

Background: The ABCG2 transporter is an ATP-dependent efflux pump that contributes to multidrug resistance.
Results: Curcumin in combination with gramicidin or ouabain reduces intracellular ATP levels in ABCG2-expressing cells and selectively kills these cells over parental cells.
Conclusion: ABCG2-expressing cells display collateral sensitivity toward these combinations of compounds.
Significance: Understanding ABCG2-mediated collateral sensitivity is helpful in finding ways to combat multidrug resistance.
Keywords: ABC Transporter; ATPase; Drug Resistance; Drug Transport; Na+,K+-ATPase; ABCG2 Transporter; Collateral Sensitivity; Curcumin; Gramicidin A; Ouabain
Abstract
This paper introduces a strategy to kill selectively multidrug-resistant cells that express the ABCG2 transporter (also called breast cancer resistance protein, or BCRP). The approach is based on specific stimulation of ATP hydrolysis by ABCG2 transporters with subtoxic doses of curcumin combined with stimulation of ATP hydrolysis by Na+,K+-ATPase with subtoxic doses of gramicidin A or ouabain. After 72 h of incubation with the drug combinations, the resulting overconsumption of ATP by both pathways inhibits the efflux activity of ABCG2 transporters, leads to depletion of intracellular ATP levels below the viability threshold, and kills resistant cells selectively over cells that lack ABCG2 transporters. This strategy, which was also tested on a clinically relevant human breast adenocarcinoma cell line (MCF-7/FLV1), exploits the overexpression of ABCG2 transporters and induces caspase-dependent apoptotic cell death selectively in resistant cells. This work thus introduces a novel strategy to exploit collateral sensitivity (CS) with a combination of two clinically used compounds that individually do not exert CS. Collectively, this work expands the current knowledge on ABCG2-mediated CS and provides a potential strategy for discovery of CS drugs against drug-resistant cancer cells.
Introduction
This paper reports a collateral sensitivity (CS)3 strategy (1) to kill selectively resistant cells that overexpress the ABCG2 transporter. This transporter is an ATP-binding cassette (ABC) protein (2) that is located in the plasma membrane of cells in the blood-brain barrier, intestines, and placenta (2–6). Expression of ABCG2 transporters is a marker for stem cells (7) and is being discussed as a functional marker of cancer stem cells (8). ABCG2 transporters, together with multidrug resistance protein 1 (MDR1/ABCB1, also called P-glycoprotein (P-gp)) and multidrug resistance-associated protein 1 (MRP1/ABCC1), prevent accumulation of xenobiotics and steroids in the human body (3, 6). This efflux activity is also believed to render stem cells resistant to drugs and oxidative stress, as well as maintain stem cells in an undifferentiated state (7, 8). Overexpression of efflux pumps in malignant cells can, however, lead to a multidrug resistance (MDR) phenotype that results in the failure of cancer chemotherapy (4, 9). This efflux-induced drug resistance has motivated attempts to circumvent ABC transporter-mediated MDR in cancer cells. Inhibitors of MDR transporters, however, have had limited success in clinical trials because of excessive systemic side effects when administered in combination with chemotherapeutic drugs (10–12).
One alternative strategy to address MDR is the so-called ATP depletion strategy that takes advantage of increased metabolic needs of cancer cells (13, 14). This approach kills tumor cells by inhibiting ATP synthesis, which leads to apoptosis and necrosis (15) in fast growing cells (16). All previously explored approaches have in common that they achieved ATP depletion by inhibiting ATP synthesis. Despite the potential of this strategy for cancer therapy, it appears difficult to inhibit the energy metabolism of tumor cells selectively because host cells are dependent on the same ATP generating pathways (13).
An emerging strategy to address MDR is to exploit mechanisms of drug resistance to target these resistant cells (17). Several research groups reported that resistant cells were more sensitive to certain compounds than their parental cells (12, 18–20). This little known and mechanistically underexplored effect is called CS or hypersensitivity (recently reviewed by Pluchino et al. (18) and Szakács et al. (1)). Collateral sensitivity has been reported in cells expressing ABCB1 (P-gp) (12) and ABCC1 (MRP1) (21) transporters. For instance, Gatenby and co-workers (12, 20) recently presented a compelling potential approach to cancer therapy by combined administration of low doses of verapamil and low doses of 2-deoxyglucose to suppress resistant P-gp-expressing cells by adaptive administration of chemotherapy with the goal of keeping tumor burden constant in disseminated cancers that are typically fatal when treated with conventional chemotherapy regimen. With regard to cells expressing ABCG2 (BCRP) transporters, we found only two previous reports on CS; in both cases, a single compound induced CS (22, 23).
Here, we introduce an approach that exploits the MDR phenotype to achieve targeted ATP depletion in ABCG2-expressing cells by variation of a strategy described by Karwatsky et al. (24) and Silva et al. (12) in cells overexpressing P-gp: rather than inhibiting ATP synthesis, we selectively stimulate ATP hydrolysis by ABCG2 transporters and thereby induce a lethal reduction of ATP levels in MDR cells but not in parental cells.
We accomplished this selective stimulation of ATP hydrolysis in ABCG2-expressing cells by treatment with a combination of subtoxic concentration of curcumin with either gramicidin A (gA) or ouabain. Curcumin, the bioactive compound in the South Asian spice turmeric, is an effective chemosensitizer that modulates the function of ABCB1, ABCC1, and ABCG2 transporters presumably without being transported by these efflux pumps (6, 25). Instead, curcumin inhibits drug efflux and increases the efficacy of many anticancer agents in multidrug-resistant cancers (25, 26). Curcumin also stimulates ATP hydrolysis by these transporters, and we exploited this activity to increase consumption of ATP in ABCG2-expressing cells. To kill ABCG2 cells selectively over parental cells, we amplified the ATP depletion effect of curcumin with a second ATP-depleting process, the activation of the Na+,K+-ATPase. To this end, we treated cells with subtoxic (micromolar) concentrations of curcumin in combination with subtoxic (nanomolar) concentrations of gA (27) or ouabain (28, 29). Gramicidin A is a pore-forming peptide that disrupts the homeostasis of ion gradients across lipid membranes and the resulting change in transmembrane potential activates the Na+,K+-ATPase (30, 31). Ouabain is a cardiac glycoside that inhibits the Na+,K+-ATPase at micromolar concentrations (29), whereas it stimulates the Na+,K+-ATPase activity at nanomolar concentrations (28). Stimulating these two ATP-depleting processes together lowered the intracellular ATP levels in ABCG2-expressing cells sufficiently to kill them selectively over parental cells.
MATERIALS AND METHODS
Chemicals
We purchased Eagle's minimal essential medium, DMEM, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell cytotoxicity assay kits from ATCC (Manassas, VA). We obtained FBS, OPTIMEM reduced serum medium, improved minimum essential medium, Dulbecco's PBS, 0.05% (w/v) trypsin-EDTA, penicillin, streptomycin, BODIPY-FL-prazosin, annexin V-FITC and ethidium homodimer I from Invitrogen. We purchased Aprotinin from Roche Diagnostics. All other chemicals were purchased from Sigma-Aldrich.
Cell Lines and Culture Conditions
HEK-293 cells transfected with the empty pcDNA3.1 vector (HEK-293 parental cells) or pcDNA3.1 vector containing the ABCG2 gene (HEK-293 ABCG2 cells) were maintained in Eagle's minimal essential medium supplemented with 10% (v/v) FBS, penicillin (100 units/ml), streptomycin (100 μg/ml), and geneticin disulfate (G418) at a concentration of 2 mg/ml (32). The MCF-7 human breast cancer cell line was exposed to incrementally increasing concentrations of flavopiridol. The resulting resistant subline, MCF-7/FLV1, overexpressed ABCG2, whereas neither P-gp nor MRP overexpression was detected (33). Control MCF7 and MCF7/FLV1 (482R) cells overexpressing ABCG2 were cultured in RPMI with 10% (v/v) FBS in the absence or presence of 1 μg/ml flavopiridol, respectively (33).
Cytotoxicity Assay
We seeded 5,000 cells/well in 96-well plates and cultured overnight. After adding various concentrations of compounds and incubating for an additional 72 h, we replaced the medium with OPTIMEM and determined the cell viability using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell cytotoxicity assay kit, according to the manufacturer's instructions. We calculated the cell viability using the following formula: Cell survival in % = (mean absorbance in test well)/(mean absorbance in untreated well) × 100. We calculated IC50 values with their errors from best curve fits of mean values of viability as a function of the drug concentration to the following equation y = A2 + (A1 − A2)/(1 + (x/x0)p̂) using Origin software version 7.5. Fluorescence-based life versus death staining of HEK-293 cells and HEK-ABCG2 cells after 72 h of incubation with the specified compound/s was performed with calcein (green, representing live cells) and ethidium bromide (red, representing dead cells).
Flow Cytometry Analysis of Efflux Activity by ABCG2
We trypsinized cells in phenol red-free improved minimum essential medium for 30 min at 37 °C in the absence or presence of 250 nm BODIPY-prazosin alone or with various concentrations of compounds. We washed the cells twice in ice-cold PBS and incubated for an additional hour at 37 °C before washing the cells and analyzing them by flow cytometry (Beckman Coulter Quanta SC). We obtained the histograms and mean fluorescence intensity (MFI) for each condition using WinMD software. We defined 100% efflux as the difference in MFI values between the most frequently observed fluorescence value in the presence of 250 nm BODIPY-prazosin combined with 10 μm fumitremorgin C (FTC), a ABCG2 inhibitor, and that in the presence of 250 nm BODIPY-prazosin without FTC. This difference therefore quantified FTC-inhibitable efflux of BODIPY-prazosin. We calculated the BODIPY-prazosin efflux (as percentages) for each condition using the following formula: (FTC-inhibitable BODIPY-prazosin efflux in the presence of compound(s))/(FTC-inhibitable BODIPY-prazosin efflux in the absence of compound(s)) × 100%.
Isolation of Crude Membranes from ABCG2-expressing HEK-293 Cells
We prepared crude membranes from HEK-ABCG2 cells as described previously (34) with some modifications. Briefly, we scraped cells from their culture dishes into ice-cold PBS containing 1% (w/v) aprotinin and centrifuged at 500 × g for 10 min to collect the pellet. We resuspended these cells in a lysis buffer containing 10 mm Tris-HCl (pH 7.5), 10 mm NaCl, 1 mm MgCl2, and 1% (w/v) aprotinin and incubated on ice for 45 min. We disrupted cells using a Dounce homogenizer (30 strokes with pestle A and B). We removed undisrupted cells and nuclear debris by centrifugation at 500 × g for 10 min. We diluted the supernatant 2-fold in resuspension buffer containing 10 mm Tris-HCl (pH 7.5), 50 mm NaCl, 250 mm sucrose, and 1% (w/v) aprotinin. Finally, we collected the membrane vesicles by centrifugation at 100,000 × g for 60 min and resuspended in resuspension buffer. We stored the membranes in small aliquots at −70 °C. Using the Amido Black protein method described by Schaffner and Weissmann (35) with BSA as a standard, we determined the protein content of each preparation.
ATPase Assay
We used crude membranes from ABCG2-expressing HEK-293 cells (100 μg protein/ml) and incubated them with varying concentrations of compounds in the presence and absence of 300 μm vanadate in ATPase assay buffer for 10 min at 37 °C. This ATPase assay buffer contained 50 μm KCl, 5 mm NaN3, 2 mm EGTA, 10 mm MgCl2, 1 mm DTT, 2 mm ouabain, and 50 mm Tris-HCl (pH 7.5). We started the reaction by adding 5 mm ATP and incubated for 20 min at 37 °C. We terminated the reaction with the addition of SDS solution (0.1 ml of 5% (w/v) SDS) and quantified the amount of inorganic phosphate released by a sensitive colorimetric reaction as described previously (34). We recorded the specific activity of the transporter as vanadate-sensitive ATPase activity (34).
We determined the Na+,K+-ATPase activity in crude membrane vesicles from ABCG2-expressing cells treated with the ABCG2 inhibitor FTC at a concentration of 10 μm to completely inhibit the ATPase activity of the ABCG2 transporter. We determined total ATP hydrolysis in crude membrane vesicles from ABCG2 cells in the absence of the Na+,K+-ATPase inhibitor ouabain and in the absence of FTC.
Determination of Cellular ATP Levels
We determined cellular ATP levels with a luciferase assay (36) using a bioluminescence assay kit from Sigma-Aldrich. We seeded 5 × 105 cells/well in 96-well plates and cultured overnight. We incubated the cells for 1 h at 37 °C with or without drug treatments. We lysed the cells with lysis buffer (150 mm NaCl, 50 mm Tris, pH 7.5, 1% (v/v) Triton X-100, 0.1% w/v SDS, and 1% w/v deoxycholate) and mixed 100 μl (2.5 × 105 cells) of cell suspension with an equal volume of luciferase solution in ATP assay dilution buffer as indicated by the manufacturer. We mixed the plate for 3–5 s and immediately placed the plate in the luminometer (Fluoroskan Ascent FL). We compared the observed light intensity from the cell samples to those from several ATP standards to determine the ATP concentration of each sample.
Determination of Membrane Potentials by Whole Cell Current Clamp Recordings
We plated cells on poly-l-lysine-coated 35-mm glass-bottomed Petri dishes (MatTek, Ashland, MA) before performing whole cell recordings in current clamp mode. The standard pipette solution consisted of 130 mm KCl, 1 mm MgCl2, 10 mm HEPES, 5 mm ATP, 0.2 mm GTP, and 0.5 mm EGTA (pH 7.2). The bath solution consisted of 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm HEPES, and 5 mm glucose (pH 7.4). We fabricated the patch electrodes with resistances of 2.0–4.0 MΩ from borosilicate glass (Sutter Instruments, Novato, CA) using a P-87 puller (Sutter Instruments). We recorded the membrane potentials using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) and digitized using a Digidata 1322A (Molecular Devices). Data acquisition was done using pCalmp9 software (Molecular Devices).
Apoptosis/Necrosis Assay
We trypsinized and harvested cells that were incubated in the absence or presence of 35 nm gA, a combination of 35 nm gA and 2 μm curcumin, 7.5 nm ouabain, and a combination of 7.5 nm ouabain and 2 μm curcumin for 24, 48, and 72 h. We washed cells with PBS and binding buffer (140 mm NaCl, 5 mm KCl, 10 mm HEPES, and 2.5 mm CaCl2 with pH 7.4) before incubation with annexin V-FITC and ethidium homodimer I (EthD I) for 15 min at room temperature in the dark. Then we determined the percentage of early apoptotic and late apoptotic or necrotic cells by flow cytometry. To inhibit the activity of caspase-3/7, we incubated cells in the presence of 50 μm Z-VAD-FMK, a well known caspase inhibitor, for 2 h prior to the addition of other compounds.
Caspase-3/7 Activity Assay
We trypsinized and harvested cells that were incubated for 72 h in the absence or presence of 35 nm gA, a combination of 35 nm gA and 2 μm curcumin, 7.5 nm ouabain, and a combination of 7.5 nm ouabain and 2 μm curcumin. We washed cells with 0.1% (w/v) BSA-PBS and stained cells with CellEvent caspase-3/7 green detection reagent (Invitrogen) for 30 min at 37 °C in the dark. Then we determined the population of caspase-3/7-activated cells by flow cytometry. To distinguish early apoptotic cells from late apoptotic or necrotic cells, we labeled cells with both CellEvent caspase-3/7 green detection reagent and SYTOX AADvanced dead cell stain (Invitrogen) as instructed by the manufacturer. We subsequently determined the percentages of early apoptotic and late apoptotic or necrotic cells by flow cytometry.
Statistical Analysis
Unless indicated otherwise, we repeated experiments at least three times and performed each measurement in triplicate. In bar graphs, each bar represents a mean value ± standard error of the mean from at least two (typically three or more) separate experiments. We determined the statistical significance of differences by a two-tailed Student's t test, and a p value < 0.05 was considered significant. A single asterisk indicates a p value ≤ 0.05, whereas a double asterisk indicates a p value ≤ 0.01.
RESULTS AND DISCUSSION
A Combination of Curcumin with Either gA or Ouabain Selectively Kills HEK-ABCG2 Cells
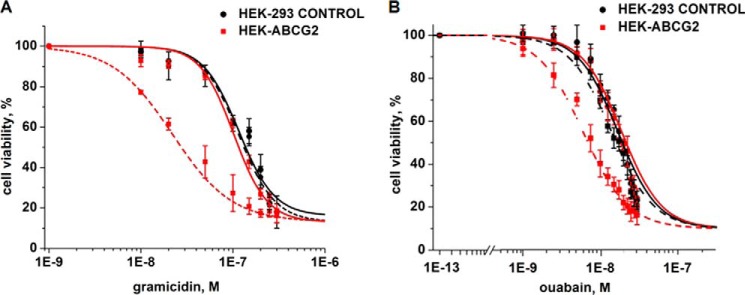
Fig. 1 shows that subtoxic concentrations (defined here as concentrations that killed ≤10% of cells, LD10) of curcumin (2 μm) in combination with subtoxic concentrations of gA (35 nm) or ouabain (7.5 nm) were able to kill most of the cells with the MDR phenotype (HEK-293 ABCG2), whereas almost all parental cells (HEK-293 control) remained viable. We observed this selective cell death only in the presence of the combination of compounds (Fig. 1 and Table 1). Curcumin, gA, or ouabain by themselves were slightly less toxic to HEK-ABCG2 cells than to the parental cells (Fig. 1 and Table 1). These results suggest that the MDR phenotype turned from an advantage that incurred a slight resistance toward these compounds individually to a disadvantage that incurred sensitivity toward combined exposure to curcumin with either gA or ouabain. These results are therefore on the one hand similar to those reported as CS (reviewed by Pluchino et al. (18) and Szakács et al. (1)), in that resistant cells are more sensitive to certain chemicals than the parental cells. On the other hand, these results are different from previous cases of CS, in that the resistant cells are actually less sensitive against each compound individually, whereas the combination of curcumin with either gA or ouabain evokes CS.
FIGURE 1.

A combination of curcumin with either gA or ouabain selectively kills cells with ABCG2-induced MDR phenotype. A, sensitivity of parental (HEK-293 control) cells (black) and HEK-ABCG2 cells (red) to gA in the absence (solid curves) or presence (dashed curves) of 2 μm curcumin. B, sensitivity of HEK-293 control (black) and HEK-ABCG2 cells (red) to ouabain in the absence (solid curves) or presence (dashed curves) of 2 μm curcumin. C and D, fluorescence-based life versus death assay of HEK-293 control (top panels) and HEK-ABCG2 cells (bottom panels) after 72 h of incubation with the specified compound(s).
TABLE 1.
IC50 values, relative resistance, and reversal of resistance of HEK-293 parental cells and HEK-ABCG2 cells towards curcumin, gA, ouabain, and mitoxantrone
| Compound | IC50 |
p value of IC50 from HEK-ABCG2 cells relative to IC50 from control cellsa | Relative resistanceb | Sensitization/reversal of resistancec | |
|---|---|---|---|---|---|
| HEK-293 control | HEK-ABCG2 | ||||
| Curcumin aloned | 5.0 ± 0.6 μm | 6.0 ± 0.9 μm | 0.2 | 1.2 ± 0.2-fold | |
| Curcumin + 10 μm FTC | 5.1 ± 0.6 μm | 6.3 ± 0.7 μm | 0.1 | 1.2 ± 0.2-fold | 1.0 ± 0.2-fold |
| Gramicidin alone | 100 ± 22 nm | 138 ± 16 nm | 0.1 | 1.4 ± 0.3-fold | |
| Gramicidin + 2 μm curcumin | 96 ± 10 nm | 26 ± 4 nm | 1E-3 | 0.3 ± 0.1-fold | 5.0 ± 1.5-fold |
| Gramicidin + 2 μm curcumin + 10 μm FTC | 102 ± 16 nm | 96 ± 15 nm | 0.7 | 1.0 ± 0.2-fold | 1.4 ± 0.5-fold |
| Ouabain alone | 14 ± 2 nm | 14 ± 3 nm | 1.0 | 1.0 ± 0.3-fold | |
| Ouabain + 2 μm curcumin | 14 ± 2 nm | 6 ± 1 nm | 1E-2 | 0.4 ± 0.1-fold | 2.5 ± 0.9-fold |
| Ouabain + 2 μm curcumin + 10 μm FTC | 15 ± 3 nm | 16 ± 3 nm | 0.7 | 1.1 ± 0.3-fold | 1.0 ± 0.2-fold |
| Mitoxantrone alone | 5.6 ± 0.8 nm | 206 ± 12 nm | 1E-4 | 37 ± 5.6-fold | |
| Mitoxantrone + 2 μm curcimun | 5.1 ± 0.9 nm | 32 ± 3 nm | 4E-3 | 6.2 ± 1.2-fold | 6.0 ± 2-fold |
| Mitoxantrone + 35 nm gramicidin | 5.2 ± 0.6 nm | 170 ± 4 nm | 1E-4 | 33 ± 4.6-fold | 1.1 ± 0.2-fold |
| Mitoxantrone + 7.5 nm ouabain | 5.3 ± 0.8 nm | 187 ± 7 nm | 1E-4 | 35 ± 5.5-fold | 1.0 ± 0.2-fold |
a To determine p values, we used a two-tailed Student's t test based on log(IC50) values. We considered p values ≤ 0.05 as statistically significant.
b Expressed as the ratio between the IC50 value of ABCG2-expressing cells and the IC50 value of control cells. This ratio is the inverse of the selectivity ratio, which is defined as IC50(parental cells)/IC50(MDR cells) (18).
c Expressed as the ratio between the relative resistance value of ABCG2-expressing cells in the absence of the second compound and the relative resistance in its presence. In the case of experiments with gA or ouabain, ABCG2-expressing cells are more sensitive to the combined treatment with curcumin than the parental cells (i.e. sensitization). In the case of experiments with mitoxantrone, the ABCG2-expressing cells are less resistant to combined treatment with curcumin or gA (i.e. reversal of resistance).
d Chearwae et al. (26) reported previously that curcumin is presumably not a substrate of ABCG2 transporters. Based on the slight curcumin resistance of ABCG2-expressing HEK cells compared to the parental HEK cells, it appears, however, also possible that the high permeability of curcumin through membranes makes it difficult to detect whether it is transported by ABCG2 transporters or not. In that sense, curcumin may actually be a substrate of ABCG2 transporters, but because of its fast passive back diffusion through membranes, it may appear not to be a substrate. We note that in general, net transport observed in transport assays is the sum of passive and active transport processes.
Table 1 shows that in the presence of curcumin, resistant cells died at four times lower gA and two times lower ouabain concentrations than parental cells. Based on these results, we tested whether curcumin, gA, or ouabain would have a similar sensitization effect in ABCG2-expressing cells when administered at subtoxic concentrations together with mitoxantrone, a well known substrate of ABCG2 transporters (5). Table 1 shows that in the presence of curcumin, HEK-ABCG2 cells were still more resistant toward mitoxantrone than parental cells. Although curcumin reduced (i.e. reversed) the resistance of these cells toward mitoxantrone significantly, it could not cause CS as we observed in Fig. 1 with the combination of curcumin with either gA or ouabain. The presence of subtoxic concentrations of gA or ouabain had only a small effect on the resistance of ABCG2 cells toward mitoxantrone. Therefore, the specific toxicity of a combination of the ABCG2 modulator curcumin with gA or ouabain toward HEK-ABCG2 cells could not be replicated with a combination of the well known ABCG2 transporter substrate mitoxantrone.
Inhibiting the Function of ABCG2 Transporters Reverses Cytotoxicity of Curcumin in Combination with gA or Ouabain toward HEK-ABCG2 Cells
To determine whether the selective cell death of HEK-ABCG2 cells by treatment with curcumin and gA or curcumin with ouabain was dependent on the activity of the ABCG2 transporter, we incubated cells with subtoxic doses of curcumin in combination with gA in the presence or absence of 10 μm FTC, a specific inhibitor of ATP hydrolysis and thus of active transport by the ABCG2 transporter (37). We found that in the presence of FTC, the selective cytotoxicity of curcumin and gA or curcumin and ouabain toward HEK-ABCG2 cells was lost, and the viability was restored to that of parental cells (Fig. 2). This result indicates that active ABCG2 transporters were indeed responsible for selective killing of HEK-ABCG2 cells in the presence of curcumin with either gA or ouabain.
FIGURE 2.
Reversal of selective cytotoxicity by curcumin in combination with either gA or ouabain in HEK-ABCG2 cells in the presence of FTC. Cell viability of HEK-293 control cells (black) and HEK-ABCG2 cells (red) exposed to increasing doses of gA (A) or ouabain (B) in the presence of 2 μm curcumin and in the absence (dashed curves) or presence (solid curves) of 10 μm FTC.
A Combination of Curcumin with Either gA or Ouabain Inhibits Efflux by ABCG2
To determine whether subtoxic concentrations of curcumin in combination with either gA or ouabain would affect the efflux activity of ABCG2 transporters, we used flow cytometry to quantify the efflux of the fluorescent substrate BODIPY-prazosin (Bp) in HEK-ABCG2 and parental cells in the presence and absence of curcumin in combination with gA or ouabain (Fig. 3) (8). Table 2 summarizes the MFI and the percentage of efflux in HEK-ABCG2 cells under various conditions. Curcumin, gA, or ouabain individually had either no effect or a small effect on efflux of BODIPY-prazosin by ABCG2. In contrast, the combination of curcumin with gA inhibited efflux by ABCG2 completely and was as effective as the inhibition by FTC. Similarly, the combination of curcumin with ouabain reduced efflux by ∼60%. For comparison, the accumulation of BODIPY-prazosin remained unaffected under all conditions in parental cells (Fig. 3). These results demonstrate a strong effect on the efflux activity of ABCG2 transporters by the combined treatments.
FIGURE 3.
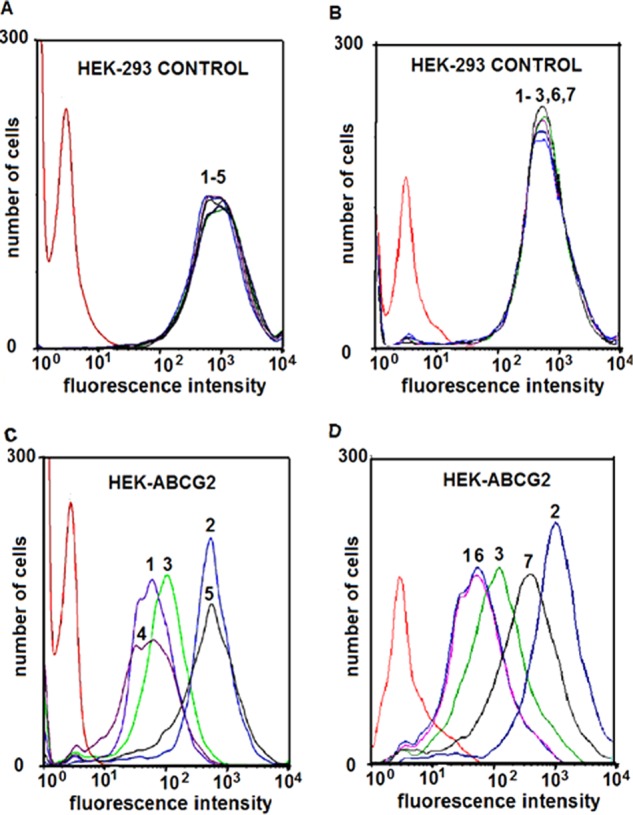
Effect of 2 μm curcumin in combination with 35 nm gA or with 7.5 nm ouabain on the accumulation of Bp in HEK-293 control and HEK-ABCG2 cells. HEK-293 control cells (A and B) and HEK-ABCG2 cells (C and D) were incubated in the absence (red curve) or presence of 250 nm Bp alone (peak 1) or in combination with 10 μm FTC (peak 2), 35 nm gA (peak 3), 2 μm curcumin (peak 4), a combination of 35 nm gA and 2 μm curcumin (peak 5), 7.5 nm ouabain (peak 6), and a combination of 7.5 nm ouabain and 2 μm curcumin (peak 7).
TABLE 2.
Mean fluorescence intensity (MFI) and percentage of efflux of Bp in HEK-ABCG2 cells treated with the specified compound(s)
| Distribution | Treatment | MFIa | Effluxb |
|---|---|---|---|
| % | |||
| 1 | 250 nm Bp (substrate) | 105 ± 66 | 100 |
| 2 | Bp + 10 μm FTC (inhibitor) | 546 ± 4 | 0 |
| 3 | Bp + 2 μm curcumin | 139 ± 60 | 92 ± 19 |
| 4 | Bp + 35 nm gA | 108 ± 71 | 99 ± 21 |
| 5 | Bp + 35 nm gA + 2 μm curcumin | 540 ± 2 | 1.3 ± 1 |
| 6 | Bp + 7.5 nm ouabain | 106 ± 42 | 100 ± 19 |
| 7 | Bp + 7.5 nm ouabain + 2 μm curcumin | 376 ± 32 | 38 ± 12 |
a Mean values of the mode (i.e. the most frequently measured fluorescence intensity) of each distribution ± standard deviation of the mean from two independent experiments.
b The difference in MFI values in the presence of 250 nm Bp alone to that in the presence of 250 nm Bp with 10 μm FTC was defined as 100% efflux activity.
A Combination of Curcumin with Either gA or Ouabain Increases ATP Turnover in HEK-ABCG2 Cells
Because simultaneous treatment with subtoxic concentrations of curcumin with gA or ouabain inhibited the transport of BODIPY-prazosin by ABCG2 transporters, we hypothesized that selective killing of ABCG2 cells after exposure to the drug combinations was not caused by transport activity (Table 2) but rather by increased ATP hydrolysis by the ABCG2 transporter and Na+,K+-ATPase. This hypothesis was based on previous observations that curcumin stimulates ATP hydrolysis by ABCG2 transporters presumably without being a substrate (26) and that nanomolar concentrations of gA and ouabain can increase Na+,K+-ATPase activity (27, 30).
To quantify ATP hydrolysis by the ABCG2 transporter, we performed experiments with crude membranes from HEK-ABCG2 cells in the presence of 2 mm ouabain (to inhibit ATP hydrolysis by the Na+,K+-ATPase) (26). Fig. 4 shows that curcumin-induced stimulation of ATP hydrolysis by ABCG2 transporters was 3.7 times higher than the basal rate of ATP hydrolysis. Gramicidin A or ouabain, in contrast, had no effect on the ATP hydrolysis rate by ABCG2 transporters. For comparison, the maximal stimulation of ATP hydrolysis by 20 μm mitoxantrone was double the basal rate. These results demonstrate that curcumin increases ATP hydrolysis by ABCG2 transporters approximately twice as strongly as mitoxantrone, whereas gA or ouabain exerts its cytotoxic effect without directly modulating ATP hydrolysis by ABCG2 transporters.
FIGURE 4.
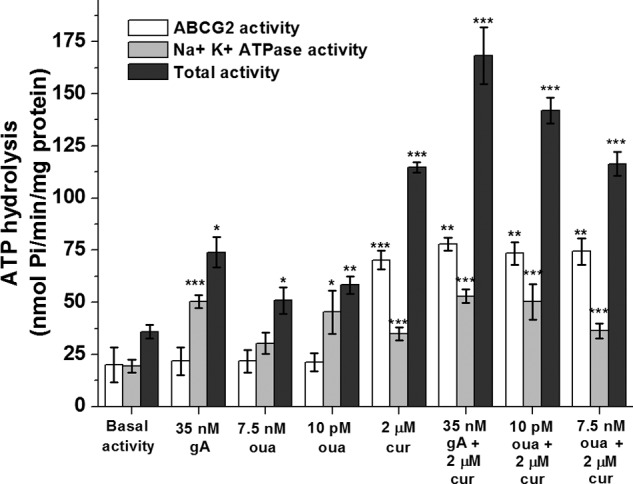
Effect of gA, curcumin (cur), ouabain (oua), and a combination of curcumin and gA, or curcumin and ouabain on ATP hydrolysis by the ABCG2 transporter, on ATP hydrolysis by the Na+,K+-ATPase, and on total ATP hydrolysis by both transporters in crude membrane vesicles from HEK-ABCG2 cells. Single, double, and triple asterisks indicate a difference compared with the basal activity with a p value ≤ 0.05, ≤ 0.01, and ≤ 0.001 respectively.
To quantify ATP hydrolysis by the Na+,K+-ATPase, we performed experiments with crude membranes from HEK-ABCG2 cells in the presence of 10 μm FTC (to inhibit all ATP hydrolysis by the ABCG2 transporter) (26). Fig. 4 shows that gA increased the rate of ATP hydrolysis by the Na+,K+-ATPase by a factor of 2.6. Because ouabain is known to have both stimulatory and inhibitory effects on the Na+,K+-ATPase (28, 29, 38, 39), we determined the effect of various concentrations of ouabain on the Na+,K+-ATPase (Fig. 5A). We found that ouabain concentrations of 1 μm and above inhibited the Na+,K+-ATPase in crude membranes from HEK-ABCG2 cells, whereas ouabain concentrations between 1 pm and 10 nm activated the Na+,K+-ATPase relative to its basal activity. A similar activation/inactivation response was reported for the ATPase activity of P-gp as a function of various verapamil concentrations (24). Maximal activation of the Na+,K+-ATPase in membrane vesicles occurred at 10 pm ouabain, leading to a 2.3 ± 0.1-fold increase in the rate of ATP hydrolysis compared with the absence of ouabain (Figs. 4 and 5A). We found that 2 μm curcumin also increased ATP hydrolysis by the Na+,K+-ATPase by a factor of 1.8 compared with basal activity (Fig. 4). The combination of gA with curcumin, however, did not exert an additive effect; ATP hydrolysis by the Na+,K+-ATPase was similar to that of gA alone.
FIGURE 5.
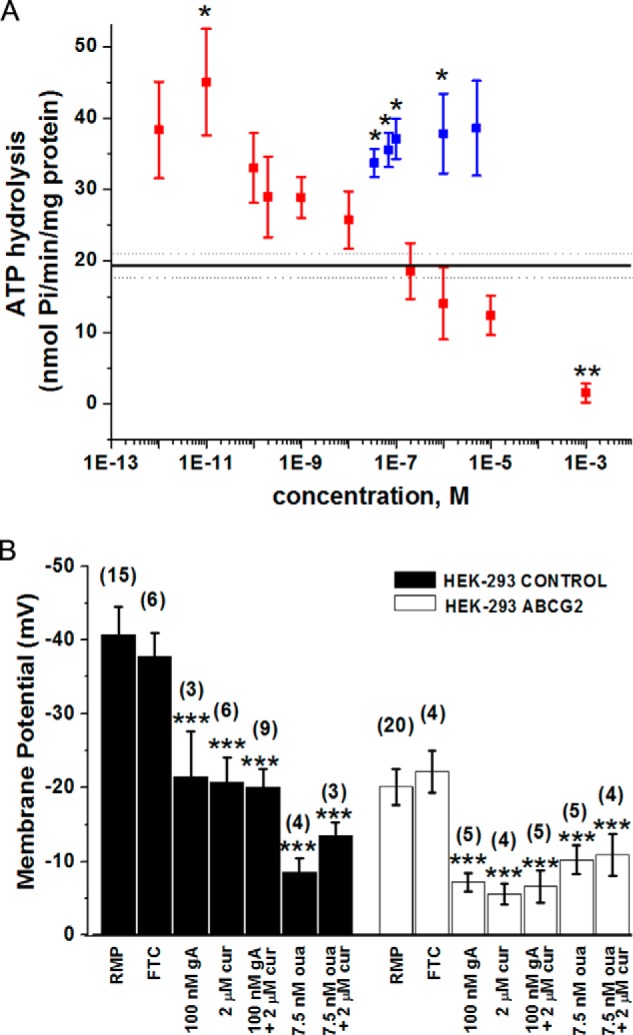
Effects of gA and ouabain on the ATP hydrolysis rate by the Na+,K+-ATPase in crude membrane fragments from HEK-ABCG2 cells and on the RMP of HEK-293 control and HEK-ABCG2 cells. A, stimulatory and inhibitory effect of various concentrations of ouabain (red) and gA (blue) on the ATP hydrolysis rate by the Na+,K+-ATPase in crude membrane fragments from HEK-ABCG2 cells. A single asterisk indicates a difference compared with the basal hydrolysis rate (black horizontal line) with a p value ≤ 0.05; a double asterisk indicates a p value ≤ 0.01. B, effect of curcumin (cur), gA, and ouabain (oua) on the RMP of HEK-293 control (black bars) and HEK-ABCG2 cells (white bars). The cells were incubated under various conditions as indicated. The bars labeled with RMP represent the control without addition of compounds. FTC was used at a concentration of 10 μm. Triple asterisks indicate a difference compared with the control (RMP) with a p value ≤ 0.001. The values in brackets indicate the numbers of measurements.
With regard to the total ATP hydrolysis rate by ABCG2 transporters and the Na+,K+-ATPase together, Fig. 4 shows that the experimentally measured total ATP hydrolysis rate induced by gA, ouabain, or curcumin by themselves was approximately the sum of the ATP hydrolysis rate by ABCG2 transporters and the Na+,K+-ATPase, as expected. In contrast, the combination of curcumin with gA or ouabain caused a synergistic effect on the rate of ATP hydrolysis, leading to 5- and 4-fold increases compared with the basal hydrolysis rate, respectively. Therefore, we hypothesized that the resulting ATP turnover of ∼140–170 nmol Pi/(min × mg protein) may be sufficiently fast to deplete intracellular ATP levels in HEK-ABCG2 cells below the viability threshold.
Gramicidin A and Ouabain Alter the Resting Membrane Potential of Cells
Gramicidin A affects energy metabolism by forming pores in cellular membranes and thereby altering their resting membrane potential (30, 31). This membrane depolarization stimulates the Na+,K+-ATPase, leading to an increased rate of ATP hydrolysis and a decrease in intracellular ATP content (30, 31). In the case of ouabain, the mechanism of activation of the Na+,K+-ATPase is not well understood. Ouabain binds to the Na+,K+-ATPase, but Oselkin et al. (40) proposed that its stimulatory effect at pico- and nanomolar concentrations may be exerted indirectly by a cascade of intracellular signaling events. To explore the effect of gA and ouabain on the resting membrane potential (RMP) of HEK-ABCG2 and parental cells, we performed whole cell current clamp experiments. Fig. 5B shows that even in the absence of curcumin, gA, or ouabain, the RMP of HEK-ABCG2 cells was approximately half that of HEK-293 parental cells. Hoffman et al. (41) have previously linked membrane depolarization to an ABC-transporter mediated phenomenon in MDR cells. Fig. 5B also shows that gA, curcumin, ouabain, or a combination of curcumin with either gA or ouabain induced further depolarization of the RMP in both cell types.
Because gA is a pore-forming peptide (27, 42), we expected a strong effect on the RMP as shown in Fig. 5B. Surprisingly, however, ouabain at concentrations in the low nanomolar range depolarized both the HEK-ABCG2 and parental cells to a similar extent as gA. In light of the result that ouabain stimulated ATP hydrolysis by the Na+,K+-ATPase (Fig. 5A), this depolarization suggests one plausible mechanism for gA- and ouabain-induced stimulation of the Na+,K+-ATPase as part of a cellular regulation response to restore the RMP. Because ouabain does not form pores in membranes (43), the mechanism for ouabain-induced depolarization remains unclear.
A Combination of Curcumin with Either gA or Ouabain Selectively Depletes Intracellular ATP Levels in HEK-ABCG2 Cells
To determine whether the increased ATP turnover may reduce the intracellular ATP level, we measured intracellular ATP concentrations in HEK-ABCG2 cells and parental cells. Fig. 6A shows that the basal intracellular ATP levels in HEK-ABCG2 and parental cells were similar. Treatments with curcumin, gA, or ouabain individually decreased the intracellular ATP levels up to ∼20%. In contrast, exposure to a combination of curcumin with gA or ouabain depleted intracellular ATP levels by 40–50% in HEK-ABCG2 cells, whereas these levels remained almost unaffected in the parental cells. These results hence demonstrate the synergistic effects by the drug combinations on intracellular ATP depletion in HEK-ABCG2 cells.
FIGURE 6.
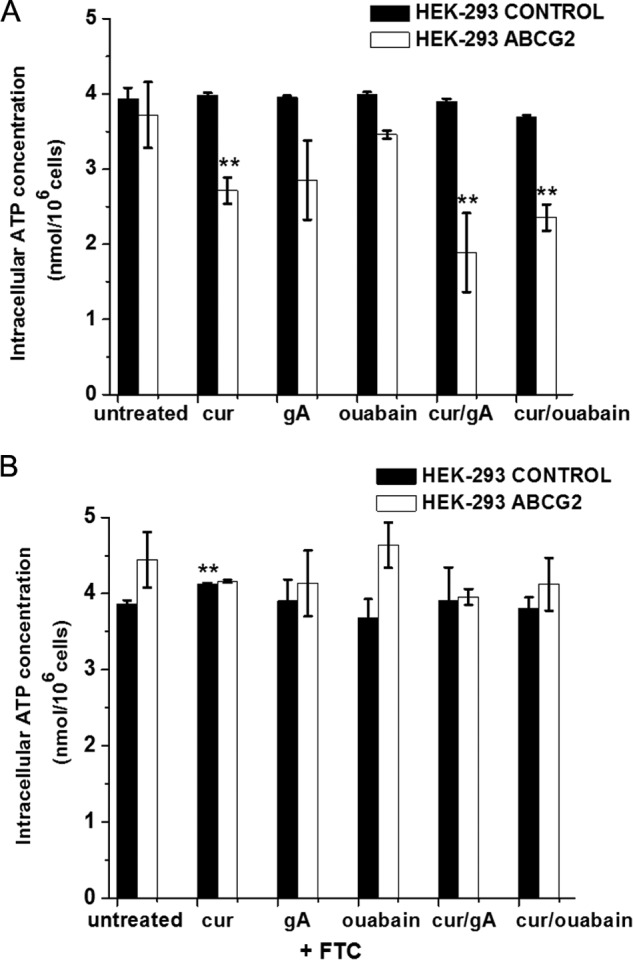
Effect of subtoxic concentrations of curcumin (2 μm), gA (35 nm), or ouabain (7.5 nm) and combinations of curcumin with either gA or ouabain on intracellular ATP levels in HEK-293 control (black bars) and HEK-ABCG2 cells (white bars) in the absence (A) and presence (B) of 10 μm FTC. A double asterisk indicates a difference compared with untreated cells with a p value ≤ 0.01.
To determine whether the selective ATP depletion in resistant cells in the presence of the drug combinations was dependent on active ABCG2 transporters, we repeated the experiments in the presence of 10 μm FTC. In this case, the intracellular ATP concentration in HEK-ABCG2 cells was the same as in untreated cells for all tested conditions (Fig. 6B). These results show that curcumin-stimulated ATPase activity of ABCG2 transporters combined with the gA- or ouabain-stimulated ATPase activity of the Na+,K+-ATPase were responsible for depleting the intracellular ATP levels in cells with ABCG2 transporters below the viability threshold (15), while leaving ATP levels in the parental cells unaffected.
Inhibitors of Oxidative Phosphorylation and Glycolysis in Combination with Curcumin Do Not Preferentially Kill HEK-ABCG2 Cells
Gramicidin A is an uncoupler of oxidative phosphorylation (44) and inhibits glycolysis in prokaryotes (45); both of these activities inhibit ATP synthesis (44). To dissect whether inhibition of oxidative phosphorylation and glycolysis might be relevant for the results reported here, we tested the effect of carbonyl cyanide meta-chlorophenylhydrazone (CCCP), an uncoupler of oxidative phosphorylation (46), and 2-deoxyglucose (2-DG), an inhibitor of glycolysis (47), in combination with curcumin. We selected these two compounds because, like gA, they were not substrates of the ABCG2 transporter in the sense that the IC50 values from cytotoxicity assays with HEK-293 parental cells and HEK-ABCG2 cells were not significantly different for these two compounds (Fig. 7 and Table 3). We found that the combination of curcumin with CCCP or 2-DG killed HEK-ABCG2 cells and parental cells alike, indicating that inhibiting oxidative phosphorylation or glycolysis was not sufficient to selectively kill resistant cells. Therefore, these results provide further evidence that gA and ouabain enhance the cytotoxicity of curcumin in HEK-ABCG2 cells by stimulating ATP turnover by the Na+,K+-ATPase rather than by inhibiting oxidative phosphorylation or glycolysis.
FIGURE 7.
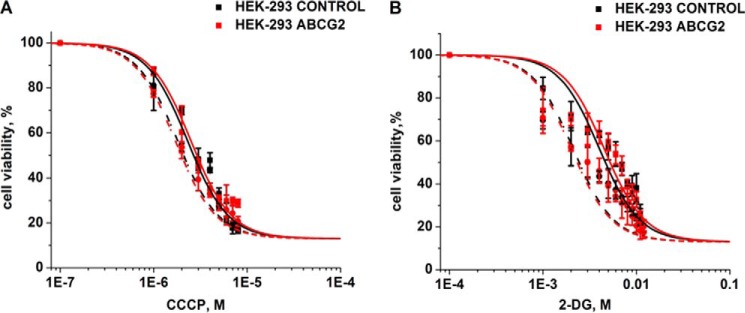
Cell viability of HEK-293 control and HEK-ABCG2 cells as a function of increasing concentrations of CCCP, and 2-DG. The sensitivity to CCCP (A) and 2-DG (B) in HEK-293 control (black) and HEK-ABCG2 (red) cells was determined in the absence (solid curves) or presence (dashed curves) of 2 μm curcumin (see Table 3 for a statistical analysis of differences in IC50 values).
TABLE 3.
IC50 values, relative resistance, and reversal of resistance of CCCP and 2-DG in HEK-293 parental cells and in HEK-ABCG2 cells
| Compound | IC50 |
p value of IC50 from HEK-ABCG2 cells relative to IC50 from control cellsa | Relative resistanceb | Reversal of resistancec | |
|---|---|---|---|---|---|
| HEK-293 control | HEK- ABCG2 | ||||
| CCCP alone | 2.3 ± 0.4 μm | 2.4 ± 0.3 μm | 0.32 | 1.0 ± 0.2-fold | |
| CCCP + 2 μm curcumin | 1.8 ± 0.2 μm | 1.7 ± 0.2 μm | 0.16 | 0.9 ± 0.2-fold | 1.1 ± 0.3-fold |
| 2-DG alone | 4.0 ± 0.6 mm | 4.2 ± 0.9 mm | 0.27 | 1.0 ± 0.3-fold | |
| 2-DG + 2 μm curcumin | 2.1 ± 0.3 mm | 2.0 ± 0.4 mm | 0.18 | 1.0 ± 0.2-fold | 1.0 ± 0.4-fold |
a To determine p values, we used a two-tailed Student's t test based on log(IC50) values. We considered p values ≤ 0.05 as statistically significant.
b Expressed as the ratio of the IC50 value of ABCG2-expressing cells to that of the control cells.
c Expressed as the ratio of the relative resistance value of ABCG2-expressing cells in the absence of the second compound to the relative resistance in its presence.
Inhibitors of the Electron Transport Chain Do Not Affect CS by Curcumin with gA or Ouabain
To further investigate the mechanism by which stimulated ATP hydrolysis and the resulting low intracellular ATP levels might kill HEK-ABCG2 cells, we investigated a previously proposed mechanism of CS. Laberge et al. (48) showed that cells expressing P-gp exhibited CS toward the P-gp substrate verapamil. In this case, verapamil by itself killed the resistant cells preferentially over the parental cells. Along with others before (49, 50), the authors showed that low concentrations of verapamil (∼10 μm) increased the ATPase activity of P-gp and that this effect could reduce intracellular ATP levels by 50%. Laberge et al. proposed that hypersensitivity of P-gp-expressing cells toward verapamil was caused by this increased demand for ATP, which caused an increased rate of oxidative phosphorylation and concomitant increased production of reactive oxygen species (ROS) (51). They hypothesized that accumulation of ROS ultimately initiated apoptosis of the resistant cells. The authors showed that two inhibitors of the electron transport chain, rotenone and antimycin A (52), amplified the collateral sensitivity to verapamil in the P-gp-expressing cell line, whereas the same concentration of these compounds had no effect on the parental cell line. Based on findings by Li et al. (52), Laberge et al. proposed that rotenone and antimycin A exerted their effect through ROS-mediated toxicity and subsequent apoptosis.
Motivated by these results, we investigated the effect of rotenone and antimycin A on HEK-ABCG2 and parental cells in the presence and absence of curcumin and gA or curcumin and ouabain. HEK-ABCG2 cells were slightly more sensitive to rotentone and antimycin A than the parental cells (SR = 1.5 for both compounds and therefore not significant). Interestingly, however, administration of rotenone or antimycin A did not lead to a significant increase in CS of HEK-ABCG2 cells that were also exposed to curcumin and gA or to curcumin and ouabain (Table 4 and Fig. 8). These experiments also indicated that rotenone and antimycin A were not substrates of ABCG2 transporters (Table 4) in the sense that ABCG2-expressing cells did not incur resistance against these compounds. The absence of a significant effect by rotenone and antimycin A indicated that ROS-mediated toxicity was insufficient to amplify CS toward the drug combinations in HEK-ABCG2 cells. Hence, mechanisms other than ROS-induced apoptosis may be involved in the selective cell death of these cells in response to intracellular ATP depletion by a combination of curcumin with gA or ouabain.
TABLE 4.
Effect of rotenone or antimycin A on the viability, relative resistance, and reversal of resistance of HEK-ABCG2 and parental cells
| Compound | IC50 |
Relative resistancea | Sensitization/reversal of resistanceb | |
|---|---|---|---|---|
| HEK-293 control | HEK-ABCG2 | |||
| Rotenone alone | 6.9 ± 1.8 nm | 4.6 ± 1.6 nm | 0.7 ± 0.3-fold | |
| Rotenone + 2 μm curcumin + 35 nm gA | 9.3 ± 3.4 nm | 4.4 ± 1.5 nm | 0.5 ± 0.2-fold | 1.4 ± 0.9-fold |
| Rotenone + 2 μm curcumin + 7.5 nm ouabain | 6.4 ± 1.9 nm | 4.5 ± 1.4 nm | 0.7 ± 0.3-fold | 0.9 ± 0.6-fold |
| Antimycin A alone | 6.0 ± 2.1 mm | 4.0 ± 2.0 mm | 0.7 ± 0.3-fold | |
| Antimycin A + 2 μm curcumin + 35 nm gA | 1.6 ± 0.7 mm | 1.3 ± 0.4 mm | 0.8 ± 0.4-fold | 0.8 ± 0.6-fold |
| Antimycin A + 2 μm curcumin + 7.5 nm ouabain | 2.0 ± 1.0 mm | 1.8 ± 1.0 mm | 0.9 ± 0.6-fold | 0.8 ± 0.7-fold |
a Expressed as the ratio of the IC50 value of ABCG2-expressing cells to that of the control cells.
b Expressed as the ratio of the relative resistance value of ABCG2-expressing cells in the absence of the second compound to the relative resistance in its presence.
FIGURE 8.

Sensitivity of HEK-293 control and HEK-ABCG2 cells to inhibitors of the electron transport chain. A, toxicity of increasing concentrations of rotenone on HEK-ABCG2 (red) and parental cells (black) in the absence (solid curve) or presence of 2 μm curcumin and 35 nm gA (dashed curve). B, toxicity of increasing concentrations of rotenone on HEK-ABCG2 (red) and parental cells (black) in the absence (solid curve) or presence of 2 μm curcumin and 7.5 nm ouabain (dashed curve). C, toxicity of increasing concentrations of antimycin A on HEK-ABCG2 (red) and parental cells (black) in the absence (solid curve) or presence of 2 μm curcumin and 35 nm gA (dashed curve). D, toxicity of increasing concentrations of antimycin A on HEK-ABCG2 (red) and parental cells (black) in the absence (solid curve) or presence of 2 μm curcumin and 7.5 nm ouabain (dashed curve). The open symbols in C and D indicate that antimycin was not soluble at this concentration. These points were not included in the best curve fits.
Selective Cell Death Occurs through Caspase-dependent Apoptosis
In a landmark paper, Leist et al. (15) showed that the intracellular ATP concentration acts as a switch between apoptosis and necrosis. At ATP concentrations significantly above 50% of basal levels, cell death by apoptosis is favored, whereas ATP concentrations below 50% trigger necrosis. In fact, caspase activation in the apoptotic pathway requires ATP, such that low ATP levels inhibit apoptosis (53). Because combined treatments with curcumin and gA or curcumin and ouabain lowered intracellular ATP in HEK-ABCG2 cells to levels close to the 50% threshold (Fig. 6), it was unclear whether ATP depletion causes selective cell death of HEK-ABCG2 cells over parental cells through apoptosis or necrosis.
To answer this question, we conducted an apoptosis/necrosis assay with both HEK-ABCG2 and parental cells that were exposed to curcumin in combination with either gA or ouabain. After only 24 h of treatment, a significant fraction (∼5%) of resistant cells was already identified as late apoptotic or necrotic. After 72 h, this fraction reached over 50% in the presence of curcumin and gA and ∼15% in the presence of curcumin and ouabain. In comparison, less than 5% of parental cells that were treated identically or ABCG2-expressing cells that were treated with a single compound (i.e. gA or ouabain) were identified as late apoptotic or necrotic after 72 h (Fig. 9).
FIGURE 9.
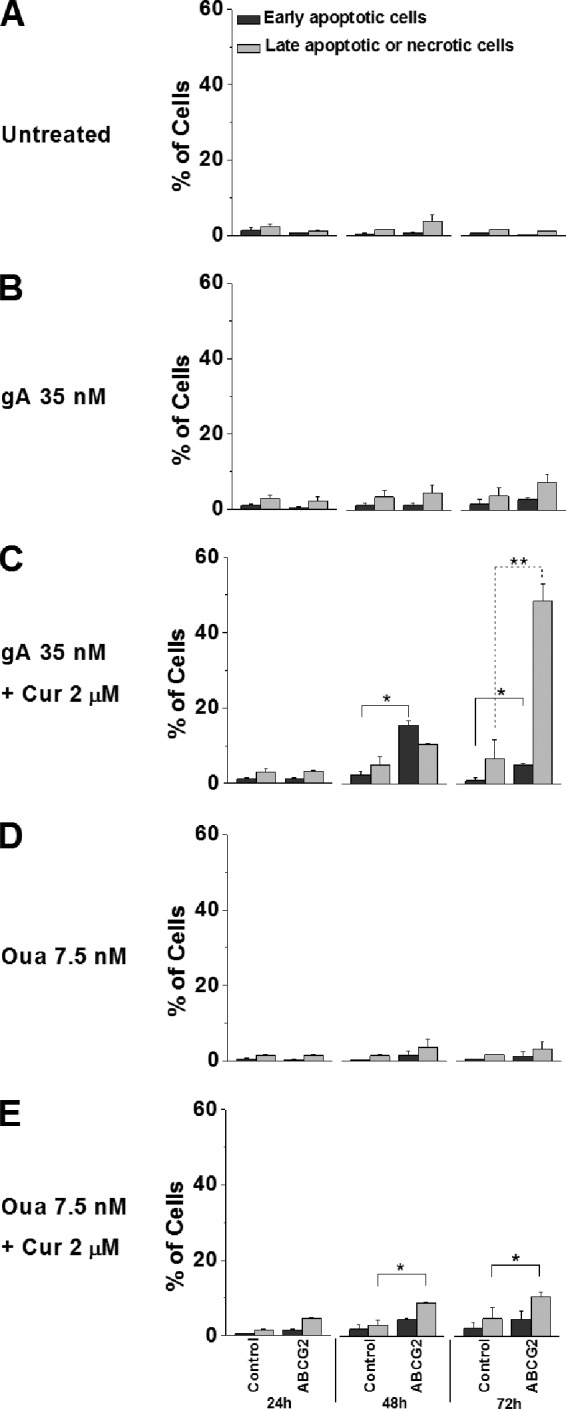
A combination of curcumin (Cur) with either gA or ouabain (Oua) stimulates selective cell death of HEK-ABCG2 cells over parental cells. Cells were incubated in the absence (A) or presence of various compounds: 35 nm gA (B), a combination of 35 nm gA and 2 μm curcumin (C), 7.5 nm ouabain (D), and a combination of 7.5 nm ouabain and 2 μm curcumin (E) for 24, 48, and 72 h and were subsequently stained with annexin V-FITC/EthD I for analysis using a flow cytometer. The dark gray bars indicate the population of early apoptotic cells (annexin V-FITC +/EthD I −), whereas the light gray bars indicate the population of late apoptotic or necrotic cells (annexin V-FITC +/EthD I +).
To clarify whether the drug combinations induce apoptosis or necrosis, we measured the activation of the effector caspases (i.e. caspase-3 and -7), the hallmark of apoptosis (54), in both HEK-ABCG2 and parental cells. After 72 h of incubation with curcumin and gA, nearly 50% of resistant cells showed a significant increase in caspase-3/7 activation. This fraction was ∼35% when these cells were treated with curcumin and ouabain (Fig. 10A). In contrast, the activity of caspase-3/7 in parental cells remained at the basal level under all conditions (Fig. 10A). In addition, we distinguished early apoptotic cells from late apoptotic or necrotic cells using the caspase-3/7 activity assay similar to the apoptosis/necrosis assay. After 72 h, ∼40% of resistant cells were late apoptotic or necrotic when treated with curcumin and gA; this fraction was ∼30% when these cells were treated with curcumin and ouabain (Fig. 10B). These results are consistent with those obtained from the apoptosis/necrosis assay (Fig. 9), suggesting that HEK-ABCG2 cells treated by the drug combinations undergo apoptosis instead of necrosis involving the activation of caspases.
FIGURE 10.
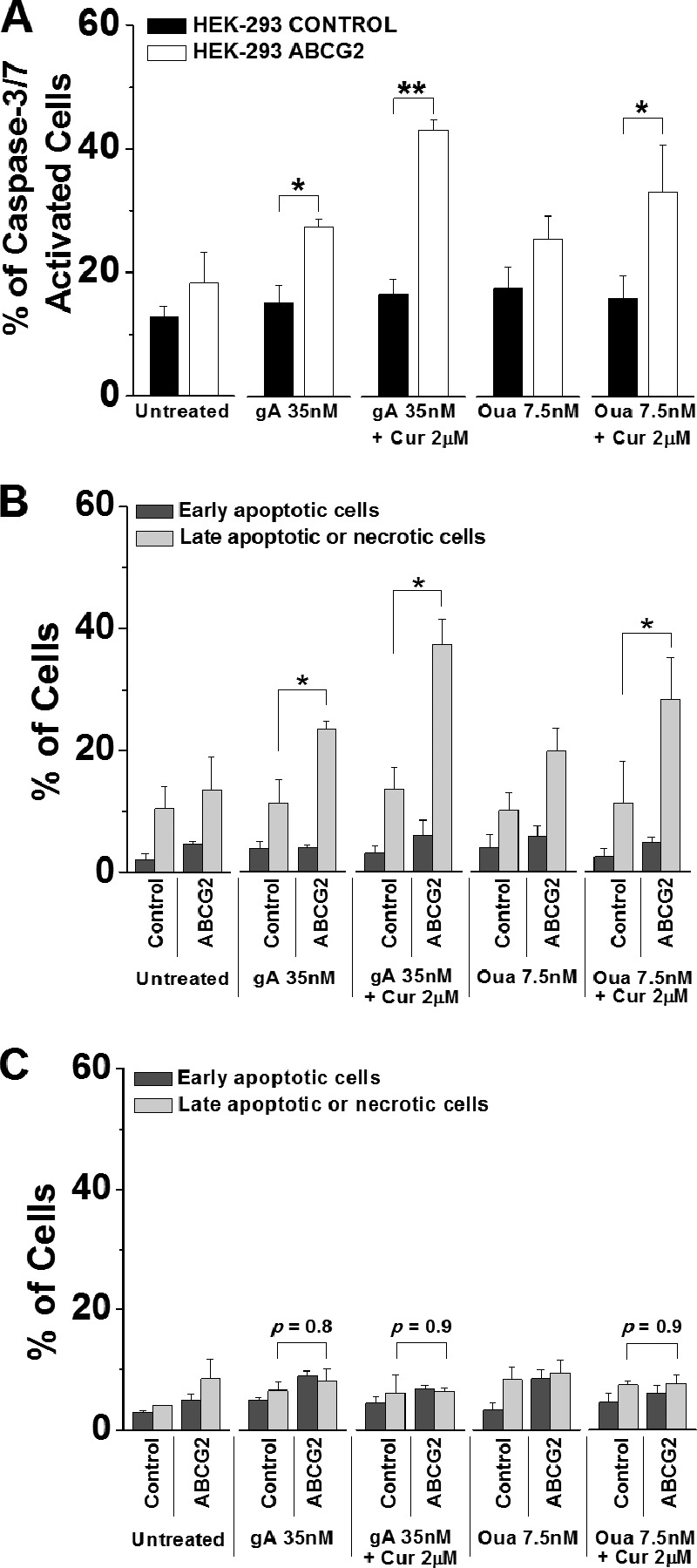
Selective cell death of HEK-ABCG2 cells occurs through caspase-3/7-dependent apoptosis. Cells were incubated in the absence or presence of 35 nm gA, a combination of 35 nm gA and 2 μm curcumin (Cur), 7.5 nm ouabain (Oua), and a combination of 7.5 nm ouabain and 2 μm curcumin for 72 h. A, cells were stained with CellEvent caspase-3/7 green detection reagent and analyzed subsequently by flow cytometry. Bars represent the percentages of caspase-3/7 activated cells of the total population. B, cells were stained with both CellEvent caspase-3/7 green detection reagent and SYTOX AADvanced dead cell stain and analyzed subsequently by flow cytometry. The dark gray bars indicate the population of early apoptotic cells (CellEvent caspase-3/7 green +/SYTOX AADvanced −), whereas the light gray bars indicate the population of late apoptotic or necrotic cells (CellEvent caspase-3/7 green +/SYTOX AADvanced +). C, cells were incubated with 50 μm Z-VAD-FMK for 2 h prior to the addition of various compound(s) for 72 h and subsequently stained with annexin V-FITC/EthD I for flow cytometry analysis. The dark gray bars indicate the population of early apoptotic cells (annexin V-FITC +/EthD I −), whereas the light gray bars indicate the population of late apoptotic or necrotic cells (annexin V-FITC +/EthD I +). p values indicate no significant difference between HEK-control and ABCG2 cells in the presence of Z-VAD-FMK.
Furthermore, we explored the effect of a caspase-3/7 inhibitor, Z-VAD-FMK, on the selective killing of HEK-ABCG2 cells using the apoptosis/necrosis assay. Interestingly, 50 μm Z-VAD-FMK completely eliminated the selective cell death of ABCG2 cells caused by the drug combinations after 72 h (Fig. 10C). Z-VAD-FMK is not a substrate of ABCG2 transporters in the sense that treatment with only the inhibitor induced no significant difference in viability between HEK-293 parental cells (IC50 = 255 μm) and ABCG2 cells (IC50 = 226 ± 3.8 μm). Consistently, results from cytotoxicity assays showed that 50 μm Z-VAD-FMK protected HEK-ABCG2 cells against the toxicity of gA in the presence of 2 μm curcumin (IC50 = 47 ± 5.6 nm in HEK-293 parental and 50 ± 3.8 nm in HEK-ABCG2 cells). Z-VAD-FMK had a similar effect on cells treated with ouabain in the presence of 2 μm curcumin (IC50 = 29 ± 1.2 nm in HEK-293 parental cells and 33 ± 4.9 nm in HEK-ABCG2 cells). Together, these results indicate that the combination of curcumin with either gA or ouabain selectively kills HEK-ABCG2 cells by a caspase-dependent pathway, which ultimately leads to apoptotic cell death.
Effect of Curcumin in Combination with gA or Ouabain on the Human Breast Adenocarcinoma Cell Lines Expressing ABCG2 Transporters (MCF-7/FLV1)
To test whether this approach would have similar effects on a clinically relevant cell line, we conducted cytotoxicity assays with human breast adenocarcinoma parental cells (MCF-7) and flavopiridol-selected daughter cells that express ABCG2 transporters (MCF-7/FLV1) (33). Surprisingly, the IC50 value of gA in MCF-7/FLV1 cells was significantly higher (4.5 ± 0.8-fold) than in the MCF-7 parental cell line (Table 5 and Fig. 11A). This result contrasts with Table 1, which shows that the IC50 values for gA in HEK-293 parental control and ABCG2 cells were not significantly different (p = 0.14). To explore whether this resistance toward gA was either a consequence of ABCG2 efflux activity or of a pleiotropic effect (55, 56) as a result of stepwise selection with flavopiridol, we exposed MCF-7/FLV1 cells to gA in the presence of FTC. Despite inhibition of ABCG2 transporters, these cells retained their resistance toward gA (IC50 = 5.9 μm in the absence of FTC and 5.2 μm in the presence of FTC), providing strong evidence for a pleiotropic effect.
TABLE 5.
Effect of gramicidin or ouabain on relative resistance and reversal of resistance of ABCG2-expressing MCF-7/FLV1 and parental cells
| Compound | IC50 |
p value of IC50 from MCF-7/FLV1 cells relative to IC50 from MCF-7 cellsa | Relative resistanceb | Reversal of resistancec | |
|---|---|---|---|---|---|
| MCF-7 control | MCF-7/FLV1 | ||||
| Curcumin alone | 7.3 ± 0.5 μm | 5.4 ± 0.5 μm | 0.12 | 1.4 ± 0.2-fold | |
| Gramicidin alone | 190 ± 20 nm | 850 ± 115 nm | 4E-3 | 4.5 ± 0.8-fold | |
| Gramicidin + 2 μm curcumin | 50 ± 11 nm | 11 ± 4 nm | 0.03 | 0.2 ± 0.1-fold | 23 ± 9-fold |
| Gramicidin + 2 μm curcumin + 10 μm FTC | 49 ± 13 nm | 51 ± 12 nm | 0.85 | 1.0 ± 0.1-fold | 4.5 ± 1.7-fold |
| Ouabain alone | 14 ± 2 nm | 15 ± 2 nm | 0.74 | 1.1 ± 0.2-fold | |
| Ouabain + 2 μm curcumin | 14 ± 1 nm | 6 ± 2 nm | 0.02 | 0.4 ± 0.1-fold | 2.8 ± 1.0-fold |
| Ouabain + 2 μm curcumin + 10 μm FTC | 15 ± 3 nm | 16 ± 3 nm | 0.70 | 1.1 ± 0.3-fold | 1 ± 0.3-fold |
a To determine p values, we used a two-tailed Student's t test based on log(IC50) values. We considered p values ≤ 0.05 as statistically significant.
b Expressed as the ratio of the IC50 value of ABCG2-expressing cells to that of the control cells.
c Expressed as the ratio of the relative resistance value of ABCG2-expressing cells in the absence of the second compound to the relative resistance in its presence.
FIGURE 11.

Cell viability of MCF-7 and MCF-7/FLV1 cells as a function of increasing gA or ouabain concentrations and effect of curcumin, gA, and ouabain on the RMP of MCF-7 and MCF-7/FLV1 cells. A, sensitivity of MCF-7 cells (black) and MCF-7/FLV1 cells (red) to gA in the absence (solid curves) or presence (dashed curves) of 2 μm curcumin. B, sensitivity of MCF-7 cells (black) and MCF-7/FLV1 cells (red) to ouabain in the absence (solid curves) or presence (dashed curves) of 2 μm curcumin. C and D, cell viability of MCF-7 cells (black) and MCF-7/FLV1 cells (red) exposed to increasing doses of gA (C) or ouabain (D) in the presence of 2 μm curcumin (cur) in the absence (dashed curves) or presence (solid curves) of 10 μm FTC (see Table 5 for a statistical analysis of differences in IC50 values). E, effect of curcumin (cur), gA, and ouabain (oua) on the RMP of MCF-7 cells (black bars) and MCF-7/FLV1 cells (white bars). Cells were incubated under various conditions as indicated. The bars labeled with RMP represent the control without addition of compound(s). FTC was used at a concentration of 10 μm. Triple asterisks indicate a difference compared with the control (RMP) with a p value ≤ 0.001. The values in brackets indicate the numbers of measurements.
Fig. 11A shows that in the presence of 2 μm curcumin, the resistance of MCF-7/FLV1 cells to gA was reversed by 22.5 ± 9-fold and slightly inversed compared with MCF-7 parental cells (Table 5). Therefore, the CS effect on ABCG2-expressing MCF-7/FLV1 cells by curcumin with gA was similar, albeit smaller, than that observed in HEK-ABCG2 cell lines (Fig. 1).
With regard to ouabain, Fig. 11B shows that in the presence of curcumin, MCF-7/FLV1 cells showed CS to increasing concentrations of ouabain and died at 2.8 times lower concentrations of ouabain than MCF-7 parental cells. Treatment with curcumin or ouabain individually showed no significant differences in viability between parental cells and the resistant MCF-7/FLV1 cells (Fig. 11B and Table 5).
Similar to results in Fig. 2, the presence of FTC restored the viability of MCF-7/FLV1 cells to that of MCF-7 parental cells when we incubated cells with varying concentrations of gA or ouabain in combination with 2 μm curcumin (Table 5 and Fig. 11, C and D). Furthermore, we found that gA, ouabain, or the combination of curcumin with gA or with ouabain induced membrane depolarization in MCF-7 and MCF-7/FLV1 similar to that observed in HEK-293 parental cells and ABCG2 cells (Fig. 11E).
These results show that the principle idea of inducing selective cytotoxicity in ABCG2-transfected cells by stimulated depletion of ATP levels could be replicated in the clinically relevant human breast adenocarcinoma cell line MCF-7/FLV1 that expresses ABCG2 transporters. This approach may therefore be generally applicable to resistant cells with significant expression of ABCG2 transporters.
In summary, the CS approach of intracellular ATP depletion by treatment with agents that stimulate ATP hydrolysis by ABC transporters and ion motive pumps presented here provides a strategy to target drug resistant cells with minimal effects on nonresistant cells. This method utilizes curcumin in combination with either gA or ouabain, all of which have previously been tested clinically (6, 57, 58). Therefore, the transporter-induced, synergistic ATP depletion (TISAD) strategy introduced here could inspire a fresh approach for discovery of CS agents against MDR cells.
For this TISAD approach to be applicable to efflux pumps other than ABCG2 transporters, it will be necessary to identify modulators or substrates of these pumps that are able to induce significantly increased ATP hydrolysis rates in MDR cells without inducing significant toxicity in nonresistant cells. In addition, co-administration of a second molecule may be necessary to induce further ATP depletion by stimulating major ATPases such as the Na+,K+-ATPase. More generally, any physical or chemical process that induces significant ATP consumption in combination with stimulated ATP hydrolysis by MDR transporters might have the potential to induce CS.
In contrast to the ATP starvation therapy, the TISAD approach introduced here provides targeting of MDR cells by selectively stimulating ATP hydrolysis rather than indiscriminately inhibiting its synthesis. The results presented here suggest that severe ATP depletion and the resulting induction of cellular apoptosis may be one of the mechanisms by which compounds induce CS in ABC-transporter-expressing drug-resistant cells. The novel aspect of the CS phenomenon reported here is that two compounds, which are individually neither toxic to the resistant nor to the parental cells, can evoke CS in resistant cells when they act synergistically. The work presented here therefore reveals a mechanism of CS that can be targeted rationally because it uses a known phenotypic difference (i.e. inducible ATP depletion by overexpressed MDR transporters) to kill resistant cells.
Acknowledgments
We thank Dr. Susan Bates (NCI) for the generous gift of the ABCG2-expressing cell lines. We also thank Dr. Suneet Shukla, and Joseph Koos for guidance in membrane preparation and ATPase assays and Gowthamy Venkidasubramonian for assistance in maintaining cell lines. Finally, we thank Dr. Gary Luker for excellent suggestions and a clinical perspective of the relevance of MDR efflux pumps.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01GM081705 (to M. M.) and by the Intramural Research Program of the NCI National Institutes of Health Center for Cancer Research (to S. V. A.).
- CS
- collateral sensitivity
- MDR
- multidrug resistance
- MFI
- mean fluorescence intensity
- Bp
- BODIPY-prazosin
- gA
- gramicidinA
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- 2-DG
- 2-deoxyglucose
- FTC
- fumitremorgin C
- Z-VAD-FMK
- benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone
- ABC
- ATP-binding cassette
- P-gp
- P-glycoprotein
- EthD I
- ethidium homodimer I
- RMP
- resting membrane potential
- ROS
- reactive oxygen species
- TISAD
- transporter-induced, synergistic ATP depletion.
REFERENCES
- 1. Szakács G., Hall M. D., Gottesman M. M., Boumendjel A., Kachadourian R., Day B. J., Baubichon-Cortay H., Di Pietro A. (2014) Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 114, 5753–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardwick L. J., Velamakanni S., van Veen H. W. (2007) The emerging pharmacotherapeutic significance of the breast cancer resistance protein (ABCG2). Br. J. Pharmacol. 151, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krishnamurthy P., Schuetz J. D. (2006) Role of ABCG2/BCRP in biology and medicine. Annu. Rev. Pharmacol. Toxicol. 46, 381–410 [DOI] [PubMed] [Google Scholar]
- 4. Doyle L. A., Yang W., Abruzzo L. V., Krogmann T., Gao Y., Rishi A. K., Ross D. D. (1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95, 15665–15670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doyle L., Ross D. D. (2003) Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22, 7340–7358 [DOI] [PubMed] [Google Scholar]
- 6. Kusuhara H., Furuie H., Inano A., Sunagawa A., Yamada S., Wu C., Fukizawa S., Morimoto N., Ieiri I., Morishita M., Sumita K., Mayahara H., Fujita T., Maeda K., Sugiyama Y. (2012) Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br. J. Pharmacol. 166, 1793–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dean M., Fojo T., Bates S. (2005) Tumour stem cells and drug resistance. Nat. Rev. Cancer 5, 275–284 [DOI] [PubMed] [Google Scholar]
- 8. Ding X. W., Wu J. H., Jiang C. P. (2010) ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 86, 631–637 [DOI] [PubMed] [Google Scholar]
- 9. Haimeur A., Conseil G., Deeley R. G., Cole S. P. (2004) The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr. Drug Metab. 5, 21–53 [DOI] [PubMed] [Google Scholar]
- 10. Shukla S., Wu C. P., Ambudkar S. V. (2008) Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges. Expert. Opin. Drug. Metab. Toxicol. 4, 205–223 [DOI] [PubMed] [Google Scholar]
- 11. Leonard G. D., Polgar O., Bates S. E. (2002) ABC transporters and inhibitors: new targets, new agents. Curr. Opin. Investig. Drugs 3, 1652–1659 [PubMed] [Google Scholar]
- 12. Silva A. S., Kam Y., Khin Z. P., Minton S. E., Gillies R. J., Gatenby R. A. (2012) Evolutionary approaches to prolong progression-free survival in breast cancer. Cancer Res. 72, 6362–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moreno-Sánchez R., Rodríguez-Enríquez S., Saavedra E., Marín-Hernández A., Gallardo-Pérez J. C. (2009) The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? BioFactors 35, 209–225 [DOI] [PubMed] [Google Scholar]
- 14. Shim H., Chun Y. S., Lewis B. C., Dang C. V. (1998) A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. U.S.A. 95, 1511–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leist M., Single B., Castoldi A. F., Kühnle S., Nicotera P. (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fearon K. C., Plumb J. A., Burns H. J., Calman K. C. (1987) Reduction of the growth rate of the Walker 256 tumor in rats by rhodamine 6G together with hypoglycemia. Cancer Res. 47, 3684–3687 [PubMed] [Google Scholar]
- 17. Gottesman M. M., Fojo T., Bates S. E. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 [DOI] [PubMed] [Google Scholar]
- 18. Pluchino K. M., Hall M. D., Goldsborough A. S., Callaghan R., Gottesman M. M. (2012) Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 15, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Szakács G., Annereau J. P., Lababidi S., Shankavaram U., Arciello A., Bussey K. J., Reinhold W., Guo Y., Kruh G. D., Reimers M., Weinstein J. N., Gottesman M. M. (2004) Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 6, 129–137 [DOI] [PubMed] [Google Scholar]
- 20. Gatenby R. A., Silva A. S., Gillies R. J., Frieden B. R. (2009) Adaptive therapy. Cancer Res. 69, 4894–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laberge R. M., Karwatsky J., Lincoln M. C., Leimanis M. L., Georges E. (2007) Modulation of GSH levels in ABCC1 expressing tumor cells triggers apoptosis through oxidative stress. Biochem. Pharmacol. 73, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 22. van der Heijden J., de Jong M. C., Dijkmans B. A., Lems W. F., Oerlemans R., Kathmann I., Scheffer G. L., Scheper R. J., Assaraf Y. G., Jansen G. (2004) Acquired resistance of human T cells to sulfasalazine: stability of the resistant phenotype and sensitivity to non-related DMARDs. Ann. Rheum. Dis. 63, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heffeter P., Jakupec M. A., Körner W., Chiba P., Pirker C., Dornetshuber R., Elbling L., Sutterlüty H., Micksche M., Keppler B. K., Berger W. (2007) Multidrug-resistant cancer cells are preferential targets of the new antineoplastic lanthanum compound KP772 (FFC24). Biochem. Pharmacol. 73, 1873–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karwatsky J., Lincoln M. C., Georges E. (2003) A mechanism for P-glycoprotein-mediated apoptosis as revealed by verapamil hypersensitivity. Biochemistry 42, 12163–12173 [DOI] [PubMed] [Google Scholar]
- 25. Shishodia S., Chaturvedi M. M., Aggarwal B. B. (2007) Role of curcumin in cancer therapy. Curr. Probl. Cancer 31, 243–305 [DOI] [PubMed] [Google Scholar]
- 26. Chearwae W., Shukla S., Limtrakul P., Ambudkar S. V. (2006) Modulation of the function of the multidrug resistance-linked ATP-binding cassette transporter ABCG2 by the cancer chemopreventive agent curcumin. Mol. Cancer Ther. 5, 1995–2006 [DOI] [PubMed] [Google Scholar]
- 27. Hladky S. B., Haydon D. A. (1970) Discreteness of conductance change in bimolecular lipid membranes in the presence of certain antibiotics. Nature 225, 451–453 [DOI] [PubMed] [Google Scholar]
- 28. Noble D. (1980) Mechanism of action of therapeutic levels of cardiac glycosides. Cardiovasc. Res. 14, 495–514 [DOI] [PubMed] [Google Scholar]
- 29. Schatzmann H. J. (1953) Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane. Helv. Physiol. Pharmacol. Acta 11, 346–354 [PubMed] [Google Scholar]
- 30. Nobes C. D., Lakin-Thomas P. L., Brand M. D. (1989) The contribution of ATP turnover by the Na+/K+-ATPase to the rate of respiration of hepatocytes: effects of thyroid status and fatty acids. Biochim. Biophys. Acta 976, 241–245 [DOI] [PubMed] [Google Scholar]
- 31. James A. M., Sheard P. W., Wei Y. H., Murphy M. P. (1999) Decreased ATP synthesis is phenotypically expressed during increased energy demand in fibroblasts containing mitochondrial tRNA mutations. Eur. J. Biochem. 259, 462–469 [DOI] [PubMed] [Google Scholar]
- 32. Robey R. W., Shukla S., Finley E. M., Oldham R. K., Barnett D., Ambudkar S. V., Fojo T., Bates S. E. (2008) Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)). Biochem. Pharmacol. 75, 1302–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robey R. W., Medina-Pérez W. Y., Nishiyama K., Lahusen T., Miyake K., Litman T., Senderowicz A. M., Ross D. D., Bates S. E. (2001) Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin. Cancer Res. 7, 145–152 [PubMed] [Google Scholar]
- 34. Ambudkar S. V. (1998) Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292, 504–514 [DOI] [PubMed] [Google Scholar]
- 35. Schaffner W., Weissmann C. (1973) A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56, 502–514 [DOI] [PubMed] [Google Scholar]
- 36. Leach F. R., Webster J. J. (1986) Commercially available firefly luciferase reagents. Methods Enzymol. 133, 51–70 [DOI] [PubMed] [Google Scholar]
- 37. Shukla S., Robey R. W., Bates S. E., Ambudkar S. V. (2006) The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry 45, 8940–8951 [DOI] [PubMed] [Google Scholar]
- 38. Godfraind T., Ghysel-Burton J. (1977) Binding sites related to ouabain-induced stimulation or inhibition of the sodium pump. Nature 265, 165–166 [DOI] [PubMed] [Google Scholar]
- 39. Hagen P. (1939) The effects of digilanid C in varying dosage upon the potassium and water content of rabbit heart muscle. J. Pharmacol. Exp. Ther. 67, 50–55 [Google Scholar]
- 40. Oselkin M., Tian D., Bergold P. J. (2010) Low-dose cardiotonic steroids increase sodium-potassium ATPase activity that protects hippocampal slice cultures from experimental ischemia. Neurosci. Lett. 473, 67–71 [DOI] [PubMed] [Google Scholar]
- 41. Hoffman M. M., Wei L. Y., Roepe P. D. (1996) Are altered pHi and membrane potential in huMDR 1 transfectants sufficient to cause MDR protein-mediated multidrug resistance? J. Gen. Physiol. 108, 295–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stankovic C. J., Heinemann S. H., Delfino J. M., Sigworth F. J., Schreiber S. L. (1989) Transmembrane channels based on tartaric acid-gramicidin A hybrids. Science 244, 813–817 [DOI] [PubMed] [Google Scholar]
- 43. Anner B. M., Moosmayer M. (1994) Na,K-ATPase characterized in artificial membranes. 2. Successive measurement of ATP-driven Rb-accumulation, ouabain-blocked Rb-flux and palytoxin-induced Rb-efflux. Mol. Membr. Biol. 11, 247–254 [DOI] [PubMed] [Google Scholar]
- 44. Rottenberg H., Koeppe R. E., 2nd. (1989) Mechanism of uncoupling of oxidative phosphorylation by gramicidin. Biochemistry 28, 4355–4360 [DOI] [PubMed] [Google Scholar]
- 45. Harold F. M., Baarda J. R. (1967) Gramicidin, valinomycin, and cation permeability of Streptococcus faecalis. J. Bacteriol. 94, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wallace K. B., Starkov A. A. (2000) Mitochondrial targets of drug toxicity. Annu. Rev. Pharmacol. Toxicol. 40, 353–388 [DOI] [PubMed] [Google Scholar]
- 47. Woodward G. E., Hudson M. T. (1954) The effect of 2-desoxy-d-glucose on glycolysis and respiration of tumor and normal tissues. Cancer Res. 14, 599–605 [PubMed] [Google Scholar]
- 48. Laberge R. M., Ambadipudi R., Georges E. (2009) P-glycoprotein (ABCB1) modulates collateral sensitivity of a multidrug resistant cell line to verapamil. Arch. Biochem. Biophys. 491, 53–60 [DOI] [PubMed] [Google Scholar]
- 49. al-Shawi M. K., Senior A. E. (1993) Characterization of the adenosine triphosphatase activity of Chinese hamster P-glycoprotein. J. Biol. Chem. 268, 4197–4206 [PubMed] [Google Scholar]
- 50. Litman T., Zeuthen T., Skovsgaard T., Stein W. D. (1997) Structure-activity relationships of P-glycoprotein interacting drugs: kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta 1361, 159–168 [DOI] [PubMed] [Google Scholar]
- 51. Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry 70, 200–214 [DOI] [PubMed] [Google Scholar]
- 52. Li N., Ragheb K., Lawler G., Sturgis J., Rajwa B., Melendez J. A., Robinson J. P. (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278, 8516–8525 [DOI] [PubMed] [Google Scholar]
- 53. Golstein P., Kroemer G. (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43 [DOI] [PubMed] [Google Scholar]
- 54. Shi Y. (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9, 459–470 [DOI] [PubMed] [Google Scholar]
- 55. Fanciulli M., Bruno T., Giovannelli A., Gentile F. P., Di Padova M., Rubiu O., Floridi A. (2000) Energy metabolism of human LoVo colon carcinoma cells: correlation to drug resistance and influence of lonidamine. Clin. Cancer Res. 6, 1590–1597 [PubMed] [Google Scholar]
- 56. Linn S. C., Giaccone G. (1995) MDR1/P-glycoprotein expression in colorectal cancer. Eur. J. Cancer 31A, 1291–1294 [DOI] [PubMed] [Google Scholar]
- 57. Carroll R. E., Benya R. V., Turgeon D. K., Vareed S., Neuman M., Rodriguez L., Kakarala M., Carpenter P. M., McLaren C., Meyskens F. L., Jr., Brenner D. E. (2011) Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev. Res. (Phila.) 4, 354–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen J. Q., Contreras R. G., Wang R., Fernandez S. V., Shoshani L., Russo I. H., Cereijido M., Russo J. (2006) Sodium/potassium ATPase (Na+,K+-ATPase) and ouabain/related cardiac glycosides: A new paradigm for development of anti-breast cancer drugs? Breast Cancer Res. Treat. 96, 1–15 [DOI] [PubMed] [Google Scholar]