

Background: Neurotrophins regulate transcription factor NFAT and NFAT-mediated neuronal functions, but the underlying mechanisms are poorly defined.
Results: NGF facilitated depolarization-induced NFAT activation in sensory neurons, which depended on PI3K, Akt, and GSK3β but not on PLC.
Conclusion: NGF-dependent facilitation of NFAT activation is mediated by the PI3K-Akt-GSK3β pathway.
Significance: This novel mechanism may represent an important component of NFAT-dependent gene regulation in neurons.
Keywords: Calcineurin, Calcium, Glycogen Synthase Kinase 3 (GSK-3), Neurotrophin, NFAT Transcription Factor, DRG Neurons, NGF
Abstract
The Ca2+/calcineurin-dependent transcription factor nuclear factor of activated T-cells (NFAT) plays an important role in regulating many neuronal functions, including excitability, axonal growth, synaptogenesis, and neuronal survival. NFAT can be activated by action potential firing or depolarization that leads to Ca2+/calcineurin-dependent dephosphorylation of NFAT and its translocation to the nucleus. Recent data suggest that NFAT and NFAT-dependent functions in neurons can also be potently regulated by NGF and other neurotrophins. However, the mechanisms of NFAT regulation by neurotrophins are not well understood. Here, we show that in dorsal root ganglion sensory neurons, NGF markedly facilitates NFAT-mediated gene expression induced by mild depolarization. The effects of NGF were not associated with changes in [Ca2+]i and were independent of phospholipase C activity. Instead, the facilitatory effect of NGF depended on activation of the PI3K/Akt pathway downstream of the TrkA receptor and on inhibition of glycogen synthase kinase 3β (GSK3β), a protein kinase known to phosphorylate NFAT and promote its nuclear export. Knockdown or knockout of NFATc3 eliminated this facilitatory effect. Simultaneous monitoring of EGFP-NFATc3 nuclear translocation and [Ca2+]i changes in dorsal root ganglion neurons indicated that NGF slowed the rate of NFATc3 nuclear export but did not affect its nuclear import rate. Collectively, our data suggest that NGF facilitates depolarization-induced NFAT activation by stimulating PI3K/Akt signaling, inactivating GSK3β, and thereby slowing NFATc3 export from the nucleus. We propose that NFAT serves as an integrator of neurotrophin action and depolarization-driven calcium signaling to regulate neuronal gene expression.
Introduction
Activity-dependent regulation of gene expression plays a crucial role in sculpting neural circuits during development and in controlling neuronal plasticity in adulthood (1, 2). Among the various Ca2+-dependent transcription factors, nuclear factor of activated T-cells (NFAT)3 has emerged as an important component of excitation-transcription coupling in neurons (3–8). Numerous neuronal functions are controlled by NFAT, including excitability, axonal growth, synaptic plasticity, neuronal development, and survival (4, 6, 8–13). Moreover, recent studies have linked aberrant NFAT activation to pain sensitization (14–16) and the neurotoxicity associated with Alzheimer disease, ischemia, and traumatic brain injury (17–20).
NFAT activation is regulated by reversible phosphorylation (21, 22). Ca2+ entering neurons via voltage- or ligand-gated Ca2+ channels activates the Ca2+- and calmodulin-dependent protein phosphatase calcineurin (CaN), which induces CaN-mediated dephosphorylation of NFAT at multiple serine residues within its regulatory domain (21–24). This, in turn, unmasks the nuclear localization signal of NFAT and facilitates its import to the nucleus, enabling it to initiate transcription (21, 22). Several protein kinases, such as glycogen synthase kinase 3β (GSK3β), casein kinase 1, and dual specificity tyrosine phosphorylation-regulated kinase 1, phosphorylate nuclear NFAT, which facilitates its binding by the nuclear exportin Crm1 and, consequently, its deactivation and nuclear export (3, 10, 22, 23, 25–27). Thus, NFAT-mediated transcriptional responses are determined by the balance between the activities of the Ca2+/CaN pathways driving nuclear import of NFAT and the NFAT kinases stimulating NFAT export from the nucleus.
Recent studies have demonstrated that not only electrical activity and intracellular Ca2+, but also neurotrophins, in particular nerve growth factor (NGF), potently regulate NFAT function in neurons (4, 15, 28). For example, NGF and brain-derived neurotrophic factor (BDNF) stimulate NFAT-dependent expression of inositol 1,4,5-trisphosphate receptor 1, BDNF, cyclooxygenase-2, and plasminogen activation inhibitor-1 in peripheral and central neurons (15, 28, 29). BDNF-dependent survival of adult hippocampal neurons and the formation of spatial memory have also been reported to require NFAT activation (13). Furthermore, NFAT is essential for NGF-dependent axonal growth, and deletion of NFAT isoforms NFATc2, NFATc3, and NFATc4 disrupts neurite outgrowth (4). Despite the growing evidence that NFAT proteins are important effectors of neurotrophin signaling, the mechanisms of NFAT regulation by neurotrophins are not well understood. It is also unclear whether and how electrical activity and neurotrophin signaling interact to regulate NFAT activity.
Here, by using genetic and pharmacological tools, we demonstrate that NGF facilitates depolarization-induced activation of NFAT in dorsal root ganglion (DRG) sensory neurons. Although Ca2+ is a critical regulator of NFAT, NGF had no significant effect on the intracellular Ca2+ concentration ([Ca2+]i) in DRG neurons, and the potentiating actions of NGF were independent of phospholipase C (PLC) activity. Instead, the NGF potentiation of NFAT activation required the PI3K-Akt signaling pathway and inhibition of the NFAT kinase, GSK3β. Furthermore, the silencing or deletion of specifically the NFATc3 isoform abolished the potentiating effect of NGF on NFAT-mediated transcription, implicating NFATc3 as a key NGF effector in sensory neurons.
EXPERIMENTAL PROCEDURES
DRG Cell Cultures and Transfection
Cultured DRG neurons were prepared as described previously (7, 30). Briefly, newborn (postnatal day 1–2) Sprague-Dawley rats or adult (2–4-month-old) wild-type and NFATc3 knock-out mice (BALB/c background) were sacrificed, and DRG were isolated from cervical, thoracic, and lumbar segments. Suspensions of DRG neurons were plated onto 25-mm glass coverslips precoated with poly-l-ornithine and laminin. Approximately 30 min after the cells were plated, Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, and penicillin (100 units/ml)/streptomycin (100 g/ml) was added to the plates (hereafter referred to as complete DMEM). The DRG neurons were maintained in culture in a 10% CO2 incubator at 37 °C and were used within 2–3 days. All surgical protocols were approved by the University of Iowa Institutional Animal Care and Use Committee. Rats were purchased from Charles River Laboratories (Wilmington, MA), and NFATc3 knock-out mice were generously provided by Dr. Santana (University of Washington, Seattle, WA) (31, 32).
Prior to plating, DRG neurons were transfected using the Amaxa nucleofection system (Program G-013; Lonza, Switzerland) as described previously (7, 30, 33). Typically, five rat pups or three mice were required to obtain a sufficient number of cells for performing a single transfection. EGFP-NFATc3 was generated by ligating NFATc3 (gift of Dr. Iino, Tokyo Metropolitan Institute of Medical Science) (34) into pEGFP-C1 (Clontech). A constitutively active form of the catalytic subunit PI3Kα (caPI3K; p110 mutant (35)) was a gift from Dr. Steven Green (University of Iowa). The shRNA constructs targeting NFATc3, NFATc4, and GSK3β were kindly provided by Dr. Michal Hetman (University of Kentucky) (12). We previously validated all of these constructs (33). A constitutively active form of GSK3β (GSK3β S9A) (36) was purchased from Addgene (plasmid 14754; Cambridge, MA).
NFAT Reporter Assays
NFAT reporter expression was assessed using a Dual-Luciferase assay as described previously (7, 33). In brief, DRG neurons were co-transfected with the NFAT-luciferase (NFAT-luc) reporter plasmid (which encodes firefly luciferase under the control of three copies of the NFAT-binding motif; pNFAT-TA-luciferase; Clontech) and the Renilla reniformis luciferase (TK-luc) reporter (which encodes Renilla luciferase under the control of constitutively active HSV-TK promoter; pRL-TK; Promega, Madison, WI). In some experiments, the investigated signaling pathways were modulated by co-transfecting GSK3β S9A or caPI3K with the luciferase reporter constructs. Initially, transfected cells were cultured in complete DMEM containing 25 ng/ml NGF (50 ng/ml in the case of mouse DRG cultures). Approximately 20 h later, the culture medium was replaced with fresh complete DMEM supplemented with B27 (Invitrogen) and ITS-A (Invitrogen) but devoid of NGF. Twenty-four hours later, the cultured DRG neurons were stimulated with 20 mm KCl medium (15 mm KCl for mouse cultures), which was prepared by mixing complete DMEM with 150 mm KCl stock solution. The L-type Ca2+ channel agonist BayK8644 (1 μm) was added to the 20 mm (15 mm) KCl medium to stabilize [Ca2+]i at the elevated levels for the duration of stimulation (7). In some cultured DRG plates, the KCl medium was supplemented with 25 ng/ml NGF (50 ng/ml for mouse DRG cultures). The cultured DRG neurons were stimulated with 20 mm (15 mm) KCl medium for either 6 or 12 h. In the case of the 6-h KCl stimulation protocol, cells were cultured in complete DMEM supplemented with B27 and ITS-A for an additional 6 h in the presence or absence of 25 ng/ml NGF (50 ng/ml NGF for mouse DRG cultures). All chemical inhibitors were applied at least 30 min prior to stimulation with 20 mm KCl. Twelve hours after the beginning of KCl stimulation, cells were lysed, and Dual-Luciferase assays were performed according to the manufacturer's protocol (DLRTM Assay System, Promega) using a Sirius luminometer (Berthold, Spain). NFAT-mediated transcription was quantified by normalizing the expression of NFAT-luciferase to that of constitutively active TK-luc (NFAT-luc/TK-luc). This approach minimizes variations in transfection efficiency and neuronal viability among various DRG cultures and culture conditions used in this study.
Western Blotting
Control and NGF-treated (25 ng/ml) DRG cultures were lysed in the presence of protease inhibitors (HaltTM protease inhibitor mixture kit, Pierce) and phosphatase inhibitors (HaltTM phosphatase inhibitor mixture, Pierce). Loading volumes for SDS-polyacrylamide gels were calculated based on BCA assays (Thermo Scientific, Rockford, IL) measuring the protein concentration in lysate. Lysates were mixed with SDS-PAGE sample buffer and boiled at 90 °C for 10 min to completely denature the proteins. Samples were loaded onto 8–20% gradient SDS-polyacrylamide gels (Bio-Rad) and run at constant voltage (100 V) for 2–3 h in a chamber that contained running buffer (3.0 g of Tris-HCl, 14.3 g of glycine, and 1 g of SDS dissolved in 1000 ml of H2O). Proteins in SDS-polyacrylamide gels were transferred to PVDF membranes (Millipore, Billerica, MA) using transfer buffer (1.5 g of Tris-HCl, 7.2 g of glycine, and 150 ml of MeOH dissolved in 1000 ml of H2O). Then the membrane was incubated with a blocking solution composed of 5% skim milk in TBS (20 mm Tris-HCl, pH 7.5, and 150 mm NaCl) for 1 h and then washed briefly with TBS prior to incubation with rabbit polyclonal anti-Ser(P)-9 GSK3β (1:10,000; catalog no. ab30619, Abcam, Cambridge, MA) dissolved in 5% skim milk in TBS-T (0.05% Tween 20 in TBS) for 2 h. The membrane was then washed with TBS-T and incubated with HRP-conjugated bovine anti-rabbit IgG (1:5000) dissolved in TBS-T for 1 h and then thoroughly washed with TBS-T. Phosphorylated GSK3β (Ser(P)-9 GSK3β) was visualized using the ECL Plus detection kit (GE Healthcare). Total GSK3β was measured using the same membrane, following a 30-min incubation in stripping buffer (0.7% β-mercaptoethanol, 2% SDS, and 62.7 mm Tris-HCl, pH 6.8) at 50 °C, two washes with TBS-T, confirmation that there was no residual signal from Ser(P)-9 GSK3β antibody, and immunoblotting using the steps described above but using rabbit monoclonal anti-GSK3β (1:5000–20,000, catalog no. 27C10, Cell Signaling, Danvers, MA) as the primary antibody. Similar procedures were used for Western blotting of tropomyosin-related kinase A (TrkA) and Tyr-490-phosphorylated TrkA, except for the following modifications. After running SDS-PAGE, proteins were transferred onto nitro-pure supported nitrocellulose membrane (Fisher) using CAPS buffer. The membranes were blocked with 5% BSA in TBS-T for 1 h. Total TrkA and phospho-TrkA (Tyr-490) were detected using anti-TrkA (1:500; catalog no. sc-118, Santa Cruz Biotechnology, Inc.) and anti-phospho-TrkA (Tyr-490) (1:500; catalog no. 9141, Cell Signaling) antibodies, respectively.
[Ca2+]i Imaging and EGFP-NFATc3 Nuclear Export/Import Assays
Cultured rat DRG neurons were prepared and treated as described under “NFAT Reporter Assays” with the exception that cells were transfected with EGFP-NFATc3. The [Ca2+]i changes and EGFP-NFATc3 movement were simultaneously recorded as described previously (7, 33). In brief, DRG neurons were loaded with the ratiometric Ca2+ indicator dye Fura-2/AM (2 μm) for 30 min. The cells were then placed in a flow-through perfusion chamber that was mounted on an inverted IX-71 microscope (Olympus, Japan) and perfused with standard extracellular HEPES buffered Hanks' salt solution (HH buffer) composed of 140 mm NaCl, 5 mm KCl, 1.3 mm CaCl2, 0.4 mm MgSO4, 0.5 mm MgCl2, 0.4 mm KH2PO4, 0.6 mm NaHPO4, 3 mm NaHCO3, 10 mm glucose, 10 mm HEPES, pH 7.4, with NaOH (310 mosm/kg with sucrose). For the nuclear export assay, the cells were perfused with 15 mm KCl medium (mixture of HH buffer and 150 mm KCl stock solution with added 1 μm BayK8644) for 40–60 min to allow EGFP-NFATc3 to translocate into the nucleus, and then cells were returned to HH buffer. [Ca2+]i changes and EGFP-NFATc3 movement were continuously recorded by alternately exciting fluorescence at 340 nm (12-nm bandpass), 380 nm (12-nm bandpass), and 475 nm (12-nm bandpass) using a Polychrome IV monochromator (TILL Photonics, Munich, Germany) and focusing on the cells via a ×40 oil immersion objective (numerical aperture = 1.35, Olympus). Fluorescence emission was collected at 530 nm (50-nm bandpass) using an IMAGO charge-coupled device camera (640 × 480 pixels; TILL Photonics). A 2 × 2 binning was used for acquisition (1 pixel ∼500 nm). Series of 340, 380, and 475 nm images were acquired at 0.05 Hz. [Ca2+] was calculated by converting the fluorescence ratio (R = F340/F380) using the formula, [Ca2+] = Kdβ(R − Rmin)/(Rmax − R). A dissociation constant (Kd) value of 275 nm was used, as provided by Shuttleworth and Thompson (37). Rmin, Rmax, and β were calculated by applying 10 μm ionomycin in either Ca2+-free buffer (1 mm EGTA) or HH buffer (1.3 mm Ca2+) and were found to be as follows: Rmin = 0.22, Rmax = 2.75, and β = 5.9. [Ca2+]i data were analyzed using TILLvisION version 4.0.12 software (TILL Photonics). Background-corrected nuclear EGFP fluorescence was plotted as a function of time, and the trace was fitted with a monoexponential decay function using pCLAMP version 9.0 software (Axon Instruments, Sunnyvale, CA); the time constant (τ) was used for quantifying nuclear export of EGFP-NFATc3. For the EGFP-NFATc3 nuclear import assay, cells were stimulated with 10 mm KCl + 1 μm BayK8644 for 15–30 min, with [Ca2+]i and nuclear EGFP fluorescence recorded simultaneously. Nuclear import of NFATc3 was quantified by calculating the average rate (slope) of EGFP-NFATc3 translocation into the nucleus during the first 5 min of translocation.
For the experiments shown in Figs. 2 (A and B) and 3, untransfected cultured rat DRG neurons were prepared, treated, and loaded with Fura-2/AM as described above. Fluorescent images (λex = 340 and 380 nm) were collected at 0.05 or 0.5 Hz using a ×20 objective (numerical aperture = 0.75, Olympus, Japan) and a 2 × 2 binning set for an IMAGO charge-coupled device camera at room temperature. For the experiments shown in Fig. 2C, the cells were stimulated with 20 mm KCl for 6 h in a 10% CO2 incubator at 37 °C, loaded with Fura-2/AM, and placed in a temperature-controlled flow-through chamber (T = 37 °C). Experimentally determined calibration constants for ×20 objective were Rmin = 0.21, Rmax = 3.45, and β = 6.97, which were used for the conversion to [Ca2+]i. The dissociation constants (Kd) of Fura-2 used for the conversion were 275 nm (experiments performed at room temperature) and 225 nm (experiments performed at 37 °C) (37).
FIGURE 2.
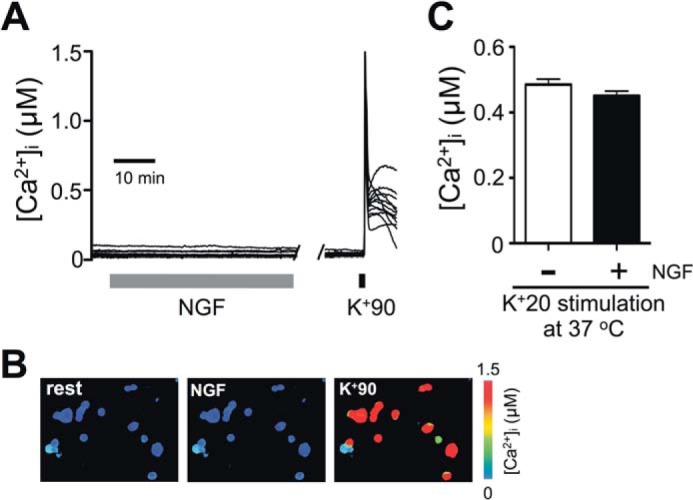
NGF does not affect [Ca2+]i in DRG neurons. Cultured DRG neurons were prepared using the protocol described in Fig. 1A. A, representative traces of [Ca2+]i from Fura-2/AM-loaded DRG neurons during treatment with 25 ng/ml NGF (gray bar). Cell viability and responsiveness to depolarization were confirmed at the end of the experiment by applying 90 mm KCl (30 s) to the cells. Ca2+ imaging was performed at room temperature (n = 64 cells, 4 independent experiments). Each trace represents an individual cell. B, [Ca2+]i in cells is indicated by pseudocolor images taken at rest (left), during the NGF treatment (middle) and at the peak of K+90-induced depolarization (right) for the same experiment as described in A. C, comparison of the effects of prolonged (6-h) K+20 depolarization on [Ca2+]i in the absence (−NGF; white) or presence (+NGF; black) of 25 ng/ml NGF. The K+20 solutions were supplemented with 1 μm BayK8644. [Ca2+]i was measured at the end of the 6-h treatment, in cells loaded with Fura-2/AM. All procedures were conducted at 37 °C. p > 0.05, Student's t test; n = 99 cells (−NGF) and 121 cells (+NGF); 3 independent experiments/condition. All data are presented as mean ± S.E. (error bars).
FIGURE 3.

The PLC inhibitor U73122 blocks bradykinin- and ATP-induced Ca2+ mobilization in DRG neurons. [Ca2+]i measurements were carried out in neonatal rat DRG neurons using Fura-2/AM as described under “Experimental Procedures.” [Ca2+]i increases were induced by either 300 nm bradykinin (A–C; 1 min; orange bars) or 100 μm ATP (D–F; 1 min; yellow bars). To isolate Ca2+ release from intracellular Ca2+ stores, cells were perfused with a Ca2+-free extracellular buffer (supplied with 0.1 mm EGTA) during the applications of bradykinin and ATP. Shown are representative [Ca2+]i traces in response to repeated applications of bradykinin (A and B) and in response to ATP (D and E). The [Ca2+]i recordings were simultaneously made from several different DRG neurons, and each color trace corresponds to an individual DRG neuron. Approximately 27% (n = 122) and 62% (n = 74) of tested DRG neurons responded to bradykinin and ATP, respectively, which is consistent with the previous reports (14, 39–41). Depolarization stimuli using 50 mm KCl (K+50, 15 s; black bar) were applied to ensure refilling of the intracellular Ca2+ stores prior to the second application of an agonist. With this approach, the amplitudes of the second bradykinin (ATP)-induced [Ca2+]i responses were >90% of the first ones (A, C, D, and F). Treating cells for 20 min with the PLC inhibitor U73122 (1 μm) prior to the second agonist application markedly reduced the amplitudes of the second [Ca2+]i responses. C and F, effects of U73122 (B and E) and vehicle/control (A and D) on the ratio of the second [Ca2+]i response amplitude to the first one (A2/A1) for bradykinin (C; n = 22 neurons for vehicle/control, and n = 11 neurons for U73122) and ATP (F; n = 29 neurons for vehicle/control, and n = 17 neurons for U73122). ***, p < 0.001, Student's t test. Error bars, S.E.
Analysis of EGFP-NFATc3 Nuclear Translocation
Cultured rat DRG neurons were prepared, treated, and stimulated as described under “NFAT Reporter Assays” with the exception that they were transfected with EGFP-NFATc3. DRG neurons were stimulated with 20 mm KCl for various times (0 min, 20 min, 1 h, 3 h, and 6 h), fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min, and then washed three times with PBS. They were counterstained with DAPI (Invitrogen) to mark the nucleus and mounted on glass slides using Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL). The distribution of EGFP-NFATc3 was analyzed using a ×60 oil immersion objective (numerical aperture = 1.4) on an Olympus BX61 microscope equipped with the Fluoview 300 laser-scanning confocal imaging system as described previously (33). Data were acquired and analyzed by a blinded experimenter using the Fluoview software (Olympus). Average EGFP fluorescence intensity values were obtained for regions of interest in both the nucleus (overlap with DAPI staining) and the cytosol. Background fluorescence was corrected for using a region of interest devoid of cells.
Reagents
The TrkA inhibitor GSK-Trk (catalog no. 648450), K252A, Akt inhibitor IV (catalog no. 124011), wortmannin, and LY294002 were obtained from Calbiochem/EMD Millipore. U73122 was purchased from R&D Systems/Tocris (Minneapolis, MN). Purified mouse nerve growth factor 2.5s was from AbD Serotec (Raleigh, NC). Pronase E and collagenase A were from Roche Applied Science. All other reagents were purchased from Sigma.
Statistical Analysis
Data were analyzed by using Student's t test for comparing two groups and by using one-way analysis of variance (ANOVA) for comparing more than two groups, followed by Bonferroni's post hoc test. The Kruskal-Wallis test was used to compare kinetics of NFAT nuclear export (Fig. 7). The statistical tests were performed using GraphPad Prism version 5.0 software (San Diego, CA). All data are expressed as mean ± S.E.
FIGURE 7.
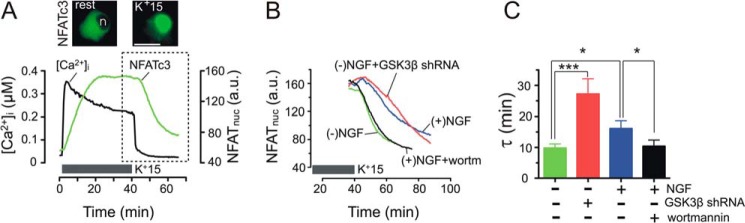
NGF slows the nuclear export of NFATc3 in DRG neurons. A, DRG neurons were transfected with EGFP-NFATc3 and subsequently loaded with Fura-2 for simultaneous measurements of changes in [Ca2+]i (black trace) and the nuclear transport of EGFP-NFATc3 nuclear transport (green trace), as described previously (7, 33). DRG cultures were deprived of NGF for at least 24 h before the recordings were initiated. Depolarization using K+15 + 1 μm BayK8644 induced a [Ca2+]i elevation and translocation of EGFP-NFATc3 into the nucleus (quantified as EGFP fluorescence intensity in the nucleus). Once nuclear EGFP-NFATc3 fluorescence reached steady state, depolarization was terminated, which led to rapid [Ca2+]i recovery to the resting level and to the initiation of EGFP-NFATc3 export from the nucleus. Images above the traces show NFATc3-EGFP distribution (background subtracted) in the same DRG neuron under resting conditions (prior to K+15 stimulation; left) and after 20 min of stimulation with K+15 (right). Scale bar (white), 20 μm; n, cell nucleus. B, superimposition of EGFP-NFATc3 nuclear export traces recorded under control conditions (green; −NGF), in cells treated with 25 ng/ml NGF (blue; +NGF), NGF and the PI3K inhibitor wortmannin (300 nm; black; NGF+wortm), or in neurons transfected with GSK3β shRNA (red). All experiments were performed as in A. C, quantification of EGFP-NFATc3 nuclear export kinetics under different treatment conditions. The NFATc3 export process was fitted with a monoexponential function using pCLAMP version 9 software (Molecular Devices), and the time constant (τ) was calculated for each experiment (mean ± S.E. (error bars)). NGF treatment (blue) significantly increased τ, and this effect was blocked by wortmannin (black). Knocking down GSK3β also significantly increased τ (red). *, p < 0.05; ***, p < 0.001, Kruskal-Wallis test, n = 15–25 cells. a.u., arbitrary units.
RESULTS
NGF Facilitates Depolarization-induced NFAT-dependent Gene Expression in DRG Neurons
NGF regulates NFAT activity in neurons, which is essential for axonal growth, neuronal survival, and pain signaling (4, 15). However, the mechanisms underlying the NGF-dependent regulation of NFAT are not well understood. To address this question, we examined the effects of NGF on NFAT-mediated transcription in DRG neurons using an NFAT-luciferase expression reporter (Dual-Luciferase assay) as described previously (7, 33) (also see “Experimental Procedures”). For this assay, the NFAT-mediated expression of firefly luciferase (NFAT-luc) was normalized to the expression of Renilla luciferase driven by the constitutively active TK-HSV promoter (TK-luc) and quantified as NFAT-luc/TK-luc. The experimental timeline is shown in Fig. 1A. DRG cultures were initially maintained in the presence of 25 ng/ml NGF to maximize transfection efficiency and cell viability. Prior to the beginning of the experiment (t = 0 h; timeline in Fig. 1A), the DRG cultures were deprived of NGF for 24 h. In the absence of depolarization, a 12-h treatment with either 25 or 100 ng/ml NGF did not affect the NFAT-dependent luciferase expression in DRG neurons (Fig. 1, B and C). However, mild depolarization using 20 mm KCl (K+20) increased NFAT-dependent luciferase expression, and this effect of depolarization was strongly potentiated by the addition of 25 ng/ml of NGF (Fig. 1B). We also found that the effect of NGF was somewhat stronger for a 6-h K+20 stimulation (2.9 ± 0.3-fold increase in luciferase expression) than for a 12-h stimulation protocol (2.4 ± 0.3-fold increase). An increase in NGF concentration from 25 to 100 ng/ml did not significantly change the magnitude of the NGF effect (Fig. 1C). Thus, in further experiments investigating the mechanisms by which NGF regulates NFAT activity in DRG neurons, we used 6-h K+20 stimulation in combination with the 25 ng/ml NGF treatment protocol. Collectively, the described data suggest that NGF facilitates NFAT-mediated transcription induced by depolarization in sensory neurons.
FIGURE 1.

NGF facilitates depolarization-induced NFAT-dependent transcription in DRG neurons. A, timeline describing the experimental protocols. NFAT-luc (NFAT reporter; firefly luciferase) and TK-luc (constitutively active Renilla luciferase reporter) constructs were co-transfected into cultured DRG neurons. Cultures were deprived of NGF for 24 h prior to initiation of treatments (t = 0 h). Cultures were left untreated or were treated with NGF (white bar) in the absence or presence of K+20 applied either for 6 h (gray bar) or 12 h (black bar) to induce depolarization. The K+20 solution was supplemented with the L-type Ca2+ channel agonist BayK8644 (1 μm) to stabilize [Ca2+]i elevation for the duration of treatments (6 and 12 h, respectively) (7). Cells were lysed 12 h after the beginning of stimulation, and NFAT-dependent transcription was quantified by Dual-Luciferase assay (NFAT-luc/TK-luc) as described previously (7, 33) (also see “Experimental Procedures”). B, quantification of the effects of NGF on NFAT-dependent transcription in DRG neurons induced by K+20 stimulation for 6 or 12 h compared with that in unstimulated cells (rest). Treatment with 25 ng/ml NGF (black bars) significantly increased K+20-induced NFAT-luciferase expression compared with that in untreated cells (white bars). ***, p < 0.001; one-way ANOVA with Bonferroni's post hoc test (n = 9–42 experiments). C, quantification of the effects of NGF at various concentrations (0, 25 and 100 ng/ml) on NFAT-dependent transcription in the absence (left) or presence (right) of K+20 (6-h stimulation). ***, p < 0.001; N.S., not significant; one-way ANOVA with Bonferroni's post hoc test (n = 6–42 experiments). All data are presented as mean ± S.E. (error bars).
NGF Does Not Affect [Ca2+]i, and the Facilitatory Effect of NGF on NFAT Activation Is Independent of PLC in Sensory Neurons
Previous studies suggested that another neurotrophin, BDNF, activates NFAT in central neurons via TrkB-dependent stimulation of phospholipase C (PLC), inositol 1,4,5-trisphosphate (IP3) synthesis, Ca2+ mobilization from intracellular IP3-sensitive Ca2+ stores, and resulting CaN activation (28). To test the possibility that NGF acts via a similar signaling pathway in DRG neurons, we first examined the effects of NGF on [Ca2+]i in DRG neurons. DRG cultures were deprived of NGF for 24 h prior to recordings. Treatment with 25 ng/ml NGF did not produce any changes in [Ca2+]i in DRG neurons (Fig. 2, A and B; n = 64). All of the tested neurons demonstrated a normal [Ca2+]i response to depolarization evoked by 90 mm KCl (K+90; Fig. 2, A and B), consistent with Ca2+ signaling in these cells being undisturbed (7, 38). Bradykinin and ATP are known to evoke Ca2+ release from IP3-sensitive Ca2+ stores in DRG neurons in a manner dependent on PLC activation (14, 39–42). Thus, as a positive control for normally functioning IP3-sensitive stores in our system, we tested the effects of bradykinin (300 nm) and ATP (100 μm) on [Ca2+]i signaling in DRG neurons. We found that both agonists were able to induce Ca2+ release from intracellular Ca2+ stores in subsets of DRG neurons and that these effects were blocked by the PLC inhibitor U73122 (1 μm; Fig. 3).
Next, we examined whether K+20-induced [Ca2+]i elevations were affected by NGF under the same conditions as those used in the Dual-Luciferase experiments. We found that both NGF-treated and untreated DRG neurons showed similar [Ca2+]i levels in the presence of 20 mm KCl (Fig. 2C). Thus, the facilitatory effect of NGF on NFAT activation is probably independent of any alteration in Ca2+ handling in DRG neurons.
To further examine the possibility that NGF recruits the PLC-IP3 signaling to regulate NFAT activation, we performed additional experiments monitoring NFAT-luciferase expression in DRG neurons. NGF produces its biological effects via two receptors, the high-affinity TrkA receptor and the low-affinity p75 receptor (43, 44). We found that NGF treatment significantly increased phosphorylation of TrkA at Tyr-490, which is consistent with NGF-induced TrkA activation in our system (43, 45, 46) (Fig. 4B). We also showed that both a potent TrkA inhibitor, GSK-Trk (47, 48), and a potent inhibitor of tyrosine kinase commonly used to study TrkA signaling, K252A (49–51), blocked the potentiating effect of NGF on K+20-induced NFAT-luciferase expression in DRG neurons (Fig. 4C). However, the PLC inhibitor U73122 (1 μm) did not affect NGF-dependent facilitation of NFAT-luciferase expression (Fig. 4D). The experiments shown in Fig. 3 and our previous work demonstrate the effectiveness of this inhibitor in DRG neurons (14, 41). Collectively, these data suggest that NGF potentiates NFAT activation via a TrkA-dependent but PLC-independent pathway in sensory neurons.
FIGURE 4.

Facilitation of NFAT-dependent transcription by NGF requires activation of the PI3K-Akt signaling pathway. A, timeline describing the experimental protocol. Horizontal bars, treatments with 25 ng/ml NGF and K+20. BayK8644 (1 μm) was added to K+20 solution. All inhibitors were applied 30 min prior to stimulation with NGF/K+20. Dual-Luciferase assays were performed to quantify NFAT-dependent transcription (NFAT-luc/TK-luc). caPI3K is a construct that expresses a constitutively active form of PI3K; it was co-transfected with NFAT-luciferase and TK-luciferase in the experiments shown in E. B, Western blot analysis of TrkA phosphorylation at Tyr-490. DRG neurons were deprived of NGF for 24 h and subsequently treated with either vehicle control (−) or 25 ng/ml NGF (+) for 90 min (n = 5 experiments). A shorter time (compared with the 6 h commonly used) was chosen because TrkA phosphorylation is expected to be an early event in the studied signaling. C, effects of the TrkA inhibitor GSK-Trk (2 μm; n = 11 experiments) and tyrosine kinase inhibitor K252A (1–2 μm, n = 11 experiments) on NGF-dependent potentiation of NFAT-luciferase expression. D, the PLC inhibitor U73122 (1 μm; n = 5 experiments) had no effect on NGF-dependent potentiation of NFAT-luciferase expression. E, effects of the PI3K inhibitors wortmannin (wortm; 100 nm, n = 15 experiments) and LY294002 (LY; 25 μm, n = 15 experiments) as well as those of caPI3K (n = 6 experiments) on NGF-dependent potentiation of NFAT-luciferase expression in the presence or absence of K+20 stimulation. Both inhibitors blocked the NGF effects in DRG neurons. Conversely, caPI3K mimicked the NGF effect in the presence but not in the absence of K+20 stimulation. F, effects of the Akt inhibitor, Akt IV (0.5, 1, or 2 μm) on NGF-dependent potentiation of NFAT-luciferase expression. Akt IV blocked the NGF effect in a concentration-dependent manner (n = 3–8 experiments). *, p < 0.05; **, p < 0.01; ***, p < 0.001. Student's t test (B) and one-way ANOVA with Bonferroni's post hoc test (C-F) were used. N.S., not significant. All data are presented as mean ± S.E. (error bars).
NGF Recruits the PI3K-Akt-GSK3β Signaling Pathway Downstream of the TrkA Receptor to Facilitate NFAT-dependent Transcription
Another common signaling pathway initiated by NGF binding to the TrkA receptor in sensory neurons involves the activation of PI3K and Akt (also known as protein kinase B), followed by Akt-dependent phosphorylation and inactivation of GSK3β (43, 44, 52, 53). Notably, GSK3β is a well established negative regulator of NFAT that phosphorylates SP motifs in NFAT, thereby promoting its export from the nucleus (3, 25). Therefore, we hypothesized that NGF facilitates depolarization-induced activation of NFAT through PI3K-Akt signaling, inactivation of GSK3β, and inhibition of the nuclear export of NFAT.
To test this hypothesis, we first examined the effects of two structurally distinct inhibitors of PI3K, wortmannin and LY294002. As shown in Fig. 4E, applying either wortmannin or LY294002 significantly reduced the effect of NGF. Conversely, overexpressing caPI3K (35) mimicked the potentiating effect of NGF (Fig. 4E). Finally, the effect of NGF was blocked by the Akt inhibitor, Akt IV, in a concentration-dependent manner (Fig. 4F). Akt IV was reported to inhibit Akt activity indirectly by blocking a protein kinase upstream of Akt and downstream of PI3K (54).
Next, we examined the role of GSK3β in NGF-dependent facilitation of NFAT activation. Transfecting DRG neurons with a constitutively active form of GSK3β (GSK3β S9A; phosphorylation-deficient form) (36), abolished the facilitatory effect of NGF on NFAT-luciferase expression (Fig. 5B). We performed reciprocal experiments in which we knocked down GSK3β expression using an shRNA construct that we previously showed reduced GSK3β expression by over 90% (33). Knockdown of GSK3β markedly enhanced NFAT activation in DRG neurons that had not been treated with NGF (Fig. 5B). Notably, this dramatic increase in NFAT activity was not observed in the absence of K+20 stimulation, consistent with the inability of NGF to enhance NFAT activation in the absence of depolarization (Fig. 1, B and C). In complementary experiments, we found that NGF treatment (25 ng/ml for 6 h) led to a significant increase in the phosphorylation of GSK3β at Ser-9 (Fig. 5C), which is known to inhibit GSK3β (55). Interestingly, the enhancement of NFAT activation by GSK3β knockdown was much greater than that resulting from NGF treatment (Fig. 5B). This difference may be explained by the fact that only ∼60% of postnatal DRG neurons express TrkA (56, 57), whereas GSK3β knockdown would affect the majority of cells co-transfected with luciferase reporter constructs. Overall, the described data are consistent with the hypothesis that NGF facilitates depolarization-induced NFAT activation by stimulating the PI3K-Akt pathway and inhibiting GSK3β.
FIGURE 5.

The facilitatory effect of NGF on NFAT-mediated transcription strongly depends on GSK3β activity in DRG neurons. A, timeline describing the experimental protocols. The GSK3β shRNA or GSK3β S9A construct was co-transfected with NFAT-luciferase and TK-luciferase into DRG neurons. Horizontal bars, treatments using 25 ng/ml NGF and K+20. The L-type Ca2+ channel agonist BayK8644 (1 μm) was added to K+20 solution to stabilize [Ca2+]i elevation. B, expression of constitutively active GSK3β (GSK3β S9A; n = 9 experiments) blocked the facilitatory effects of NGF on NFAT-luciferase expression, whereas GSK3β knockdown mimicked the effect of NGF (n = 5 experiments). *, p < 0.05; ***, p < 0.001, one-way ANOVA with Bonferroni's post hoc test; mean ± S.E. (error bars). C, effects of NGF (25 ng/ml) on GSK3β phosphorylation at Ser-9, as determined by Western blotting. Cultured DRG neurons either remained untreated (−NGF; white bar) or were treated with 25 ng/ml NGF (+NGF; black bar) for 6 h and subsequently processed for Western blotting. Values were quantified as the ratio of Ser(P)-9 GSK3β/total GSK3β (n = 3 experiments); *, p < 0.05, Student's t test.
NFATc3 Is Required for the Potentiating Effect of NGF on NFAT-dependent Transcription in Sensory Neurons
We and others previously identified NFATc3 and NFATc4 as the main NFAT isoforms that are expressed and functional in DRG neurons and showed that depolarization-induced NFAT-dependent gene expression is strongly dependent on the NFATc3 isoform in both DRG and hippocampal neurons (7, 15, 16, 33). We tested whether NFATc3 or NFATc4 mediated NGF-dependent enhancement of NFAT transcriptional response. This question was addressed by using previously validated shRNA plasmids to knock down NFATc3 and NFATc4 in DRG neurons (33). Knocking down NFATc3 expression in cultured rat DRG neurons resulted in an almost complete elimination of the NGF effect, whereas knockdown of NFATc4 had virtually no effect (Fig. 6B). To further validate the role of NFATc3 in the NGF effect, we examined the expression of NFAT-luciferase in DRG neurons prepared from NFATc3 knock-out (KO) (58) and wild-type adult mice (Fig. 6C). As in the case of DRG neurons from neonatal rats, NGF treatment of DRG neurons obtained from adult mice produced a marked enhancement of depolarization-induced NFAT-luciferase expression but did not produce an increase of NFAT activity in the absence of depolarization (Fig. 6C). Notably, the NGF effect was abolished in DRG neurons from NFATc3 KO mice. Knocking out NFATc3 also markedly reduced depolarization-induced expression of NFAT-luciferase, consistent with our previous report demonstrating the key role of NFATc3 in DRG neurons (33). Collectively, these results suggest that NFATc3 is the main isoform that is responsible for the potentiating effect of NGF on the NFAT-mediated gene expression in DRG neurons.
FIGURE 6.
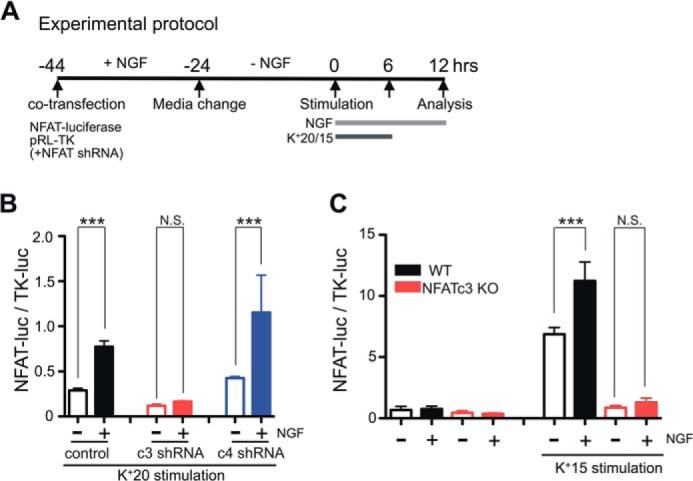
The facilitatory effect of NGF on NFAT-mediated gene expression in DRG neurons relies on the NFATc3 isoform. A, timeline describing the experimental protocols. For NFATc3 and NFATc4 knockdown experiments, the corresponding shRNA constructs were co-transfected with NFAT-luciferase and TK-luciferase. K+15 and K+20 solutions were supplemented with the L-type Ca2+ channel agonist BayK8644 (1 μm) for stabilizing [Ca2+]i at elevated levels (7). B, effects of NFATc3 and NFATc4 knockdown on NGF-dependent potentiation of NFAT-luciferase expression in DRG neurons. Knocking down NFATc3 (c3, red; n = 7 experiments) but not NFATc4 (c4, blue; n = 6 experiments) significantly reduced the effect of NGF (25 ng/ml) on depolarization (K+20)-induced NFAT-luciferase expression in DRG neurons. C, effects of NGF (50 ng/ml) on depolarization (K+15)-induced expression of NFAT-luciferase in DRG neurons from adult WT and NFATc3 KO mice. NGF treatment significantly enhanced K+15-induced NFAT-luciferase expression in DRG neurons from WT (black; n = 7 experiments) but not from NFATc3 KO mice (red; n = 4 experiments). ***, p < 0.001; N.S., not significant, one-way ANOVA with Bonferroni's post hoc test. All data are presented as mean ± S.E. (error bars).
NGF Slows the Nuclear Export but Does Not Affect the Nuclear Import of NFATc3 in DRG Neurons
The nuclear translocation and retention of NFAT upon its activation are determined by the relative rates of nuclear NFAT import and export (21, 22). Having established the importance of NFATc3 for the potentiating effect of NGF on NFAT-mediated transcription, we next tested whether NGF inhibits the nuclear export of NFATc3, facilitates the nuclear import of NFATc3, or affects both processes. To address this question, we used simultaneous imaging of EGFP-tagged NFATc3 dynamics and [Ca2+]i changes in DRG neurons, as described previously (7, 33). This method allows monitoring nuclear translocation of EGFP-NFATc3 in neurons in real time while controlling for the effects of NGF on Ca2+ signaling. EGFP-NFATc3-transfected and Fura-2-loaded DRG neurons were depolarized using 15 mm KCl (K+15) for 40–60 min, which led to an increase in [Ca2+]i (Fig. 7A, black trace) and EGFP-NFATc3 translocation to the nucleus (Fig. 7A, green trace and EGFP-NFATc3 images). Once the nuclear localization of EGFP-NFATc3 reached a steady state, the extracellular solution was changed to normal HH buffer ([K+] = 5 mm), resulting in a rapid decrease in [Ca2+]i and initiating export of EGFP-NFATc3 from the nucleus (Fig. 7A). The kinetics of NFATc3 export could be well approximated by a monoexponential function, with τ of ∼10 min (Fig. 7). In the absence of NGF, EGFP-NFATc3 rapidly returned to the cytoplasm upon termination of K+15-evoked depolarization (Fig. 7, B and C). Treating cells with 25 ng/ml NGF or knocking down GSK3β resulted in a significant slowing of EGFP-NFATc3 export from the nucleus, whereas treatment with the PI3K inhibitor wortmannin blocked the NGF effect (Fig. 7, B and C).
Because Ca2+/CaN-dependent NFAT nuclear import is constantly opposed by NFAT kinases, it is possible that NGF-mediated inhibition of GSK3β enhances the nuclear import of NFAT. This possibility was tested by mildly depolarizing cells using 10 mm KCl (K+10). We reasoned that submaximal activation of Ca2+/CaN by mild depolarization would be optimal for detecting any contribution that suppression of GSK3β might make to the nuclear import of NFATc3. As in the case of the NFATc3 nuclear export experiments, the translocation of EGFP-NFATc3 was simultaneously monitored with [Ca2+]i in response to depolarization (Fig. 8A). The nuclear import of EGFP-NFATc3 was quantified as the initial rate of increase of EGFP fluorescence intensity in the nucleus. We found that neither NGF treatment nor GSK3β knockdown produced a detectable change in the rate of EGFP-NFATc3 import into the nuclei of DRG neurons (Fig. 8, B and C). Collectively, our findings suggest that NGF markedly slows the rate of NFATc3 export from the nuclei of DRG neurons without significantly affecting the rate of NFATc3 nuclear import.
FIGURE 8.
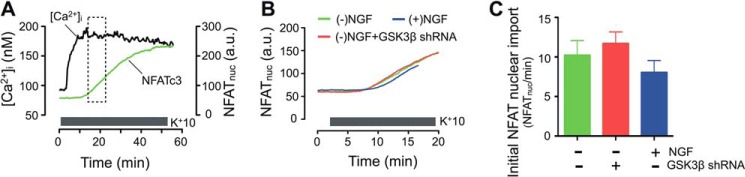
NGF does not affect the rate of nuclear import of NFATc3 in DRG neurons. A, nuclear transport of EGFP-NFATc3 (green) was simultaneously monitored with [Ca2+]i changes (black) in DRG neurons as described previously (7, 33) (also see “Experimental Procedures”). DRG cultures were deprived of NGF for 24 h before the experiment was initiated. [Ca2+]i elevations and nuclear import of EGFP-NFATc3 were induced by mild depolarization with K+10 (supplemented with 1 μm BayK8644). The nuclear import of EGFP-NFATc3 was quantified by measuring EGFP fluorescence intensity in the nuclear region and is expressed as the average rate (slope) of translocation into the nucleus during the first 5 min of translocation (dotted rectangular box). B, superimposition of representative traces showing EGFP-NFATc3 nuclear import in the absence (green) or presence (blue) of 25 ng/ml NGF or in neurons transfected with GSK3β shRNA (red). C, quantification of the rate of EGFP-NFATc3 nuclear import in experiments like those described in A and B. No significant differences were observed under the conditions described (n = 4–10 cells). a.u., arbitrary units. Error bars, S.E.
To better understand how the slowing of nuclear export of NFATc3 affects its overall nuclear-cytosolic distribution during prolonged depolarization, we examined the translocation of EGFP-NFATc3 under conditions similar to those used in the NFAT-luciferase experiments (Figs. 1 and 4–6). Specifically, EGFP-NFATc3-transfected DRG neurons deprived of NGF for 24 h were depolarized using K+20 in the presence or absence of NGF, fixed at various time points, and analyzed for the distribution of EGFP-NFATc3 as described previously (33) (see also “Experimental Procedures”). K+20-evoked depolarization produced a rapid and robust nuclear translocation of NFATc3 that was similar between the NGF-treated and untreated cells at 20 min and 1 h after the beginning of depolarization (Fig. 9A). Notably, in the absence of NGF, NFATc3 reached its maximal nuclear translocation at 1 h and was then steadily exported from the nucleus to the cytosol despite the continuing depolarization. In contrast, DRG neurons treated with NGF not only retained NFATc3 in the nucleus but also showed that NFATc3 continued to slowly accumulate in the nucleus throughout the period of depolarization (Fig. 9). These observations are consistent with the prominent potentiating effects of NGF on depolarization-induced expression of NFAT-luciferase and highlight the importance of NGF signaling for retaining activated NFATc3 in the nucleus, probably via the suppression of its nuclear export.
FIGURE 9.
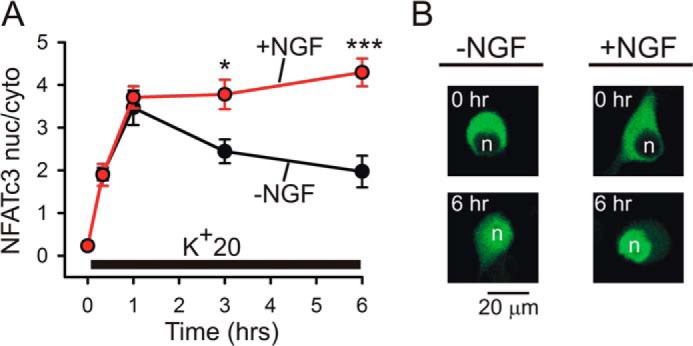
NGF increases depolarization-induced nuclear translocation of NFATc3 in DRG neurons. A, DRG neurons transfected with EGFP-NFATc3 were deprived of NGF for 24 h prior to stimulation. K+20 (supplemented with 1 μm BayK8644) was applied in the absence (black) or presence (red) of 25 ng/ml NGF under conditions similar to those described for the NFAT-luciferase experiments (Figs. 1 and 4–6). The translocation of EGFP-NFATc3 was imaged and quantified as described previously (33). In the absence of NGF, EGFP-NFATc3 was gradually exported from the nucleus despite continuous depolarization, whereas in NGF-treated DRG neurons, the levels of EGFP-NFATc3 in the nucleus continued to increase slowly throughout the period of depolarization. *, p < 0.05; ***, p < 0.001, one-way ANOVA with Bonferroni's post hoc test (12–47 cells). Data are presented as mean ± S.E. (error bars). B, representative images showing distribution of EGFP-NFATc3 in DRG neurons at rest and after 6 h of K+20 stimulation in the absence (left) or presence (right) of 25 ng/ml NGF. n, cellular nuclei.
DISCUSSION
Electrical activity and neurotrophins are important regulators of gene expression in the nervous system and are responsible for long term structural and functional changes in neurons during development, synaptic plasticity, and adaptation to environmental conditions (1, 43, 44, 59). The work presented here identifies mechanisms of interaction between electrically driven Ca2+ signaling and neurotrophin signaling in regulating the transcription factor NFAT. Based on our results, we propose a model in which Ca2+/CaN-dependent nuclear import of NFAT is driven by electrical activity/depolarization in neurons (Fig. 10). The parallel NGF-induced activation of the TrkA-PI3K-Akt signaling pathway inhibits GSK3β, the protein kinase that rephosphorylates NFAT and promotes its export from the nucleus (3, 25, 60), thereby prolonging retention of activated NFAT in the nucleus. Thus, depolarization-driven Ca2+ signaling and NGF signaling act in concert to stimulate NFAT-dependent gene expression by concurrently inducing the nuclear import of NFAT and inhibiting its nuclear export (Fig. 10).
FIGURE 10.

Model of concerted regulation of NFATc3 by electrical activity and NGF in sensory neurons. Electrical stimulation (depolarization in this study) induces Ca2+ influx into the cell via voltage-gated Ca2+ channels, which leads to CaN-dependent dephosphorylation of NFATc3 and initiation of its import into the nucleus. NGF facilitates NFATc3 activation and retention in the nucleus by stimulating the PI3K-Akt signaling pathway, which leads to phosphorylation-dependent inhibition of GSK3β and thereby to the slowing of NFATc3 export from the nucleus.
The finding that NGF potentiates NFAT activation by attenuating NFAT kinase activity is novel, with previous studies having focused on the importance of a neurotrophin-induced increase in [Ca2+]i for NFAT activation (15, 28). Indeed, Groth et al. (15, 28) showed that another major neurotrophin, BDNF, activates NFAT-mediated transcription in hippocampal and spinal cord neurons and that these effects can be abolished by inhibiting either PLC or the endoplasmic reticulum Ca2+-ATPase. Based on these findings, the authors proposed that BDNF activates NFAT in neurons via PLC- and IP3-dependent Ca2+ release from intracellular Ca2+ stores, although the direct effects of BDNF on [Ca2+]i were not tested in these studies (15, 28). Ozaki et al. (61) recently reported that DRG neurons cultured for 4–14 days in the presence of NGF can subsequently produce spontaneous [Ca2+]i transients when recorded in the absence of NGF. However, the amplitudes of these [Ca2+]i fluctuations were rather small (<50 nm) and below the [Ca2+]i levels required to activate NFAT in DRG neurons (200–300 nm) (7). In contrast, we and others observed no evidence of [Ca2+]i fluctuations under resting conditions in the majority of DRG neurons cultured either with or without NGF (7, 30, 41, 62–69). One possible explanation for these differences is that the study by Ozaki et al. (61) used cells that had been maintained in culture for an extended period prior to the experimentation (4–14 days) and also a relatively high concentration of NGF (100 ng/ml).
Overall, our data argue against the possibility that the PLC-IP3-Ca2+ pathway downstream of TrkA contributed to the NGF-dependent enhancement of NFAT activation in DRG neurons described here. First, NGF had no detectable effect on [Ca2+]i in DRG neurons, either under resting conditions or during depolarization (Fig. 2). Second, the potentiating effect of NGF on NFAT activity was not sensitive to the potent PLC inhibitor U73122 (Fig. 4D). Note that in our hands, U73122 is capable of effectively blocking bradykinin- and ATP-evoked Ca2+ release from the intracellular Ca2+ stores (Fig. 3) as well as bradykinin-induced NFAT activation in DRG neurons (14, 41). Third, the facilitation of NFAT activity by NGF required concomitant mild depolarization of neurons to induce nuclear import of NFAT (7). Indeed, in DRG neurons from both rats and mice, treatment with NGF alone had no effect on NFAT-mediated transcription (Figs. 1 and 6); this further argues against the involvement of an NGF-induced [Ca2+]i increase in the described phenomena. Instead, NGF-induced potentiation of the NFAT response in DRG neurons was blocked by inhibitors of PI3K and Akt (Fig. 4) as well as by constitutively active GSK3β (Fig. 5), suggesting recruitment of the PI3K-Akt-GSK3β pathway downstream of TrkA. Moreover, we found that the NGF effect was mimicked by the introduction of constitutively active PI3K (Fig. 4) and by GSK3β knockdown (Fig. 5). It was reported that insulin-like growth factor induced potentiation of L-type Ca2+ channels via Akt in central neurons (70). However, it is unlikely that a similar mechanism played role in our system because depolarization-induced [Ca2+]i elevations were not affected by NGF (Fig. 2C). Interestingly, in studies using cortical neurons that lack endogenous TrkA as a model to recapitulate TrkA-NFAT signaling by transfecting them with wild-type TrkA or signaling-deficient TrkA mutants, both the PI3K and PLC signaling pathways were found to be required for NGF/TrkA-dependent activation of NFAT (4). Collectively, our findings and work by others suggest that at least two distinct pathways, the PI3K-Akt-GSK3β and/or PLC-IP3-Ca2+ signaling cascades, can mediate neurotrophin-dependent regulation of NFAT in neurons and that their relative contribution depends on the type of neurotrophin involved, the type of neurons, and the status of neuronal activity.
GSK3β is one of the first NFAT kinases to have been identified and is known to regulate nuclear export of all of the canonical NFAT isoforms (NFATc1–c4) (3, 10, 25, 27, 71). Also, recent work has demonstrated that GSK3β suppresses NFAT-mediated gene expression in neurons (3, 72). However, it remained unclear whether endogenous neuromodulators affect GSK3β-NFAT signaling. Here, we demonstrate that neuronal GSK3β-NFAT signaling is modulated by NGF, resulting in the potentiation of NFAT transcriptional activity. Notably, GSK3β knockdown resulted in a stronger enhancement of NFAT activation (Fig. 5B) and a more prominent slowing of nuclear export of NFATc3 (Fig. 7C) than did NGF treatment. This could potentially be explained by the fact that only ∼60% (or fewer) of postnatal DRG neurons express the NGF receptor TrkA (56) and thus are likely to be affected by NGF. A relatively modest but significant effect of NGF on GSK3β phosphorylation at Ser-9 (Fig. 5C), which is known to inhibit GSK3β (55, 73), is in agreement with this estimation. GSK3β is not the only NFAT kinase regulated by the PI3K-Akt signaling. For example, mammalian target of rapamycin (mTOR) kinase is known to phosphorylate and inhibit NFATc4 (74), and mTOR kinase is activated by Akt signaling (75). However, its involvement would in theory result in NGF-dependent inhibition rather than the enhancement of NFAT activity described here (Figs. 1 and 4–6) and thus is unlikely to account for the observed outcomes.
Our knockdown and knockout approaches revealed that the NFAT isoform NFATc3 is absolutely crucial for NGF-induced potentiation of NFAT-mediated gene expression in DRG neurons (Fig. 6). These findings are in good agreement with our previous molecular and functional data indicating that NFATc3 is the predominant NFAT isoform in DRG neurons (7, 33). However, given that the expression and roles of specific NFAT isoforms vary among different types of neurons (4, 8, 12, 13, 28), it is likely that other NFAT isoforms are also regulated by neurotrophins. For example, NFATc4 is targeted by BDNF in hippocampal neurons (12, 13, 28). By monitoring the nuclear translocation of EGFP-NFATc3 in DRG neurons in real time, we found that NGF and GSK3β inhibited nuclear export of NFATc3 but had no detectable effect on the rate of nuclear import of NFATc3 (Figs. 7 and 8). The latter is consistent with our previous finding that the rate of depolarization-induced nuclear import of NFATc3 is not affected by GSK3β knockdown in DRG neurons (33). Our data also further support the view that GSK3β is the major NFAT export kinase in neurons (3, 25, 60).
In summary, we have identified a novel mechanism of NFAT activation by NGF and suggest that neurotrophins act in concert with depolarization-driven Ca2+ signaling to regulate NFATc3-dependent gene expression in sensory neurons. Although the functional significance of this mechanism remains to be determined, the new model proposed in this study (Fig. 10) will guide further research directed at better understanding of how electrical activity and neurotrophic factors cooperate in long term regulation of neuronal excitability and synaptic function and how they orchestrate the wiring of neuronal networks.
Acknowledgments
We thank Dr. Fernando Santana for providing NFATc3 KO mice; Dr. Michal Hetman for the NFATc3-shRNA, NFATc4-shRNA, and GSK3β-shRNA plasmids; and Dr. Steven Green for the caPI3K plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grants NS072432 and NS087068. This work was also supported by the Fraternal Order of Eagles Diabetes Research Center.
- NFAT
- nuclear factor of activated T-cells
- ANOVA
- analysis of variance
- BDNF
- brain-derived neurotrophic factor
- CaN
- calcineurin
- DRG
- dorsal root ganglion/ganglia
- EGFP
- enhanced GFP
- GSK3β
- glycogen synthase kinase 3β
- IP3
- inositol 1,4,5-trisphosphate
- mTOR
- mammalian target of rapamycin
- NFAT-luc
- NFAT luciferase reporter
- PLC
- phospholipase C
- TK-luc
- R. reniformis luciferase reporter controlled by HSV-TK promoter
- caPI3K
- constitutively active PI3Kα
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- K+10
- K+15, K+20, and K+90, 10, 15, 20, and 90 mm KCl, respectively.
REFERENCES
- 1. Deisseroth K., Mermelstein P. G., Xia H., Tsien R. W. (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol. 13, 354–365 [DOI] [PubMed] [Google Scholar]
- 2. Cohen S., Greenberg M. E. (2008) Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 24, 183–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Graef I. A., Mermelstein P. G., Stankunas K., Neilson J. R., Deisseroth K., Tsien R. W., Crabtree G. R. (1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401, 703–708 [DOI] [PubMed] [Google Scholar]
- 4. Graef I. A., Wang F., Charron F., Chen L., Neilson J., Tessier-Lavigne M., Crabtree G. R. (2003) Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113, 657–670 [DOI] [PubMed] [Google Scholar]
- 5. Oliveria S. F., Dell'Acqua M. L., Sather W. A. (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55, 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen T., Di Giovanni S. (2008) NFAT signaling in neural development and axon growth. Int. J. Dev. Neurosci. 26, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim M. S., Usachev Y. M. (2009) Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J. Neurosci. 29, 12101–12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J., Shapiro M. S. (2012) Activity-dependent Transcriptional Regulation of M-type (Kv7) K+ channels by AKAP79/150-mediated NFAT actions. Neuron 76, 1133–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benedito A. B., Lehtinen M., Massol R., Lopes U. G., Kirchhausen T., Rao A., Bonni A. (2005) The transcription factor NFAT3 mediates neuronal survival. J. Biol. Chem. 280, 2818–2825 [DOI] [PubMed] [Google Scholar]
- 10. Arron J. R., Winslow M. M., Polleri A., Chang C. P., Wu H., Gao X., Neilson J. R., Chen L., Heit J. J., Kim S. K., Yamasaki N., Miyakawa T., Francke U., Graef I. A., Crabtree G. R. (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600 [DOI] [PubMed] [Google Scholar]
- 11. Schwartz N., Schohl A., Ruthazer E. S. (2009) Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 62, 655–669 [DOI] [PubMed] [Google Scholar]
- 12. Vashishta A., Habas A., Pruunsild P., Zheng J. J., Timmusk T., Hetman M. (2009) Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J. Neurosci. 29, 15331–15340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quadrato G., Benevento M., Alber S., Jacob C., Floriddia E. M., Nguyen T., Elnaggar M. Y., Pedroarena C. M., Molkentin J. D., Di Giovanni S. (2012) Nuclear factor of activated T cells (NFATc4) is required for BDNF-dependent survival of adult-born neurons and spatial memory formation in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 109, E1499–E1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson J. G., Usachev Y. M., Thayer S. A. (2007) Bradykinin-induced nuclear factor of activated T-cells-dependent transcription in rat dorsal root ganglion neurons. Mol. Pharmacol. 72, 303–310 [DOI] [PubMed] [Google Scholar]
- 15. Groth R. D., Coicou L. G., Mermelstein P. G., Seybold V. S. (2007) Neurotrophin activation of NFAT-dependent transcription contributes to the regulation of pro-nociceptive genes. J. Neurochem. 102, 1162–1174 [DOI] [PubMed] [Google Scholar]
- 16. Cai Y. Q., Chen S. R., Pan H. L. (2013) Upregulation of nuclear factor of activated T-cells by nerve injury contributes to development of neuropathic pain. J. Pharmacol. Exp. Ther. 345, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shioda N., Han F., Moriguchi S., Fukunaga K. (2007) Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J. Neurochem. 102, 1506–1517 [DOI] [PubMed] [Google Scholar]
- 18. Abdul H. M., Sama M. A., Furman J. L., Mathis D. M., Beckett T. L., Weidner A. M., Patel E. S., Baig I., Murphy M. P., LeVine H., 3rd, Kraner S. D., Norris C. M. (2009) Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hudry E., Wu H. Y., Arbel-Ornath M., Hashimoto T., Matsouaka R., Fan Z., Spires-Jones T. L., Betensky R. A., Bacskai B. J., Hyman B. T. (2012) Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer's disease. J. Neurosci. 32, 3176–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan H. Q., Shin S. S., Ma X., Li Y., Dixon C. E. (2014) Differential effect of traumatic brain injury on the nuclear factor of activated T cells C3 and C4 isoforms in the rat hippocampus. Brain Res. 1548, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crabtree G. R., Olson E. N. (2002) NFAT signaling: choreographing the social lives of cells. Cell 109, S67–S79 [DOI] [PubMed] [Google Scholar]
- 22. Hogan P. G., Chen L., Nardone J., Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 [DOI] [PubMed] [Google Scholar]
- 23. Okamura H., Aramburu J., García-Rodríguez C., Viola J. P., Raghavan A., Tahiliani M., Zhang X., Qin J., Hogan P. G., Rao A. (2000) Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6, 539–550 [DOI] [PubMed] [Google Scholar]
- 24. Li H., Pink M. D., Murphy J. G., Stein A., Dell'Acqua M. L., Hogan P. G. (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat. Struct. Mol. Biol. 19, 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beals C. R., Sheridan C. M., Turck C. W., Gardner P., Crabtree G. R. (1997) Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 275, 1930–1934 [DOI] [PubMed] [Google Scholar]
- 26. Zhu J., McKeon F. (1999) NF-AT activation requires suppression of Crm1-dependent export by calcineurin. Nature 398, 256–260 [DOI] [PubMed] [Google Scholar]
- 27. Gwack Y., Sharma S., Nardone J., Tanasa B., Iuga A., Srikanth S., Okamura H., Bolton D., Feske S., Hogan P. G., Rao A. (2006) A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441, 646–650 [DOI] [PubMed] [Google Scholar]
- 28. Groth R. D., Mermelstein P. G. (2003) Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J. Neurosci. 23, 8125–8134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stefos G. C., Soppa U., Dierssen M., Becker W. (2013) NGF upregulates the plasminogen activation inhibitor-1 in neurons via the calcineurin/NFAT pathway and the Down syndrome-related proteins DYRK1A and RCAN1 attenuate this effect. PLoS One 8, e67470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schnizler K., Shutov L. P., Van Kanegan M. J., Merrill M. A., Nichols B., McKnight G. S., Strack S., Hell J. W., Usachev Y. M. (2008) Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J. Neurosci. 28, 4904–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rossow C. F., Minami E., Chase E. G., Murry C. E., Santana L. F. (2004) NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ. Res. 94, 1340–1350 [DOI] [PubMed] [Google Scholar]
- 32. Nieves-Cintrón M., Amberg G. C., Nichols C. B., Molkentin J. D., Santana L. F. (2007) Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J. Biol. Chem. 282, 3231–3240 [DOI] [PubMed] [Google Scholar]
- 33. Ulrich J. D., Kim M. S., Houlihan P. R., Shutov L. P., Mohapatra D. P., Strack S., Usachev Y. M. (2012) Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J. Biol. Chem. 287, 37594–37609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomida T., Hirose K., Takizawa A., Shibasaki F., Iino M. (2003) NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 22, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu Q., Klippel A., Muslin A. J., Fantl W. J., Williams L. T. (1995) Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 268, 100–102 [DOI] [PubMed] [Google Scholar]
- 36. Hur E. M., Zhou F. Q. (2010) GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shuttleworth T. J., Thompson J. L. (1991) Effect of temperature on receptor-activated changes in [Ca2+]i and their determination using fluorescent probes. J. Biol. Chem. 266, 1410–1414 [PubMed] [Google Scholar]
- 38. Thayer S. A., Miller R. J. (1990) Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J. Physiol. 425, 85–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thayer S. A., Perney T. M., Miller R. J. (1988) Regulation of calcium homeostasis in sensory neurons by bradykinin. J. Neurosci. 8, 4089–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svichar N., Shmigol A., Verkhratsky A., Kostyuk P. (1997) ATP induces Ca2+ release from IP3-sensitive Ca2+ stores exclusively in large DRG neurones. Neuroreport 8, 1555–1559 [DOI] [PubMed] [Google Scholar]
- 41. Usachev Y. M., DeMarco S. J., Campbell C., Strehler E. E., Thayer S. A. (2002) Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron 33, 113–122 [DOI] [PubMed] [Google Scholar]
- 42. Verkhratsky A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279 [DOI] [PubMed] [Google Scholar]
- 43. Patapoutian A., Reichardt L. F. (2001) Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol. 11, 272–280 [DOI] [PubMed] [Google Scholar]
- 44. Chao M. V. (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309 [DOI] [PubMed] [Google Scholar]
- 45. Stephens R. M., Loeb D. M., Copeland T. D., Pawson T., Greene L. A., Kaplan D. R. (1994) Trk receptors use redundant signal transduction pathways involving SHC and PLC-γ1 to mediate NGF responses. Neuron 12, 691–705 [DOI] [PubMed] [Google Scholar]
- 46. Marsh H. N., Dubreuil C. I., Quevedo C., Lee A., Majdan M., Walsh G. S., Hausdorff S., Said F. A., Zoueva O., Kozlowski M., Siminovitch K., Neel B. G., Miller F. D., Kaplan D. R. (2003) SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. J. Cell Biol. 163, 999–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wood E. R., Kuyper L., Petrov K. G., Hunter R. N., 3rd, Harris P. A., Lackey K. (2004) Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg. Med. Chem. Lett. 14, 953–957 [DOI] [PubMed] [Google Scholar]
- 48. Martin K. J., Shpiro N., Traynor R., Elliott M., Arthur J. S. (2011) Comparison of the specificity of Trk inhibitors in recombinant and neuronal assays. Neuropharmacology 61, 148–155 [DOI] [PubMed] [Google Scholar]
- 49. Knüsel B., Hefti F. (1992) K-252 compounds: modulators of neurotrophin signal transduction. J. Neurochem. 59, 1987–1996 [DOI] [PubMed] [Google Scholar]
- 50. Watson J. J., Allen S. J., Dawbarn D. (2008) Targeting nerve growth factor in pain: what is the therapeutic potential? BioDrugs 22, 349–359 [DOI] [PubMed] [Google Scholar]
- 51. Eibl J. K., Strasser B. C., Ross G. M. (2012) Structural, biological, and pharmacological strategies for the inhibition of nerve growth factor. Neurochem. Int. 61, 1266–1275 [DOI] [PubMed] [Google Scholar]
- 52. Zhou F. Q., Zhou J., Dedhar S., Wu Y. H., Snider W. D. (2004) NGF-induced axon growth is mediated by localized inactivation of GSK-3β and functions of the microtubule plus end binding protein APC. Neuron 42, 897–912 [DOI] [PubMed] [Google Scholar]
- 53. Jones D. M., Tucker B. A., Rahimtula M., Mearow K. M. (2003) The synergistic effects of NGF and IGF-1 on neurite growth in adult sensory neurons: convergence on the PI 3-kinase signaling pathway. J. Neurochem. 86, 1116–1128 [DOI] [PubMed] [Google Scholar]
- 54. Kau T. R., Schroeder F., Ramaswamy S., Wojciechowski C. L., Zhao J. J., Roberts T. M., Clardy J., Sellers W. R., Silver P. A. (2003) A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 4, 463–476 [DOI] [PubMed] [Google Scholar]
- 55. Stambolic V., Woodgett J. R. (1994) Mitogen inactivation of glycogen synthase kinase-3β in intact cells via serine 9 phosphorylation. Biochem. J. 303, 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Molliver D. C., Wright D. E., Leitner M. L., Parsadanian A. S., Doster K., Wen D., Yan Q., Snider W. D. (1997) Ib4-binding Drg neurons switch from Ngf to Gdnf dependence in early postnatal life. Neuron 19, 849–861 [DOI] [PubMed] [Google Scholar]
- 57. Meyer R. A., Ringhamp M., Campbell J. N., Raja S. N. (2006) in Textbook of Pain (McMahon S. B., Koltzenburg M., eds) pp 3–34, Elsevier, Amsterdam [Google Scholar]
- 58. Wilkins B. J., De Windt L. J., Bueno O. F., Braz J. C., Glascock B. J., Kimball T. F., Molkentin J. D. (2002) Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol. Cell Biol. 22, 7603–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. West A. E., Griffith E. C., Greenberg M. E. (2002) Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 3, 921–931 [DOI] [PubMed] [Google Scholar]
- 60. Müller M. R., Rao A. (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10, 645–656 [DOI] [PubMed] [Google Scholar]
- 61. Ozaki Y., Kitamura N., Tsutsumi A., Dayanithi G., Shibuya I. (2009) NGF-induced hyperexcitability causes spontaneous fluctuations of intracellular Ca2+ in rat nociceptive dorsal root ganglion neurons. Cell Calcium 45, 209–215 [DOI] [PubMed] [Google Scholar]
- 62. Usachev Y. M., Thayer S. A. (1999) Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J. Physiol. 519, 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kress M., Distler C. (2004) Differences in calcium signalling in rat peripheral sensory neurons. Neurosci. Lett. 354, 127–130 [DOI] [PubMed] [Google Scholar]
- 64. Linhart O., Obreja O., Kress M. (2003) The inflammatory mediators serotonin, prostaglandin E2 and bradykinin evoke calcium influx in rat sensory neurons. Neuroscience 118, 69–74 [DOI] [PubMed] [Google Scholar]
- 65. Stucky C. L., Thayer S. A., Seybold V. S. (1996) Prostaglandin E2 increases the proportion of neonatal rat dorsal root ganglion neurons that respond to bradykinin. Neuroscience 74, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 66. Khasabova I. A., Simone D. A., Seybold V. S. (2002) Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience 115, 613–625 [DOI] [PubMed] [Google Scholar]
- 67. Khasabova I. A., Harding-Rose C., Simone D. A., Seybold V. S. (2004) Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J. Neurosci. 24, 1744–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Oh S. B., Tran P. B., Gillard S. E., Hurley R. W., Hammond D. L., Miller R. J. (2001) Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 21, 5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Malin S. A., Molliver D. C., Koerber H. R., Cornuet P., Frye R., Albers K. M., Davis B. M. (2006) Glial cell line-derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. J. Neurosci. 26, 8588–8599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blair L. A., Bence-Hanulec K. K., Mehta S., Franke T., Kaplan D., Marshall J. (1999) Akt-dependent potentiation of L channels by insulin-like growth factor-1 is required for neuronal survival. J. Neurosci. 19, 1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van der Velden J. L., Schols A. M., Willems J., Kelders M. C., Langen R. C. (2008) Glycogen synthase kinase 3 suppresses myogenic differentiation through negative regulation of NFATc3. J. Biol. Chem. 283, 358–366 [DOI] [PubMed] [Google Scholar]
- 72. Gómez-Sintes R., Lucas J. J. (2010) NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J. Clin. Invest. 120, 2432–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fang X., Yu S. X., Lu Y., Bast R. C., Jr., Woodgett J. R., Mills G. B. (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 97, 11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang T. T., Yu R. Y., Agadir A., Gao G. J., Campos-Gonzalez R., Tournier C., Chow C. W. (2008) Integration of protein kinases mTOR and extracellular signal-regulated kinase 5 in regulating nucleocytoplasmic localization of NFATc4. Mol. Cell Biol. 28, 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sugden P. H., Fuller S. J., Weiss S. C., Clerk A. (2008) Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 153, S137–S153 [DOI] [PMC free article] [PubMed] [Google Scholar]