalB increases p53 protein stability. A, H460 cells were treated with nontargeting (NT), K-Ras, RalA, or RalB siRNAs for 48 h followed by cycloheximide (CHX) treatment for the indicated lengths of time. Cells were harvested and processed for Western blot analysis as described under “Experimental Procedures.” B, H460 cells were treated with nontargeting (NT), K-Ras, RalA, or RalB siRNAs for 48 h. The cells were lysed, the ATM was immunoprecipitated (IP) and processed for ATM kinase assay using GST-p53 as a substrate, and the reaction products were subjected to immunoblotting with antibodies to phospho-Ser-15-p53, GST, and ATM as described under “Experimental Procedures.” Data are representative of two independent experiments.
FIGURE 5.

Depletion of K-Ras, RalA, and RalB increases p53 but not MDM2 half-life in H460 cells. Densitometer values represent the expression levels of p53 and MDM2 following depletion of K-Ras, RalA, or RalB. The expression levels of p53 and actin were measured at the indicated time points, and the relative p53 expression level was determined by dividing p53 by actin densitometry values. The value at the 0-h time point was set as 1, and the rest of the values at different time points were normalized against the 0-h value. Data are representative of two independent experiments. NT, nontargeting; CHX, cycloheximide.
FIGURE 6.

Depletion of RalB increases p53 stability in A549 cells. A, A549 cells were transfected with the indicated siRNA for 48 h followed by cycloheximide (CHX) treatment for the indicated length of time and processed for Western blot analysis as indicated under “Experimental Procedures.” Data are representative of two independent experiments. B, p53 and MDM2 from A were quantified as described in Fig. 5 legend. C, A549 cells were transfected with the indicated siRNA for 48 h and then processed for Western blot analysis. Data are representative of three independent experiments. NT, nontargeting.
Phosphorylation of p53 at Ser-15 has been extensively used as a marker for stability of p53 (45, 46). Therefore, we next determined the effects of RalA, RalB, and K-Ras depletion on p53 Ser-15 phosphorylation. Fig. 4A shows that deletion of RalA and RalB in H460 cells resulted in increased p53 Ser-15 phosphorylation. Similar results were seen with A549 cells where depletion of RalB also increased p53 Ser-15 phosphorylation. ATM kinase phosphorylates p53 on Ser-15; therefore, we reasoned that depletion of these GTPases might activate ATM to phosphorylate p53. To this end, we transfected H460 cells with either nontargeting siRNA or siRNA to K-Ras, RalA, or RalB for 48 h, lysed the cells, immunoprecipitated ATM, performed in vitro ATM kinase assays using GST-p53 as a substrate, and processed the samples for Western blotting as described under “Experimental Procedures.” Fig. 4B shows that depletion of K-Ras, RalA, and RalB activated ATM as documented by increased phosphorylation of GST-p53 on Ser-15. Quantitation by densitometry shows that the amounts of Ser-15-phosphorylated p53 per amount of GST-p53 per amount of ATM immunoprecipitated were 1, 2.94, 3.91, and 2.85 for nontargeted, K-Ras, RalA, and RalB siRNAs, respectively (Fig. 4B). Therefore, depletion of K-Ras, RalA, and RalB enhances the ability of ATM to phosphorylate p53 by 2.94-, 3.91-, and 2.85-fold, respectively.
Depletion of RalB Inhibits Cell Cycle Progression in a p53- and p21-dependent Manner by Decreasing the Proportion of Cells in S Phase
Cells exposed to DNA-damaging agents induce p53, which in turn induces, depending on the extent of the damage, either cell cycle arrest or apoptosis by regulating the transcription of specific genes (41–44). Because p53 was induced and up-regulated p21 expression following depletion of RalA, RalB, or K-Ras, we next determined whether this increase in p53 has functional consequences on cell cycle progression. To this end, we transfected A549 cells with siRNA to K-Ras, RalA, or RalB and processed the cells for cell cycle analysis as described under “Experimental Procedures.” Figs. 4B and 7A show that depletion of RalA had little effect on cell cycle progression. In contrast, depletion of RalB decreased the percentage of cells in S phase from 57% (Fig. 7A and Table 1) to 70% (Fig. 7B and Table 2), whereas K-Ras increased the percentage of cells in G2M phase from 4 to 6.7-fold (Fig. 7A and Tables 1 and 2). To determine whether these effects were p53-dependent, we co-transfected the cells with siRNA to p53 along with siRNAs to K-Ras, RalA, or RalB and determined the effects on cell cycle progression as described under “Experimental Procedures.” Fig. 7A and Table 1 show that depletion of p53 inhibited RalB and K-Ras siRNAs from affecting cell cycle progression. Unlike A549 cells, in H460 cells, depletion of RalA and RalB had little effect on cell cycle progression (Fig. 8). Interestingly, in these cells, depletion of K-Ras decreased the percentage of cells in S phase (Fig. 8).
FIGURE 7.

Effects of depletion of K-Ras, RalA, and RalB on cell cycle progression. A549 cells were treated with indicated siRNAs for 48 h and processed for cell cycle analysis as described under “Experimental Procedures.” The percentage of cells in each cell cycle phase relative to nontargeting (NT) is shown. A, depletion of p53 rescue. B, depletion of p21 rescue.
TABLE 1.
Effects of depletion of K-Ras, RalA, and RalB on cell cycle progression
A549 cells were treated with the indicated siRNAs for 48 h and processed for cell cycle analysis as described under “Experimental Procedures” and in the legend to Fig. 7. The percentage of cells in each cell cycle phase is shown. This table shows p53 rescue.
| G1 | G2M | S | |
|---|---|---|---|
| Nontargeting | 71 | 5 | 23 |
| K-Ras | 63 | 20 | 17 |
| RalA | 72 | 7 | 21 |
| RalB | 84 | 6 | 10 |
| NT + p53 | 61 | 15 | 24 |
| K-Ras + p53 | 68 | 13 | 19 |
| RalA + p53 | 59 | 13 | 27 |
| RalB + p53 | 69 | 6 | 25 |
TABLE 2.
Effects of depletion of K-Ras, RalA, and RalB on cell cycle progression
A549 cells were treated with the indicated siRNA for 48 h and processed for cell cycle analysis as described under “Experimental Procedures” and in the legend to Fig. 7. The percentage of cells in each cell cycle phase is shown.. This table shows p21 rescue. NT means nontargeting.
| G1 | G2M | S | |
|---|---|---|---|
| Nontargeting | 77 | 3 | 20 |
| K-Ras | 65 | 20 | 15 |
| RalA | 74 | 6 | 20 |
| RalB | 88 | 7 | 6 |
| p21 | 69 | 5 | 26 |
| NT + NT | 81 | 2 | 18 |
| K-Ras + p21 | 60 | 12 | 28 |
| RalA + p21 | 69 | 5 | 26 |
| RalB + p21 | 63 | 8 | 29 |
| NT + p21 | 68 | 3 | 29 |
FIGURE 8.
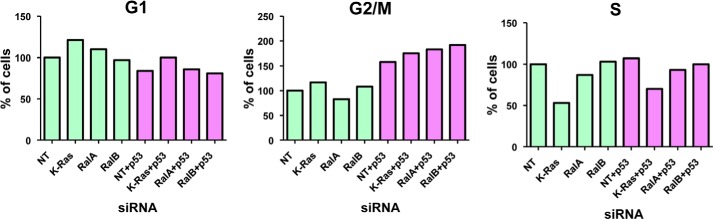
Effects of depletion of K-Ras, RalA, and RalB on cell cycle progression in H460 cells. H460 cells were treated with the indicated siRNA for 48 h and processed for cell cycle analysis as described under “Experimental Procedures.” The percentage of cells in each cell cycle phase relative to nontargeting (NT) is shown.
Because p21 is known to be up-regulated by p53 and to be involved in G1/S transition, we next determined whether the p21 induction by p53 following depletion of RalB is required for RalB siRNA to decrease the percentage of cells in S phase. To this end, we co-transfected the cells with siRNA to p21 along with siRNAs to either K-Ras, RalA, or RalB and determined the effects on cell cycle progression as described under “Experimental Procedures.” Fig. 7B and Table 2 show that co-depletion of p21 was completely rescued from the effects of RalB siRNA on cell cycle progression, suggesting that the ability of RalB siRNA to decrease the proportion of cells in S phase depends on p53-induced p21.
Effects of Depleting RalA and RalB on Apoptosis
As mentioned above, another important function of p53 is to promote apoptosis. Furthermore, depletion of RalB but not RalA has been shown to induce apoptosis (34), but the molecular mechanism by which this occurs is not known. Therefore, we next determined whether depletion of RalB induces apoptosis and whether this is p53-dependent. To this end, we transfected A549 and H460 cells with siRNA to K-Ras, RalA, or RalB in the presence or absence of siRNA to p53 and processed the cells for Western blotting and for caspase-3 activation by a fluorescence assay as described under “Experimental Procedures.” Fig. 9 shows that, in A549 cells, depletion of K-Ras and RalB but not RalA induced apoptosis, with K-Ras depletion having significantly higher effects (7-fold from 200 to 1400) than RalB depletion (3-fold from 200 to 600). Fig. 9 also shows that siRNA to p53 inhibited K-Ras and RalB siRNA from inducing apoptosis. These results suggest that depletion of K-Ras and RalB induced apoptosis by a p53-dependent mechanism in A549 cells. In H460 cells, however, depletion of K-Ras but not that of RalA and RalB significantly induced apoptosis, and depletion of p53 rescued apoptosis (Fig. 9).
FIGURE 9.

Effects of depletion of K-Ras, RalA, and RalB on apoptosis. H460 and A549 cells were transfected with siRNA for 48 h and then harvested for Western blot analysis (left panel) or caspase-3 activation assay (right panel) as described under “Experimental Procedures.” Data are representative of two independent experiments. NT, nontargeting.
Depletion of RalB and to a Lesser Extent RalA Inhibits Anchorage-independent Growth and Invasion in a p53-dependent Manner
The involvement of RalA and RalB in malignant transformation has been well documented (47). Although RalA is believed to be involved in promoting anchorage-independent growth in soft agar, RalB is believed to promote invasion and migration, although this is cell type-dependent (29–33). Because p53 is a potent tumor suppressor, we reasoned that Ral proteins may affect some of these hallmarks of transformation in a p53-dependent manner. To this end, we depleted K-Ras, RalA, or RalB in the presence or absence of p53 siRNA and processed the cells for soft agar and invasion assays as described under “Experimental Procedures.” Fig. 10 shows that depleting K-Ras or RalB but not RalA inhibited soft agar growth of H460 and HCT116 cells. The inhibition of soft agar growth following RalB depletion was rescued fully by siRNA to p53 (Fig. 10). In contrast, siRNA to p53 only partially rescued from the inhibition of soft agar growth following K-Ras depletion (Fig. 10). Another critical hallmark of cancer is invasion; we therefore investigated the effects of depleting these three proteins on invasion. Fig. 11 shows that depletion of K-Ras, RalB, and to a lesser extent RalA inhibited invasion of all three cell lines (A549, HCT116, and H460). Co-depletion of these proteins along with p53 rescued from this inhibition of invasion. These results suggest that depletion of K-Ras, RalA, and RalB requires p53 to inhibit invasion of A549, H460, and HCT116 cells.
FIGURE 10.

Effects of depletion of K-Ras, RalA, and RalB on anchorage-independent growth. H460 or HCT116 cells were transfected with indicated siRNAs for 48 h before plating in soft agar as described under “Experimental Procedures.” Results are shown as average percent inhibition of four independent experiments (H460) or three independent experiments (HCT116). Nontargeting (NT) and nontargeting + p53 siRNAs were set as 100%. For H460, mean number of colonies for nontargeting was 323 ± 8.5 and for nontargeting + p53 siRNA was 232 ± 66.22. For HCT116, mean number of colonies for nontargeting was 469 ± 7.2 and for nontargeting + p53 siRNA was 484 ± 6. Error bars show S.E. Student's t test was used to compare the means; p values are shown for tests that show significant differences (*, p < 0.05). Top panel shows representative soft agar pictures for HCT116 cells.
FIGURE 11.

Effects of depletion of K-Ras, RalA, and RalB on invasion. A549, H460, or HCT116 cells were transfected with the indicated siRNAs for 48 h, and then equal numbers of cells were seeded for invasion assays as described under “Experimental Procedures.” The cells were then photographed and counted to determine the mean number of invading cells. Top panel shows representative image of invading cells of H460, and bottom panels show average number of invading cells. p values for H460 are K-Ras = 0.02, RalA = 0.02, and RalB = 0.03. p values for HCT116 are K-Ras = 0.006 and RalB = 0.007. p values for A549 are K-Ras = 0.004 and RalB = 0.0006. NT, nontargeting.
DISCUSSION
Among the three most studied downstream effector pathways of Ras, Raf/MEK/ERK, PI3K/AKT, and RalGDS/Ral, the latter has gained more attention as it has been shown to be more critical in mediating Ras-driven malignant transformation in several cancers (28, 48). Specifically, RalA has been implicated in anchorage-independent growth, whereas RalB has been implicated in apoptosis evasion, migration, and invasion, although this is cell type-dependent (29–33, 41). The mechanism by which RalA and RalB mediate these effects is not well understood. Furthermore, mutant K-Ras suppresses p53 levels, but it is not known which of its downstream effectors mediate this effect. In this study, we have demonstrated that the expression of K-Ras and RalB but not c-Raf and AKT1/2 is required for maintaining low levels of p53 in human cancer cells that harbor mutant K-Ras and wild-type p53. Depletion of RalA also increases p53 levels except in A549 cells. The observation that depletion of K-Ras increased p53 levels is novel and consistent with the observation that K-Ras can suppress p53 levels (16). Indeed, although mutant H-Ras and mutant N-Ras were shown to induce p53 levels and senescence, mutant K-Ras was shown to suppress p53 levels (11, 14, 16–20). Therefore, it is not surprising that, unlike cells that harbor mutant H-Ras and mutant N-Ras, many human cancer cells that harbor mutant K-Ras also express wild-type p53, suggesting that mutant K-Ras triggers pathways capable of overcoming the tumor-suppressive activity of p53. Our studies suggest that RalA and RalB could be among those pathways that may be used by mutant K-Ras to suppress p53 levels. In contrast, pathways mediated by c-Raf and AKT1/2 do not appear to be required because their depletion did not increase p53 levels. Although our studies demonstrated that depletion of Raf or AKT does not increase p53 levels, others have shown that forced expression of Raf or AKT induces p53 degradation (22, 23). This suggests that, although Raf and AKT can induce degradation of p53, their expression is not required to maintain low levels of p53 in some human cancer cells. It is important to note that depletion of RalA (except in A459 cells) and RalB increased p53 levels only in human cancers cells that harbor mutant K-Ras and wild-type p53, suggesting that RalA and RalB may mediate the ability of mutant K-Ras to suppress p53. However, the fact that depletion of RalB was more potent at increasing the levels of p53 than depletion of K-Ras (Fig. 1) coupled with the fact that depletion of RalB and K-Ras affected cell cycle progression differently (Fig. 7) suggest that RalB may also regulate p53 in a K-Ras-independent manner.
In addition to increasing p53 levels, depletion of RalA and RalB also increased the levels of p21 and MDM2, two genes known to be transcriptionally regulated by p53. The p53 siRNA rescue from these effects suggests that the ability of siRNA for RalA and RalB to increase p21 and MDM2 levels is dependent on their ability to increase the levels of p53. This also indicates that the p53 induced following RalA or RalB depletion was functional and able to regulate the transcription of its target genes. Furthermore, the ability of Ral siRNAs to regulate the levels of the p21 and MDM2 genes in a p53-dependent manner has important consequences on the ability of Ral GTPases to contribute to malignant transformation (see more below). In cells that lack both p53 alleles such as Calu-1 cells, depletion of RalA or RalB failed to affect p21 levels, further confirming the dependence of the Ral proteins on p53 to regulate the levels of this gene. Our results also suggest that K-Ras regulates the expression of p21 in a p53-dependent manner.
Depletion of RalA and RalB as well as K-Ras resulted in a significant increase in p53 half-life in H-460 cells. This increased p53 stability is specific in that depletion of K-Ras, RalA, and RalB increased the MDM2 protein levels with little effects on its half-life. Consistent with this, depletion of RalA and RalB resulted in ATM kinase activation and increased p53 Ser-15 phosphorylation levels, which have been associated with p53 stability (45, 46). The observation that depletion of Ral proteins leads to activation of ATM suggests that Ral proteins may maintain low levels of p53 by inactivating ATM. Several Ral effectors such as RalBP1/CDC42, Sec5, ZONAB, and PLD have been identified (47), and some of these may mediate these effects of Ral on ATM and p53. For example, RalBP1 is known to inactivate CDC42, which could in turn lead to suppression of ATM. Clearly, investigating the mechanism by which the Ral effectors may inactivate ATM is warranted. Taken together, our observations suggest that depletion of RalA and RalB results in increased p53 Ser-15 phosphorylation and a more stable p53 that was able to induce p21 and MDM2 levels. Depletion of these GTPases could also affect other phosphorylation sites on p53 that affect its stability. Indeed, cellular stress and DNA damage induce p53 phosphorylation at Ser-15 as well as Ser-20 and Ser-37, which affect p53 stability by preventing its ubiquitination and degradation (45, 46). Therefore, investigations determining the effects of RalA and/or RalB depletion on p53 post-translational modifications other than Ser-15 are also warranted to further understand the mechanism by which depletion of RalA and RalB stabilizes p53. Finally, mutant K-Ras was shown to decrease p53 stability by activating the E3 ligase SNAIL, which ubiquitinates p53 and targets it to proteasomal degradation (16). It is therefore possible that depletion of RalA and/or RalB inhibits SNAIL as well as MDM2 E3 ligases, leading to less ubiquitination of p53.
The effects of depletion of the Ral GTPases on anchorage-dependent and -independent growth, invasion, apoptosis, and proliferation have previously been studied (30, 34). Furthermore, Ral proteins have also been implicated in regulating cell cycle progression, possibly through increased cyclin D1 transcription (49, 50) and FOXO4/AFX-dependent decreased p27 transcription (51). However, the contribution of p53 to the effects of Ral proteins on cell cycle progression is not known. Here, we show that depletion of RalB inhibited cell cycle progression by decreasing the proportion of cells reaching S phase and by increasing the proportion of cells in both G1 and G2/M phases of the cell cycle. The ability of RalB depletion to decrease the proportion of cells in S phase is rescued by p53 siRNA as well as by p21 siRNA, suggesting that RalB depletion induces p53 and p21, which contribute to inhibition of cell cycle progression. Furthermore, depletion of K-Ras induces G2/M cell cycle arrest in a p53-independent manner. Therefore, the fact that depletions of K-Ras or RalB resulted in increased p53 levels with different effects on cell cycle progression suggests that, in addition to affecting common genes such as p53, each specific depletion also differentially affected other genes. This is not surprising as it is known that Ral GTPases are regulated in a Ras-dependent and -independent manner and that RalA and RalB themselves mediate different cellular effects. It is interesting to note that depletion of RalA does not lead to the same effects in all cell lines. For example, depletion of RalA in A549 cells does not increase p53 levels. The reasons for this are not known, but it is conceivable that RalA-specific effectors are not expressed in A549 cells. However, as of yet, no Ral isoform-specific effector proteins have been identified. In addition to the cell cycle effects, depletion of RalB and to a lesser degree RalA inhibits anchorage-independent tumor cell growth and invasion, which were also rescued by p53 siRNA. This suggests that, in the absence of RalB, the stabilized p53 not only inhibits cell cycle progression and invasion but also contributes to inhibition of anchorage-independent tumor cell growth. Unlike cell cycle progression, depletion of K-Ras induces apoptosis and inhibits anchorage-independent tumor cell growth and invasion in a p53-dependent manner.
In summary, we have uncovered a novel molecular mechanism by which Ral GTPases affect transformation. Furthermore, we have provided a biochemical link between Ral GTPases and the tumor suppressor p53 by demonstrating that the expression of Ral GTPases is required for maintaining low levels of p53 and that down-regulation of the Ral proteins results in the stabilization and reactivation of p53.
Acknowledgments
We thank members of the Sebti laboratory, Maria Balasis, Norbert Berndt, Aslamuzzaman Kazi, and Hua Yang as well as Rasa Hamilton (Moffitt Cancer Center), for carefully reviewing the manuscript and for their comments. We thank Dr. Jiandong Chen (Moffitt Cancer Center) for valuable comments. We also thank members of the Flow Cytometer Core Facility at the Moffitt Cancer Center and Research Institute for assisting with the cell cycle experiments. We also thank the Dr. David Shibata laboratory for providing us with the isogenic cell lines HCT116 and HKH2. The core facilities at the Moffitt Cancer Center are supported partially by the Cancer Center support grant NIH P30-CA76292.
Footnotes
- ATM
- ataxia telangiectasia-mutated
- mt
- mutant.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., Futreal P. A. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Downward J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 [DOI] [PubMed] [Google Scholar]
- 4. Brown C. J., Lain S., Verma C. S., Fersht A. R., Lane D. P. (2009) Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873 [DOI] [PubMed] [Google Scholar]
- 5. Ferbeyre G., de Stanchina E., Lin A. W., Querido E., McCurrach M. E., Hannon G. J., Lowe S. W. (2002) Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 22, 3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirao A., Kong Y. Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., Liu D., Elledge S. J., Mak T. W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287, 1824–1827 [DOI] [PubMed] [Google Scholar]
- 7. Sakaguchi K., Herrera J. E., Saito S., Miki T., Bustin M., Vassilev A., Anderson C. W., Appella E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Riley T., Sontag E., Chen P., Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 [DOI] [PubMed] [Google Scholar]
- 9. Hollstein M., Sidransky D., Vogelstein B., Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 [DOI] [PubMed] [Google Scholar]
- 10. Wu X., Bayle J. H., Olson D., Levine A. J. (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7, 1126–1132 [DOI] [PubMed] [Google Scholar]
- 11. Honda R., Yasuda H. (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18, 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ries S. J., Brandts C. H., Chung A. S., Biederer C. H., Hann B. C., Lipner E. M., McCormick F., Korn W. M. (2000) Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015). Nat. Med. 6, 1128–1133 [DOI] [PubMed] [Google Scholar]
- 13. Tao W., Levine A. J. (1999) P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl. Acad. Sci. U.S.A. 96, 6937–6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braig M., Lee S., Loddenkemper C., Rudolph C., Peters A. H., Schlegelberger B., Stein H., Dörken B., Jenuwein T., Schmitt C. A. (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665 [DOI] [PubMed] [Google Scholar]
- 15. Collado M., Serrano M. (2010) Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S. H., Lee S. J., Jung Y. S., Xu Y., Kang H. S., Ha N. C., Park B. J. (2009) Blocking of p53-Snail binding, promoted by oncogenic K-Ras, recovers p53 expression and function. Neoplasia 11, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 18. Ha L., Ichikawa T., Anver M., Dickins R., Lowe S., Sharpless N. E., Krimpenfort P., Depinho R. A., Bennett D. C., Sviderskaya E. V., Merlino G. (2007) ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc. Natl. Acad. Sci. U.S.A. 104, 10968–10973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin A. W., Lowe S. W. (2001) Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc. Natl. Acad. Sci. U.S.A. 98, 5025–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lowe S. W. (1999) Activation of p53 by oncogenes. Endocr. Relat. Cancer 6, 45–48 [DOI] [PubMed] [Google Scholar]
- 21. Guerra C., Mijimolle N., Dhawahir A., Dubus P., Barradas M., Serrano M., Campuzano V., Barbacid M. (2003) Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4, 111–120 [DOI] [PubMed] [Google Scholar]
- 22. Ries S., Biederer C., Woods D., Shifman O., Shirasawa S., Sasazuki T., McMahon M., Oren M., McCormick F. (2000) Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell 103, 321–330 [DOI] [PubMed] [Google Scholar]
- 23. Ogawara Y., Kishishita S., Obata T., Isazawa Y., Suzuki T., Tanaka K., Masuyama N., Gotoh Y. (2002) Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 277, 21843–21850 [DOI] [PubMed] [Google Scholar]
- 24. Wolthuis R. M., Zwartkruis F., Moen T. C., Bos J. L. (1998) Ras-dependent activation of the small GTPase Ral. Curr. Biol. 8, 471–474 [DOI] [PubMed] [Google Scholar]
- 25. Ferro E., Trabalzini L. (2010) RalGDS family members couple Ras to Ral signalling and that's not all. Cell. Signal. 22, 1804–1810 [DOI] [PubMed] [Google Scholar]
- 26. de Bruyn K. M., de Rooij J., Wolthuis R. M., Rehmann H., Wesenbeek J., Cool R. H., Wittinghofer A. H., Bos J. L. (2000) RalGEF2, a pleckstrin homology domain containing guanine nucleotide exchange factor for Ral. J. Biol. Chem. 275, 29761–29766 [DOI] [PubMed] [Google Scholar]
- 27. Rebhun J. F., Chen H., Quilliam L. A. (2000) Identification and characterization of a new family of guanine nucleotide exchange factors for the ras-related GTPase Ral. J. Biol. Chem. 275, 13406–13410 [DOI] [PubMed] [Google Scholar]
- 28. Hamad N. M., Elconin J. H., Karnoub A. E., Bai W., Rich J. N., Abraham R. T., Der C. J., Counter C. M. (2002) Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 16, 2045–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim K. H., Brady D. C., Kashatus D. F., Ancrile B. B., Der C. J., Cox A. D., Counter C. M. (2010) Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol. Cell. Biol. 30, 508–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim K. H., O'Hayer K., Adam S. J., Kendall S. D., Campbell P. M., Der C. J., Counter C. M. (2006) Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 16, 2385–2394 [DOI] [PubMed] [Google Scholar]
- 31. Martin T. D., Der C. J. (2012) Differential involvement of RalA and RalB in colorectal cancer. Small GTPases 3, 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin T. D., Mitin N., Cox A. D., Yeh J. J., Der C. J. (2012) Phosphorylation by protein kinase Cα regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J. Biol. Chem. 287, 14827–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shipitsin M., Feig L. A. (2004) RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol. Cell. Biol. 24, 5746–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chien Y., White M. A. (2003) RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 4, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oxford G., Owens C. R., Titus B. J., Foreman T. L., Herlevsen M. C., Smith S. C., Theodorescu D. (2005) RalA and RalB: antagonistic relatives in cancer cell migration. Cancer Res. 65, 7111–7120 [DOI] [PubMed] [Google Scholar]
- 36. Kodama M., Otsubo C., Hirota T., Yokota J., Enari M., Taya Y. (2010) Requirement of ATM for rapid p53 phosphorylation at Ser46 without Ser/Thr-Gln sequences. Mol. Cell. Biol. 30, 1620–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker S. J., Fearon E. R., Nigro J. M., Hamilton S. R., Preisinger A. C., Jessup J. M., vanTuinen P., Ledbetter D. H., Barker D. F., Nakamura Y., White R., Vogelstein B. (1989) Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244, 217–221 [DOI] [PubMed] [Google Scholar]
- 38. Hollstein M. C., Metcalf R. A., Welsh J. A., Montesano R., Harris C. C. (1990) Frequent mutation of the p53 gene in human esophageal cancer. Proc. Natl. Acad. Sci. U.S.A. 87, 9958–9961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nigro J. M., Baker S. J., Preisinger A. C., Jessup J. M., Hostetter R., Cleary K., Bigner S. H., Davidson N., Baylin S., Devilee P. (1989) Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708 [DOI] [PubMed] [Google Scholar]
- 40. Takahashi T., Nau M. M., Chiba I., Birrer M. J., Rosenberg R. K., Vinocour M., Levitt M., Pass H., Gazdar A. F., Minna J. D. (1989) p53: a frequent target for genetic abnormalities in lung cancer. Science 246, 491–494 [DOI] [PubMed] [Google Scholar]
- 41. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 42. Raj D., Liu T., Samadashwily G., Li F., Grossman D. (2008) Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 29, 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sionov R. V., Haupt Y. (1999) The cellular response to p53: the decision between life and death. Oncogene 18, 6145–6157 [DOI] [PubMed] [Google Scholar]
- 44. Vousden K. H., Lu X. (2002) Live or let die: the cell's response to p53. Nat. Rev. Cancer 2, 594–604 [DOI] [PubMed] [Google Scholar]
- 45. Ashcroft M., Kubbutat M. H., Vousden K. H. (1999) Regulation of p53 function and stability by phosphorylation. Mol. Cell. Biol. 19, 1751–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shieh S. Y., Ikeda M., Taya Y., Prives C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 [DOI] [PubMed] [Google Scholar]
- 47. Bodemann B. O., White M. A. (2008) Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140 [DOI] [PubMed] [Google Scholar]
- 48. Rangarajan A., Hong S. J., Gifford A., Weinberg R. A. (2004) Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6, 171–183 [DOI] [PubMed] [Google Scholar]
- 49. Gille H., Downward J. (1999) Multiple ras effector pathways contribute to G1 cell cycle progression. J. Biol. Chem. 274, 22033–22040 [DOI] [PubMed] [Google Scholar]
- 50. Henry D. O., Moskalenko S. A., Kaur K. J., Fu M., Pestell R. G., Camonis J. H., White M. A. (2000) Ral GTPases contribute to regulation of cyclin D1 through activation of NF-κB. Mol. Cell. Biol. 20, 8084–8092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Ruiter N. D., Burgering B. M., Bos J. L. (2001) Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol. Cell. Biol. 21, 8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]