

Background: lynx1 reduces sensitivity of α4β2 nAChRs in vitro and also reduces nicotinic responses in vivo.
Results: The GPI protein, lynx1, affects α4/α4 dimer formation in the ER, altering plasma membrane α4β2 stoichiometry.
Conclusion: nAChR modulation can occur as early as the ER, by biasing the starting material for receptor assembly.
Significance: Acute pharmacology and behavior caused by PM nAChRs may be modified by molecules governing nAChR assembly.
Keywords: Cys Loop Receptor, Glycosylphosphatidylinositol (GPI Anchor), Nicotinic Acetylcholine Receptors (nAChR), Snake Venom, Toxin
Abstract
Glycosylphosphatidylinositol-anchored neurotoxin-like receptor binding proteins, such as lynx modulators, are topologically positioned to exert pharmacological effects by binding to the extracellular portion of nAChRs. These actions are generally thought to proceed when both lynx and the nAChRs are on the plasma membrane. Here, we demonstrate that lynx1 also exerts effects on α4β2 nAChRs within the endoplasmic reticulum. Lynx1 affects assembly of nascent α4 and β2 subunits and alters the stoichiometry of the receptor population that reaches the plasma membrane. Additionally, these data suggest that lynx1 shifts nAChR stoichiometry to low sensitivity (α4)3(β2)2 pentamers primarily through this interaction in the endoplasmic reticulum, rather than solely via direct modulation of activity on the plasma membrane. To our knowledge, these data represent the first test of the hypothesis that a lynx family member, or indeed any glycosylphosphatidylinositol-anchored protein, could act within the cell to alter assembly of a multisubunit protein.
Introduction
Cholinergic systems are involved in diverse functions including mood, anxiety, learning and memory, addiction, and movement disorders (1). The nicotinic acetylcholine receptors (nAChRs)2 of the cholinergic system are comprised of 15 different subunits (α1–10 and β1–4) that assemble into both homopentameric receptors, such as (α7)5, or (α9)5, and heteropentameric receptors, such as α4β2 (2). Subunit composition, and even the position of specific subunits within pentamers, can influence the biophysical, pharmacological, and cell biological properties of the receptor, with profound effects on information processing in neurons that express these nAChRs (3).
Subunit composition influences the properties of the two possible pentameric stoichiometries of α4β2 nAChRs, the most abundant heteromeric nAChR subtype in the brain. In a terminology that emphasizes their differences with respect to acute pharmacology, the (α4)3(β2)2 receptors are known as low sensitivity (LS) receptors, whereas (α4)2(β2)3 receptors are known as high sensitivity (HS) receptors (3, 4). These two stoichiometries exhibit different desensitization kinetics and respond differently to chronic nicotine treatment. HS receptors are up-regulated in response to nicotine, whereas LS receptors are unaffected. This is thought to play an important role in mechanisms of nicotine addiction (5, 6). Additionally, subunit composition could possibly influence subcellular localization of receptors on the plasma membrane (PM).
The lynx family proteins are a subset of the Ly6/uPAR superfamily. This superfamily is related to the elapid snake venom toxin genes, such as α-bungarotoxin, consisting of a three-fingered folding motif and multiple internal disulfide bonds (7, 8). α-Bungarotoxin and other such toxins exemplify a highly conserved receptor binding motif that apparently evolved from lynx proteins (7–9). α-Bungarotoxin and other toxins have become widely used probes for the investigation of the properties of nAChR. Snakes inject these toxins into their prey; in contrast, the role of a toxin-like molecule expressed within a cell together with nAChRs has not been widely studied. Since the discovery of lynx1, many other lynx family members have been found, and several include GPI anchors (1, 9, 10). The GPI-linked lynx1 and lynx2 molecules are presumably present in membranes early in the exocytotic pathway, such as the endoplasmic reticulum (ER). Therefore, the existence of the GPI anchor on lynx1 and lynx2 proteins could contribute to the structure, composition, or function of nascent nAChRs as well.
The differential contributions of lynx genes to brain function are influenced to a large degree by the regions of expression. For instance, lynx1 is widely expressed throughout the brain, with high levels in the cerebral cortex and hippocampus, playing a role in learning and neuronal plasticity (11–13). The lynx2 gene, in contrast, has a more restricted expression with high levels in the fear/anxiety structure of the amygdala, and lynx2 KO mice display elevated fear and anxiety-related behaviors (14). Selectivity for nAChR subunits (α7 and β2 nAChRs) has also been found for lynx1 proteins and could provide another level of regulation not yet well explored.
GPI-anchored neurotoxin-like receptor binding proteins, such as lynx modulators, are confined to a volume just a few nanometers above the extracellular face of the membrane. This topology positions them to exert effects by binding to the extracellular portion of nAChRs. These actions have previously been thought to proceed on the PM. Among the pharmacological effects of small molecule ligands is pharmacological chaperoning of α4β2 nAChRs; this proceeds when the ligands enter the ER (6) and, perhaps, the Golgi (15). Thus far, there have been no reports of lynx modulators exerting an effect inside cells. Here, we investigated the hypothesis that lynx1 also exerts an effect on α4β2 nAChRs within the ER. To our knowledge, these data represent the first report of a lynx family member, or indeed any GPI-anchored protein, acting within the cell to alter assembly of multisubunit proteins.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Mouse lynx1 cDNAs including an N-terminally fused fluorescent tag (eGFP, mCherry, or SEP) were constructed by PCR amplification using the forward primer 5′-gcctgcctgtggcccaggctatggtgagcaagggcgag-3′ and the reverse primer 5′-aggcacacacgtggcactccagcttgtacagctcgtccatgcc-3′, which overlap with both sequences before and after the N-terminal signal sequence of the lynx1-coding sequence (see Fig. 1A). Four XFP-lynx1 constructs were made with four different linkers separating the fluorescent tag from mature lynx1 (these are available from Addgene): 1) no additional sequences, 2) Gly-Ser, 3) Gly-Gly-Ser-Gly, and 4) Tyr-Ser-Asp-Leu (YSDL). All four variants are fluorescent, indicating that the mCherry fluorophore folds properly. All experiments presented in this paper utilized the YSDL linker version of the construct because it has been used previously with another GPI-linked fusion protein, GFP-CD59 (16, 17),3 and it was the first construct we validated via electrophysiology. This PCR product was then cloned directly into the vector containing the lynx1 gene using Pfu-Turbo DNA polymerase. We used mouse α4-WT, a4-GFP, α4-mCherry, β2-WT, β2-GFP, and β2-mCherry cDNA clones inserted into pCI-NEO vectors as described previously (18).
FIGURE 1.

Lynx1 is present in ER in addition to the PM. A, schematic of GFP, mCherry, or SEP insertion after the N-terminal signal sequence of lynx1. B–E, cultured mouse neurons transfected with various fluorescently tagged proteins. B, both GFP-lynx1 and mCherry-lynx1. C, GFP-lynx1 and tau1-mCherry, a marker for microtubules, partially co-localize. D, GFP-lynx1 and GalT-mCherry, indicating that the majority of lynx1 is outside the Golgi. E, SEP-lynx1 and ER-dsRED, at both pH 7.4 and 5.5. Partial quenching of SEP at pH 5.5 indicates that lynx1 is present in the ER as well as on the plasma membrane. F, topology of an α4- or β2-GFP construct. α4- or β2-GFP nAChRs were labeled with SEP at the extracellular N-terminus in a previous study (24), and with GFP inserted into the intracellular M3-M4 loop (present study). The cartoon of nAChR derives from PDB 2BG9, and the cytoplasmic portions are incomplete. G, co-immunoprecipitation of GFP-tagged α4 subunits with an anti-GF antibody indicates that lynx1 binds and forms stable complexes with α4-GFP both with β2 present (lane 1) and with no other nAChR subunits present (lane 2). α4 subunits alone are unable to leave the ER; thus lynx1 must bind in the ER. lynx1 was not detectable when either co-transfected with β2-GFP (lane 3) or alone (lane 4).
Tissue Culture and Transfection
Mouse neuroblastoma-2a (N2a) cells were cultured using standard techniques and maintained in DMEM (Invitrogen) supplemented with 10% FBS. N2a cells were plated by adding 90,000 cells to poly-d-lysine-coated 35-mm glass bottom imaging dishes (MatTek, Ashland, MA). The next day, plasmid DNA was mixed with cationic lipids by adding 500–750 ng of each nAChR plasmid and lynx1 construct to 4 μl of Lipofectamine 2000 transfection reagent in 200 μl of OptiMEM (Invitrogen). After 20 min at room temperature, the transfection mixture was added to N2a cells in 1 ml of plating medium and incubated at 37 °C for 24 h. Dishes were rinsed twice with plating medium and incubated at 37 °C for an additional 24 h before imaging or electrophysiological experiments. HEK293 cells were also cultured using standard cell techniques as detailed above. N2a cells and HEK293 cells were obtained from ATCC (Manassas, VA).
Primary mouse embryonic day 16.5 neurons were extracted from mouse embryos and plated on 35-mm polylysine-coated glass bottom culture dishes (MatTek) in a neuronal medium containing Neurobasal, B27 (Invitrogen), and GlutaMAX supplemented with 3% equine serum. We defined this initial plating as day in vitro zero (DIV 0). Neurons were plated at a density of 60,000 cells/dish. On day 4 in vitro, neurons were treated with 1 μm cytosine arabinoside to halt the division of glial cells. Neurons were maintained via 50% exchange with feeding medium (Neurobasal, B27, and GlutaMAX) twice per week. On day 9 in vitro, plasmids were mixed in 100 μl of OptiMEM, and 4 μl of Lipofectamine-2000 was mixed with a separate 100-μl aliquot of OptiMEM. After 5 min at room temperature, the separate solutions were mixed together and kept at room temperature for an additional 25 min. Neurons were transfected with 750 ng of each nAChR plasmid and of XFP-lynx1 plasmid. After 3 h at 37 °C, transfection medium was replaced with neuronal feeding medium. Neurons were imaged on day 10 in vitro.
Co-immunoprecipitation
HEK293 cells were plated (on day 1) at a density of 4 million cells/10-cm dish. The cells were transfected on day 2 with 5 μg of each construct DNA using ExpressFect reagent (Denville Scientific, South Plainfield, NJ) according to the manufacturer's protocol. On day 3, medium was aspirated gently and replaced with fresh growth medium. Cells were harvested on day 4 by removing cells from 37 °C incubator, placing on ice, scraping cells from dish with a cell scraper (Corning Inc., Corning, NY), and transferring cells to a 15-ml conical tube for centrifugation and lysis. Cells were lysed in 1 ml of ice-cold Nonidet P-40 extraction buffer (50 mm Tris, pH 7.4, 50 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 1 mm EGTA, 1% P8340, and 4 mm PMSF) by gently pipetting up and down 25 times. Cell lysate was centrifuged for 5 min at 4 °C and then combined with preformed complexes of protein A Dynabeads (Invitrogen) and rabbit-anti-GFP antibody (A11122; Molecular Probes, Invitrogen) for immunoprecipitation at room temperature on a rocker for 1 h. Dynabead-antibody-antigen complexes were removed from the supernatant via magnet and washed. Complexes were dissociated by incubating in reducing buffer (50% Laemmli sample buffer; Bio-Rad) for 10 min at 70 °C. Dissociated samples were removed from beads via magnet, allowed to cool on ice, and loaded into an SDS-PAGE gel for Western blot analysis.
Western Blot
Samples were run on an SDS-PAGE protein gel, transferred to a nitrocellulose membrane, blocked with blocking buffer (TBS with 0.1% Tween 20 + 5% dry milk), and probed with goat-anti-lynx primary antibody (sc-23060; Santa Cruz Biotechnology, Santa Cruz, CA). Of three antibody lots purchased from the supplier, only one gave satisfactory results; unfortunately that lot is no longer available. Membranes were probed with secondary IR800 anti-goat antibody (926–32214; Li-Cor Biosciences, Lincoln, NE). The membranes were then imaged in an Odyssey near infrared imager (Li-Cor Biosciences).
Confocal Microscopy
N2a cells and neurons were imaged as described previously (19, 20) in L15 buffer (150 mm NaCl, 4 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm HEPES, and 10 mm d-glucose, pH 7.4), a CO2-independent medium. Briefly, cells were imaged with a Nikon (Nikon Instruments, Melville, NY) C1 laser-scanning confocal microscope system equipped with spectral imaging capabilities and a Prior (Rockland, ME) remote focus device, and a Nikon Plan Apo 60× 1.40 NA oil objective. Pinhole diameter was 30–60 μm, and cells were imaged at 12-bit intensity resolution over 512 × 512 pixels at a pixel dwell time 6.72 μs. GFP was excited using a 488-nm modulated diode laser, and mCherry was excited with an argon laser at 561 nm. In most cases, imaging was carried out using the Nikon C1si DEES grating and spectral detector with 32 parallel photomultiplier tubes. This allowed us to collect spectral images (λ stacks). In such images, each pixel of the X-Y image contains a list of emission intensity values across a range of wavelengths. We collected light at wavelength intervals between 485 and 650 nm, at steps of 5 nm. We used the Nikon EZC1 linear unmixing algorithm to reconstruct GFP and mCherry images. For each pixel of a spectral image, intensity of GFP and mCherry was determined from fluorescence intensity values at the peak emission wavelength derived from the reference spectra.
Phosphotidylinositol phospholipase C (PI-PLC) (catalog no. 37288-19-0, product no. P8804; Sigma-Aldrich) was used to cleave the GPI anchor of lynx1 and thus cleave the protein from the PM. After addition of 0.25 units/ml of PI-PLC to cell culture dish, cells were incubated for 1 h at 37 °C either in a mammalian cell incubator or within a climate-controlled chamber on the microscope stage (for before and after images). After PI-PLC treatment, cells were briefly washed twice with 2 ml of fresh medium and subsequently imaged. An aqueous glycerol solution buffer of 60% v/v glycerol, 10 mm Tris-HCl, 10 mm EDTA, at a pH of 8.0 was used as a control with no PI-PLC enzyme.
Measuring FRET with Donor Recovery after Photobleaching (DRAP)
To examine FRET between various nAChR subunits, the acceptor photobleaching method was used with a modified fluorescence recovery after photobleaching macro built into the Nikon EZC1 imaging software, as described previously (19). In this method, FRET was detected by recording GFP signal increases during incremental photodestruction of mCherry. A spectral image was acquired once before mCherry bleaching and at six time points during mCherry bleaching at 561 nm. Laser power during bleaching was constant at 80%. One bleach scan per cycle was used. This bleaching protocol was optimized to achieve 75–90% photodestruction of mCherry while still enabling us to record incremental increases in GFP emission at each time point. To measure FRET, spectral images were unmixed into their GFP and mCherry components as described above. We measured GFP and mCherry mean intensity throughout the entire cell by selecting the cell perimeter as the boundary of a region of interest in Nikon's EZC1 software. GFP and mCherry components were saved in Excel format, and fluorescence intensities were normalized to the prebleach time point (100%). FRET efficiency (E) was calculated as E = 1 − (IDA/ID), where IDA represents the normalized fluorescence intensity of GFP (100%) in the presence of both donor (GFP) and acceptor (mCherry), and ID represents the normalized fluorescence intensity of GFP in the presence of donor only (complete photo destruction of mCherry). The ID value was extrapolated from a scatter plot of the fractional increase of CFP versus the fractional decrease of YFP. The E values were averaged from several cells per condition.
Mammalian Cell Electrophysiology
N2a cells were co-transfected with nAChR subunits tagged with eGFP to facilitate visual identification of transfected cells. We used an inverted fluorescence microscope (IX71; Olympus) equipped with a mercury lamp (HB-10103AF; Nikon) to visualize the fluorescent cells. Whole cell voltage-clamp currents were recorded using an Axopatch-1D amplifier (Axon Instruments) and digitized with a Digidata 1440A analog to digital converter (Axon) under the control of pClamp 10.0 software (Axon). The pipette solution contained 135 mm potassium gluconate, 5 mm KCl, 5 mm EGTA, 0.5 mm CaCl2, 10 mm HEPES, 2 mm Mg-ATP, and 0.1 mm GTP (pH adjusted to 7.2 with Tris-base, osmolarity to 280–300 mOsm with sucrose). Electrode resistance was 2–6 MΩ. All recordings were obtained at ambient temperature. Data were low pass filtered at 2 kHz and sampled at 10 kHz. ACh was dissolved in an extracellular solution containing 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 10 mm glucose (320 mOsm, pH to 7.3 with Tris-base) and applied to cells using a Picospritzer (20 p.s.i.) or a commercially available microperfusion system (Octaflow II; ALA Scientific Instruments). The holding potential was −50 mV. To minimize agonist-induced desensitization, we applied ACh at ∼3-min intervals and continually perfused the recording chamber with extracellular solution.
PI-PLC was used to cleave lynx1 from the PM. We exposed cells to 0.25 units/ml for 1 h at 37 °C incubation prior to electrophysiology assays.
Statistical Analysis
Concentration-response relations for the ACh-induced fluorescent membrane potential changes and voltage-clamp currents were fit to the Hill equation using nonlinear least squares regression (OriginLab software). The errors reported for the mean values are ± S.E. Statistical significance was determined by either Student's t test or by one-way analysis of variance where multiple pairwise comparisons were conducted. Significant differences are reported at the level of p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
RESULTS
Observations of XFP-lynx1 Reveal Localization Both in the Endoplasmic Reticulum and on the Plasma Membrane
We initially studied an mCherry-lynx1 construct. The mCherry was inserted after the N-terminal signal sequence, followed by a linker with sequence YDSL, and by the mature lynx1 protein sequence as shown in Fig. 1A. This linker was previously characterized on an analogous fluorescent GPI-linked fusion protein, GFP-CD59 (16). After electrophysiological validation of the mCherry-linked construct (see Figs. 3 and 5 below), we made two additional constructs by replacing the mCherry sequence with that of GFP or superecliptic pHluorin (SEP).
FIGURE 3.

Electrophysiology in N2a cells. lynx1-WT and mCherry-lynx1 have similar effects on α4β2. A–C, representative traces from N2a cells transfected with α4-GFP, β2-WT, and either empty vector (A), lynx1-WT (B), or mCherry-lynx1 (C) where ACh was puffed for 100 ms. D, both lynx1-WT and mCherry-lynx1 decrease peak currents at 300 μm ACh when compared with empty vector. E, traces from N2a cells when transfected with α4β2 and either lynx1-WT or mCherry-lynx1. F and G, addition of lynx1 variants shifts the concentration-response curve of ACh on α4β2 nAChRs to the right, toward a greater proportion of the low sensitivity (α4)3(β2)2 stoichiometry. F, concentration-response curves of α4β2 nAChRs when co-transfected with empty vector, lynx1, mCherry-lynx1, or lynx1 and treatment with PLC for 1 h at 37 °C. G, comparison of normalized current between α4β2 and lynx1 variant conditions at 10 μm ACh. 10 μm was selected to maximize activation of high-sensitivity (EC50 ≈ 3 μm) stoichiometry (α4)2(β2)3 receptors, while minimizing activation of the lower sensitivity (EC50 ≈ 50 μm) stoichiometry (α4)3(β2)2 receptors. The maximal response is significantly reduced for the three lynx1 variants. There is no significant difference between lynx1, mCherry-lynx, and lynx1 plus PLC treatment. The numbers within parentheses in the bars represent number of cells per condition.
FIGURE 5.

PLC treatment does not affect ACh-induced currents of cells transfected with α4β2 and lynx1. A–C, representative traces from N2a cells transfected with α4, β2, and either empty vector (A), lynx1 (B), or lynx1 with subsequent PLC treatment (C). Cells were subjected to 100 μm ACh for 10 s to measure both peak current and desensitization (quantified here as the time between 90 and 10% of max current). D, overlay of traces comparing lynx1 with or without PLC treatment. E and F, PLC does not alter peak current (E) or decay time (F) of N2a cells transfected with α4, β2, and lynx1. However, lynx1 does decrease the decay time compared with α4β2 alone both without and with PLC treatment. F presents a shift in 90% to 10% decay time from 6.14 ± 0.55 s for empty vector to 2.35 ± 0.37 s and 2.68 ± 0.46 s for lynx-WT and lynx1-WT + PLC, respectively
To test the similarity of the GFP and mCherry constructs, we co-transfected them together into cultured mouse embryonic day 16.5 cortical neurons (Fig. 1B). The two constructs are co-localized in much of the cytoplasm, both in somata and dendrites. We compared this distribution with that of two other proteins. We co-transfected GFP-lynx1 with tau1-mCherry (Fig. 1C). The tau1-mCherry localizes to microtubules. In many neurons, GFP-lynx1 fluorescence displayed streaks, discontinuities, and open areas not displayed by tau1-mCherry fluorescence (Fig. 1C), suggesting that lynx1 may normally be present in restricted regions. GPI-anchored proteins, including those in the Ly6/uPAR family, are often found in cholesterol-rich membrane domains; as explained in the next paragraph, restriction to cholesterol-rich domains does not fully explain the distribution of lynx1. When co-transfected with GalT-mCherry, GFP-lynx1 showed little overlap (Fig. 1D), indicating that most lynx1 molecules do not accumulate preferentially in the trans-Golgi network.
SEP is a pH-sensitive variant of GFP with a pKa of ∼7.0. SEP is fluorescent at pH 7.4 but is quenched at pH < 7.0 (22). Previously, when linked to the extracellular portion of Cys loop receptors and other membrane proteins, SEP has provided evidence that a portion of the tagged protein is present in intracellular compartments, as well as on the PM (15, 23–25). For the present study, once the SEP construct was verified electrophysiologically (see Figs. 3 and 5 below), the SEP method presented several advantages over conventional studies using immune reagents and cell permeabilization. First, the entire experiment was performed on live neurons or clonal cells. Second, not all neurons or clonal cells express the intended protein after transfection; therefore, it was essential to use each cell as its own control. Because the quenching and dequenching of PM fluorescence is reversible, this was accomplished by imaging the neurons or clonal cells before, during, and after perfusion with low pH. Third, the SEP method, but not the permeabilization/immune method, can distinguish between the presence of the molecule in neutral versus acidic organelles. These considerations rendered the SEP construct the reagent of choice
We used this pH dependence of SEP to visualize and discriminate between SEP-lynx1 molecules residing intracellularly versus those residing on the PM, as shown in Fig. 1E. Five to six minutes after the pH was adjusted to 5.5 by perfusion, we observed that fluorescence decreased only partially in each of 10 cells examined, suggesting that a population of SEP-lynx1 resides in an organelle with pH > 7.0. The two candidate organelles are the ER and the cis-Golgi. The Golgi compartment is much smaller than the ER and does not appear to accumulate appreciable lynx1 (Fig. 1D). Furthermore, the fluorescent image remaining at pH 5.5 overlaps substantially with the image from co-transfected ER-dsRED (Fig. 1E). We conclude that SEP-lynx1 resides at least partially in the ER. Analogous experiments have previously shown that the extracellular face of nAChRs resides partially in the ER (15, 24).
lynx1 Binds α4-GFP in the ER
Previous assays on HEK293T cells transfected with α4, β2 (both FLAG-tagged constructs), and lynx1, immunoprecipitation of either α4 with anti-α4 antibody (mAb299), or immunoprecipitation of both α4 and β2 with anti-FLAG antibodies showed co-immunoprecipitation of nAChRs with lynx1 (26). Here, we demonstrate that lynx1 and α4 also co-immunoprecipitate when the β2 subunit is omitted (Fig. 1G). Because α4 subunits alone do not form a complete, pentameric receptor, they do not efficiently leave the ER (27).
To test whether lynx1 could bind the individual subunits of the α4β2 nAChR and potentially affect assembly, we co-transfected either: 1) α4-GFP, β2-WT, and lynx1; 2) α4-GFP and lynx1; 3) β2-GFP and lynx1; or 4) lynx1 alone into HEK293 cells (Fig. 1, F and G). We probed for lynx1 after precipitating the nAChR subunits with an anti-GFP antibody. We found that when α4 is present, either alone or with β2, lynx1 was pulled down. We could not detect lynx1 when β2-GFP alone was co-transfected with lynx1 and immunoprecipitated or in the control where there was no nAChR subunit co-transfected (Fig. 1G). Because it appears that lynx1 can bind the α4 subunit without the presence of β2, we conclude that lynx1 is able to bind to α4 before pentamers are formed, suggesting that association of lynx1 with α4 occurs in the ER.
lynx1 Stabilizes α4/α4 Dimers, but Not β2/β2 Dimers, in the ER
To measure whether the presence of lynx1 affects receptor formation, we conducted four types of DRAP FRET experiments on mouse N2a cells transfected with nAChR subunits plus or minus lynx1 (Fig. 2). We co-transfected α4-GFP, α4-mCherry, and either empty vector or lynx1 (we did not co-transfect a β subunit). In this case, we measured DRAP between α4/α4 dimers in the ER, because α4 without a β subunit cannot assemble into a full pentamer and subsequently cannot efficiently exit the ER. The addition of lynx1 increased the FRET efficiency between these dimers from 6.7 ± 1.13% (26 cells) to 14.59 ± 1.51% (32 cells), suggesting that lynx1 binds α4 subunits in the ER, increasing dimerization and potentially assisting the formation of the LS stoichiometry (Fig. 2, A, B, and E). In another set of experiments, we obtained FRET measurements between α4-GFP and α4-mCherry in the presence of β2-WT of 9.33 ± 0.85% (40 cells) and 13.09 ± 1.11% (36 cells) with empty vector and lynx, respectively. When we co-expressed fluorescently labeled β2 subunits, β2-GFP and β2-mCherry, either in the presence or absence of α4-WT, the addition of lynx1 did not significantly alter the FRET efficiency (Fig. 2, B1, B2, C1, C2, and E). For β2-GFP/β2-mCherry with no α4-WT or lynx, the FRET efficiency was 25.1 ± 2.79% (31 cells); when lynx was added, the FRET efficiency was 27.5 ± 2.4% (24 cells). For β2-GFP/β2-mCherry with α4-WT, the FRET efficiencies were 18.4 ± 1.3% (29 cells) and 18.5 ± 1.7% (33 cells) for co-transfected empty vector and lynx, respectively. The data are reported here as mean ± S.E. (number of cells per condition). The contrast in FRET efficiencies between the lynx1 condition and the control condition persisted when the control transfection employed GAT1 cDNA instead of empty vector.
FIGURE 2.

DRAP. N2a cells were transfected with either α4-GFP and α4-mCherry (A1, A2, B1, and B2 show exemplar data) or β2-GFP and β2-mCherry (C1, C2, D1, and D2 show exemplar data). The acceptor fluorophore (mCherry) was photobleached, causing an increase in fluorescence of the donor fluorophore (GFP). α4-GFP and α4-mCherry were co-transfected either with empty vector (A1, A2) or with lynx1 (B1, B2). β2-GFP and β2-mCherry were co-transfected either with empty vector (C1 and C2), or with lynx1 (D1 and D2). Lynx1 increases FRET efficiency between α4 subunits, but not between β2 subunits. E, lynx1 increases FRET efficiency between α4-GFP and α4-mCherry subunits when no other subunits are present (top left) and also when co-transfected with β2 (bottom left). lynx1 does not increase FRET efficiency between fluorescently tagged β2 subunits either without α4 present (top right) or with co-transfected α4 (bottom right). F and G, lynx1 has no effect on the ratio of signals, GFP/mCherry for fluorescently tagged α4 (F) or β2 (G). The numbers within parentheses in the bars represent number of cells per condition.
These data show that lynx1 increases the assembly of α4/α4 dimers and does not alter β2/β2 dimer assembly. To assess the effect of lynx1 on fully assembled α4β2 nAChR pentamers, we co-expressed α4-GFP, α4-mCherry, β2-WT, and either empty vector (pcDNA3.1) or lynx1. The addition of lynx1 increased the FRET between the fluorescently labeled α4 subunits, indicating that lynx1 increases the extent to which α4 subunits assemble together in complexes (Fig. 2E). This shift could be due to increased incorporation of the α4 subunit into the auxiliary position of α4β2 fully formed pentamers (LS, (α4)3(β2)2 stoichiometry). It could also be due to increased α4/α4 dimers in the ER. These two outcomes are not mutually exclusive. In fact, the latter (increased α4/α4 dimers in the ER) would likely lead to the former by biasing assembly toward the formation of (α4)3(β2)2 (LS) nAChRs. This would presumably result in more LS receptors being exported from the ER and eventually trafficked to the PM.
A high baseline level of acceptor or low post-acceptor bleaching level of donor could distort the measurements of FRET efficiency. To rule this out, we calculated the ratio between GFP and mCherry expression (Fig. 2, F and G). We achieved ratios consistently between 0.6 to 2.0, which are within the acceptable range for DRAP measurements.
lynx1 Causes a Shift in Population Stoichiometry of the α4β2 nAChR on the Plasma Membrane
Previous studies on lynx1 showed that lynx1 reduces the sensitivity of α4β2 receptors to ACh in heterologous expression systems and also to nicotine responses in vivo (7, 12, 26). However, it has not been previously demonstrated that lynx1 shifts the stoichiometry of α4β2 receptors on the PM by biasing assembly of nascent subunits within the ER. To test this hypothesis, we completed a series of electrophysiological assays (Fig. 3).
These electrophysiological experiments examined the effect of lynx1 and fluorescently tagged XFP-lynx1 on α4β2. We transfected N2a cells with α4-GFP and β2-WT nAChR subunits and either lynx1 (WT or mCherry) or empty vector. The addition of lynx1-WT or mCherry-lynx1 reduced peak current sizes, reduced the proportion of HS receptors, and decreased the decay time (Fig. 3 and Table 1). Addition of lynx1-WT reduced α4β2 nAChR peak current amplitude to ∼40% of the maximal response (from 3280 ± 1320 pA (5 cells) to 1,060, ± 402 pA (6 cells)) for lynx1-WT (Fig. 3, A, B, and D). Addition of mCherry-lynx1 similarly reduced the α4β2 nAChR peak current amplitude to ∼30% of the maximal response (539.4 ± 115 pA (10 cells)) (Fig. 3, A, C, and D). There was no significant difference between lynx1-WT and mCherry-lynx1 in regards to mean peak current amplitude, decay time, or waveform (Fig. 3, D and E). This suggests that the addition of the mCherry fluorophore to lynx1 does not significantly alter its function.
TABLE 1.
Comparison of two-component fits to α4β2 concentration-response relations in N2a cells when co-transfected with empty vector, or one of three lynx1 conditions
lynx1, mCherry-lynx1, or lynx1 plus PLC treatment was used. Fits were generated under two assumptions: 1) all HS receptors, and no LS receptors, are activated at 10 μm ACh, and 2) Hill coefficients were constrained to ≤1.5. Fitted parameters for ACh concentration-response relations of α4β2 when co-transfected with empty vector, lynx1-wt, mCherry-lynx1, and lynx1-wt after treatment with PI-PLC. Parameters correspond to ACh concentration-response relations in Fig. 3. The EC50 and Hill coefficient values represent the means ± S.E. for the number of measurements (n) obtained. The relative fraction of the high sensitivity component (first component) for the biphasic relations is included.
| α4β2 ACh | HS |
LS |
HS | R2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 | EC50 error | n | n error | EC50 | EC50 error | n | n error | |||
| % | ||||||||||
| WT +pcDNA | 2.34 | 1.38 | 0.86 | 0.43 | 36.9 | 20.1 | 1.5 | 0.76 | 50.3 | 0.97 |
| +lynx-pTracer | 5.42 | 3.36 | 1.5 | 1.18 | 61.4 | 14.1 | 1.5 | 0.44 | 27.0 | 0.99 |
| + mC-lynx | 2.49 | 1.41 | 1.05 | 0.76 | 58.6 | 12.8 | 1.5 | 0.37 | 28.4 | 0.99 |
| Lynx+PLC | 6.55 | 9.47 | 0.54 | 0.41 | 41.9 | 14.7 | 1.5 | 0.37 | 24.9 | 0.99 |
Next we collected concentration-response data to assess changes in α4β2 nAChR stoichiometry. Previous assays have validated that the LS and HS α4β2 nAChR stoichiometries have distinct pharmacological profiles: LS nAChRs and HS nAChRs have different EC50 values with ACh (∼30 and 3 μm, respectively) (5, 28). Without lynx1, α4β2 nAChRs have a two-component concentration-response relation, with a pronounced HS-like behavior at [ACh] ≤ 1 μm (Fig. 3F). The addition of lynx1 (WT or mCherry) produced less pronounced responses at lower [ACh] (Fig. 3F). This suggests that in the presence of lynx1, the HS (α4)2(β2)3 nAChR population is decreased. Therefore, PM α4β2 nAChRs are composed mainly of LS α4β2 nAChRs. The Hill equation fits (Table 1) are consistent with a shift in components. In the absence of lynx1, ∼50% of the response is the HS component (as often observed in α4β2 experiments); but in each of the lynx1 conditions this is reduced to ∼25–28% HS. A compelling contrast is shown for the normalized data at 10 μm ACh (Fig. 3G), which was chosen because this dose produces near maximal responses at HS receptors, with minimal contribution by LS receptor (4, 5).
Ric3, another protein that affects nAChR expression, appears to act catalytically within the ER (29, 30). It has previously been shown that lynx1 does not affect the function of GABAA receptors in oocytes (26); we have not studied this point systematically in the present experiments.
PI-PLC Treatment Cleaves lynx1 from the Plasma Membrane
We cleaved the GPI anchor of lynx1, releasing lynx1 from the PM via treatment with PI-PLC. This allowed us to determine whether acute removal of surface lynx1 affects the response of α4β2 nAChRs. Using confocal imaging, we demonstrate that we can remove lynx1 from the PM of N2a cells expressing mCherry-lynx1 (Fig 4A), or co-expressing α4β2 nAChRs and mCherry-lynx1 (Fig. 4B). Before treatment with PI-PLC, “railroad tracks” of red fluorescence are clear on the borders of processes, indicating mCherry-lynx1 on the PM, and they are quantified by intensities in typical line profiles where the peaks align with the borders. After PI-PLC treatment for 1 h, the membrane fluorescence is decreased and becomes undetectable above background (Fig. 4, A and B). This can be seen clearly through line profiles (Fig. 4, C and D). In control experiments, cells were treated with identical solution lacking PI-PLC, and we observed no reduction in PM railroad track-like appearance or line profile quantification (data not shown).
FIGURE 4.
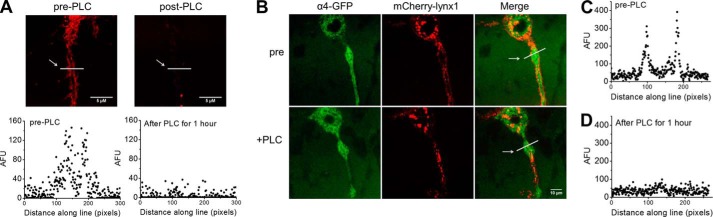
PI-PLC treatment cleaves mCherry-lynx1 from the plasma membrane. A, images of N2a cells transfected with mCherry-lynx1 before (left) and after (right) treatment with PI-PLC. Lower, line profiles quantifying fluorescence levels, demarcated as a white bar in upper images. AFU, arbitrary fluorescence units. B, images of N2a cells transfected with α4-GFP, β2-WT, and mCherry-lynx1 taken with a confocal microscope before (B, upper) and after (B, lower) treatment with PI-PLC. C and D, line profiles quantifying fluorescence levels across a line demonstrate the presence of the railroad track morphology of mCherry-lynx1 on the membrane of N2a cells before treatment with PI-PLC (C) and after (D). The arrows point to the lines used for quantification. 100 pixels = 10 μm. These data are typical of five experiments.
PI-PLC Treatment Does Not Affect Concentration-Response Relations to ACh, Peak Current, or Decay Kinetics of α4β2 + lynx1
Before and after treatment with PI-PLC to cleave lynx1, we performed electrophysiological experiments to measure concentration-response relations to ACh (Fig. 3F). After PI-PLC treatment, the peak currents and concentration response curve were not different when compared with lynx1-WT and mCherry-lynx1 conditions (Fig. 3F). Peak current amplitude at saturating concentrations of ACh (300 μm) and desensitization kinetics (90% to 10% of maximal current) were similar between lynx1-WT plus PI-PLC and lynx1-WT without PI-PLC (Fig. 5). We confirmed earlier observations that the expression of lynx does accelerate desensitization of α4β2 currents (12, 26). However, this acceleration is not reversed by PI-PLC treatment. Overlay of α4β2 nAChRs with lynx1 (treated or not treated with PI-PLC) shows a close overlap and little difference among waveforms (Fig. 5D). These results suggest that the major effects of lynx1 on α4β2 occur before the nAChRs appear on the PM. The data together demonstrate that continued expression of lynx on the PM is not necessary for maintenance of its effects on α4β2 and suggest that lynx1 may exert effects intracellularly by stabilizing α4/α4 dimers in the ER, thus favoring the LS (α4)3(β2)2 stoichiometry.
DISCUSSION
Our data lead to three conclusions. First, lynx1 binds α4 nAChR subunits in the ER and increases α4/α4 dimer formation. Second, lynx1 biases the stoichiometry of α4β2 receptors toward LS (α4)3(β2)2 nAChR pentamers. Finally, the minimal effects of removing lynx1 at the PM suggest that lynx1 exerts its effects on nascent receptors within the ER, thereby influencing receptor assembly and the stoichiometry of the final receptor exported to the PM. To our knowledge, this is the first report of a GPI-anchored protein playing a role in regulating assembly of a multimeric protein through an intracellular mechanism.
It has been previously demonstrated that the expression of lynx1 reduces the sensitivity of α4β2 nAChRs, and knock-out of the lynx1 gene increases sensitivity (12, 26). The molecular mechanisms for these actions, however, had yet to be addressed. Taking into account the relationship between lynx genes and snake venom toxins, a straightforward mechanism of lynx binding to the extracellular portion of the receptor on the cell surface to influence gating has been the assumed mode of action for lynx proteins (7, 8). In this study, we applied new tools to demonstrate that lynx1 is resident both on the cell surface and in the interior of the cell. Apparently, the internal lynx1/α4 interactions are dominant, because removing surface lynx had little effect on receptor function. The most likely mechanism is that lynx1 controls the functional properties of nAChRs by influencing the type and number of receptors during receptor assembly in the ER.
If lynx1 does exert a functional gating effect on PM α4β2 nAChRs, it may be effective after cleavage of the GPI anchor and/or at very high concentrations. Indeed, exogenously applied lynx1 (minus the GPI anchor) alters the peak response of α4β2 nAChRs to nicotine and ACh in vitro (7). Further, previous studies in vivo show that overexpression of secreted lynx1 in mouse cerebellum, a region involved in motor learning, results in enhanced motor learning (13), a behavioral phenotype similar to the lynx1 KO mice. Our results cannot definitively exclude a mechanism by which the lynx1 proteins that were previously bound to receptors prior to PI-PLC treatment remained bound after GPI anchor cleavage. In this scenario, these interactions may have continued to influence gating function, but the concentration of bound lynx molecules was below the detection threshold of our confocal imaging assay.
Single-channel studies of α4β2 and lynx1 in CHO cells indicate a shift in the proportion of channel openings toward higher amplitude, faster inactivating species (26). The two different amplitudes of single channel openings may arise from the two different stoichiometries, where the (α4)3(β2)2 stoichiometry mediates larger amplitude openings (28). The interactions of lynx1 and α4/α4 subunits are likely to increase the internal concentration of total α4/α4 interfaces, and because an α4/α4 interface occurs only in the (α4)3(β2)2 nAChR stoichiometry, this could explain the greater expression of (α4)3(β2)2 nAChRs when lynx1 is present. Because (α4)3(β2)2 nAChRs are also thought to inactivate more rapidly, the accelerated decay of the macroscopic current in cells co-transfected with lynx1 might be explained by this stoichiometry shift. A lynx1-based model of pentameric receptor assembly might be slightly different from those previously put forth. It is generally thought that α4/β2 dimers are formed first, and the fifth position is filled by either an α4 or β2 subunit. The lynx1-nAChR interaction might affect this final step, in favor of incorporating α4 subunits. Increasing the α4/α4 population may influence the expression of fully formed receptors, resulting in both greater selectivity (shift in stoichiometry) and less efficiency (reduction in the total number of receptors).
The ratio of α4 and β2 subunits has been a well documented determinant for α4β2 stoichiometry (24). With the data presented here, it is likely that a preferential affinity for α4 monomers by lynx1 underlies stoichiometry shifts, although we cannot rule out the possibility that α4 and β2 subunit levels are altered by the presence of lynx1. Indeed, if lynx1 is acting as a catalyst for dimer formation, it could be acting on other subunits (e.g. β2), but if those other subunits are at sufficiently high concentrations, the effect of lynx1 may not be detected. There are several reasons we do not believe this to be happening. First, in our system, we do not see a demonstrably higher level of β2 subunit transcription, as compared with α4, or changes in receptor protein levels caused by lynx. Also, lynx1 does not bind when only β2 is overexpressed with lynx in cells, nor does it add to β2 FRET efficiency. These data indicate affinity differences of lynx1 toward α4 over β2 subunits. All things being equal, the addition of lynx1 to the system appears to drive the formation of α4/α4 dimers with a shift in stoichiometry as a consequence of that affinity difference.
Indeed, although we have not detected differences in receptor subunit levels, we do find that β2 subunits appear to make more natural pairs than α4 subunits do; in Fig. 2E, the FRET efficiency is higher in the β2 only condition (irrespective of lynx presence) than the α4 only condition. It appears that α4 subunits benefit from a helper molecule to aid the formation—hence increasing the concentration—of α4/α4 dimers. This might provide a teleology for the lynx1 gene, in its ability to shape nAChR responsiveness through subunit affinity differences.
The elapid neurotoxins, such as α-bungarotoxin and α-cobratoxin, apparently evolved from a lynx gene (9). The selective advantage of the neurotoxins arises from binding as antagonists, to α/non-α interfaces of muscle nAChRs. α-Bungarotoxin also binds to α7 nAChRs via α7/α7 interfaces. This study shows that lynx1 binds to α4/α4 interfaces. We do not yet know at which interfaces and in which subcellular compartment lynx1 binds to other homomultimeric and heteromultimeric nAChRs. It is interesting to note that nicotine binds to α/β interfaces, and prolonged nicotine binding within the ER is thought to up-regulate α4β2 nAChRs by stabilizing the (α4)2(β2)3 stoichiometry (15, 31–33). Our data suggest that lynx1 could oppose some of the effects of chronic nicotine by shifting the dimer concentration toward the α4/α4 species (26). The differential up-regulation by chronic nicotine exposure in the brain may be due in part to the differential expression of lynx1 genes in different neuronal cell types. In the future, we would like to study interactions between lynx1 and nicotine effects.
As noted above, our data remain consistent with the possibility that nAChR-lynx1 interactions, which begin in the ER, could continue throughout the lifetime of the receptor. The diminution of peak ACh-mediated currents caused by lynx1 is somewhat greater than would be explained by loss of the HS component; perhaps lynx1 also affects receptor turnover at the PM. This presents the possibility of a multimodal action of lynx1 on several aspects of cholinergic signaling. This should be taken into account when one considers models of lynx1 action on the function of the cholinergic system in the brain. The control that lynx1 exerts on cholinergic tone through nAChR interactions (1, 9, 11) makes lynx1 a reasonable target for pharmaceutical drug development. Several drug candidates targeting nAChRs are in development (34–37), and some may act by potentiating nAChRs (37–41). One potential drawback of strategies involving agonists or partial agonists is the uncertainty of either receptor activation or desensitization, or both (42, 43). The lynx family proteins appear to increase the rate of desensitization of nAChRs (14, 21), and their removal results in nAChRs exhibiting less receptor desensitization. Therefore, inhibition of the endogenous action of lynx proteins may achieve the possibly desirable effect of increasing cholinergic tone while reducing desensitization. Pharmaceutical development now focuses on compounds acting on PM-localized targets. Our data suggest that it may be important to consider intracellular mechanisms of action as well.
Acknowledgments
We thank Sheri McKinney for providing hippocampal neuron cultures. We also thank R. Srinivasan and M. Starbird for comments and technical advice.
This work was supported, in whole or in part, by National Institutes of Health Grants DA033831, NS034407, EY018502, and MH088550. This work was also supported by Tobacco-related Disease Research Program (TRDRP) Grant 19KT-0032.
P. Satpute and J. Lippincott-Schwartz, personal communication.
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- ER
- endoplasmic reticulum
- N2a
- neuroblastoma 2a
- SEP
- super ecliptic pHluorin
- GPI
- glycosylphosphatidylinositol
- LS
- low sensitivity
- HS
- high sensitivity
- PM
- plasma membrane
- PI-PLC
- phosphotidylinositol-specific phospholipase C
- DRAP
- donor recovery after photobleaching
- PI-PLC
- phosphoinositide phospholipase C.
REFERENCES
- 1. Miwa J. M., Freedman R., Lester H. A. (2011) Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron 70, 20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jensen A. A., Frølund B., Liljefors T., Krogsgaard-Larsen P. (2005) Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 48, 4705–4745 [DOI] [PubMed] [Google Scholar]
- 3. Gotti C., Clementi F., Fornari A., Gaimarri A., Guiducci S., Manfredi I., Moretti M., Pedrazzi P., Pucci L., Zoli M. (2009) Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 78, 703–711 [DOI] [PubMed] [Google Scholar]
- 4. Moroni M., Zwart R., Sher E., Cassels B. K., Bermudez I. (2006) α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 70, 755–768 [DOI] [PubMed] [Google Scholar]
- 5. Nelson M. E., Kuryatov A., Choi C. H., Zhou Y., Lindstrom J. (2003) Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol. 63, 332–341 [DOI] [PubMed] [Google Scholar]
- 6. Srinivasan R., Henderson B. J., Lester H. A., Richards C. I. (2014) Pharmacological chaperoning of nAChRs: A therapeutic target for Parkinson's disease. Pharmacol. Res. 83, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miwa J. M., Ibanez-Tallon I., Crabtree G. W., Sánchez R., Sali A., Role L. W., Heintz N. (1999) lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron 23, 105–114 [DOI] [PubMed] [Google Scholar]
- 8. Lyukmanova E. N., Shenkarev Z. O., Shulepko M. A., Mineev K. S., D'Hoedt D., Kasheverov I. E., Filkin S. Y., Krivolapova A. P., Janickova H., Dolezal V., Dolgikh D. A., Arseniev A. S., Bertrand D., Tsetlin V. I., Kirpichnikov M. P. (2011) NMR structure and action on nicotinic acetylcholine receptors of water-soluble domain of human LYNX1. J. Biol. Chem. 286, 10618–10627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miwa J. M., Lester H. A., Walz A. (2012) Optimizing cholinergic tone through lynx modulators of nicotinic receptors: implications for plasticity and nicotine addiction. Physiology 27, 187–199 [DOI] [PubMed] [Google Scholar]
- 10. Chimienti F., Hogg R. C., Plantard L., Lehmann C., Brakch N., Fischer J., Huber M., Bertrand D., Hohl D. (2003) Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum. Mol. Genet. 12, 3017–3024 [DOI] [PubMed] [Google Scholar]
- 11. Morishita H., Miwa J. M., Heintz N., Hensch T. K. (2010) Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 330, 1238–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miwa J. M., Stevens T. R., King S. L., Caldarone B. J., Ibanez-Tallon I., Xiao C., Fitzsimonds R. M., Pavlides C., Lester H. A., Picciotto M. R., Heintz N. (2006) The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron 51, 587–600 [DOI] [PubMed] [Google Scholar]
- 13. Miwa J. M., Walz A. (2012) Enhancement in motor learning through genetic manipulation of the Lynx1 gene. PLoS One 7, e43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tekinay A. B., Nong Y., Miwa J. M., Lieberam I., Ibanez-Tallon I., Greengard P., Heintz N. (2009) A role for LYNX2 in anxiety-related behavior. Proc. Natl. Acad. Sci. U.S.A. 106, 4477–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henderson B. J., Srinivasan R., Nichols W. A., Dilworth C. N., Gutierrez D. F., Mackey E. D., McKinney S., Drenan R. M., Richards C. I., Lester H. A. (2014) Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. J. Gen. Physiol. 143, 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nichols B. J., Kenworthy A. K., Polishchuk R. S., Lodge R., Roberts T. H., Hirschberg K., Phair R. D., Lippincott-Schwartz J. (2001) Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153, 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cha B., Kenworthy A., Murtazina R., Donowitz M. (2004) The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J. Cell Sci. 117, 3353–3365 [DOI] [PubMed] [Google Scholar]
- 18. Nashmi R., Dickinson M. E., McKinney S., Jareb M., Labarca C., Fraser S. E., Lester H. A. (2003) Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain lneurons. J. Neurosci. 23, 11554–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drenan R. M., Nashmi R., Imoukhuede P., Just H., McKinney S., Lester H. A. (2008) Subcellular trafficking, pentameric assembly and subunit stoichiometry of neuronal nicotinic ACh receptors containing fluorescently-labeled α6 and β3 subunits. Mol. Pharmacol. 73, 27–41 [DOI] [PubMed] [Google Scholar]
- 20. Moss F. J., Imoukhuede P. I., Scott K., Hu J., Jankowsky J. L., Quick M. W., Lester H. A. (2009) GABA transporter function, oligomerization state, and anchoring: correlates with subcellularly resolved FRET. J. Gen. Physiol. 134, 489–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ibañez-Tallon I., Wen H., Miwa J. M., Xing J., Tekinay A. B., Ono F., Brehm P., Heintz N. (2004) Tethering naturally occurring peptide toxins for cell-autonomous modulation of ion channels and receptors in vivo. Neuron 43, 305–311 [DOI] [PubMed] [Google Scholar]
- 22. Miesenböck G., De Angelis D. A., Rothman J. E. (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 [DOI] [PubMed] [Google Scholar]
- 23. Jacob T. C., Bogdanov Y. D., Magnus C., Saliba R. S., Kittler J. T., Haydon P. G., Moss S. J. (2005) Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J. Neurosci. 25, 10469–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richards C. I., Srinivasan R., Xiao C., Mackey E. D., Miwa J. M., Lester H. A. (2011) Trafficking of α4* nicotinic receptors revealed by superecliptic phluorin: effects of a β4 amyotrophic lateral sclerosis-associated mutation and chronic exposure to nicotine. J. Biol. Chem. 286, 31241–31249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khiroug S. S., Pryazhnikov E., Coleman S. K., Jeromin A., Keinänen K., Khiroug L. (2009) Dynamic visualization of membrane-inserted fraction of pHluorin-tagged channels using repetitive acidification technique. BMC Neurosci. 10, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ibañez-Tallon I., Miwa J. M., Wang H. L., Adams N. C., Crabtree G. W., Sine S. M., Heintz N. (2002) Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron 33, 893–903 [DOI] [PubMed] [Google Scholar]
- 27. Whiteaker P., Sharples C. G., Wonnacott S. (1998) Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol. Pharmacol. 53, 950–962 [PubMed] [Google Scholar]
- 28. Tapia L., Kuryatov A., Lindstrom J. (2007) Ca2+ permeability of the (α4)3(β2)2 stoichiometry greatly exceeds that of (α4)2(β2)3 human acetylcholine receptors. Mol. Pharmacol. 71, 769–776 [DOI] [PubMed] [Google Scholar]
- 29. Wang Y., Yao Y., Tang X. Q., Wang Z. Z. (2009) Mouse RIC-3, an endoplasmic reticulum chaperone, promotes assembly of the α7 acetylcholine receptor through a cytoplasmic coiled-coil domain. J. Neurosci. 29, 12625–12635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander J. K., Sagher D., Krivoshein A. V., Criado M., Jefford G., Green W. N. (2010) Ric-3 promotes α7 nicotinic receptor assembly and trafficking through the ER subcompartment of dendrites. J. Neurosci. 30, 10112–10126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lester H. A., Xiao C., Srinivasan R., Son C. D., Miwa J., Pantoja R., Banghart M. R., Dougherty D. A., Goate A. M., Wang J. C. (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry: implications for drug discovery. AAPS J. 11, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nashmi R., Xiao C., Deshpande P., McKinney S., Grady S. R., Whiteaker P., Huang Q., McClure-Begley T., Lindstrom J. M., Labarca C., Collins A. C., Marks M. J., Lester H. A. (2007) Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J. Neurosci. 27, 8202–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Srinivasan R., Pantoja R., Moss F. J., Mackey E. D., Son C. D., Miwa J., Lester H. A. (2011) Nicotine upregulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J. Gen. Physiol. 137, 59–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gault L., Ritchie C., Robieson W., Pritchett Y., Othman A., Lenz R. (2013) Efficacy and safety of the alpha7 agonist ABT-126 in mild-to-moderate Alzheimer's dementia. Alzheimer's Dementia 9, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hauser T. A., Kucinski A., Jordan K. G., Gatto G. J., Wersinger S. R., Hesse R. A., Stachowiak E. K., Stachowiak M. K., Papke R. L., Lippiello P. M., Bencherif M. (2009) TC-5619: An alpha7 neuronal nicotinic receptor-selective agonist that demonstrates efficacy in animal models of the positive and negative symptoms and cognitive dysfunction of schizophrenia. Biochem. Pharmacol. 78, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lieberman J. A., Dunbar G., Segreti A. C., Girgis R. R., Seoane F., Beaver J. S., Duan N., Hosford D. A. (2013) A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology 38, 968–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prickaerts J., van Goethem N. P., Chesworth R., Shapiro G., Boess F. G., Methfessel C., Reneerkens O. A., Flood D. G., Hilt D., Gawryl M., Bertrand S., Bertrand D., König G. (2012) EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology 62, 1099–1110 [DOI] [PubMed] [Google Scholar]
- 38. Timmermann D. B., Grønlien J. H., Kohlhaas K. L., Nielsen E. Ø., Dam E., Jørgensen T. D., Ahring P. K., Peters D., Holst D., Christensen J. K., Malysz J., Briggs C. A., Gopalakrishnan M., Olsen G. M. (2007) An allosteric modulator of the α7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J. Pharmacol. Exp. Ther. 323, 294–307 [DOI] [PubMed] [Google Scholar]
- 39. Harvey A., Fluck A., Giethlen B., Paul D., Schaeffer L. (January 30, 2014) Positive allosteric modulators of the alpha 7 nicotinic acetylcholine receptor and uses thereof. U. S. Patent 20,140,031,395 A1
- 40. Kanner R. (July 29, 2010) α-7 nicotinic acetylcholine receptor allosteric modulators, their derivatives and uses thereof. U. S. Patent 20,100,190,819 A1
- 41. Putman D., Dasse O. (November 14, 2013) Alpha 7 nicotinic acetylcholine receptor allosteric modulators, their derivatives and uses thereof. WO 2013169889 A1 [Google Scholar]
- 42. Lester H. A., Dibas M. I., Dahan D. S., Leite J. F., Dougherty D. A. (2004) Cys-loop receptors: new twists and turns. Trends Neurosci. 27, 329–336 [DOI] [PubMed] [Google Scholar]
- 43. Buisson B., Bertrand D. (2001) Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J. Neurosci. 21, 1819–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]