

Background: GnT-V levels are implicated in regulating cancer stem cells and tumor development.
Results: GnT-V levels via altered Wnt signaling regulate the compartment of colon cancer stem cells and tumor formation.
Conclusion: GnT-V levels modulate Wnt signaling that regulate colon adenoma progression.
Significance: A specific post-translational modification regulates Wnt signaling and colon cancer progression.
Keywords: Cancer Stem Cells, Colon Cancer, Gene Knockout, N-Linked Glycosylation, Wnt Signaling, Apcmin/+ Mice, GnT-V
Abstract
Deletion of GnT-V (MGAT5), which synthesizes N-glycans with β(1,6)-branched glycans, reduced the compartment of cancer stem cells (CSC) in the her-2 mouse model of breast cancer, leading to delay of tumor onset. Because GnT-V levels are also commonly up-regulated in colon cancer, we investigated their regulation of colon CSC and adenoma development. Anchorage-independent cell growth and tumor formation induced by injection of colon tumor cells into NOD/SCID mice were positively associated with GnT-V levels, indicating regulation of proliferation and tumorigenicity. Using Apcmin/+ mice with different GnT-V backgrounds, knock-out of GnT-V had no significant effect on the number of adenoma/mouse, but adenoma size was significantly reduced and accompanied increased survival of Apcmin/+ mice with GnT-V deletion (p < 0.01), suggesting an inhibition in the progression of colon adenoma caused by deletion of GnT-V. Decreased expression levels of GnT-V down-regulated the population of colon (intestine) CSC, affecting their ability for self-renewal and tumorigenicity in NOD/SCID mice. Furthermore, altered nuclear translocation of β-catenin and expression of Wnt target genes were positively associated with expression levels of GnT-V, indicating the regulation of canonical Wnt/β-catenin signaling. By overexpressing the Wnt receptor, FZD-7, in colon cancer cells, we found that FZD-7 receptors expressed N-linked β(1,6) branching, indicating that FZD-7 can be modified by GnT-V. The aberrant Wnt signaling observed after modulating GnT-V levels is likely to result from altered N-linked β(1,6) branching on FZD-7, thereby affecting Wnt signaling, the compartment of CSC, and tumor progression.
Introduction
There is a growing body of evidence that a variety of cancers, including breast and colon carcinoma, may be initiated and maintained by a small subset of cells termed tumor initiating cells or cancer stem cells (CSC)3 that share many features with normal stem cells, including self-renewal and differentiation (1, 2). Small numbers of these cells have the capacity to rapidly produce tumors upon transplantation in immune-compromised mice, whereas the same numbers of non-CSC produce tumors very slowly; CSC may also contribute to tumor recurrence, metastasis, and treatment resistance in some tumors (3–5). Therefore, the characterization of key signaling pathways that regulate CSC is crucial for understanding how CSC regulate tumorigenesis and progression as well as the development of novel treatment strategies that target the CSC. Colon CSC (CCSC) have been identified and isolated from both human colon cancer (6–8) and mouse intestine adenoma (4, 9).
Aberrant N-linked glycosylation of cell surface receptors as a consequence of oncogenic transformation has been widely implicated in regulating tumorigenicity and tumor progression. A family of N-glycans whose expression is controlled by oncogenic signaling pathways and is often up-regulated during malignant transformation of epithelial cells is synthesized by the glycosyltransferase, N-acetylglucosaminyltransferase V (GnT-V or Mgat5, EC 2.4.1.155) (Fig. 1A) (10). Studies have implicated GnT-V levels in regulating tumorigenesis and invasiveness in many types of human and mouse tumors via the modulation of the function of various cell surface receptors and their intracellular signaling pathways (11–16). Our recent results (17) show that deletion of GnT-V delayed her-2-induced mouse mammary tumor onset via down-regulating the size of the compartment of mammary CSC.
FIGURE 1.

Expression levels of GnT-V regulate tumorigenesis-related phenotypes in human colon cancer cell lines. A, depiction of the GnT-V enzymatic reaction and glycan product. The GlcNAc residue transferred by GnT-V is highlighted in red. Swainsonine indirectly blocks GnT-V via inhibition of mannosidase II. B, 2 days after tumor cells were transfected with GnT-V (transient) or with GnT-V siRNA (stable), total RNA was isolated for detection of levels of GnT-V mRNA. For each transcript, the values were normalized to control (GAPDH) and expressed as mean ± S.D. from three independent experiments. C, levels of N-linked β(1,6) branching were detected by L-PHA staining in control and GnT-V-transfected LS180 stable cell lines. Bar, 50 μm. D, both LS180 and HT-29 cells were transfected with GnT-V cDNA and grown on plastic in selection medium for 2–3 weeks, and colony formation was quantified. The number of colonies in six random fields was counted and expressed as the mean ± S.D. *, p < 0.001. Parental: cells without transfection. E, transfected LS180 cells were grown in soft agar for 2–3 weeks, and the number of colonies in six random fields was counted and expressed as the mean ± S.D. *, p < 0.05. F, GnT-V-transfected LS180 cells (1 × 106) were injected subcutaneously into the backs of NOD/SCID mice (n = 6), and tumor growth was observed for up to 8 weeks. *, p < 0.05. Representative H&E-stained sections of tumors at week 8 are shown on the right. Note that the neoplastic cells often form acini (circled) as often seen in colorectal tumors. NECR denotes necrotic area. G, GnT-V-suppressed HT-29 cells (1 × 106) expressing siRNA directed against GnT-V transcripts were injected subcutaneously into the backs of NOD/SCID mice (n = 5), and tumor growth was observed for up to 8 weeks (*, p < 0.05; **, p < 0.01), and representative sections of tumors stained with anti-Ki-67 antibody (recognizing a nuclear protein which is marker of cell proliferation) are shown on the right. Bar, 20 μm.
Although normal colonic epithelia do not stain with L-phytohemagglutinin (L-PHA), a lectin that specifically recognizes N-linked glycan structures catalyzed by GnT-V, staining is observed in adenomas and increases as they progress to carcinoma (18, 19). Patients with colorectal carcinomas that show GnT-V glycan products bound by L-PHA have lowered 5-year survival rates (20). Another study using a glycomics approach identified 30 proteins, including tissue inhibitor of metalloproteinase-1, as an acceptor substrate for GnT-V in human colon carcinoma cells that may be associated with invasiveness (21). So far, the effect of GnT-V levels on colon cancer development and progression in vivo as well as the molecular mechanisms that are involved in this regulation, particularly in regard to CSC, has not been investigated. A mouse model for the study of colon cancer, Apcmin/+ mice, has been utilized to study the oncogenesis and progression of adenomas. This model carries an ethylnitrosourea-induced missense mutation of the adenomatous polyposis coli (Apc) gene at stop codon 850 that leads to truncation of the APC protein (22). A defective APC protein results in the cytoplasmic accumulation and translocation of β-catenin to the nucleus to form a complex with T cell factor/lymphocyte enhancer factor-1 (TCF/LEF-1) transcription factors, consequently activating Wnt target genes (23, 24). The Apcmin/+ mice develop multiple intestinal adenomas relatively rapidly (12 weeks), and in many ways this mouse model mimics human familial adenomatosis polyposis coli, in which the mutated APC gene can often be detected.
In the present study the Apcmin/+ colon cancer model along with colon carcinoma cultured cells were employed to investigate the regulation of GnT-V in colon tumorigenesis and colon adenoma progression. It is likely that expression levels of GnT-V regulate the canonical Wnt/β-catenin signaling pathway by affecting the N-glycosylation of Wnt receptors, which further leads to altered adenoma progression in the Apcmin/+ mice by modulating the population of CCSC.
EXPERIMENTAL PROCEDURES
Cell Lines and Materials
Human colon cancer cell lines LS180, HT-29, and SW480 were from the American Type Culture Collection (Manassas, VA). CHO Lec4 cells were kindly provided by Dr. P. Stanley, Albert Einstein College of Medicine. pSuper vectors containing scrambled and GnT-V siRNA were constructed as described (15). pCMV6-FZD-7 plasmid was from OriGene Technologies; human Wnt-3a was from R&D system, and CCT036477 was purchased from Enzo Life Sciences. Rabbit anti-Ki-67 (Invitrogen), anti-human CD44-PE (CD44 conjugated with R-phycoerythrin) and CD24-FITC (eBioscience), anti-Ascl2 (Millipore), rabbit monoclonal anti-c-Myc (Epitomics), anti-β-actin (Sigma), anti-c-Myc and antin-Axin2 (Cell Signaling), mouse anti-β-catenin (BD), anti-DDK tag monoclonal antibody (OriGene), and antibodies against ERK and phospho-ERK (Santa Cruz Biotechnology) were used in the different experiments.
Mouse Breeding and Adenoma Tumor Tissue Isolation
All procedures used for this study were approved by the Institutional Animal Care and Use Committee of the University of Georgia. Apcmin/+ mice were purchased from The Jackson Laboratory. GnT-V+/− female mice in the C57BL6/J background were described (11, 17) and were mated to C57BL/6J Apcmin/+ male mice to generate C57BL6/J Apcmin/+ mice with GnT-V−/−, GnT-V+/−, and GnT-V+/+ backgrounds. The Apcmin/+ locus was detected by PCR with the primers 5′-TTCCACTTTGGCATAAGGC-3′ and 5′-TTCTGAGAAAGACAGAAGTTA-3′. GnT-V knock-out mice were genotyped as described (17). Mice from different groups were sacrificed at 18–22 weeks and examined for intestinal tumors. The location, number, and size of adenomas in each mouse were determined, and tissue specimens were prepared. Adenomas from each group were sectioned and stained with H&E for histological examination. To evaluate adenoma progression, a survival study of Apcmin/+ mice in different GnT-V backgrounds was performed and evaluated by using Kaplan-Meier survival curve with 28 weeks as the cut-off time for observation (25).
Transfection
Cell transfections were performed with LipofectamineTM 2000 according to the manufacturer's instructions using pMMTVGnT-V or retroviral pSUPER GnT-V siRNA plasmids. 24 h after transfection, cells were placed under G418 (800 μg/ml) selection for 3 weeks. Cells with the highest GFP expression were isolated using fluorescence-activated cell sorting. The human pcDNA3/GnT-V and pCMV6-FZD-7 plasmids were used for transient expression of GnT-V and FZD-7. Cells were incubated for 48 h after transfection and used for the indicated experiments.
Colony Formation and Anchorage-independent Growth Assay
For colony formation assay, cells were transfected with 4 μg of expression plasmids in 6-well plates and transferred into 100-mm culture dishes the next day. Selection was performed 2 days after transfection with G418 (800 μg/ml) for 2 weeks. Colonies were stained with crystal violet and counted under a phase contrast microscope from 5–10 random fields. Assay of cell growth in soft agar was performed using 24-well culture plates coated with two layers of agar at different concentrations (lower layer, 0.7% agar in 0.9% sodium chloride; upper layer, 0.35% soft agar in complete culture medium). 3 × 104 cells were added onto the upper layers of the wells and incubated at 37 °C for 2–3 weeks. The numbers of the colonies that developed in soft agar were counted under a microscope in 5–10 random fields. The data were expressed by the mean value of cells per field in triplicate with two independent experiments.
Wnt Luciferase Reporter Assay
TCF/LEF-1 transcriptional activity was determined by using TOPFLASH and FOPFLASH reporter plasmid kit (Millipore). Tumor cells were cultured on 24-well plates for 24 h and co-transfected with TOPFLASH or FOPFLASH reporter plasmids, a Renilla luciferase plasmid (pGL4.74[hRluc/TK] vector, Promega), and pcDNA3/GnT-V or empty pcDNA3 plasmids using the LipofectamineTM 2000 transfection reagent. Cells were harvested 48 h after transfection, and luciferase activity was measured using the Dual Luciferase Assay kit (Promega). Luciferase activity was normalized for transfection efficiency by renilla activity. Relative TCF/LEF activity is defined as the ratio of TOPFLASH/FOPFLASH.
Hoechst 33342 Staining
Single cell suspensions were prepared in DMEM/F-12 medium (10% FBS, 10 μg/ml insulin, and 5 ng/ml EGF) at the density of 1 × 105/ml and incubated at 37 °C for 60 min. Hoechst 33342 (Sigma) was then added at a final concentration of 2.5 μg/ml, and cells were incubated at 37 °C for another hour. Verapamil (Sigma) was pre-added at a final concentration of 50 μg/ml in to inhibit dye transport in the control reactions (26). After washing once with HBSS, cells were resuspended in HBSS with 5% FBS, stained with propidium iodide (1 μg/ml), and analyzed using flow cytometry.
Flow Cytometry Analysis
Tumor cells were suspended in flow cytometry buffer (PBS containing 1% BSA and 0.01% sodium azide) at 5 × 106/ml and incubated with anti-CD24 conjugated with FITC (CD24-FITC, 1 μg/106 cells) and anti-CD44 conjugated with R-phycoerythrin (CD44-PE, 1 μg/106 cells) for 30 min on ice. After washing 3 times with flow cytometry buffer, cells were resuspended in the same buffer (0.5 ml) and subjected to flow cytometry analysis (27).
Aldefluor Assay
The Aldefluor assay was performed using the ALDEFLUOR kit from Stemcell technologies (28). In brief, dissociated single cells (1 × 106 cells/ml) were incubated in Aldefluor assay buffer containing aldehyde dehydrogenase substrate, bodipyaminoacetaldehyde (1.5 μm) at 37 °C for 30 min. In each experiment a fraction of cells was stained under identical conditions with a specific aldehyde dehydrogenase inhibitor, diethylaminobenzaldehyde (15 μm), as a negative control. After staining with propidium iodide, Alde-positive (CSC) and -negative cells (non-CSC) were analyzed using flow cytometry.
Self-renewal of Colon Cancer Stem cells in Vitro
Single cell suspensions were plated in 6-well ultralow attachment plates (Costar) at a density of 3 × 103 to 2 × 104/ml. Tumorspheroid cultures were grown in a serum-free epithelial growth medium containing B27 (Invitrogen), EGF (20 ng/ml), bovine FGF (20 ng/ml), and heparin (4 μg/ml) for 7–10 day. Tumorspheres were collected and dissociated with trypsin (0.05%)/EDTA (0.53 mm) into single cells for the next suspension culture (26, 29). The size and number of the spheres were evaluated for each experiment.
Limiting Dilution Assay
Spheres were dissociated and seeded in ultra-low adhesion 96-well plates in 0.2 ml of stem cell culture media at a density that ranged from 400 to 1 cells/well. At each cell density 8 wells were plated. After growth for 7–10 days, the percentage of wells not containing spheres at each cell plating density was calculated and plotted against the number of cells per well. Using linear regression analysis, stem cell frequency was calculated and expressed as the number of cells required to form at least one sphere/well (30).
Implantation of Tumor Cells in NOD/SCID Mice
NOD/SCID mice were purchased from The Jackson Laboratory. Subconfluent tumor cells were harvested and resuspended in serum-free HBSS media in a 70-μl volume containing 1–3 × 106 cells. After mice were anesthetized with isofluorane, 70 μl of the single cell suspension mixed with 30 μl of Matrigel was injected subcutaneously into the back of NOD/SCID mice of 6–8 weeks age using a 27-gauge needle (27). Tumor growth and tumor size were measured using calipers once a week.
Immunochemical and Fluorescent Staining
Immunochemical staining was performed using VECTASTAIN® Elite ABC kit (Vector laboratories) following the manufacturer's instructions. For fluorescent staining, cells were cultured on chamber slides, fixed with 4% paraformaldehyde in PBS for 10 min, and permeabilized with 0.05% Triton X-100. After blocking with 10% goat serum, cells were stained with primary antibodies followed by incubation with secondary fluorescence-conjugated anti-mouse or rabbit IgG (1:250). After washing with PBS, the chamber slides were mounted, and the cells were subjected to fluorescence microscopy. Aldefluor-sorted cell staining was performed as previously described (29).
Quantitative PCR Analysis
Total RNA from both human tumor cells and Apcmin/+ tissues was isolated using TRIzol (Invitrogen). The reverse transcription reactions and qRT-PCR analyses were performed using iScriptTM cDNA Synthesis kit and iQTMSYBR®Green Supermix (Bio-Rad), respectively. Primers used in the qRT-PCR analysis are listed in Table 1.
TABLE 1.
Sequences of primers used in qRT-PCR
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Human | ||
| MGAT5 | gagcagatcctggacctcag | gctgtcatgactccagcgta |
| GAPDHa | tggacctgacctgccgtctag | cctccgacgcctgcttcac |
| CCND1 | aactacctggaccgcttcct | ccacttgagcttgttcacca |
| CMYC | ttcgggtagtggaaaaccag | cagcagctcgaatttcttcc |
| LGR5 | ctcttcctcaaaccgtctgc | gatcggaggctaagcaactg |
| ASCL2 | gtgaagctggtgaacttgggc | cagcgtctccaccttgctca |
| AXIN2 | ctccttatcgtgtgggcagt | cttcatcctctcggatctgc |
| APCDD1 | catccagacagcaggtctca | gggcctgaccttacttcaca |
| DKK1 | gccccgggaattactgcaaaaatg | ccggagacaaacagaaccttcttgt |
| ALDH1 | ttggaatttcccgttggtta | ctgtaggcccataaccagga |
| CD133 | agtggcatcgtgcaaacctg | ctccgaatccattcgacgatagta |
| BMI1 | gagggtacttcattgatgccacaac | gctggtctccaggtaacgaacaata |
| SOX2 | tgagcgccctgcagtacaa | gctgcgagtaggacatgctgtag |
| OCT4 | ctggagaaggagaagctgga | caaattgctcgagttctttctg |
| Mouse | ||
| Mgat5 | ccctggaagttgtcctctca | tcctctgccagtgccttaat |
| Gapdha | tgcgacttcaacagcaactc | atgtaggccatgaggtccac |
| Cmyc | ctagtgctgcatgaggagacac | gtagttgtgctggtgagtggag |
| Lgr5 | ccttggccctgaacaaaata | atttctttcccagggagtgg |
| Ascl2 | caggagctgcttgacttttcca | gggctagaagcaggtaggtcca |
| Axin2 | gcagctcagcaaaaagggaaat | tacatggggagcactgtctcgt |
| Keratin20 | gcacagattaaagagctgcaaa | cactgggcattttgtacttgag |
a Normalization control gene.
Glycosidase Treatment of Cell Lysates
Deglycosylation of FZD-7 was performed using whole cell lysates. Cell extracts containing 30 μg of protein in denaturing buffer were incubated at 37 °C with 500 milliunits of endoglycosidase H for 2 h or peptide N-glycosidase F (PNGase F) overnight. The deglycosylated proteins were subjected to SDS-PAGE and immunoblotting with the anti-DDK tag antibody.
Statistical Analysis
Statistical analysis was performed using statistical software R. Significant differences between two groups were determined by the unpaired t test or non-parametric Wilcoxon rank-sum test. p values <0.05 were considered significant.
RESULTS
Regulation of Tumorigenesis-related Phenotypes by Expression Levels of GnT-V in Cultured Colon Cancer Cell Lines
To determine the effects of altering GnT-V activity on colon tumorigenesis in vitro, we first employed human colon cancer cells LS180, HT-29, and SW480, all of which are established cell lines with similar, moderate gene expression of GnT-V detected by qRT-PCR (data not shown). Expression levels of GnT-V were either increased or inhibited in these cells by transfection with GnT-V cDNA or GnT-V siRNA, respectively. Altered expression of GnT-V in these tumor cells was confirmed by detection of GnT-V mRNA transcripts by qRT-PCR; GnT-V glycan products were measured by lectin staining using L-PHA (Fig. 1, B and C). As shown in Fig. 1, D and E, colony formation and anchorage-independent cell growth (grown in soft agar) were significantly enhanced after GnT-V overexpression, indicating increased cell proliferation and anchorage-independent growth caused by GnT-V overexpression. Tumors formed by LS180 cells overexpressing GnT-V injected into NOD/SCID mice grew significantly faster than the tumors formed by control cells (Fig. 1F), and these tumors were accompanied by more necrotic areas compared with tumors formed from non-transfected, controls cells (Fig. 1F). Conversely, tumors formed by injection of cells with GnT-V suppression due to siRNA against GnT-V grew significantly slower than the tumors formed by control cells (Fig. 1G). Consistent with reduced tumor size, tumors with GnT-V suppression showed decreased Ki-67 staining (Fig. 1G), indicating a reduced proliferation of tumor cells due to inhibition of GnT-V expression. These results suggest that GnT-V and its glycan products are implicated as a positive regulator of cancer cell proliferation in vitro and of tumorigenicity in vivo using xenografts.
GnT-V Expression Levels Regulate Colon Adenoma Progression in Apcmin/+ mice
To investigate the significance of GnT-V expression in colon tumor development and tumor progression in vivo, a mouse model of colon cancer, Apcmin/+ mice, was utilized. L-PHA staining was increased in intestinal crypts that showed histological evidence of early adenoma formation (dysplastic crypts) compared with normal crypts (Fig. 2A), indicating increased GnT-V activity during adenoma development in Apcmin/+ mice, consistent with previous reports (18, 19). Apcmin/+ mice that differ in GnT-V backgrounds were generated by breeding Apcmin/+ mice with GnT-V null mice and confirmed by genotyping and L-PHA staining, respectively (Fig. 2, B and C). As shown in Fig. 3A, the survival times, shown by Kaplan-Meier analysis, were significantly increased in Apcmin/+ mice with GnT-V deletion compared with GnT-V wild-type (WT) mice (median >28 weeks versus 21 weeks, p = 0.001), indicating reduced adenoma progression and increased survival times. Although adenomas were formed throughout the intestinal tract of Apcmin/+mice in both WT and GnT-V null backgrounds, no significant differences in the number of adenomas between different genotypes were observed at either 18 (Fig. 3B) or 22 weeks of age (data not shown). However, the size of the adenomas was significantly smaller in Apcmin/+ mice that lacked GnT-V expression compared with WT mice. The percentage of adenomas with sizes larger than 2.5 mm was lower in GnT-V inhibition groups (19.5% for GnT-V−/−, 18.6% for GnT-V+/−, and 28.6% for GnT-V+/+) (Fig. 3C). A box plot analysis revealed a distribution of significantly smaller adenomas in GnT-V deletion groups (Fig. 3D). The average size of adenomas was 1.8, 1.9, and 2.2 mm for GnT-V−/−, -+/−, and -+/+ groups, respectively (p < 0.05). Reduced tumor size was consistent with enhanced survival observed in Apcmin/+ mice with GnT-V deletion (Fig. 3A), supporting a reduced tumor progression after inhibition of GnT-V expression. Concomitant with these results, immunohistochemical studies further showed reduced Ki-67 staining of adenomas with GnT-V deletion (Fig. 3E), indicating an inhibition of adenoma cell proliferation. These results together implicate GnT-V in the regulation of intestinal adenoma progression rather than tumor formation in vivo.
FIGURE 2.
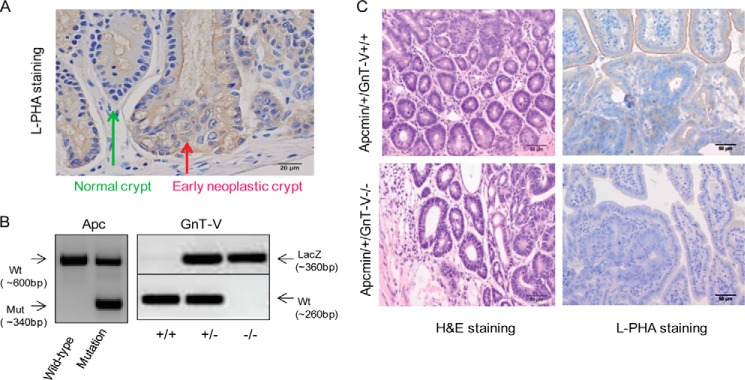
Generation of Apcmin/+ mice with different GnT-V genotypes. A, normal and neoplastic crypts of Apcmin/+ mice were immunostained with L-PHA. Arrows indicate normal and early neoplastic crypts. Bar, 20 μm. B, after genomic DNA was extracted from tails of Apcmin/+ mice, genotyping was performed to detect the expression of mutated Apc, wild-type GnT-V, and LacZ genes by PCR. C, adenoma sections from different GnT-V genotypes using 18-week-old adenoma tissues prepared and stained with hematoxylin/eosin and L-PHA. Bar, 50 μm.
FIGURE 3.

GnT-V regulates colon adenoma progression in Apcmin/+ mice. A, Kaplan-Meier curves were used to display the survival of Apcmin/+ mice in different GnT-V backgrounds (p = 0.001). t½ represents the time point at which 50% of mice in that group survive. B, Apcmin/+ mice in different GnT-V backgrounds were grown to the age of 18–20 weeks, and the number of adenoma formed in intestine was examined. Horizontal bars represent the median value of adenomas per mouse. C, distribution of adenomas of Apcmin/+ mice in different GnT-V backgrounds based on their size. D, Box Plot analysis of the adenoma size of Apcmin/+ mice with different GnT-V genotypes. E, intestines were dissected at week 18, and immunochemical staining was performed using antibody against Ki-67. Bar, 20 μm.
GnT-V Regulates the Population of CCSC
We recently reported that deletion of GnT-V delayed her-2-induced mouse mammary tumor onset via down-regulating the size of the compartment of CSC (17). Similar to mammary carcinoma, studies have suggested that colon carcinoma is initiated by a subset of crypt cells termed colon cancer stem cells (6, 7, 31, 32). Because increased activity of aldehyde dehydrogenase 1, detected by the Aldefluor assay, is a characteristic of CCSC, this assay has been used for both measurement and enrichment of CCSC (7, 33). To confirm that the Aldefluor-positive population from colon cancer cell lines has stem cell-like properties, both Aldefluor-positive and -negative cells were sorted using flow cytometry and collected for analysis of different intestinal stem cell markers (34, 35) by qRT-PCR. In LS180 cells, both intestine-specific stem cell markers (ASCL2 and BMI1) (36, 37) and primitive stem cell markers (OCT-4 and SOX4) were enriched in Aldefluor-positive cells (Fig. 4A), whereas increased ASCL2 and CD133 were observed in Aldefluor-positive cells isolated from the HT-29 line (Fig. 4B). Interestingly, no significant change in the expression of another intestine-specific stem cell marker, LGR5 (37), was observed in Aldefluor-positive cells from either LS180 and HT-29 cells. Consistent with increased stem cell gene expression, the Aldefluor-positive cells from HT-29 cells showed an increased ability to form tumorspheres (both the number and the size of spheres) when grown in suspension culture for 7–10 days (Fig. 4C) compared with the Aldefluor-negative cells, indicating increased self-renewal, a fundamental characteristic of stem cells (38, 39). These results suggested that CCSC were enriched in the Aldefluor-positive cell population and confirmed the Aldefluor assay as an effective method to be used for detection and isolation of CCSC. Compared with Aldefluor-negative cells, Aldefluor-positive cells showed a significant increase in the expression of GnT-V that was accompanied by increased L-PHA staining (Fig. 4D). These results suggested that GnT-V may be involved in the regulation of the CCSC compartment.
FIGURE 4.

Aldefluor-positive cells show increased tumorsphere formation and GnT-V expression. A, Aldefluor-positive and -negative were sorted and “stemness”-associated genes were detected by qRT-PCR from LS180 cells (A) and HT-29 cells (B), respectively. ALDH, aldehyde dehydrogenase. C, sorted Aldefluor-positive and -negative HT-29 cells (4000 cells/well) were grown in stem cell culture media in suspension for 7–10 days, and the number of tumorspheres was counted in 5–6 random fields. Bar, 50 μm. D, GnT-V mRNA levels (left) and N-linked β(1,6) branching (right) were determined by qRT-PCR and L-PHA staining, respectively, in both Aldefluor-positive and -negative LS180 cells. For each transcript, the values was normalized to control (GAPDH) and expressed as the mean ± S.D. of three independent experiments. Images are representative of two independent experiments. Bar, 50 μm.
To further test the hypothesis that expression levels of GnT-V regulate CCSC, GnT-V was either overexpressed or knocked-down by siRNA. The Aldefluor-positive population (CCSC) was remarkably increased in the GnT-V overexpressing cell population compared with control cells (Fig. 5A, left and middle panels). A reduced proportion of Aldefluor-positive cells was observed for tumor cells transfected with GnT-V siRNA (Fig. 5A, right panel). Consistent with these in vitro findings, the Aldefluor-positive cell population was remarkably reduced in adenoma tissues from Apcmin/+ mice with GnT-V deletion compared with GnT-V WT adenomas (Fig. 5B). The increased CSC population in GnT-V overexpressing tumor cells was further confirmed by Hoechst 33342 staining. Cell surface transporters are observed to exclude vital dyes such as Hoechst 33342 from CSC but not from non-CSC (differentiated) cells (40). Using this assay, the control tumor cells displayed a CSC population of 0.64% (1.09% minus the control value of 0.45%); by contrast, the GnT-V overexpressing cells showed a significantly increased population of 2.18% (2.58% minus control value of 0.4%) (Fig. 5C). CCSC have also been shown to be enriched in the CD44+CD24+ population of colon cancer cell lines, and this population of cells showed an ability of both self-renewal and differentiation as well as showing the highest efficiency in production of tumors in NOD/SCID mice (41). GnT-V overexpressing tumor cells displayed an increase in the CD44+CD24+ population compared with control cells (data not shown), consistent with an increased CCSC population after overexpression of GnT-V. To further confirm these results, we treated the colon cancer cell lines with swainsonine, an inhibitor that results in a block in GnT-V glycan product formation (Fig. 1A). A reduced proportion of Aldefluor-positive cells was observed in three different colon cancer cell lines, HT-29, LS180 (Fig. 5D), and SW480 (data not shown) after treatment with swainsonine, indicating that the population of CCSC was affected by inhibition of N-glycan branching that includes β(1,6) branching, supporting the effect of GnT-V expression levels on regulating the CCSC population. Because studies have shown that only Aldefluor-positive tumor cells (CSC) can form outgrowths in NOD/SCID mice (7, 42), the altered tumorigenicity observed in our study most likely resulted, at least in part, from the altered proportion of CCSC resulting from altered GnT-V expression levels.
FIGURE 5.

Expression levels of GnT-V regulate the population of CCSC. A, Aldefluor assays were performed using colon tumor cells transfected with either GnT-V cDNA (left and middle panels) or GnT-V siRNA (right panel), and the percentage of Aldefluor-positive cells was determined under similar gating criteria and is noted in the top right of each sorting profile. B, Aldefluor assays were performed using adenomas of Apcmin/+ mice in different GnT-V backgrounds, and the percentage of Aldefluor-positive cells was determined under similar gating criteria. C, control and GnT-V transfected LS180 cells were stained with Hoechst 33342 with and without verapamil treatment, which blocks dye exclusion. Side populations, representative of CCSC, were determined by flow cytometry and are indicated by arrows. D, Aldefluor assays were performed using tumor cells treated with swainsonine (1 μg/ml) for 24 h, and the percentage of Aldefluor-positive cells was determined using similar gating criteria. In all Aldefluor experiments, diethylaminobenzaldehyde, inhibitor of aldehyde dehydrogenase, was used to determine the background of the assay. Data are representative of two independent experiments.
GnT-V Expression Levels Regulate Self-renewal and Tumorigenicity of CCSC
To identify the mechanisms by which GnT-V levels affect the population of CCSC, we first focused on the growth of tumorspheres in culture. When grown in serum-free stem cell culture media for 7–10 days, HT-29 cells formed typical tumorspheres, a measurement of the self-renewal of stem cells (38, 39) as shown in Fig. 6A. Increased gene expression of both aldehyde dehydrogenase-1 and GnT-V were observed in tumorspheres compared with parental cells grown on plastic plates, supporting further the enrichment of CCSC in tumorspheres and involvement of GnT-V in their regulation. To examine the role of GnT-V in regulating self-renewal, the ability of cells to form tumorspheres in suspension culture was measured for HT-29 cells in which GnT-V expression was suppressed by GnT-V siRNA. Although both control and GnT-V knockdown HT-29 cells showed the ability to form tumorspheres in suspension culture for 7–10 days, both the number and size of tumorspheres were significantly decreased in GnT-V knockdown cells compared with the controls (Fig. 6B). The number of tumorspheres generated upon serial passage provides a measurement of self-renewal of stem cells (26). To examine the ability of tumorspheres (primary tumorspheres formed from cultured tumor cells) to form new tumorspheres (secondary tumorspheres formed after trypsinization of primary tumorspheres), primary tumorspheres were collected, trypsinized, and further cultured in suspension. Similarly, the number of secondary tumorspheres formed from the GnT-V-suppressing cells was significantly lower than that from the control cells (Fig. 6B, right panel), indicating a reduced self-renewal of cells after inhibition of GnT-V. Limiting dilution assays were then performed to estimate the tumorsphere-forming frequency (43). As shown in Fig. 6C, the number of the cells required to generate at least one tumorsphere/well was higher in CCSC from GnT-V knockdown HT-29 cells (∼101 cells) than those from control cells (∼33 cells), supporting a decreased self-renewal capacity in GnT-V suppressing cells. These results indicated that GnT-V expression levels were implicated in the regulation of self-renewal of CCSC, affecting the population of CCSC.
FIGURE 6.

GnT-V regulates self-renewal and tumorigenicity of CCSC. A, HT-29 cells (4000 cells/well) were grown in stem cell culture media in suspension for 7–10 days, and tumorspheres were formed (left panel, top). Bar, 50 μm. Both aldehyde dehydrogenase-1 (ALDH1) and GnT-V mRNA levels were determined by qRT-PCR in both tumorspheres and parental cells on plastic plates (right panel). B, GnT-V-suppressed HT-29 cells (4000 cells/well) were grown in suspension for 7–10 days, and the number of tumorspheres was counted in 5–6 random fields. After tumorspheres were collected and single-cell suspension prepared, they (4000 cells/well) were grown in stem cell culture media in suspension for another 7–10 days for secondary sphere formation. Bar, 50 μm. C, primary tumorspheres formed from GnT-V suppressed HT-29 cells were dissociated and seeded in 96-well plates in stem cell culture media at densities ranging from 400 to 1 cell/well. After growth for 7–10 days, the percentage of wells that did not contain spheres at each cell plating density was calculated and plotted against the number of cells per well. D, Aldefluor-positive cells (3 × 103) isolated from GnT-V overexpressing LS180 cells were injected subcutaneously into the backs of NOD/SCID mice (n = 5), and secondary tumor growth was observed for up to 8 weeks. *, p < 0.05. E, tumors were dissected at week 8, and H&E and other immunochemical staining were performed as indicated. The arrow indicates local invasion, whereas cytokeratin staining confirms the epithelial origin of the tumor cells.
Rapid rate of tumor formation in NOD/SCID mice is another fundamental characteristic of CSC. To characterize the tumor forming ability of CCSC purified from both control and GnT-V overexpressing tumor cells, the same number of CCSC (103) was sorted from cultures using the Aldefluor assay and injected into NOD/SCID mice. Although CCSC isolated from both control and GnT-V overexpressing cells formed epithelial tumors, confirmed by H&E and cytokeratin staining, tumor growth was significantly increased in those mice that received CCSC with GnT-V overexpression (Fig. 6, D and E). The tumors formed by injection of CCSC from the GnT-V overexpressing cells still retained high expression of GnT-V, detected by L-PHA staining and were found to have local invasion into adjacent connective tissues (Fig. 6E, indicated by the arrow). By contrast, no local invasion was observed in the tumors formed by CCSC from the control cells, indicating an enhanced invasiveness and progression caused by GnT-V overexpression. Supporting these results, immunohistochemical staining showed an increase in expression of the proliferation marker Ki-67 in some areas of tumors generated by CCSC with GnT-V overexpression compared with control cells (Fig. 6E). These results are consistent with those from the in vitro experiments, demonstrating that GnT-V expression levels affected the CSC population and implicated GnT-V in regulating the cancer stem cell pool in vivo via affecting their self-renewal and tumorigenicity.
GnT-V Expression Levels Regulate Canonical Wnt Signaling Pathway
The canonical Wnt signaling pathway that regulates cell fate and proliferation has a crucial role in colorectal cancer development in both mouse and humans (44). Recent studies have also implicated Wnt/β-catenin signaling as the key regulator of CSC in colon cancer (45–47). To investigate if altered Wnt/β-catenin signaling was involved in the regulation of CCSC population and colon adenoma development by the expression levels of GnT-V, both Wnt target gene expression and nuclear β-catenin localization were analyzed. As expected, adenoma tissues from Apcmin/+ mice with both GnT-V WT and KO backgrounds showed a remarkable increase in expression of Wnt target genes, including c-myc, Lgr5, Ascl2, and Axin-2, compared with adjacent normal tissues (Fig. 7A, left panel). These results were consistent with the conclusion that activation of the Wnt signaling pathway is an early event in colon cancer development (46). However, significant decreases in these Wnt target gene expression were observed in adenoma tissues with GnT-V deletion compared with those with WT GnT-V (Fig. 7A, left and middle panels), indicating an inhibited Wnt activity in adenomas due to deletion of GnT-V expression. Interestingly, the terminal differentiation epithelial marker, cytokeratin 20, was significantly increased in both adjacent normal tissues and adenomas of Apcmin/+ mice with GnT-V deletion (Fig. 7A, right panel), likely indicating a reduced malignancy of adenomas after deletion of GnT-V. Consistent with these results from Apcmin/+ mice, human LS180 (Fig. 7B, left panel) and SW480 (data not shown) colon cancer lines with GnT-V overexpression showed increased expression of some Wnt target genes, such as CMYC, ASCL2, and AXIN2, compared with control cells. Interestingly, the Wnt target gene LGR5, also an intestinal stem cell marker, was found significantly suppressed in human colon cancer cells with GnT-V overexpression but increased in the cells with GnT-V knock-down (Fig. 7B, right panel). This result indicated an inverse regulation of LGR5 gene transcription by GnT-V expression levels and supports LGR5 as a negative regulator of the canonical Wnt signaling pathway in human colon cancer cells (48, 49). Altered expression of Wnt target genes by GnT-V expression was further confirmed by immunoblotting (Fig. 7, C and D) and immunohistochemistry (Fig. 7E) where the protein expression levels of the Wnt target genes CMYC, AXIN-2, and Ascl2 showed concomitant changes consistent with their transcript changes.
FIGURE 7.

GnT-V regulates canonical Wnt/β-catenin signaling pathway. A, total RNA was pooled from four adjacent and adenoma tissues of GnT-V wild-type and knock-out Apcmin/+ mice, and expression of Wnt target genes (two left panels) and cytokeratin 20 (right panel) were detected by qRT-PCR. B, expression of Wnt target genes was detected by qRT-PCR using total RNA from control and GnT-V transfected LS180 (left) and GnT-V siRNA transfected HT-29 (right) cells. For each transcript the values were normalized to control (GAPDH) and expressed as the mean ± S.D. from three independent experiments. C, adjacent (control) and adenoma tissues were collected and used to detect protein expression by immunoblotting; Adj, adjacent; Tu, adenoma. D, GnT-V transfected colon cancer cell lines were used to detect protein expression by immunoblotting. C, control cells; G, GnT-V transfected cells. β-Actin was used as a loading control. E, c-myc expression and nuclear translocation of β-catenin were visualized by immunochemical staining of tumor tissues formed from injection of CCSC isolated from GnT-V transfected LS180 cells (middle panel) and adenomas of Apcmin/+ mice with different GnT-V genotypes (left panel); bar, 20 μm. Quantification of differences in nuclear β-catenin-positive cells (percentage of positive cells in six random fields of control or GnT-V transfected cells or per adenoma in Apcmin/+ mice with wild-type or GnT-V knock-out genotypes) (right panel). F, TOPFLASH/FOPFLASH analysis of Wnt activity of cultured colon cancer cell line with and without GnT-V transfection. The results are expressed as the mean ± S.D. of three independent experiments. *, p < 0.05. G, Aldefluor assays were performed using HT-29 cells treated with Wnt inhibitor (CCT036477, 20 μm, left panel) or LS180 cells with Wnt ligand (Wnt3a, 100 ng/ml, right panel) for 24 h, and the percentage of Aldefluor-positive cells was determined using similar gating criteria. Data are representative of two independent experiments.
Nuclear translocation of β-catenin is a hallmark for active Wnt signaling, leading to the formation of β-catenin/TCF/LEF that triggers a complex transcriptional expression of Wnt target genes (45). Compared with adjacent normal tissues, adenomas showed a significant increase in β-catenin levels in Apcmin/+ mice as expected (Fig. 7C), indicating a reduced degradation of β-catenin during the development of adenomas. However, reduced β-catenin levels were observed in adenomas from GnT-V KO mice (Fig. 7C). Decreased staining of nuclear β-catenin was observed in adenoma tissues of Apcmin/+ mice in the GnT-V null background compared with the mice with WT GnT-V (Fig. 7E, left panel). Nuclear β-catenin staining was significantly increased in adenocarcinoma tissues formed by injection into SCID mice of CCSC from GnT-V-overexpressing LS180 cells compared with those adenocarcinomas formed by injection of CCSC from control cells (Fig. 7E, middle panel). These results were consistent with altered Wnt target gene transcription caused by altered GnT-V expression and support a positive role of GnT-V in regulating Wnt signaling activity. To strengthen our hypothesis that Wnt signaling was regulated by GnT-V, the effect of GnT-V levels on Wnt activity was measured using Topflash luciferase reporter plasmids. As shown in Fig. 7F, significantly increased Wnt activity (determined by the ratio of TOPFLASH/FOPFLASH) was observed in GnT-V-transfected LS180 cells, indicating an increase in TCF/LEF-1 transcriptional activity. SW480 showed, although significant, less of a change than LS180 (data not shown) likely due to higher activation of Wnt activity by an APC mutation in these cells (50).
To provide additional evidence that CCSC were regulated by GnT-V levels via the Wnt signaling pathway, colon cancer cells were treated by Wnt-3a, an active ligand for Wnt signaling in colon carcinoma (50), and the Wnt inhibitor CCT036477 (51). As expected, the CCSC population was significantly increased in LS180 cells after treatment with Wnt-3a (Fig. 7G, right panel) but reduced in HT-29 cells with CCT036477 treatment (left panel). These results confirmed the involvement of Wnt signaling in the regulation of CCSC and indicated that the effect of GnT-V expression on CCSC population was, at least in part, through the Wnt signaling pathway.
Wnt Receptors Express N-Glycans with β(1,6) Branching
GnT-V modulates the function of various cell surface receptors, leading to altered receptor clustering and ligand binding as well as regulating endocytosis of signaling complexes, resulting in aberrant downstream signaling mediated by these receptors (14, 17). The binding of Wnt ligand to the Wnt co-receptors, Frizzled-7 (FZD-7) and low density lipoprotein receptor-related protein-5 or -6 (Lrp-5/6), forms the Wnt·FZD·Lrp-5/6 complex, leading to Wnt-target gene expression (52). To determine if altered Wnt signaling caused by aberrant expression of GnT-V resulted from altered N-glycan modification of the Wnt (co)receptor, FZD-7, this protein was exogenously expressed in LS180 cell line by transfection and detected by Western blotting (Fig. 8A). Interestingly, we observed that denaturing samples by boiling at 95 °C in the presence of β-mercaptoethanol, common conditions for reducing SDS-PAGE, caused extensive aggregation of expressed proteins after blotting with anti-FZD-7 antibody. If the samples were simply mixed with SDS-PAGE loading buffer, however, and subjected to electrophoresis, a band at a molecular mass of ∼63 kDa, the molecular mass of FZD-7, was resolved after blotting with the antibody, although aggregation was still observed. Similar results were obtained from SW480 cells after transfection with FZD-7 cDNA (data not shown). Incubation of membrane extracts from colon carcinoma cells with PNGase F, a glycosidase that cleaves diverse vertebrate N-glycans, including high mannose, hybrid, and complex structures, in the presence of low amounts of SDS without boiling resulted in a shift of the size of expressed FZD-7 after SDS-PAGE to that of ∼55 kDa, indicating that the expressed receptors were N-glycosylated (Fig. 8B). Compared with PNGase F, endoglycosidase H digestion caused little, if any, change in mobility of overexpressed FZD-7 (Fig. 8B), suggesting that this receptor in LS180 cells contains complex N-glycans. Similar results with PNGase-F and Endo H digestion were seen with SW480 cells (data not shown). To determine if this receptor was an acceptor substrate of GnT-V, cell lysates of FZD-7-overexpressing cells were pulled down by L-PHA to enrich for glycoproteins that contained N-linked β(1,6)-branched glycans, products of GnT-V. As shown in Fig. 8C, after L-PHA pulldown, significant amounts of aggregated receptors were detected in both LS180 and SW480 cells (data not shown) expressing FZD-7, indicating that these glycoprotein receptors contained GnT-V glycan products. Experiments to quantify L-PHA binding to bands on these blots, however, were problematic as enrichment by anti-FZD-7 or L-PHA pulldown was required but was unsuccessful due to the necessity for denaturation, resulting in the inability to visualize the FZD-7 band at 63 kDa as seen in Fig. 8, A and B. To simplify the experiment, we instead utilized CHO Lec4 cells that have no GnT-V activity to express FDZ-7 before and after transfection with GnT-V cDNA. Large amounts of FZD-7, observed at 63 kDa after blotting with anti FZD-7 antibody, were precipitated by L-PHA after expression of GnT-V in these cells (Fig. 8D), demonstrating N-linked β(1,6) branching on these receptors caused by GnT-V expression. For unknown reasons, however, using the identical experimental protocol with either colon carcinoma cell line did not resolve the 63-kDa FZD-7 band after L-PHA pulldown, precluding quantification in these cancer cell lines. In addition, making a secreted protein of FZD-7 turned out to be very difficult (data not shown), which unfortunately prevented us from full N-glycan analysis of this protein using mass spectrometry. Nevertheless, our results show that the Wnt receptor, FZD-7, expresses N-glycans that are bound by L-PHA and reflect levels of GnT-V activity. Taken together, it is very likely that the altered Wnt signaling observed after modulating the expression of GnT-V activity results, at least in part, from aberrant expression of GnT-V-modified N-glycans on FZD-7. Moreover, the evidence that altered Wnt signaling resulting from altered glycosylation of Wnt by GnT-V expression levels strongly supports the conclusion that posttranslational modifications of Wnt can significantly regulate tumor progression in vivo.
FIGURE 8.

Wnt receptor FZD-7 is N-glycosylated with β(1,6) branching. A, after human pCMV6-FZD7 plasmid was transiently expressed in LS180 cells, tagged FZD-7 proteins were detected by western-blotting (WB) using anti-DDK tag antibody. Note: denaturing samples by boiling at 95 °C caused aggregation of proteins, resulting in poor resolution. β-Actin was used as a loading control. B, LS180 cell lysates expressing FZD-7 were digested either with PNGase F or with Endo-H followed by SDS-PAGE and Western blotting using anti-DDK antibody. The arrowheads indicate a deglycosylated band of FZD-7 by PNGase F digestion. C, FZD-7 transfected LS180 cell lysates were used for precipitation with biotinylated L-PHA followed by the addition of streptavidin-agarose and elution by boiling in SDS-buffer followed by SDS-PAGE and Western blotting with anti-DDK tag antibody. D, after human pCMV6-FZD-7 plasmid was transiently expressed in Lec4 cells, tagged FZD-7 proteins were detected by Western blotting using anti-DDK tag antibody (left); after mouse GnT-V cDNA along with FZD-7 cDNA were transiently expressed in Lec4 cells, levels of N-linked β(1,6) branching were detected by L-PHA staining (middle panel); FZD-7 and GnT-V co-transfected Lec4 cell lysates were used for precipitation with biotinylated L-PHA followed by Western blotting with anti-DDK tag antibody (right panel).
DISCUSSION
During human colon cancer progression, an increase in the activity of GnT-V, whose expression is controlled by several oncogenes, such as H-ras, v-sis, and her-2/neu (53, 54), has been shown to occur (18, 19). In our study we chose human colon cancer lines with different mutations as our in vitro models, including LS180 (with a β-catenin mutation), SW480, and HT-29 (with APC mutations). Results from these cultured cell lines strongly suggest that colon tumorigenesis is regulated by GnT-V expression levels. First, both anchorage-independent cell growth in soft agar and colony formation were remarkably enhanced when GnT-V was overexpressed in colon cancer cells. Second, in vivo tumor growth in NOD/SCID mice that resulted from injection of tumor cells overexpressing GnT-V was significantly increased, but suppressed when injection of tumor cells with inhibited GnT-V expression was used, compared with that observed from injection of mock-transfected cells. Based on these observations, GnT-V expression levels are implicated in the regulation of tumor-related phenotypes.
To further confirm the effect of GnT-V expression on colon tumorigenesis, Apcmin/+ mice were used to breed with GnT-V knock-out mice to generate Apcmin/+ mice with different GnT-V backgrounds. We found that the survival of Apcmin/+ mice with reduced GnT-V expression was significantly enhanced. Interestingly, the number of adenoma formed in each Apcmin/+ mice was not affected by GnT-V expression levels. However, a reduction in the size of the adenomas was observed after ablation of GnT-V, consistent with increased survival of these mice. These results suggested that GnT-V expression primarily affected the rate of tumor growth rather than tumor formation (the number of adenomas) in Apcmin/+ mice. This observation was in synchrony with a recent study where the regulation of tumor load, but not the number of adenomas, was reported in Apcmin/+ mice after treatment with a PKA antagonist (Rp-8Br-cAMPS) (55). The inhibitory effect of GnT-V deletion on tumor growth observed in Apcmin/+ mice was consistent with the results obtained from NOD/SCID mice injected with tumor cells with GnT-V knockdown. These results strongly suggested that GnT-V expression promoted colon adenoma progression in vivo.
Recent studies have highlighted the importance of cancer stem cells in regulating colon tumorigenesis and progression. CCSC have been isolated from both human and mouse colon tumors (4, 6, 7, 9). Consistent with these reports, CCSC were identified from both human colon cancer lines and colon adenomas in Apcmin/+ mice, demonstrated by using different markers for stem cells. Interestingly, we found that the population of CCSC was significantly increased in GnT-V-overexpressing tumor cells, whereas reduced expression of GnT-V in tumor cells or in adenomas down-regulated the proportion of CCSC, indicating the regulation of CCSC by GnT-V expression levels. These results suggest the hypothesis that the altered proportion of CCSC was involved in GnT-V-regulated tumorigenesis and tumor progression. Like other CSC (26), CCSC can form tumorspheres in suspension culture, reflecting their ability for stem cell self-renewal. We found that self-renewal of CCSC was deregulated by GnT-V, determined by stem cell suspension culture, and further confirmed by limiting dilution assay, consistent with a deregulated proportion of CCSC by GnT-V. Coincident with these results, the tumor-forming ability in NOD/SCID mice injected with CCSC isolated from GnT-V-suppressing tumor cells was significantly decreased compared with those from wild-type tumor cell injection. These results indicated that GnT-V expression levels regulated both self-renewal and tumorigenicity of the CCSC resulting in the aberrant proportion of CCSC observed both in colon cancer cells and mouse adenomas due to aberrant expression of GnT-V. Based on the fact that CSC play a pivotal role in tumor initiation and progression (1, 2), a decrease in the CCSC compartment might, therefore, contribute to inhibited adenoma progression observed in Apcmin/+ mice after deletion of GnT-V. Adenoma initiation (onset) was not affected in these mice, reflecting the regulatory role of CCSC in tumor progression and not in initial tumor formation (5).
The canonical Wnt/β-catenin signaling pathway is one of the most important pathways to regulate CCSC and colon cancer development (44). Altered Wnt signaling is mainly attributive to inactivation of the tumor suppressor gene, Apc, or activation of the oncogene β-catenin (Ctnnb1) (44). The binding of the Wnt ligand to Wnt receptors FZD-7 and Lrp-5 or -6 promotes the nuclear translocation of cytoplasmic β-catenin, the stability of which is regulated by a destruction complex containing Apc, Axin, and glycogen synthase kinase 3β. Translocated β-catenin is associated with TCF/LEF-family transcription factors, thus activating the transcription of target genes (56). Studies have shown that Wnt activity defines colon CSC and regulates growth and maintenance of tumorspheres (29, 45). Our findings strongly support the hypothesis that Wnt/β-catenin signaling pathway was implicated in the regulation of CCSC and colon tumor progression by GnT-V. First, the expression of Wnt target genes was changed by aberrant GnT-V expression levels. Second, the nuclear translocation of β-catenin, detected by immunochemical staining was regulated by GnT-V expression levels. Third, Wnt activity detected by TCF-luciferase (TOPFLASH/FOPFLASH) assay was shown to be regulated by GnT-V expression. Moreover, we found an increased or decreased population of CCSC after exposure of cells to a Wnt ligand, Wnt-3a, or a Wnt inhibitor, CCT036477, respectively. Therefore, the altered relative population of CCSC caused by altered GnT-V expression levels resulted, at least in part, from altered Wnt/β-catenin signaling, therefore, regulating Wnt-mediated colon tumor development.
Interestingly, the regulation of one Wnt target gene, Lgr5, by GnT-V expression appeared to differ between human colon cancer cells and mouse adenoma tissues, although GnT-V expression consistently regulated their Wnt activity (Fig. 7). This differential regulation of Lgr5 by GnT-V may reflect different roles of Lgr5 in the Wnt signaling pathway. Lgr5 along with Ascl2, serving as downstream target genes of Wnt signaling (51, 57), have been identified as normal and cancer stem cell markers of mouse intestine (colon) (36, 58). Therefore, the reduced gene expression of both Lgr5 and Ascl2 observed in Apcmin/+ mice with GnT-V deletion not only indicated an inhibited Wnt activity, but also supported the reduced proportion of CCSC observed in these mice. By contrast, LGR5 expression was found to be negatively regulated by GnT-V in human colon cancer cells, suggesting that LGR5 may function as a Wnt inhibitor in these cells, consistent with a report showing LGR5 is a negative regulator of tumorigenicity by antagonizing Wnt signaling in colon cancer cell lines (49). This regulation might reflect a feature of Wnt signaling in which multiple Wnt targets have a role in the negative feedback inhibition of signaling by blocking Wnt receptor function and reducing the stabilization of the destruction complex (59, 60). In addition, this negative regulation of LGR5 by GnT-V in colon cancer cells was consistent with our observation that only ASCL2, and not LGR5, was found to be increased significantly in stem cell-enriched Aldefluor-positive population, although studies have suggested that LGR5 can serve as a human CCSC marker (39, 61).
On the other hand, activated Wnt signaling by GnT-V action may, in turn, stimulate GnT-V expression further, strengthening the effect of GnT-V on Wnt signaling, since reciprocal regulation between Wnt signaling and glycosyltransferase action has been demonstrated. For example, stimulation of the Wnt signaling pathway by addition of exogenous Wnt3a or BIO, a glycogen synthase kinase 3β inhibitor, consistently and significantly inhibited GnT-III expression and its products, indicating a regulation of GnT-III by Wnt signaling pathway (62).
In addition to the canonical Wnt/β-catenin signaling pathway, the involvement of other signaling pathways in regulating the CCSC population cannot be ruled out because GnT-V can modify glycoprotein receptors other than FZD-7. Indeed, we noticed reduced levels of phosphorylated ERK (p-ERK) in adenomas of Apcmin/+ mice with GnT-V deletion, evident by decreased expression level of phosphorylation of ERK (p-ERK) (Fig. 7C), consistent with a previous report that the ERK pathway is activated in Apcmin/+ mice (63).
The function of various cell surface receptors has been shown to be modulated by GnT-V and this modulation often causes aberrant downstream signaling mediated by these receptors (14, 15, 17, 64). The binding of the Wnt ligand to Wnt receptors triggers canonical Wnt signaling, leading to downstream Wnt-target gene expression (52). Members of the Wnt receptor family FZD are seven-pass transmembrane glycoproteins with either 1 or 2 putative N-glycosylation sites in their extracellular N-terminal cysteine-rich domain (CRD) that serves as the Wnt binding domain (52). A conserved N-glycosylation site in the CRD domain observed in the mouse FZD-8 crystal structure has recently been reported to express N-glycans (65). Although FZD-7 is one of the Wnt receptors predominantly expressed in colon cancer cells that harbor APC or Ctnnb1 mutations, little is known about its N-glycosylation status in these cells. By overexpressing FZD-7 in colon cancer cells, we found that FZD-7 receptors were clearly N-glycosylated, evident by altered mobility after PNGase F treatment. N-Linked β(1,6) branching on these receptors was further characterized by L-PHA pulldown experiments, indicating that FZD-7 is modified by GnT-V. Results using GnT-V-defective Lec4 cells transfected with GnT-V further confirmed the fact that FZD-7, when expressed in cells, is an acceptor for GnT-V. Technical difficulties, due to substantial aggregation of FZD-7 under the conditions required to release these receptors from the affinity beads before SDS-PAGE, did not allow us to resolve the 63-kDa band of FZD-7 observed without denaturation and, consequently, precluding accurate quantification of increased N-linked β(1,6) branching on FZD-7 after GnT-V overexpression. Many examples of glycoproteins that are cellular acceptors for GnT-V do show quantitative changes in β(1,6) branching when levels of GnT-V are altered; for example, integrins (12, 66), cadherins (67, 68), growth factor receptors (14, 15, 17), matriptase (69), tissue inhibitor of metalloproteinase-1 (21), and receptor-like protein tyrosine phosphatases (70). It is reasonable to conclude, therefore, that increased GnT-V expression leads to increased β(1,6) branching on FZD-7. If so, then the altered Wnt signaling observed in colon cancer cells overexpressing FZD-7 is likely to be attributed, at least in part, to aberrant N-glycosylation of FZD-7 due to modulated GnT-V expression.
It is possible that the altered expression of β(1,6) branching on Wnt receptors may cause altered ligand affinity and/or receptor stability, thereby affecting the formation of the Wnt·Frizzled·Lrp-5/6 complex and thus leading to altered downstream Wnt-target gene activation. This possibility is substantiated by a recent study (71) showing that partial inhibition of DPAGT1, an N-glycosylation gene, in human squamous carcinoma cell lines reduced canonical Wnt signaling, likely via altered glycan modification of Wnt3a and Lrp-5/6. On the other hand, the regulation of Wnt signaling by GnT-V may be indirectly mediated by other cell surface proteins whose functions are regulated by GnT-V; for example, E-cadherin. Hypoglycosylated E-cadherin has been shown to significantly inhibit canonical Wnt activity in tumor cells (71); therefore, it is possible that altered GnT-V activity may have caused altered N-glycosylation of E-cadherin, a key acceptor substrate of GnT-V, also leading to aberrant Wnt activity.
In conclusion, we have provided evidence that the expression level of GnT-V regulates canonical Wnt/β-catenin signaling pathway, most likely via modulating the N-glycosylation of Wnt receptors, which results in changes in the relative size of the population of CCSC in colorectal tumors, ultimately causing altered colon tumorigenesis and adenoma progression in Apcmin/+ mice (Fig. 9). These findings shed new light on the molecular mechanisms of how altered GnT-V expression levels can modulate colon tumorigenesis and progression, and they support the hypothesis that inhibitors that target increased N-linked glycan branching could have therapeutic utility in the treatment of human colon cancer.
FIGURE 9.
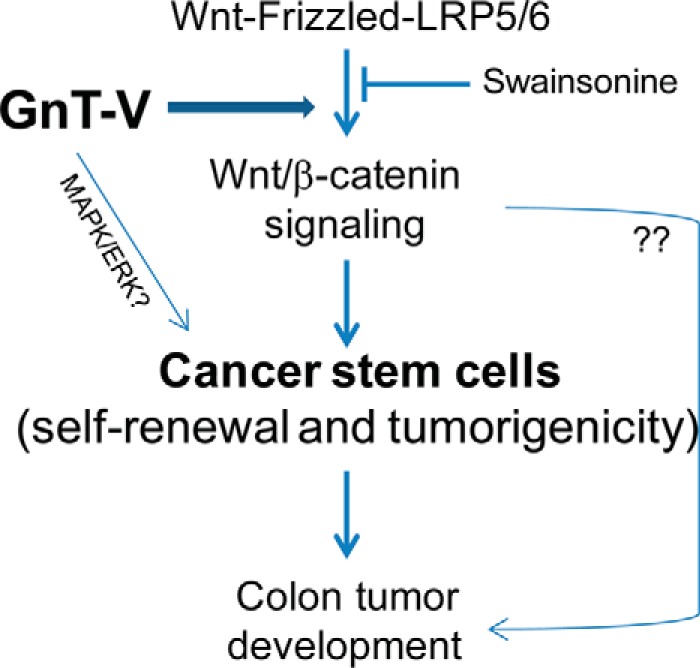
Schematic diagram indicating a hypothesis of the mechanism of GnT-V expression regulating colon tumor development. Pathways implicated in the regulation of colon tumor development are depicted based on the observations presented in this study. The expression level of GnT-V regulates the canonical Wnt/β-catenin signaling pathway through N-glycan modulation of Wnt receptors, resulting in abnormal colon tumorigenesis and affecting adenoma progression in Apcmin/+ mice. The regulation of colon tumor development by GnT-V is mediated at least in part by alterations in the cancer stem cell population.
Acknowledgments
We thank Julie Nelson, Heather Johnson, Danielle Sambo, Sarah Dolisca, and Kimberly Yau for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants U01CA128454 and P41-GM103490 (to M. P.). This work was also supported by Georgia Research Alliance Cancer Research Award 2012 and University of Georgia Faculty Research Grant FRG 064-417 (to H. G.).
- CSC
- cancer stem cells
- GnT-V
- N-acetylglucosaminyltransferase V
- CCSC
- colon cancer stem cells
- FZD
- frizzled
- L-PHA
- phytohemagglutinin
- Apc
- adenomatous polyposis coli
- TCF
- T cell factor
- LEF-1
- lymphocyte enhancer factor-1
- PNGase F
- peptide N-glycosidase F
- Lrp
- LDL receptor-related protein
- qRT
- quantitative real-time.
REFERENCES
- 1. Wicha M. S., Liu S., Dontu G. (2006) Cancer stem cells: an old idea: a paradigm shift. Cancer Res. 66, 1883–1890 [DOI] [PubMed] [Google Scholar]
- 2. Ghiaur G., Gerber J., Jones R. J. (2012) Concise review: cancer stem cells and minimal residual disease. Stem Cells 30, 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bao S., Wu Q., McLendon R. E., Hao Y., Shi Q., Hjelmeland A. B., Dewhirst M. W., Bigner D. D., Rich J. N. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 [DOI] [PubMed] [Google Scholar]
- 4. Merlos-Suárez A., Barriga F. M., Jung P., Iglesias M., Céspedes M. V., Rossell D., Sevillano M., Hernando-Momblona X., da Silva-Diz V., Muñoz P., Clevers H., Sancho E., Mangues R., Batlle E. (2011) The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8, 511–524 [DOI] [PubMed] [Google Scholar]
- 5. Weng D., Penzner J. H., Song B., Koido S., Calderwood S. K., Gong J. (2012) Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 14, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ricci-Vitiani L., Lombardi D. G., Pilozzi E., Biffoni M., Todaro M., Peschle C., De Maria R. (2007) Identification and expansion of human colon cancer-initiating cells. Nature 445, 111–115 [DOI] [PubMed] [Google Scholar]
- 7. Huang E. H., Hynes M. J., Zhang T., Ginestier C., Dontu G., Appelman H., Fields J. Z., Wicha M. S., Boman B. M. (2009) Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 69, 3382–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yeung T. M., Gandhi S. C., Bodmer W. F. (2011) Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4382–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeilstra J., Joosten S. P., Dokter M., Verwiel E., Spaargaren M., Pals S. T. (2008) Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 68, 3655–3661 [DOI] [PubMed] [Google Scholar]
- 10. Hakomori S. (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U.S.A. 99, 10231–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Granovsky M., Fata J., Pawling J., Muller W. J., Khokha R., Dennis J. W. (2000) Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 6, 306–312 [DOI] [PubMed] [Google Scholar]
- 12. Guo H. B., Lee I., Kamar M., Akiyama S. K., Pierce M. (2002) Aberrant N-glycosylation of β1 integrin causes reduced α5β1 integrin clustering and stimulates cell migration. Cancer Res. 62, 6837–6845 [PubMed] [Google Scholar]
- 13. Vagin O., Tokhtaeva E., Yakubov I., Shevchenko E., Sachs G. (2008) Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na,K-ATPase β1 subunit. J. Biol. Chem. 283, 2192–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Partridge E. A., Le Roy C., Di Guglielmo G. M., Pawling J., Cheung P., Granovsky M., Nabi I. R., Wrana J. L., Dennis J. W. (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306, 120–124 [DOI] [PubMed] [Google Scholar]
- 15. Guo H. B., Randolph M., Pierce M. (2007) Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J. Biol. Chem. 282, 22150–22162 [DOI] [PubMed] [Google Scholar]
- 16. Guo H. B., Johnson H., Randolph M., Lee I., Pierce M. (2009) Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 19, 547–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo H. B., Johnson H., Randolph M., Nagy T., Blalock R., Pierce M. (2010) Specific posttranslational modification regulates early events in mammary carcinoma formation. Proc. Natl. Acad. Sci. U.S.A. 107, 21116–21121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Handerson T., Pawelek J. M. (2003) β1,6-Branched oligosaccharides and coarse vesicles: a common, pervasive phenotype in melanoma and other human cancers. Cancer Res. 63, 5363–5369 [PubMed] [Google Scholar]
- 19. Murata K., Miyoshi E., Kameyama M., Ishikawa O., Kabuto T., Sasaki Y., Hiratsuka M., Ohigashi H., Ishiguro S., Ito S., Honda H., Takemura F., Taniguchi N., Imaoka S. (2000) Expression of N-acetylglucosaminyltransferase V in colorectal cancer correlates with metastasis and poor prognosis. Clin. Cancer Res. 6, 1772–1777 [PubMed] [Google Scholar]
- 20. Seelentag W. K., Li W. P., Schmitz S. F., Metzger U., Aeberhard P., Heitz P. U., Roth J. (1998) Prognostic value of β1,6-branched oligosaccharides in human colorectal carcinoma. Cancer Res. 58, 5559–5564 [PubMed] [Google Scholar]
- 21. Kim Y. S., Hwang S. Y., Kang H. Y., Sohn H., Oh S., Kim J. Y., Yoo J. S., Kim Y. H., Kim C. H., Jeon J. H., Lee J. M., Kang H. A., Miyoshi E., Taniguchi N., Yoo H. S., Ko J. H. (2008) Functional proteomics study reveals that N-acetylglucosaminyltransferase V reinforces the invasive/metastatic potential of colon cancer through aberrant glycosylation on tissue inhibitor of metalloproteinase-1. Mol. Cell Proteomics 7, 1–14 [DOI] [PubMed] [Google Scholar]
- 22. Su L. K., Kinzler K. W., Vogelstein B., Preisinger A. C., Moser A. R., Luongo C., Gould K. A., Dove W. F. (1992) Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670 [DOI] [PubMed] [Google Scholar]
- 23. Bienz M., Clevers H. (2000) Linking colorectal cancer to Wnt signaling. Cell 103, 311–320 [DOI] [PubMed] [Google Scholar]
- 24. He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998) Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- 25. McConnell B. B., Bialkowska A. B., Nandan M. O., Ghaleb A. M., Gordon F. J., Yang V. W. (2009) Haploinsufficiency of Kruppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res. 69, 4125–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo M., Fan H., Nagy T., Wei H., Wang C., Liu S., Wicha M. S., Guan J. L. (2009) Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 69, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ginestier C., Hur M. H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C. G., Liu S., Schott A., Hayes D., Birnbaum D., Wicha M. S., Dontu G. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanwar S. S., Yu Y., Nautiyal J., Patel B. B., Majumdar A. P. (2010) The Wnt/β-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer 9, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang Y., Boije M., Westermark B., Uhrbom L. (2011) PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia 13, 492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Brien C. A., Pollett A., Gallinger S., Dick J. E. (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110 [DOI] [PubMed] [Google Scholar]
- 32. Dalerba P., Dylla S. J., Park I. K., Liu R., Wang X., Cho R. W., Hoey T., Gurney A., Huang E. H., Simeone D. M., Shelton A. A., Parmiani G., Castelli C., Clarke M. F. (2007) Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin L., Liu A., Peng Z., Lin H. J., Li P. K., Li C., Lin J. (2011) STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 71, 7226–7237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu R., Yang Y., Tian Y., Bai J., Zhang X., Li X., Peng Z., He Y., Chen L., Pan Q., Fang D., Chen W., Qian C., Bian X., Wang R. (2012) Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS ONE 7, e32170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ziskin J. L., Dunlap D., Yaylaoglu M., Fodor I. K., Forrest W. F., Patel R., Ge N., Hutchins G. G., Pine J. K., Quirke P., Koeppen H., Jubb A. M. (2013) In situ validation of an intestinal stem cell signature in colorectal cancer. Gut 62, 1012–1023 [DOI] [PubMed] [Google Scholar]
- 36. van der Flier L. G., van Gijn M. E., Hatzis P., Kujala P., Haegebarth A., Stange D. E., Begthel H., van den Born M., Guryev V., Oving I., van Es J. H., Barker N., Peters P. J., van de Wetering M., Clevers H. (2009) Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 136, 903–912 [DOI] [PubMed] [Google Scholar]
- 37. Tian H., Biehs B., Warming S., Leong K. G., Rangell L., Klein O. D., de Sauvage F. J. (2011) A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zucchi I., Sanzone S., Astigiano S., Pelucchi P., Scotti M., Valsecchi V., Barbieri O., Bertoli G., Albertini A., Reinbold R. A., Dulbecco R. (2007) The properties of a mammary gland cancer stem cell. Proc. Natl. Acad. Sci. U.S.A. 104, 10476–10481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vermeulen L., Todaro M., de Sousa Mello F., Sprick M. R., Kemper K., Perez Alea M., Richel D. J., Stassi G., Medema J. P. (2008) Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. U.S.A. 105, 13427–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Charafe-Jauffret E., Monville F., Ginestier C., Dontu G., Birnbaum D., Wicha M. S. (2008) Cancer stem cells in breast: current opinion and future challenges. Pathobiology 75, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeung T. M., Gandhi S. C., Wilding J. L., Muschel R., Bodmer W. F. (2010) Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. U.S.A. 107, 3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Korkaya H., Paulson A., Iovino F., Wicha M. S. (2008) HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 27, 6120–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rota L. M., Lazzarino D. A., Ziegler A. N., LeRoith D., Wood T. L. (2012) Determining mammosphere-forming potential: application of the limiting dilution analysis. J. Mammary Gland Biol. Neoplasia 17, 119–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anastas J. N., Moon R. T. (2013) WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 [DOI] [PubMed] [Google Scholar]
- 45. de Sousa E. M., Vermeulen L., Richel D., Medema J. P. (2011) Targeting Wnt signaling in colon cancer stem cells. Clin. Cancer Res. 17, 647–653 [DOI] [PubMed] [Google Scholar]
- 46. de Sousa E Melo F., Colak S., Buikhuisen J., Koster J., Cameron K., de Jong J. H., Tuynman J. B., Prasetyanti P. R., Fessler E., van den Bergh S. P., Rodermond H., Dekker E., van der Loos C. M., Pals S. T., van de Vijver M. J., Versteeg R., Richel D. J., Vermeulen L., Medema J. P. (2011) Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 9, 476–485 [DOI] [PubMed] [Google Scholar]
- 47. Vermeulen L., De Sousa E Melo F., van der Heijden M., Cameron K., de Jong J. H., Borovski T., Tuynman J. B., Todaro M., Merz C., Rodermond H., Sprick M. R., Kemper K., Richel D. J., Stassi G., Medema J. P. (2010) Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 [DOI] [PubMed] [Google Scholar]
- 48. Garcia M. I., Ghiani M., Lefort A., Libert F., Strollo S., Vassart G. (2009) LGR5 deficiency deregulates Wnt signaling and leads to precocious Paneth cell differentiation in the fetal intestine. Dev. Biol. 331, 58–67 [DOI] [PubMed] [Google Scholar]
- 49. Walker F., Zhang H. H., Odorizzi A., Burgess A. W. (2011) LGR5 is a negative regulator of tumourigenicity, antagonizes Wnt signalling and regulates cell adhesion in colorectal cancer cell lines. PLoS ONE 6, e22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu Z. Q., Brabletz T., Fearon E., Willis A. L., Hu C. Y., Li X. Y., Weiss S. J. (2012) Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. U.S.A. 109, 11312–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jardé T., Evans R. J., McQuillan K. L., Parry L., Feng G. J., Alvares B., Clarke A. R., Dale T. C. (2013) In vivo and in vitro models for the therapeutic targeting of Wnt signaling using a Tet-OδN89β-catenin system. Oncogene 32, 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. MacDonald B. T., He X. (2012) Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo H. B., Zhang Q. S., Chen H. L. (2000) Effects of H-ras and v-sis overexpression on N-acetylglucosaminyltransferase V and metastasis-related phenotypes in human hepatocarcinoma cells. J Cancer Res. Clin. Oncol. 126, 263–270 [DOI] [PubMed] [Google Scholar]
- 54. Chen L., Zhang W., Fregien N., Pierce M. (1998) The her-2/neu oncogene stimulates the transcription of N-acetylglucosaminyltransferase V and expression of its cell surface oligosaccharide products. Oncogene 17, 2087–2093 [DOI] [PubMed] [Google Scholar]
- 55. Brudvik K. W., Paulsen J. E., Aandahl E. M., Roald B., Taskén K. (2011) Protein kinase A antagonist inhibits β-catenin nuclear translocation, c-Myc and COX-2 expression and tumor promotion in Apc(Min/+) mice. Mol. Cancer 10, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tetsu O., McCormick F. (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- 57. Jubb A. M., Chalasani S., Frantz G. D., Smits R., Grabsch H. I., Kavi V., Maughan N. J., Hillan K. J., Quirke P., Koeppen H. (2006) Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene 25, 3445–3457 [DOI] [PubMed] [Google Scholar]
- 58. Barker N., Ridgway R. A., van Es J. H., van de Wetering M., Begthel H., van den Born M., Danenberg E., Clarke A. R., Sansom O. J., Clevers H. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 [DOI] [PubMed] [Google Scholar]
- 59. Niida A., Hiroko T., Kasai M., Furukawa Y., Nakamura Y., Suzuki Y., Sugano S., Akiyama T. (2004) DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 23, 8520–8526 [DOI] [PubMed] [Google Scholar]
- 60. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 61. Kemper K., Prasetyanti P. R., De Lau W., Rodermond H., Clevers H., Medema J. P. (2012) Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 30, 2378–2386 [DOI] [PubMed] [Google Scholar]
- 62. Xu Q., Akama R., Isaji T., Lu Y., Hashimoto H., Kariya Y., Fukuda T., Du Y., Gu J. (2011) Wnt/β-catenin signaling down-regulates N-acetylglucosaminyltransferase III expression: the implications of two mutually exclusive pathways for regulation. J. Biol. Chem. 286, 4310–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee S. H., Hu L. L., Gonzalez-Navajas J., Seo G. S., Shen C., Brick J., Herdman S., Varki N., Corr M., Lee J., Raz E. (2010) ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat. Med. 16, 665–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Johswich A., Longuet C., Pawling J., Abdel Rahman A., Ryczko M., Drucker D. J., Dennis J. W. (2014) N-Glycan remodeling on glucagon receptor is an effector of nutrient sensing by the hexosamine biosynthesis pathway. J. Biol. Chem. 289, 15927–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Janda C. Y., Waghray D., Levin A. M., Thomas C., Garcia K. C. (2012) Structural basis of Wnt recognition by Frizzled. Science 337, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao Y., Nakagawa T., Itoh S., Inamori K., Isaji T., Kariya Y., Kondo A., Miyoshi E., Miyazaki K., Kawasaki N., Taniguchi N., Gu J. (2006) N-Acetylglucosaminyltransferase III antagonizes the effect of N-acetylglucosaminyltransferase V on α3β1 integrin-mediated cell migration. J. Biol. Chem. 281, 32122–32130 [DOI] [PubMed] [Google Scholar]
- 67. Guo H. B., Lee I., Kamar M., Pierce M. (2003) N-Acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J. Biol. Chem. 278, 52412–52424 [DOI] [PubMed] [Google Scholar]
- 68. Pinho S. S., Reis C. A., Paredes J., Magalhães A. M., Ferreira A. C., Figueiredo J., Xiaogang W., Carneiro F., Gärtner F., Seruca R. (2009) The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum. Mol. Genet. 18, 2599–2608 [DOI] [PubMed] [Google Scholar]
- 69. Ihara S., Miyoshi E., Ko J. H., Murata K., Nakahara S., Honke K., Dickson R. B., Lin C. Y., Taniguchi N. (2002) Prometastatic effect of N-acetylglucosaminyltransferase V is due to modification and stabilization of active matriptase by adding β1–6 GlcNAc branching. J. Biol. Chem. 277, 16960–16967 [DOI] [PubMed] [Google Scholar]
- 70. Qi J., Li N., Fan K., Yin P., Zhao C., Li Z., Lin Y., Wang L., Zha X. (2014) β1,6 GlcNAc branches-modified PTPRT attenuates its activity and promotes cell migration by STAT3 pathway. PLoS ONE 9, e98052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jamal B., Sengupta P. K., Gao Z. N., Nita-Lazar M., Amin B., Jalisi S., Bouchie M. P., Kukuruzinska M. A. (2012) Aberrant amplification of the crosstalk between canonical Wnt signaling and N-glycosylation gene DPAGT1 promotes oral cancer. Oral oncology 48, 523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]