

Background: Highly conserved residues of DNA polymerase β alter their interactions as the enzyme transitions from open and closed conformations.
Results: Site-directed mutagenesis coupled with kinetic/structural characterization of key mutants defines the role(s) in enzyme activation.
Conclusion: These residues both hasten and deter correct and incorrect nucleotide insertion.
Significance: Conformational equilibrium(s) of the precatalytic ternary substrate complex influences the observed rate of nucleotide insertion.
Keywords: Conformational Change, DNA Polymerase, Enzyme Mechanism, Mutagenesis, X-ray Crystallography
Abstract
DNA polymerases and substrates undergo conformational changes upon forming protein-ligand complexes. These conformational adjustments can hasten or deter DNA synthesis and influence substrate discrimination. From structural comparison of binary DNA and ternary DNA-dNTP complexes of DNA polymerase β, several side chains have been implicated in facilitating formation of an active ternary complex poised for chemistry. Site-directed mutagenesis of these highly conserved residues (Asp-192, Arg-258, Phe-272, Glu-295, and Tyr-296) and kinetic characterization provides insight into the role these residues play during correct and incorrect insertion as well as their role in conformational activation. The catalytic efficiencies for correct nucleotide insertion for alanine mutants were wild type ∼ R258A > F272A ∼ Y296A > E295A > D192A. Because the efficiencies for incorrect insertion were affected to about the same extent for each mutant, the effects on fidelity were modest (<5-fold). The R258A mutant exhibited an increase in the single-turnover rate of correct nucleotide insertion. This suggests that the wild-type Arg-258 side chain generates a population of non-productive ternary complexes. Structures of binary and ternary substrate complexes of the R258A mutant and a mutant associated with gastric carcinomas, E295K, provide molecular insight into intermediate structural conformations not appreciated previously. Although the R258A mutant crystal structures were similar to wild-type enzyme, the open ternary complex structure of E295K indicates that Arg-258 stabilizes a non-productive conformation of the primer terminus that would decrease catalysis. Significantly, the open E295K ternary complex binds two metal ions indicating that metal binding cannot overcome the modified interactions that have interrupted the closure of the N-subdomain.
Introduction
DNA polymerases and their ligands (dNTP, DNA, metals) undergo conformational adjustments upon complex formation. These conformational changes may be global in scope as exemplified by protein subdomain repositioning (1) or subtle, such as a change in protein side chain hydrogen bonding or metal coordination (2). Although great attention has focused on subdomain motions and their role in substrate discrimination, crystallographic studies have revealed that active site geometry is particularly sensitive to the identity of the conformational state of the complex (i.e. correct opposed to incorrect bound nucleotide) (3).
DNA polymerase β (pol β)2 contributes two enzymatic activities during the repair of simple base lesions in genomic DNA; that is, template-directed DNA synthesis (nucleotidyltransferase) and deoxyribose 5′-phosphate removal (lyase) (4). These activities reside in separate domains: a 31-kDa polymerase domain and an 8-kDa amino-terminal lyase domain. The polymerase domain is structurally organized into functionally distinct subdomains referred to as DNA binding (D, residues 90–150), catalytic (C, 151–260), and nascent base pair binding (N, residues 261–335) subdomains (5). These are referred to as thumb, palm, and fingers subdomains, respectively, for right-handed polymerases that exhibit a non-homologous catalytic (palm) subdomain (6).
DNA binding (7), crystallography (2, 3, 8–10), NMR (11–13), and fluorescence studies (14–18) indicate that pol β and substrates undergo several conformational transitions upon binding substrates and metals during catalytic cycling. These transitions are believed to play a critical role during substrate discrimination, i.e. facilitate selecting right from wrong dNTPs. This is generally referred to as “induced fit” where binding of the correct nucleoside triphosphate results in an optimal alignment of catalytic residues that promotes binding and catalysis. In contrast, an incorrect dNTP leads to alternate protein/substrate conformations that discourage insertion. The kinetic result is that an incorrect substrate exhibits a lower apparent binding affinity (Kd) and decreased rate of nucleotide insertion (kpol) relative to those for the correct incoming nucleotide.
Crystallographic structures of binary DNA and ternary substrate complexes of pol β suggest that DNA and several protein side chains alter their position and/or interactions upon nucleotide binding (8) (Fig. 1). These interactions may provide a means by which the polymerase active site (i.e. metal binding ligands) detects whether the N-subdomain is able to form a stable closed complex. Alanine substitution for Arg-283 (>15 Å from the active site) results in a catalytically compromised enzyme that exhibits diminished fidelity (19–21). This loss in fidelity is completely due to the inability to insert the correct nucleotide. We now examine the role of several other residues that alter their interactions during formation of the closed ternary complex in modulating catalytic activation and fidelity.
FIGURE 1.
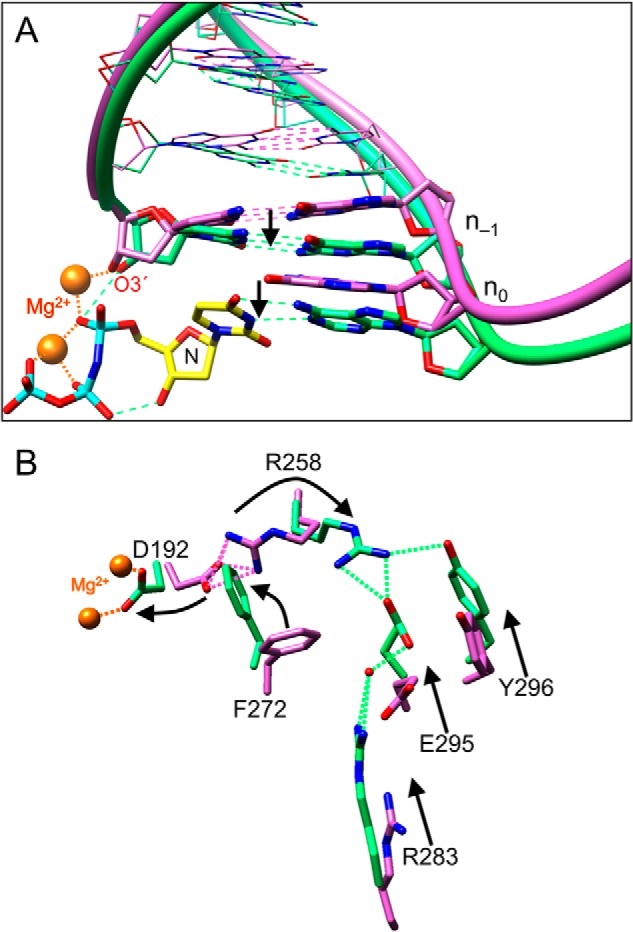
Open and closed conformations of key pol β and DNA residues. The crystallographic structures of the catalytic subdomains of the open binary DNA (PDB accession code 1BPX; pink carbon atoms) and closed ternary substrate complex (PDB accession code 2FMS; green, protein and DNA carbon atoms; yellow, dNTP carbon atoms) were superimposed (root mean square deviation = 1.0 Å). A, a view of the DNA major groove edge of the nascent base pair and primer terminus (O3′, stick representation) illustrating repositioning of the template strand and primer bases upon forming the closed ternary complex. The arrows represent a shift in the template strand associated with the closing transition where the template strand moves toward the N-subdomain. Base pairs upstream of the primer terminus are illustrated in a wire representation. The polymerase is omitted for clarity. The incoming nucleotide and its coding template base of the ternary complex are labeled N and n0, respectively. B, the position of the N-subdomain can be monitored in the active site through a series of altered interactions between Asp-192 (D192), which coordinates both active site metals (orange), and Arg-283 (R283), that is situated in the N-subdomain and interacts with the minor groove edge of the templating strand (not shown). The arrows represent motions associated with the closing transition. N-subdomain closing is associated with Arg-258 (R258) releasing Asp-192 and forming hydrogen bonds with Glu-295 (E295) and Tyr-296 (Y296). Phe-272 (F272) is repositioned in the closed complex to insulate Asp-192 from Arg-258. The DNA is omitted for clarity.
EXPERIMENTAL PROCEDURES
Materials
Ultrapure deoxynucleoside triphosphates, [γ-32P]ATP, and MicroSpin G-25 columns were from Amersham Biosciences-GE Healthcare.
Mutagenesis of the Human Pol β Gene
Oligonucleotide site-directed mutagenesis was performed using a procedure described previously (19). The codons for Asp-192, Arg-258, Phe-272, Glu-295, or Tyr-296 were altered to generate single alanine substitutions. Additionally, a double alanine mutant was generated at residues 258 and 272 (R258A/F272A), and an alternate lysine substitution was created at position 295 (E295K). To ensure that the resulting pol β gene contained the desired change(s), the entire coding sequence of the mutant was confirmed by DNA sequence analysis. Each mutant was cloned into pWL-11 (22), a bacterial expression plasmid containing the λ PL promoter and overexpressed in Escherichia coli TAP56 cells.
Protein Purification
Wild-type and mutant proteins were purified as described previously (23). Enzyme concentrations were determined by Coomassie dye binding using purified pol β as the standard. The concentration of purified pol β was determined by total amino acid analysis.
DNA Preparation
A 34-mer oligonucleotide DNA substrate containing a single nucleotide gap was prepared by annealing 3 gel-purified oligonucleotides (IDT, Coralville, IA) to create a single nucleotide gap at position 16. Each oligonucleotide was resuspended in 10 mm Tris-HCl, pH 7.4, and 1 mm EDTA, and the concentration was determined from their UV absorbance at 260 nm. The annealing reactions were carried out by incubating a solution of 10 μm primer with 12 μm concentrations each of downstream and template oligonucleotides at 90–100 °C for 3 min followed by 30 min at 65 °C and then slow cooling to room temperature. The sequence of the gapped DNA substrate was: primer, 5′-CTG CAG CTG ATG CGC-3′; downstream oligonucleotide, 5′-GTA CGG ATC CCC GGG TAC-3′; template, 3′-GAC GTC GAC TAC GCG XCA TGC CTA GGG GCC CAT G-5′, where the X represents T or G. The primer was 5′-labeled with [γ-32P]ATP using T4 polynucleotide kinase (New England Biolabs), and radioactive ATP was removed with a MicroSpin G-25 column. The downstream oligonucleotide was synthesized with a 5′-phosphate.
Kinetic Assays
Steady-state kinetic parameters for single nucleotide gap-filling reactions were determined by initial velocity measurements as described previously (24). Unless noted otherwise, enzyme activities were determined using a standard reaction mixture containing 50 mm Tris-HCl, pH 7.4 (37 °C), 100 mm KCl, 5 mm MgCl2, and 200 nm single nucleotide-gapped DNA. Due to the lower stability of the R258A mutant at 37 °C, the reaction mixtures were supplemented with 50 μg/ml BSA. BSA does not influence the activity of the wild-type enzyme. Enzyme concentrations and reaction time intervals were chosen so that substrate depletion or product inhibition did not influence initial velocity measurements. Reactions were stopped with 20 μl of 0.5 m EDTA and mixed with an equal volume of formamide dye, and the products were separated on 12% denaturing polyacrylamide gels. The dried gels were analyzed using a PhosphorImager (Amersham Biosciences) to quantify product formation.
To directly measure the rate of the first insertion (kpol) and the apparent equilibrium nucleotide dissociation constant (Kd), single-turnover kinetic assays (enzyme/DNA = 10) were performed as outlined previously (25) employing a KinTek Model RQF-3 chemical quench-flow apparatus (KinTek Corp., Austin, TX). Typically, a solution of pol β (1 μm) was preincubated with single nucleotide-gapped DNA (100 nm). This solution was rapidly mixed (2-fold dilution) with various concentrations of dNTP/Mg2+. Final conditions (pH, temperature) and salt concentrations were like those described for the steady-state assay. After various time periods, the reactions were stopped with 0.25 m EDTA, and the quenched samples were mixed with an equal volume of formamide dye. Products were separated and quantified as described above. Under these conditions the first-order rate constant of the exponential time-courses was dependent on the concentration of dNTP. A secondary plot of the concentration dependence of kobs was hyperbolic and fitted by a non-linear least-squares method to Equation 1 where kpol is the intrinsic rate constant for the step limiting the first insertion.
Crystallization of Mutant Pol β Substrate Complexes
The DNA substrate consisted of a 16-mer template, a complementary 10-mer primer strand, and a 5-mer downstream oligonucleotide. The annealed 10-mer primer creates a one-nucleotide gap with a templating A residue. The downstream oligonucleotide is 5′-phosphorylated. The template sequence was 5′-CCG ACA GCG CAT CAG C-3′ (the underlined base is the coding nucleotide). Oligonucleotides were dissolved in 20 mm MgCl2 and 100 mm Tris-HCl, pH 7.5. Each set of template, primer, and downstream oligonucleotide was mixed in a 1:1:1 ratio and annealed using a PCR thermocycler by heating 10 min at 90 °C and cooling to 4 °C (1 °C/min) resulting in a 1 mm mixture of gapped duplex DNA. This solution was then mixed with an equal volume of mutant (R258A or E295K) pol β at 4 °C, and the mixture warmed to 35 °C and gradually cooled to 4 °C.
Pol β-DNA complexes were crystallized by sitting-drop vapor diffusion. The crystallization buffer was 16% PEG-3350, 350 mm sodium acetate, and 50 mm imidazole, pH 7.5. Drops were incubated at 18 °C and streak-seeded after 1 day. Crystals grew in ∼2–4 days after seeding. The ternary complex was obtained by soaking crystals of binary 1-nucleotide-gapped DNA complexes in artificial mother liquor with 100 mm MgCl2 or 50 mm MnCl2 and with 2 mm dUMPNPP, 20% PEG-3350, and 12% ethylene glycol and then flash-frozen to 100 K in a nitrogen stream. All crystals belong to the space group P21.
Data Collection and Structure Determination
X-ray crystal diffraction data were collected on a Saturn 92 CCD detector system mounted on a MicroMax-007HF (Rigaku Corp.) rotating anode generator. Data were integrated and reduced for structure refinement with HKL2000 software (26).
Structures were determined by molecular replacement with previously determined structures of pol β complexed with a one-nucleotide gapped DNA (binary complex, PDB ID 3ISB) (27) or a ternary complex with an incoming dUMPNPP (ternary complex, PDB ID 2FMS) (9). The crystal structures have similar lattices and are sufficiently isomorphous to determine the molecular replacement model using PHENIX (28). Further refinement and model building were carried out using O (29). The molecular graphic images were prepared in Chimera (30).
Accession Codes
Protein Data Bank coordinates and structure factors for the pol R258A pol β/DNA binary and ternary (+dUMPNPP) and E295K pol β/DNA binary and ternary (+dUMPNPP) complexes have been deposited with accession codes 4R63, 4R65, 4R64, and 4R66, respectively.
RESULTS
Site-directed Mutagenesis
Comparing crystallographic structures of different liganded states of pol β indicate that DNA around the active site and several side chains of the C- and N-subdomains alter their interactions upon going from an open binary DNA complex to a closed ternary substrate complex (8, 31) (Fig. 1). These altered interactions result from repositioning of the N-subdomain upon dNTP binding. Specifically, when the N-subdomain is in the open “inactive” conformation, Asp-192 forms a salt bridge with Arg-258 thereby diverting a critical metal coordinating ligand (Fig. 1B). A conservative glutamate substitution at Asp-192 (D192E) results in a dramatic loss of activity highlighting the critical role of this residue (32). Asp-190 and Asp-192 coordinate both Mg2+ ions necessary for catalysis. In the closed conformation, the phenylalanine ring of residue 272 is positioned between Asp-192 and Arg-258, thereby insulating these residues. Asp-192 and Arg-258 have rotated away from one another to coordinate active site metals or form hydrogen bonds with Glu-295 and Tyr-296, respectively. Interestingly, a lysine mutant at residue 295 has been detected in some human gastric cancers and a role in modulating base excision repair suggested (33). Molecular modeling of the N-subdomain movement has suggested that side-chain motions do not occur in a concerted manner but follow a sequential path (34–36). To probe the functional significance of these residues, alanine has been substituted for Asp-192 (D192A), Arg-258 (R258A), Phe-272 (F272A), Glu-295 (E295A), or Tyr-296 (Y296A). Additionally, lysine was substituted for Glu-295 (E295K) to address the functional consequences that may lead to the observed biological repercussions (33). The mutant proteins were expressed in E. coli and purified. The purified mutant pol β proteins were >95% homogeneous and devoid of contaminating exonuclease activity (data not shown). The catalytic consequences of these protein modifications were assessed.
Single Nucleotide Gap-filling DNA Synthesis
To analyze the effect of the altered side chain on DNA synthesis, we determined steady-state kinetic parameters for insertion of a correct nucleotide (dATP or dCTP) into a single nucleotide-gapped heteropolymeric DNA substrate (templating thymine or guanine, respectively) (Table 1; Fig. 2). For most mutants, kcat for correct insertion was minimally affected by alanine substitution. The R258A mutant exhibited a modest increase in the respective turnover numbers for correct insertion. However, it should be noted that the steady-state rate of correct nucleotide insertion is partially limited by at least two kinetic steps, chemistry and product dissociation (25). In contrast, kcat is significantly decreased for incorrect insertion relative to wild-type enzyme for the alanine mutants of Phe-272, Glu-295, and Tyr-296. In these cases, Km,incorrect is hardly affected. It is generally believed that the rate of incorrect nucleotide insertion is limited by chemistry so that Km,incorrect represents the apparent equilibrium dissociation constant, Kd, for the incorrect nucleotide.
TABLE 1.
Steady-state kinetic summary for single nucleotide gap-filling DNA substrates
The results represent the mean (S.E.) of at least two independent determinations. ND, not determined.
| Enzyme | Template dNTPa | kcat | Km,dNTP | kcat/Km |
|---|---|---|---|---|
| 10−2 s−1 | μm | 10−4 μm−1s−1 | ||
| Wild type | dT-dATP | 90 (10) | 1.1 (0.04) | 8200 (1000) |
| dT-dGTP | 9 (1) | 480 (110) | 1.9 (0.5) | |
| dG-dCTP | 58 (3) | 0.40 (0.10) | 14500 (3700) | |
| dG-dTTP | 4.1 (0.3) | 1300 (170) | 0.31 (0.05) | |
| D192A | dG-dCTP | Ib | ||
| D192E | dG-dCTP | NDc | ||
| R258A | dT-dATP | 180 (20) | 7.9 (0.4) | 2300 (300) |
| dT-dGTP | 4 (0.9) | 1635 (98) | 0.24 (0.06) | |
| dG-dCTP | 120 (6) | 3.2 (0.9) | 3750 (1070) | |
| dG-dTTP | 2.1 (0.1) | 1150 (100) | 0.18 (0.02) | |
| F272A | dT-dATP | 20 (1) | 7.2 (1.6) | 280 (60) |
| dT-dGTP | 0.22 (0.06) | 1200 (40) | 0.018 (0.005) | |
| dG-dCTP | 6.5 (0.2) | 1.3 (0.2) | 500 (80) | |
| dG-dTTP | 0.076 (0.001) | 1448 (46) | 0.0052 (0.0002) | |
| E295A | dT-dATP | 15.0 (1.0) | 70 (6) | 21 (2) |
| dT-dGTP | ND | |||
| dG-dCTP | 14.2 (0.7) | 20 (2) | 71 (8) | |
| dG-dTTP | ND | |||
| E295K | dG-dCTP | ND | ||
| Y296A | dT-dATP | 86 (20) | 75 (8) | 115 (30) |
| dT-dGTP | 0.05 (0.01) | 450 (50) | 0.011 (0.002) | |
| dG-dCTP | 66 (8) | 29 (3) | 230 (40) | |
| dG-dTTP | 0.049 (0.003) | 1023 (120) | 0.0048 (0.0006) |
a Template nucleotide-incoming nucleoside triphosphate.
b Inactive.
c Under steady-state conditions, reliable kinetic parameters activity could not be determined.
FIGURE 2.
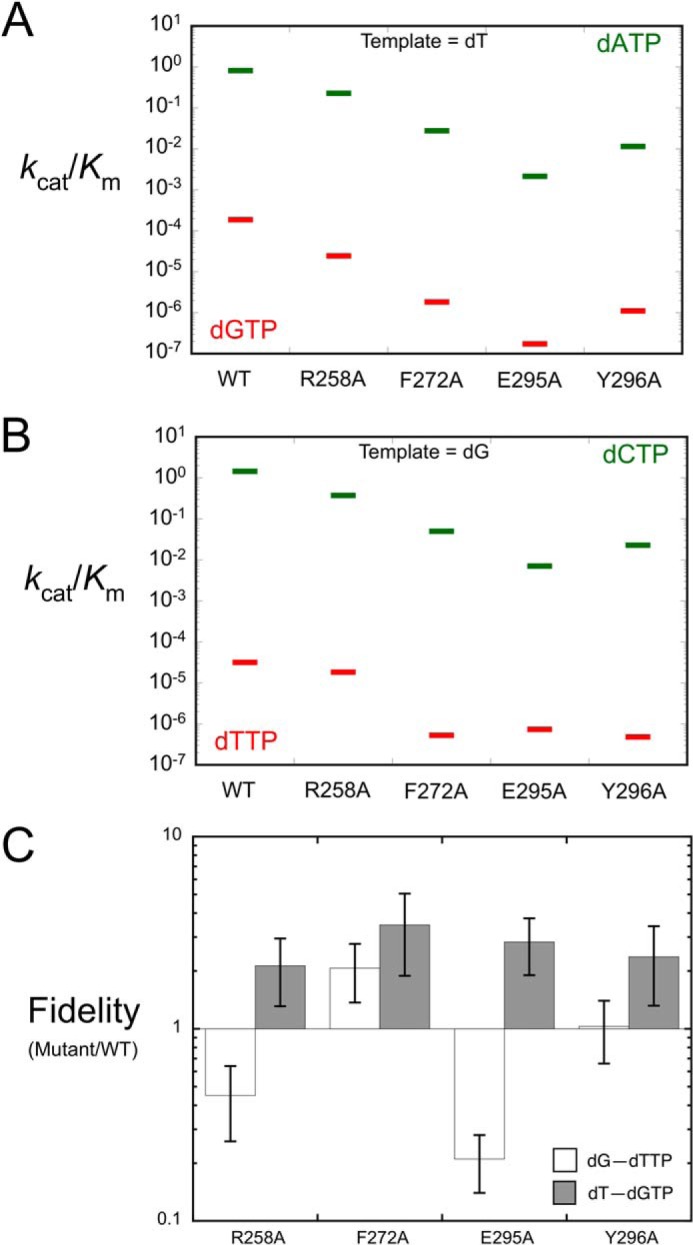
Effect of alanine substitution for Arg-258, Phe-272, Glu-295, and Tyr-296 on catalytic efficiency and substrate discrimination. A, a discrimination (57) plot for 1-nucleotide gap filling (templating dT). The catalytic efficiencies (kcat/Km, μm−1s−1) for correct (green) dATP insertion are compared with those for incorrect (red) dGTP insertion. The distance between these points on the ordinate is a measure of substrate discrimination. B, a discrimination plot for one-nucleotide gap filling (templating dG). The catalytic efficiencies (kcat/Km, μm−1s−1) for correct (green) dCTP insertion are compared with those for incorrect (red) dTTP insertion. C, the relative (mutant/wild-type) fidelity (efficiency-correct/efficiency-incorrect) of the mutants was calculated from the data tabulated in Table 1. The bars above 1 represent an increase in fidelity, and those below 1 exhibit a decrease in fidelity.
The catalytic efficiency of correct insertion for the E295A mutant was reduced by >200-fold, and the activity of E295K enzyme was too low to determine steady-state kinetic parameters. As described below, kinetic parameters for these mutants were estimated by single-turnover analyses (Table 2). As reported previously (32), D192A has no activity even under single-turnover conditions where the enzyme exceeds substrate DNA. The single-turnover rate of the glutamate mutant (i.e. D192E) was decreased 10,000-fold relative to wild-type enzyme (Table 2).
TABLE 2.
Single-turnover kinetic summary for single nucleotide gap-filling DNA substrates
The results represent the best-fit parameters (S.E.) to Equation 1 as described under “Experimental Procedures.”
| Enzyme | Template-dNTPa | kpol | Kd,dNTP | kpol/Kd |
|---|---|---|---|---|
| s−1 | μm | 10−4 μm−1s−1 | ||
| Wild type | dT-dATP | 7.6 (0.3) | 4.6 (0.7) | 16,500 (2,600) |
| dG-dCTP | 3.3 (0.2) | 1.6 (0.4) | 20,625 (5,300) | |
| D192E | dG-dCTP | 2.0 × 10−4 (6 × 10−6)b | ||
| R258A | dT-dATP | 18.0 (0.3) | 25 (2) | 7,200 (590) |
| dG-dCTP | 7.2 (0.2) | 8.4 (0.8) | 8,570 (850) | |
| E295A | dG-dTTP | 9.55 × 10−5 (1.2 × 10−5) | 550 (30) | 0.0017 (0.0002) |
| dT-dGTP | 1.58 × 10−4 (2.0 × 10−6) | 215 (5) | 0.0073 (0.0002) | |
| E295K | dG-dCTP | 3.1 × 10−3 (2.0 10−4) | 5 (2) | 6.2 (2.5) |
| dG-dTTP | 1.35 × 10−5 (1.6 × 10−6) | 250 (90) | 0.00054 (0.0002) |
a Template nucleotide-incoming nucleoside triphosphate.
b Observed rate constant at 250 μm dCTP.
Role of the Mutants in Substrate Discrimination
Substrate specificity is determined by comparing the respective catalytic efficiencies (kcat/Km) for alternate substrates. The fidelity of nucleotide insertion is generally expressed as the ratio of catalytic efficiencies for correct and incorrect nucleotides (i.e. eff.correct/eff.incorrect). A discrimination plot for single nucleotide gap-filling DNA synthesis by wild-type and mutant enzymes is shown in Fig. 2 (panels A and B). In these plots the distance between the respective catalytic efficiencies (correct and incorrect nucleotides) is a measure of substrate discrimination; i.e. the greater the distance, the greater the discrimination. For the alanine mutants, the catalytic efficiencies, correct and incorrect, were decreased relative to wild-type enzyme; R258A < F272A < Y296A < E295A. Because both correct and incorrect nucleotide insertion were decreased to approximately the same extent, the effects on fidelities were modest (<5-fold relative to wild-type enzyme; Fig. 2C).
Single-turnover Kinetic Analysis of R258A
The turnover number, kcat, is a composite of different rate-limiting steps for insertion of correct and incorrect nucleotides and, therefore, should not be compared directly. Likewise, there may be a change in the rate-limiting step for correct nucleotide insertion when analyzing a mutant enzyme. For wild-type pol β, catalytic cycling for correct nucleotide DNA synthesis is limited by insertion (kpol) and product release (koff,DNA) (25). This precludes direct determination of the intrinsic rate constant for insertion by presteady-state methods as the observed exponential rate constant of the burst phase includes a contribution from product release. Alternatively, a single-turnover analysis (E ≫ DNA) can be used to eliminate catalytic cycling and directly measure kpol and Kd,dNTP (37). Under this condition, the single-exponential time courses for correct nucleotide gap-filling DNA synthesis is dependent on dATP concentration (Fig. 3A). A secondary plot of the nucleotide concentration dependence of kobs provides estimates of kpol and Kd,dNTP (Equation 1; Fig. 3B; Table 2).
FIGURE 3.

dATP concentration dependence of single nucleotide incorporation by wild-type pol β. A, wild-type enzyme (1 μm) was preincubated with DNA (pol/DNA = 10) and rapidly mixed with 0.2 (▾), 1.1 (▴), 2 □), 11 (■), 20 (○), or 110 μm (●) dATP. Reactions were quenched at the indicated times, and the products were isolated and quantified as described under “Experimental Procedures.” The solid lines represent the best fit of the data to a rising exponential. B, a secondary plot of the dATP concentration dependence of the observed first-order rate constants measured in panel A. The data were fitted to a hyperbola (Equation 1) to yield a Kd,dATP of 4.6 μm and kpol of 7.6 s−1 (Table 2).
Because Arg-258 forms a salt bridge with Asp-192 in the binary DNA complex, this acidic side chain is not available to provide the necessary ligands for the active site metals needed for catalytic activation (Fig. 1B). Modeling of the N-subdomain closing motion upon binding a correct nucleotide identified several conformational transitions that may occur as Arg-258 releases Asp-192 and forms hydrogen bonds with Glu-295 and Tyr-296 (36). Accordingly, the Arg-258/Asp-192 interaction represents a key barrier that must be overcome to permit formation of an active catalytic site. If the events required for this enzyme activation pathway are kinetically or thermodynamically significant, alanine substitution for Arg-258 could lower the barrier for catalytic activation and/or might augment the population of activated complex (see “Discussion”), resulting in an increase in the observed (i.e. measured) rate of nucleotide insertion.
In the presence of a saturating concentration of the correct nucleotide (dATP), the single-exponential time course provides a measure of kpol (Fig. 4A). Although the observed rate of correct nucleotide insertion was dependent on the identity of the incoming nucleotide, the rate was significantly more rapid for the R258A mutant than wild-type enzyme (Fig. 4). From the nucleotide concentration dependence of the observed rate constants for these single-exponential time courses, the binding affinity for the incoming nucleotide was 5-fold lower for the R258A mutant relative to wild type (Table 2). This results in a 2.3-fold loss in the specificity constant (kpol/Kd), similar to that determined by the steady-state kinetic approach (kcat/Km; Table 1).
FIGURE 4.

Effect of alanine substitution for Arg-258 on the rate of correct nucleotide insertion. Wild-type (WT) enzyme or the R258A mutant was preincubated with single nucleotide-gapped DNA (pol/DNA = 10) and rapidly mixed with a saturating concentration ([dNTP] > 8 Kd) of the appropriate complementary nucleotide as described under “Experimental Procedures.” The first-order time courses were fitted to a rising exponential equation. A, time course of product formation (dATP insertion) for wild-type (●) and R258A (○) enzymes. The best-fit single-order rate constants (kpol) were 5.5 and 14.9 s−1 for wild-type and R258A enzymes, respectively. B, effect of alanine substitution for Arg-258 on kpol for dATP or dCTP insertion as determined by single-turnover analysis (Table 2). The kpol for the R258A mutant is 2.4 ± 0.1 and 2.3 ± 0.2 greater than WT enzyme for formation of dT-dATP and dG-dCTP base pairs, respectively.
Single-turnover Kinetic Analysis of F272A
The decrease in the efficiency of correct nucleotide insertion observed for the F272A mutant (Table 1) suggests that nucleotide insertion rather than product release is rate determining during steady-state turnover. This was verified by directly measuring kpol employing single-turnover conditions as described above. The time course indicates that dATP insertion occurs at a similar rate as that observed for the steady-state rate (Fig. 5 and Table 1).
FIGURE 5.

Effect of alanine substitution for Arg-258 with the F272A enzyme on the rate of correct nucleotide insertion. The F272A or double alanine (R258A/F272A) mutant enzymes were preincubated with single nucleotide-gapped DNA (pol/DNA = 10) and rapidly mixed with a saturating concentration ([dNTP] > 8 Kd) of dATP as described under “Experimental Procedures.” Under these conditions, the first-order time courses were fitted to a rising exponential equation. The time courses were identical with kpol of 0.45 s−1.
These results with the single alanine mutants indicate that Arg-258 modulates wild-type insertion negatively (decreases insertion), whereas Phe-272 facilitates insertion. If the only role of Phe-272 is to insulate Arg-258 and Asp-192, then eliminating the Arg-258 and Asp-192 interaction through alanine substitution for Arg-258 should rescue the loss in activity observed with alanine substitution for Phe-272. However, if Phe-272 has a different or an additional role, then the double mutant will behave like the F272A mutant. The single-turnover time course performed with saturating dATP indicates that kpol for the double alanine mutant R258A/F272A is identical to that for single F272A mutant (Fig. 5). Thus, Phe-272 provides an additional role distinct from insulating Asp-192 and Arg-258.
Single-turnover Kinetic Analysis of E295K
The poor insertion efficiency of the gastric cancer-associated E295K mutant precluded a steady-state kinetic analysis. Using conditions adequate for single-turnover analyses that utilize high enzyme concentrations, insertion (kpol) and dNTP binding (Kd,dNTP) can be assessed. Under these conditions where catalytic cycling does not occur, events at the polymerase active site are measured. The 2400-fold loss in catalytic efficiency (kpol/Kd) with the lysine mutant is completely due to the inability to insert the correct nucleotide (kpol) as the affinity for the correct nucleotide is not diminished (Table 2). The specificity constant for misinsertion of dTTP opposite a templating guanine was 11,500-fold lower than for dCTP insertion. This suggests that the fidelity of this mutant is ∼4-fold lower than wild-type enzyme in this DNA sequence context. The poor insertion efficiency for this mutant enzyme precludes it from posing a direct mutagenic threat to the genome but could pose an indirect threat as a trans-dominant inhibitor of base excision DNA repair.
Structural Characterization of R258A and E295K
Crystallographic structures of binary single nucleotide-gapped DNA and ternary (+dUMPNPP) substrate complexes were determined for two mutants (i.e. R258A and E295K) that would be expected to alter the dynamics of the residues participating in hydrogen bonding during catalytic cycling (Table 3).
TABLE 3.
Crystallographic data and refinement statistics
| Complexa |
||||
|---|---|---|---|---|
| Binary |
Ternary |
|||
| R258A | E295K | R258A | E295K | |
| PDB code | 4R63 | 4R64 | 4R65 | 4R66 |
| Data collection | ||||
| a (Å) | 54.4 | 53.5 | 50.8 | 55.0 |
| b (Å) | 79.3 | 79.0 | 79.9 | 79.2 |
| c (Å) | 54.8 | 54.5 | 55.5 | 54.9 |
| β (°) | 105.8 | 106.1 | 107.3 | 92.9 |
| Resolution range (Å) | 50-1.85 (1.92-1.85)b | 50-2.20 (2.28-2.20) | 50-1.95 (2.02-1.95) | 50-2.25 (2.33-2.25) |
| Rmergec | 9.9 (48.4)d | 5.8 (28.9) | 7.5 (39.2) | 8.9 (25.2) |
| Completeness (%) | 99.7 (99.0) | 95.9 (93.7) | 91.6 (63.7) | 98.7 (89.9) |
| I/σI | 13.8 (2.8) | 16.6 (3.8) | 17.4 (2.6) | 14.7 (3.3) |
| No. of observed reflections | 233,174 | 79,350 | 96,563 | 79,343 |
| No. of unique reflections | 38,158 (3,750) | 21,541 (2,114) | 28,340 (1,958) | 22,091 (2,002) |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Refinement | ||||
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.007 | 0.008 | 0.007 | 0.008 |
| Bond angles (°) | 1.15 | 1.05 | 1.14 | 1.12 |
| Rworke (%) | 18.0 | 19.0 | 18.7 | 18.1 |
| Rfreef (%) | 23.2 | 24.3 | 22.2 | 24.8 |
| Average B factor (Å2) | ||||
| Protein | 21.2 | 40.5 | 23.6 | 31.7 |
| DNA | 21.4 | 34.7 | 31.4 | 27.2 |
| Solvent | 28.7 | 42.2 | 30.5 | 34.1 |
| dUMPNPP | NAg | NA | 15.5 | 39.9 |
| Ramachandran analysish | ||||
| Favored (%) | 97.5 | 95.4 | 99.1 | 96.3 |
| Allowed (%) | 100 | 99.1 | 100 | 98.8 |
a The binary single nucleotide-gapped DNA complexes have deoxyadenosine in the coding-unpaired position of the template strand. The ternary complex includes the non-hydrolysable dUMPNPP analog.
b Highest resolution shell.
c Rmerge =100ΣhΣi|Ih,i − Σh|ΣhΣiIh,i, where Ih is the mean intensity of symmetry-related reflections Ih,i.
d Numbers in parentheses refer to the highest resolution shell of data (10%).
e Rwork = 100Σ‖Fobs| − |Fcalc‖/Σ|Fobs|.
f Rfreeinsertion is partially limited by at least two kinetic steps for a 5% subset of reflections withheld from refinement.
g NA, not applicable.
h Data were determined by MolProbity (58).
The global conformation of the binary and ternary complex structures of the R258A mutant is nearly identical to that of wild-type enzyme (r.m.s.d. for all Cα = 0.16 and 0.20 Å with the open binary and closed ternary wild-type complexes). Like wild-type enzyme, the global conformation of the binary DNA complex is open, whereas the ternary complex is in a closed conformation (Fig. 6A). Except that the arginine has been replaced with alanine, the other signaling residues superimpose with their wild-type counterparts (Fig. 6B).
FIGURE 6.
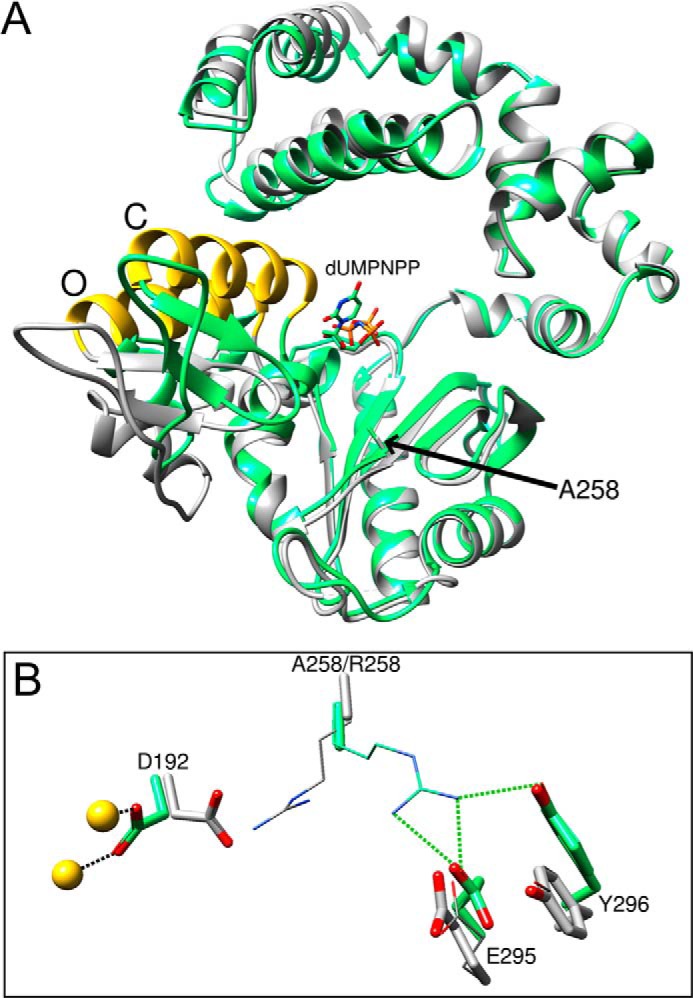
Crystallographic structures of binary and ternary substrate complexes of the R258A mutant. A, ribbon representation of the superimposed structures of the binary DNA (gray) and ternary (green; +dUMPNPP) structures. The superimposed structures (r.m.s.d. = 0.88 Å; 219 Cα) indicate that the binary DNA complex is in the open (O) conformation, whereas the ternary substrate complex is closed (C) as judged by the position of α-helix N (gold). The position of the Ala-258 (A258) side chain is indicated. The DNA is omitted for clarity. B, key active site residues of the binary DNA (gray carbons) and ternary (+dUMPNPP; green carbons) complexes of the R258A mutant (stick representation) are superimposed with those from wild-type (thin stick representation) enzyme (PDB ID 3ISB, binary complex; 2FMS, ternary complex). The r.m.s.d. for the binary and ternary complexes was 0.16 (327 Cα) and 0.20 Å (326 Cα), respectively. Active site magnesium ions in the ternary complex of the mutant are colored yellow.
In contrast to the lack of significant structural perturbation observed with the R258A mutant, the E295K mutant exhibited significant structural differences in the ternary substrate complex compared with wild-type enzyme. Although the open binary complex was similar to wild-type enzyme, the ternary substrate complex remained in the open conformation upon binding a complementary non-hydrolysable incoming nucleotide (Fig. 7A). In this case, two manganese ions coordinate the triphosphate moiety of the incoming nucleotide, whereas the Watson-Crick edge of the uracil base hydrogen binds with the templating adenine. The nascent base pair exhibits a severe buckling (Fig. 7B). As a result, O3′ of the primer terminus hydrogen bonds with Arg-258, effectively removing this essential catalytic atom from the active site (Fig. 7C).
FIGURE 7.

Crystallographic structures of binary and ternary substrate complexes of the E295K mutant. A, ribbon representation of the superimposed structures of the binary DNA (gray) and ternary (green; +dUMPNPP) structures. The superimposed structures (r.m.s.d. = 0.64 Å; 322 Cα) indicate that the ternary substrate complex is in the open conformation typically observed for binary DNA complexes of pol β. The position of the Lys-295 (K295) side chain is indicated, and the first residue (residue 10) that can be observed in the structure is labeled (10). The DNA and dUMPNPP are omitted for clarity. B, active site structural comparison of the ternary substrate complexes of E295K mutant (green stick representation) and wild-type (thin gray stick representation) enzyme (PDB ID 2FMS). The open/closed position of the α-helix N is shown for these enzymes (r.m.s.d. = 0.84 Å, 201 Cα). The templating nucleotide (t0) and its upstream neighbor (t-1) are shown for the mutant enzyme, whereas the primer terminus (p-1) is shown for both the mutant and wild-type enzymes. The magnesium ions in the wild-type enzyme are shown as gray spheres, and the manganese ions of the mutant complex are shown as purple spheres. The triphosphate moiety of the incoming dUMPNPP in the mutant structure hydrogen bonds to Arg-183 (R183), whereas the O4 (uracil) is within hydrogen bonding distance to N1 and N6 of the templating adenine. C, key active site residues of the superimposed binary (thin gray stick representation) and ternary (green stick representation) complexes of the E295K mutant are shown. The primer terminus (PT) of the ternary complex is geometrically distorted removing O3′ from a catalytically relevant position. This inactive sugar position is stabilized with a hydrogen bond with Arg-258.
DISCUSSION
Residue Conservation
The residues examined here were based on their proposed role in protein conformational adjustments as a result of substrate binding. Consequently, their functional role could be in substrate binding, substrate discrimination, and/or catalysis. The high conservation exhibited by these residues when compared with all members of the DNA polymerase X-family is consistent with a critical functional role for these residues (Table 4). Interestingly, these residues do not appear to be highly conserved when compared with the other three human X-family enzymes. This later observation is consistent with the divergent functional roles these enzymes have in the cell (38). Likewise, these other human enzymes (pol λ, pol μ, and terminal deoxynucleotidyltransferase) do not exhibit an open to closed subdomain repositioning when they form a ternary substrate complex (39–41).
TABLE 4.
X-family conservation of human pol β residues involved in active site signaling
| X-family conservationa | DNA polymerase β |
|||||
|---|---|---|---|---|---|---|
| Asp-192 | Arg-258 | Phe-272 | Glu-295 | Tyr-296 | Arg-283 | |
| Humanb | 1.00 | 0.00 | 0.33 | 0.33 | 0.00 | 1.00 |
| Overall | 0.96 | 0.50 | 0.66 | 0.64 | 0.47 | 0.93 |
a Taken from Bienstock et al. (59).
b Residue conservation of three other human X-family members (pol λ, pol μ, and terminal deoxynucleotidyltransferase) relative to pol β.
Not surprisingly, the strongest kinetic consequences were observed with the two most conserved residues, Asp-192 and Arg-283. Alanine substitution for these residues effectively eliminates activity. Whereas activity is not measurable for the metal binding ligand D192A (Table 1), catalytic efficiency is decreased >104-fold for the alanine mutant of Arg-283 (21). The high conservation of these residues among all X-family DNA polymerases indicates that they have an essential role in addition to their role in conformational changes associated with subdomain motions.
Structural Insights
The high resolution crystallographic structures of the binary and ternary precatalytic complexes of the R258A mutant are very similar to those of the wild-type enzyme. As illustrated in Fig. 6B, the side-chain conformations of the residues that propagate a hydrogen bonding cascade in the open and closed polymerase conformations are similar to that observed with the corresponding binary and ternary complex structures of wild-type enzyme. Removing the potential for a salt bridge with Asp-192 or Glu-295 apparently does not have a strong influence on the position of these side chains in the binary or ternary complexes. Because these structures represent static endpoints, intermediate events must be isolated or trapped with mutant enzymes, substrate analogs, or through computational studies.
In contrast to the structures of the R258A mutant, the ternary complex structure of the lysine mutant of Glu-295 is unique. The E295K variant of pol β has been identified in gastric (33) and colon carcinomas (42) and shown to have low activity (Table 2) (43, 44). Although the overall conformation is similar to that of the open ternary complex observed previously for the R283K mutant with a correct incoming nucleotide (2), the structure of the ternary complex of E295K exhibits two Mn2+ ions that coordinate the phosphate moiety of the incoming dUMPNPP (Fig. 7B). In contrast, binding of the second Mn2+ ion to the R283K mutant resulted in a closed ternary complex. Importantly, the trapped open complex of the E295K mutant indicates that Arg-258 can hydrogen bond with O3′ of the primer terminus that has moved to a catalytically inactive position (Fig. 7C). A similar open ternary complex of wild-type enzyme with two manganese ions has been reported for pol β inserting dCMPNPP opposite 2′-fluoro-N7-methylguanine (45). However, in this case, O3′ of the primer terminus coordinates the catalytic metal rather than interacting with Arg-258. A structure of an open mismatch E295K ternary complex structure has been reported by Eckenroth et al. (43) indicating that Arg-258 and Asp-256 can interact with O3′ of the primer terminus through a water-mediated hydrogen bond. This results in an inactive conformation as O3′ is misaligned and Asp-256 cannot coordinate the catalytic metal.
The structural results support a model where both Arg-258 and Glu-295 influence active site conformational equilibria between active and inactive conformations. Glu-295 stabilizes the closed active complex upon forming a ternary substrate complex, and altering this interaction(s) can result in an inactive conformation(s); that is, open N-subdomain and displaced primer terminus stabilized by Arg-258. Consequently, activity of this mutant is severely decreased. In contrast, Arg-258 interacts with a metal binding ligand (Asp-192) in the binary DNA complex and is observed to stabilize an inactive complex in the ternary complex of the E295K mutant. Accordingly, removal of these possible inactivating interactions by alanine substitution potentially enhances the population of active ternary substrate complex that would result in the observed apparent increase in insertion (Fig. 4). An apparent increase in the activity of the R258A mutant on a homopolymeric template-primer system had also been reported (46).
Conformational Activation and Deactivation
Because many DNA polymerases appear to utilize substrate-induced conformational changes to align catalytic groups, an induced fit mechanism has been proposed to describe substrate specificity. Good substrates optimize the active site by aligning catalytic atoms, whereas poor substrates deter catalysis through the misalignment of reactive atoms. Although protein and substrate conformational adjustments can be rapid, the equilibrium with non-catalytic complexes can influence polymerase fidelity (47, 48).
Because single nucleotide insertion appears to be limited by a chemical step rather than conformational adjustments (16, 49), the increased rate of insertion exhibited by the R258A mutant must be due to a change in the thermodynamic equilibrium with non-productive complexes rather than a kinetic change in the rate-limiting step. Scheme 1 illustrates structural adjustments that occur in the enzyme and/or substrates of the ternary substrate complex. In this oversimplified scheme, DNAEdNTP represents the active ternary substrate complex that irreversibly forms product (kpol). F represents an inactive enzyme conformation (e.g. open conformation), whereas the substrates may be bound in a catalytically active or inactive state (denoted by their subscript or superscript position; the inactive forms are highlighted in red). In this scenario the measured single-turnover rate of nucleotide insertion is kobs = kpol(DNAEdNTP/ET) where ET represents the sum of all ternary substrate complexes. Accordingly, the measured rate constant is not only dependent on the intrinsic rate constant for nucleotide insertion (kpol) but also on the relative concentration of active ternary substrate complex. If this complex is in equilibrium with non-productive complexes, then the measured rate underestimates the true intrinsic rate constant. Although the rate-limiting step for the forward reaction is the chemical step, altering the conformational equilibrium with alternate non- or less-productive forms of the ternary substrate complex would decrease the observed rate.
SCHEME 1.
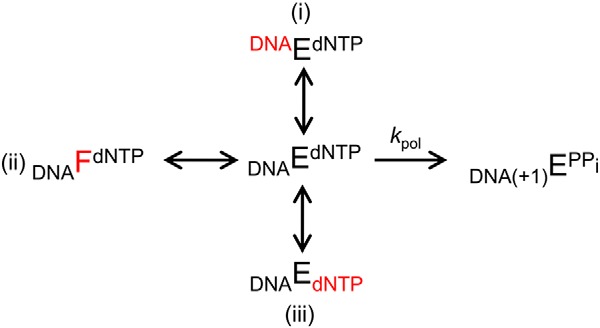
The inactive ternary complex DNAEdNTP (complex i) represents a complex where the DNA substrate is not correctly aligned (e.g. primer terminus in the incoming nucleotide binding pocket) (13), DNAFdNTP (complex ii) represents a protein conformational defect (e.g. open conformation) (2), and DNAEdNTP (complex iii) represents a complex where the incoming dNTP is not positioned correctly (e.g. poor metal coordination (Fig. 7B) (2). In reality, these substrate and protein irregularities would be expected to impact one another (i.e. protein conformation influence substrate alignment). However, it serves to illustrate the simple idea that enzyme-substrate complexes are not a simple homogeneous species. The open and closed forms of many DNA polymerases have long been recognized (1), and single-molecule studies have revealed a spectrum of polymerase populations in varying states of “openness” (50). Using a fluorescence probe attached to the N-subdomain (i.e. fingers of A-family DNA polymerases), Tsai and Johnson (48) identified a unique substrate complex conformation induced by binding an incorrect nucleotide they termed “misaligned.” Thus, DNA polymerases can prevent misinsertion through inducing an inactive complex with an irregular substrate. For example, incorrect nucleotide binding to pol β results in a closed complex where the templating nucleotide moves upstream vacating the coding position and repositioning the primer terminus away from the active site, effectively deterring misinsertion (10). For pol β, other conformational changes that occur before chemistry have been identified through protein (18) and DNA fluorescence changes (17, 51). In these later studies, the R258A mutant was shown to alter pre- and post-chemistry fluorescence changes. Interestingly, Lys-472 of human pol λ (52) and Arg-273 of Leishmania infantum pol β (53) have been observed to stabilize inactive conformations of the primer terminus in crystallographic binary and ternary substrate complexes, respectively.
Since chemistry is rate-limiting for single nucleotide insertion of a correct nucleotide for pol β determined by single-turnover analysis, the increase in the observed rate is thermodynamic rather than an intrinsic change in the rate-limiting step; i.e. a change in the concentration of DNAEdNTP rather than a change in kpol. Thus, alanine substitution for Arg-258 results in less inactive ternary complex.
Although it might seem counterproductive for a DNA polymerase to be in rapid equilibrium with “non-productive” complexes, it can be an advantage in situations where alternate substrates must be accommodated. It would be a practical strategy for enzymes that are confronted with a DNA substrate that displays structural diversity. It is well known that DNA polymerase function is dependent on DNA structure as illustrated by the strong sequence dependence of fidelity.
Fidelity
A mutant enzyme with an altered fidelity indicates that the modified side chain plays a unique role in correct and/or incorrect nucleotide insertion. If a side chain plays a similar role for both correct/incorrect insertions, the catalytic efficiency for each will be altered to the same extent and in the same direction (increase or decrease) so that fidelity will be unaffected. Although altering the residues highlighted in this study by site-directed mutagenesis has profound effects on catalytic efficiency for correct insertion, there is a similar effect on incorrect nucleotide insertion so that the mutant enzymes exhibit little or small effects on fidelity (Fig. 2).
In contrast to the low or modest effects observed with the mutants characterized here, alanine substitution for Arg-283 results in a highly error-prone but catalytically compromised DNA polymerase (19, 54, 55). For this mutant, the loss in fidelity is entirely due to the loss in correct insertion efficiency, whereas incorrect insertion was hardly affected (5). Thus, Arg-283 has a key role during correct nucleotide insertion but plays no or a limited role in incorrect nucleotide insertion.
Enzyme Side-chain Versatility
Structural comparison of the open binary DNA complex with the precatalytic ternary substrate complex suggests that the Phe-272 side chain insulates Arg-258 from Asp-192 in the ternary complex thereby promoting enzyme activation (Fig. 1B). The observation that the double alanine mutant R258A/F272A does not recover the diminished insertion rate of the single F272A mutant (Fig. 5) indicates that the primary role of Phe-272 is not to isolate Asp-192 and Arg-258 from one another. Because the aromatic ring of Phe-272 contacts the sugar of the incoming nucleotide, replacing the large hydrophobic surface with a single methyl group would be expected to alter active site interactions in the vicinity of the chemical reaction. Consistent with this interpretation, previous characterization of a leucine substitution at residue 272 did not observe a loss in the insertion rate of a correct nucleotide, indicating that the longer aliphatic side chain did not dramatically distort the active site (56). Because Phe-272 is also situated on α-helix M of the N-subdomain, it undergoes a small rotation as pol β transitions between open and closed states (Fig. 1B) (4). Thus, Phe-272 plays unique roles depending on the conformational/catalytic state of the enzyme. It appears to be involved in 1) the open/closed enzyme conformational transition, 2) precise positioning of the incoming nucleotide, and 3) insulating Asp-192 and Arg-258 from one another.
Lys-280 also appears to play disparate roles in correct nucleotide insertion depending on the identity of the templating nucleotide. Whereas this side chain contributes key interactions with templating purines, these interactions are far less important with templating pyrimidines (5). Accordingly, it should not be surprising to discover that an enzyme side chain has multi-faceted roles during DNA synthesis that depend on the identity of the incoming nucleotide and DNA sequence.
Acknowledgments
We thank Drs. K. Bebenek and B. D. Freudenthal for critical reading of the manuscript and valuable discussions. Molecular graphics images were produced using the Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 GM-103311).
This work was supported, in whole or in part, by National Institutes of Health Grants Z01-ES050158 and Z01-ES050161 (Intramural Research Program, NIEHS) and in association with the National Institutes of Health Grant U19CA105010.
The atomic coordinates and structure factors (codes 4R63, 4R64, 4R65, and 4R66) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- pol
- DNA polymerase
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviations
- UMPNPP
- 2′-deoxyuridine-5′-[(α,β)-imido]triphosphate.
REFERENCES
- 1. Doublié S., Sawaya M. R., Ellenberger T. (1999) An open and closed case for all polymerases. Structure 7, R31–R35 [DOI] [PubMed] [Google Scholar]
- 2. Freudenthal B. D., Beard W. A., Wilson S. H. (2012) Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure 20, 1829–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freudenthal B. D., Beard W. A., Shock D. D., Wilson S. H. (2013) Observing a DNA polymerase choose right from wrong. Cell 154, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beard W. A., Wilson S. H. (2014) Structure and mechanism of DNA polymerase β. Biochemistry 53, 2768–2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beard W. A., Shock D. D., Yang X.-P., DeLauder S. F., Wilson S. H. (2002) Loss of DNA polymerase β stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity. J. Biol. Chem. 277, 8235–8242 [DOI] [PubMed] [Google Scholar]
- 6. Sawaya M. R., Pelletier H., Kumar A., Wilson S. H., Kraut J. (1994) Crystal structure of rat DNA polymerase β: evidence for a common polymerase mechanism. Science 264, 1930–1935 [DOI] [PubMed] [Google Scholar]
- 7. Beard W. A., Shock D. D., Wilson S. H. (2004) Influence of DNA structure on DNA polymerase β active site function: extension of mutagenic DNA intermediates. J. Biol. Chem. 279, 31921–31929 [DOI] [PubMed] [Google Scholar]
- 8. Sawaya M. R., Prasad R., Wilson S. H., Kraut J., Pelletier H. (1997) Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36, 11205–11215 [DOI] [PubMed] [Google Scholar]
- 9. Batra V. K., Beard W. A., Shock D. D., Krahn J. M., Pedersen L. C., Wilson S. H. (2006) Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure 14, 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Batra V. K., Beard W. A., Shock D. D., Pedersen L. C., Wilson S. H. (2008) Structures of DNA polymerase β with active site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell 30, 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bose-Basu B., DeRose E. F., Kirby T. W., Mueller G. A., Beard W. A., Wilson S. H., London R. E. (2004) Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase β. Biochemistry 43, 8911–8922 [DOI] [PubMed] [Google Scholar]
- 12. Berlow R. B., Swain M., Dalal S., Sweasy J. B., Loria J. P. (2012) Substrate-dependent millisecond domain motions in DNA polymerase β. J. Mol. Biol. 419, 171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirby T. W., DeRose E. F., Cavanaugh N. A., Beard W. A., Shock D. D., Mueller G. A., Wilson S. H., London R. E. (2012) Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucleic Acids Res. 40, 2974–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dunlap C. A., Tsai M.-D. (2002) Use of 2-aminopurine and tryptophan fluorescence as probes in kinetic analyses of DNA polymerase β. Biochemistry 41, 11226–11235 [DOI] [PubMed] [Google Scholar]
- 15. Kim S.-J., Beard W. A., Harvey J., Shock D. D., Knutson J. R., Wilson S. H. (2003) Rapid segmental and subdomain motions of DNA polymerase β. J. Biol. Chem. 278, 5072–5081 [DOI] [PubMed] [Google Scholar]
- 16. Bakhtina M., Lee S., Wang Y., Dunlap C., Lamarche B., Tsai M.-D. (2005) Use of viscogens, dNTPαS, and rhodium(III) as probes in stopped-flow experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase β. Biochemistry 44, 5177–5187 [DOI] [PubMed] [Google Scholar]
- 17. Bakhtina M., Roettger M. P., Tsai M.-D. (2009) Contribution of the reverse rate of the conformational step to polymerase β fidelity. Biochemistry 48, 3197–3208 [DOI] [PubMed] [Google Scholar]
- 18. Towle-Weicksel J. B., Dalal S., Sohl C. D., Doublié S., Anderson K. S., Sweasy J. B. (2014) Fluorescence resonance energy transfer studies of DNA polymerase β: the critical role of fingers domain movements and a novel non-covalent step during nucleotide selection. J. Biol. Chem. 289, 16541–16550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beard W. A., Osheroff W. P., Prasad R., Sawaya M. R., Jaju M., Wood T. G., Kraut J., Kunkel T. A., Wilson S. H. (1996) Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J. Biol. Chem. 271, 12141–12144 [DOI] [PubMed] [Google Scholar]
- 20. Ahn J., Werneburg B. G., Tsai M. D. (1997) DNA polymerase β: structure-fidelity relationship from pre-steady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry 36, 1100–1107 [DOI] [PubMed] [Google Scholar]
- 21. Beard W. A., Shock D. D., Vande Berg B. J., Wilson S. H. (2002) Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J. Biol. Chem. 277, 47393–47398 [DOI] [PubMed] [Google Scholar]
- 22. Patterson T. A., Little W., Cheng X., Widen S. G., Kumar A., Beard W. A., Wilson S. H. (2000) Molecular cloning and high-level expression of human polymerase β cDNA and comparison of the purified recombinant human and rat enzymes. Protein Expr. Purif. 18, 100–110 [DOI] [PubMed] [Google Scholar]
- 23. Beard W. A., Wilson S. H. (1995) Purification and domain-mapping of mammalian DNA polymerase®. Methods Enzymol. 262, 98–107 [DOI] [PubMed] [Google Scholar]
- 24. Cavanaugh N. A., Beard W. A., Wilson S. H. (2010) DNA polymerase β ribonucleotide discrimination: insertion, misinsertion, extension, and coding. J. Biol. Chem. 285, 24457–24465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vande Berg B. J., Beard W. A., Wilson S. H. (2001) DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase β: implication for the identity of the rate-limiting conformational change. J. Biol. Chem. 276, 3408–3416 [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. Beard W. A., Shock D. D., Batra V. K., Pedersen L. C., Wilson S. H. (2009) DNA polymerase β substrate specificity: side chain modulation of the “A-rule.” J. Biol. Chem. 284, 31680–31689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 30. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 31. Wu S., Beard W. A., Pedersen L. G., Wilson S. H. (2014) Structural comparison of DNA polymerase architecture suggests a nucleotide gateway to the polymerase active site. Chem. Rev. 114, 2759–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Date T., Yamamoto S., Tanihara K., Nishimoto Y., Matsukage A. (1991) Aspartic acid residues at positions 190 and 192 of rat polymerase β are involved in primer binding. Biochemistry 30, 5286–5292 [DOI] [PubMed] [Google Scholar]
- 33. Iwanaga A., Ouchida M., Miyazaki K., Hori K., Mukai T. (1999) Functional mutation of DNA polymerase β found in human gastric cancer: inability of the base excision repair in vitro. Mutat. Res. 435, 121–128 [DOI] [PubMed] [Google Scholar]
- 34. Yang L., Beard W. A., Wilson S. H., Broyde S., Schlick T. (2002) Polymerase β simulations suggest that Arg-258 rotation is a slow step rather than large subdomain motions per se. J. Mol. Biol. 317, 651–671 [DOI] [PubMed] [Google Scholar]
- 35. Yang L., Beard W. A., Wilson S. H., Broyde S., Schlick T. (2004) Highly organized but pliant active site of DNA polymerase β: compensatory mechanisms in mutant enzymes revealed by dynamics simulations and energy analyses. Biophys. J. 86, 3392–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Radhakrishnan R., Arora K., Wang Y., Beard W. A., Wilson S. H., Schlick T. (2006) Regulation of DNA repair fidelity by molecular checkpoints: “gates” in DNA polymerase β's substrate selection. Biochemistry 45, 15142–15156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beard W. A., Wilson S. H. (2006) Structure and mechanism of DNA polymerase β. Chem. Rev. 106, 361–382 [DOI] [PubMed] [Google Scholar]
- 38. Moon A. F., Garcia-Diaz M., Batra V. K., Beard W. A., Bebenek K., Kunkel T. A., Wilson S. H., Pedersen L. C. (2007) The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair 6, 1709–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moon A. F., Pryor J. M., Ramsden D. A., Kunkel T. A., Bebenek K., Pedersen L. C. (2014) Sustained active site rigidity during synthesis by human DNA polymerase μ. Nat. Struct. Mol. Biol. 21, 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garcia-Diaz M., Bebenek K., Krahn J. M., Kunkel T. A., Pedersen L. C. (2005) A closed conformation for the Pol λ catalytic cycle. Nat. Struct. Mol. Biol. 12, 97–98 [DOI] [PubMed] [Google Scholar]
- 41. Gouge J., Rosario S., Romain F., Beguin P., Delarue M. (2013) Structures of intermediates along the catalytic cycle of terminal deoxynucleotidyltransferase: dynamical aspects of the two-metal ion mechanism. J. Mol. Biol. 425, 4334–4352 [DOI] [PubMed] [Google Scholar]
- 42. Donigan K. A., Sun K.-W., Nemec A. A., Murphy D. L., Cong X., Northrup V., Zelterman D., Sweasy J. B. (2012) Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 287, 23830–23839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eckenroth B. E., Towle-Weicksel J. B., Sweasy J. B., Doublié S. (2013) The E295K cancer variant of human polymerase β favors the mismatch conformational pathway during nucleotide selection. J. Biol. Chem. 288, 34850–34860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lang T., Dalal S., Chikova A., DiMaio D., Sweasy J. B. (2007) The E295K DNA polymerase β gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol. Cell. Biol. 27, 5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koag M.-C., Kou Y., Ouzon-Shubeita H., Lee S. (2014) Transition-state destabilization reveals how human DNA polymerase β proceeds across the chemically unstable lesion N7-methylguanine. Nucleic Acids Res. 42, 8755–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menge K. L., Hostomsky Z., Nodes B. R., Hudson G. O., Rahmati S., Moomaw E. W., Almassy R. J., Hostomska Z. (1995) Structure-function analysis of the mammalian DNA polymerase β active site: role of aspartic acid 256, arginine 254, and arginine 258 in nucleotidyl transfer. Biochemistry 34, 15934–15942 [DOI] [PubMed] [Google Scholar]
- 47. Johnson K. A. (2008) Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J. Biol. Chem. 283, 26297–26301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsai Y.-C., Johnson K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sucato C. A., Upton T. G., Kashemirov B. A., Batra V. K., Martínek V., Xiang Y., Beard W. A., Pedersen L. C., Wilson S. H., McKenna C. E., Florián J., Warshel A., Goodman M. F. (2007) Modifying the β,γ leaving-group bridging oxygen alters nucleotide incorporation efficiency, fidelity, and the catalytic mechanism of DNA polymerase β. Biochemistry 46, 461–471 [DOI] [PubMed] [Google Scholar]
- 50. Santoso Y., Joyce C. M., Potapova O., Le Reste L., Hohlbein J., Torella J. P., Grindley N. D., Kapanidis A. N. (2010) Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 107, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bakhtina M., Roettger M. P., Kumar S., Tsai M. D. (2007) A unified kinetic mechanism applicable to multiple DNA polymerases. Biochemistry 46, 5463–5472 [DOI] [PubMed] [Google Scholar]
- 52. Bebenek K., Garcia-Diaz M., Zhou R.-Z., Povirk L. F., Kunkel T. A. (2010) Loop 1 modulates the fidelity of DNA polymerase λ. Nucleic Acids Res. 38, 5419–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mejia E., Burak M., Alonso A., Larraga V., Kunkel T. A., Bebenek K., Garcia-Diaz M. (2014) Structures of the Leishmania infantum polymerase β. DNA Repair 18, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Osheroff W. P., Beard W. A., Wilson S. H., Kunkel T. A. (1999) Base substitution specificity of DNA polymerase β depends on interactions in the DNA minor groove. J. Biol. Chem. 274, 20749–20752 [DOI] [PubMed] [Google Scholar]
- 55. Osheroff W. P., Beard W. A., Yin S., Wilson S. H., Kunkel T. A. (2000) Minor groove interactions at the DNA polymerase β active site modulate single-base deletion error rates. J. Biol. Chem. 275, 28033–28038 [DOI] [PubMed] [Google Scholar]
- 56. Li S. X., Vaccaro J. A., Sweasy J. B. (1999) Involvement of phenylalanine 272 of DNA polymerase β in discriminating between correct and incorrect deoxynucleoside triphosphates. Biochemistry 38, 4800–4808 [DOI] [PubMed] [Google Scholar]
- 57. Beard W. A., Batra V. K., Wilson S. H. (2010) DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res. 703, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bienstock R. J., Beard W. A., Wilson S. H. (2014) Phylogenetic analysis and evolutionary origins of DNA polymerase X-family members. DNA Repair 22, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]