

Background: Allostery is one of the important but complicated properties of proteins.
Results: Structural and kinetic analyses indicated the simple allosteric machinery of Thermus caldophilus l-lactate dehydrogenase (TcLDH).
Conclusion: TcLDH employs a compact mobile core and electrostatic repulsion for the mediation of allosteric protein motion and allosteric equilibrium.
Significance: This allosteric machinery expands the knowledge of the allostery of proteins.
Keywords: Allosteric Regulation, Dehydrogenase, Enzyme Mutation, Enzyme Structure, Lactic Acid, Thermophile
Abstract
For Thermus caldophilus l-lactate dehydrogenase (TcLDH), fructose 1,6-bisphosphate (FBP) reduced the pyruvate S0.5 value 103-fold and increased the Vmax value 4-fold at 30 °C and pH 7.0, indicating that TcLDH has a much more T state-sided allosteric equilibrium than Thermus thermophilus l-lactate dehydrogenase, which has only two amino acid replacements, A154G and H179Y. The inactive (T) and active (R) state structures of TcLDH were determined at 1.8 and 2.0 Å resolution, respectively. The structures indicated that two mobile regions, MR1 (positions 172–185) and MR2 (positions 211–221), form a compact core for allosteric motion, and His179 of MR1 forms constitutive hydrogen bonds with MR2. The Q4(R) mutation, which comprises the L67E, H68D, E178K, and A235R replacements, increased Vmax 4-fold but reduced pyruvate S0.5 only 5-fold in the reaction without FBP. In contrast, the P2 mutation, comprising the R173Q and R216L replacements, did not markedly increase Vmax, but 102-reduced pyruvate S0.5, and additively increased the FBP-independent activity of the Q4(R) enzyme. The two types of mutation consistently increased the thermal stability of the enzyme. The MR1-MR2 area is a positively charged cluster, and its center approaches another positively charged cluster (N domain cluster) across the Q-axis subunit interface by 5 Å, when the enzyme undergoes the T to R transition. Structural and kinetic analyses thus revealed the simple and unique allosteric machinery of TcLDH, where the MR1-MR2 area pivotally moves during the allosteric motion and mediates the allosteric equilibrium through electrostatic repulsion within the protein molecule.
Introduction
Allostery is one of the important properties of proteins that plays a crucial role in the regulation of many biological processes, such as metabolism and signal transduction in the cell, but is also one of the complicated phenomena for experimental analysis. Allosteric transition of a protein is usually accompanied by a great change in the protein structure, where the separate binding sites for allosteric effectors undergo cooperative structural changes. There are two representative models, the Monod-Wyman-Changeux (MWC) model proposed by Monod et al. (1) and the Koshland-Nemethy-Filmer (KNF) model by Koshland et al. (2), to explain the allosteric transition. The former model is based on the pre-existing theory, which was expanded from the lock and key theory (3), and the latter is based on the induced fit theory (4). It is, however, generally difficult to exactly distinguish the two models on experimental analysis (5), and only a few allosteric proteins have been studied as to the detailed allosteric machinery. l-Lactate dehydrogenase (LDH; EC 1.1.1.27)2 catalyzes oxidation-reduction between pyruvate and l-lactate with a coenzyme, NAD, and usually forms a tetrameric structure comprising four identical subunits, which are related through three 2-fold axes: the P, Q, and R axes (6). LDH is one of the highly divergent enzymes in organisms and species, and many bacterial cells possess allosteric types of LDH, which are commonly activated by fructose 1,6-bisphosphate (FBP) (7). In allosteric LDHs, FBP usually induces a drastic reduction of substrate Km (S0.5) often together with a slight increase of Vmax. Although the active site (substrate-binding site) is located near the Q-axis subunit interface, the FBP binding site (allosteric site) is located at the P-axis interface area (8, 9), where Arg173 and His188 play essential roles in the FBP-induced activation (10–14). Bacterial allosteric LDHs provide relatively simple research models for the allosteric machinery of proteins (15).
The crystal structure of Bifidobacterium longum LDH (BlLDH) contains active and inactive state structures in a 1:1 ratio within the same crystal lattice, where the two state enzymes consistently form complexes with FBP (9), this being consistent with the MWC model. Hence, the active (R) and inactive (T) states of BlLDH, which exhibit high and low affinities to FBP, respectively, coexist in an allosteric equilibrium independently of FBP or a substrate. In addition, we recently determined the two state structures of Lactobacillus casei LDH (LcLDH) (16). Strikingly, the two structures were consistently unliganded with any allosteric effector, directly indicating that LcLDH undergoes the MWC-type (pre-existing) allosteric transition. In the catalytic process, LDH also changes its protein structure, and particularly the βd-αD/E loop (active site loop) area exhibits closed and open motion through the binding and releasing of ligands, respectively (17). The two state structures of LcLDH consistently have an open active site loop and clearly show net structural changes of the allosteric transition (16). BlLDH and LcLDH exhibit common structural changes, as the following. First, the quaternary structures of the two enzymes consistently change between the two states, where the P-axis-related dimers take on closed and open conformations. Second, the enzymes show a change in the orientation of the Arg171 side chain, which forms bifurcated salt bridges with a substrate, through a change in the contact between the α2F helix and the αC helix of the Q-axis-related subunit. On the other hand, Lactobacillus pentosus LDH (LpLDH) exhibits high catalytic activity independently of FBP (18) and possesses a unique intersubunit salt bridge network around the contact between the two helices (19) (Figs. 1A and 2). The Q mutant LcLDH, which mimics the Q-axis interface of LpLDH, exhibits high catalytic activity in the absence of FBP (20).
FIGURE 1.

The Q-axis intersubunit regions around Arg171 of LpLDH (A) and TtMDH (B) and the R (C) and T (D) states of TcLDH. The structures are represented by ribbon diagrams, where only the amino acids at positions 67, 68, 171, 178, and 235 are indicated by a stick model. The residues are numbered according to the N system proposed by Eventoff et al. (50). The two Q-axis-related subunits are colored red and blue, as in Fig. 3A. Salt bridges and hydrogen bonds among these amino acids are indicated by green broken lines.
FIGURE 2.

Structure-based sequence alignment of representative l-LDHs. The amino acid sequences of l-LDHs from four organisms are aligned: T. caldophilus (TcLDH), L. casei (LcLDH), L. pentosus (LpLDH), and B. longum (BlLDH). The residues are numbered according to the N system proposed by Eventoff et al. (50). Conserved amino acids important for the catalysis are colored red. The residues forming positively charged clusters in TcLDH (see Fig. 8) are colored blue, as well as conserved Arg173 and His188 of the FBP binding sites in the other enzymes. The residues forming the Q-axis intersubunit salt bridges (Fig. 1) in LpLDH are colored green. The two amino acids, Ala154 and His179, that are replaced with Gly and Tyr in T. thermophilus LDH (TtLDH), respectively, are indicated by asterisks. The residues that exhibit more than 2.0 Å deviation (RMSD at Cα) between the T and R state structures of TcLDH, LcLDH, and BlLDH are underlined. The secondary structural elements of TcLDH, which are identical in the two state structures, are indicated by bars on the sequence.
Thermus caldophilus LDH (TcLDH) is a heat-stable allosteric LDH (21) and shows less than 40% amino acid identity to LcLDH or BlLDH (Fig. 2). This enzyme is uniquely activated through chemical modifications with 2,3-butanedione (22–25), diethyl pyrocarbonate, or acetic anhydride (18) with protection of the catalytic site by NADH and oxamate. Arg173 (12) and Arg216 (25) are predominantly modified with 2,3-butandione, and the R173Q and R216L replacements (P mutations) also markedly enhance the FBP-independent activity of TcLDH (12, 25). These observations strongly suggest that the inactive state (T state) of TcLDH is stabilized by the positively charged groups within the protein.
The allosteric constant (L = [T state]/[R state]) was estimated to be 1.8 × 105 for TcLDH on kinetic analysis of the lactate oxidation reaction at pH 6.75, assuming the MWC-type allosteric transition (25). This L value is markedly greater than the value for LcLDH (3.0 × 102) (16) and is reduced to 6.8 by the P2 mutation (R173Q/R216L) (25). Recently, the apo and holo structures of Thermus thermophilus LDH (TtLDH), which only has the A154G and H179Y replacements of TcLDH, were determined (27). However, TtLDH exhibits only a 10-fold increased Km value for pyruvate in the absence of FBP at pH 7.0 and 30 °C, and the 1-Mut (R218A) and 5-Mut (R79W, R151A, E279A, E313A, and E299A) mutant TtLDHs exhibit ∼2- and 3.6-fold reduced Km values in the absence of FBP, respectively (28). TtLDH thus appears to show much lower FBP dependence than TcLDH, although TcLDH has not been assayed under the corresponding conditions.
In this study, we determined the T and R state structures of TcLDH, and evaluated the catalytic properties of the P and Q mutant enzymes, as well as the wild-type enzyme. The results revealed the simple and unique allosteric machinery of TcLDH.
EXPERIMENTAL PROCEDURES
Crystallization, Data Collection, and Model Building
An overproduction plasmid for TcLDH, p8T4 (29), which was constructed from pEXP7 (30) and the structural gene of TcLDH (31), was kindly provided by Profs. S. Koide and H. Matsuzawa. Escherichia coli MV1184 cells harboring the expression plasmid were grown at 37 °C in 2× YT medium (1.6% tryptone, 1.0% yeast extract, and 0.5% NaCl) containing 150 μg/ml ampicillin. A cell-free extract was prepared from the cells by sonication in 20 mm Tris-HCl buffer (pH 7.5) and then treated at 90 °C for 30 min. After removing the precipitate from the extract by centrifugation, the enzyme was purified by Butyl-Toyopearl (Tosoh, Tokyo, Japan) and UnoQ (Bio-Rad) or Resource Q (GE Healthcare) column chromatographies.
Crystals for unliganded (T state) TcLDH were grown as previously reported (29), the crystals being grown from a 1:1 mixture of a protein solution (4–8 mg/ml in 50 mm sodium MES buffer, pH 6.0) and a reservoir solution (50 mm sodium MES buffer, pH 6.0, containing 8–14% PEG6000) using the hanging drop vapor diffusion method at 25 °C. Crystals for liganded (R state) TcLDH were grown from a 1:1 mixture of a protein solution (4 mg/ml in 70 mm trisodium citrate buffer, pH 5.6, containing 30% glycerol, 0.2 mm NADH, 0.2 mm sodium oxamate and 0.2 mm FBP) and a reservoir solution (70 mm trisodium citrate, pH 5.6, 30% glycerol, and 0.7 m ammonium phosphate) using the hanging drop vapor diffusion method at 25 °C. Diffraction data were collected at 100 K (λ = 0.978 Å) with a CCD camera at the BL6A Station of the Photon Factory, High Energy Accelerator Research Organization (KEK). The two types of crystal were transferred to the corresponding reservoir solutions containing 30% glycerol prior to flash cooling for data collection. The diffraction images were indexed, integrated, and scaled using the DPS/MOSFLM program suite (32). The initial phases were determined by means of the molecular replacement method. For the crystal of the T state enzyme, the structure was solved by fitting the structure of Geobacillus stearothermophilus LDH (Protein Data Bank code 1LDN) to the molecular positions in the P21 crystal form previously determined by Wigley et al. (8), because the cell dimensions of this crystal were essentially identical to those of the crystal reported. The R state structure of the enzyme was solved by the molecular replacement method, using the T state structure of TcLDH obtained here as a search model. The structural models were built and refined by using program CNS (33) and program O (34).
The atomic coordinates and measured structure factor amplitudes of the T and R states of TcLDH were deposited and are available in the Protein Data Bank under accession codes 3VPG and 3VPH, respectively. For structural comparison, the T (Protein Data Bank code 2ZQY) and R (Protein Data Bank code 2ZQZ) state structures of LcLDH, the two state structures of complex BlLDH (Protein Data Bank code 1LTH), the T state of unliganded BlLDH (Protein Data Bank code 1LLD), the apo-form structure of LpLDH (Protein Data Bank code 1EZ4), the apo (Protein Data Bank code 2V6M) and holo (Protein Data Bank code 2V7P) form structures of TtLDH, and the T. thermophilus l-malate dehydrogenase structure (TtMDH) (Protein Data Bank code 1IZ9) were used. All the figures for protein structures were prepared using PyMOL (35), except for Fig. 3A, which was drawn with MOLSCRIPT (36) and Raster3D (37).
FIGURE 3.

Quaternary and tertiary structural changes in the allosteric transition. A, ribbon diagrams of the tetrameric structures: liganded (R state) (left panel) and unliganded (T state) (center panel) structures of TcLDH and holo TtLDH structure (right panel). All the structures are viewed along the molecular R-axis. The four subunits are colored red, green, blue, and gray. The P-, Q-, and R-axes are indicated by arrows and a circle. Each of the subunits is named according to the name registered in Protein Data Bank. B, left, deviation of Cα atoms (RSMD values) between the R and T state structures of TcLDH (light blue line) and the holo and apo structures of TtLDH (pink line). MR1 and MR2 indicate mobile regions that display marked deviation (more than 2.0 Å) in the two structures of TcLDH. Right, superimpositioning of the R state of TtLDH (magenta) and R state (purple) and T state (yellow) TcLDH subunit structures. The subunits (subunits A in A) of R state TtLDH and T state TcLDH were fitted with subunit A of R state TcLDH by means of least squares deviation for Cα atoms. C, superimpositioning of the backbone structures of the T and R state structures for TcLDH (left panel), BlLDH (center panel), and LcLDH (right panel). The T and R state structures are shown in cyan and orange, respectively, and the regions that show more than 2.0 Å RMSD (at Cα) in any of the three enzymes are indicated by blue and red for the T and R states, respectively.
Preparation of the Mutant TcLDHs
The expression plasmids for the R173Q and R173Q/R216L mutant enzymes (P1 and P2 mutant TcLDHs, respectively) were kindly provided by Profs. H. Matsuzawa and T. Ohta (The University of Tokyo) (23). The following primers and the respective complementary primers were purchased from Takara Syuzo (Kyoto, Japan) and Operon Biotechnologies (Tokyo, Japan) and used for site-directed mutagenesis: L67E/H68D (for the Q2 enzyme), 5′-C CAC GCC GAG GAC ATC CTC CAC GCC ACG-3′; E178K (for the Q3 enzyme), 5′-CGG GCC CTT CTG GCG AAG CAC CTC CGG-3′; A235K (for the Q4(K) enzyme), 5′-GAC GAA GGG GTT CGC CGG GCC AAG GCC TAC CGG ATC ATT GAG-3′; A235R (for the Q4(R) enzyme), 5′-GAC GAA GGG GTT CGC CGG GCC CGC GCC TAC CGG ATC ATT GAG-3′; R173Q (for the Q4/P1 enzymes), 5′-CAG GCC CTT CTG GCG AAG CAC CTC CGG-3′; and R216L (for the Q4/P2 enzymes), 5′-CTG GGG CGG GCC CTT TCC CCG GAG-3′. Site-directed mutagenesis was performed with a QuikChange II in vitro mutagenesis kit (Stratagene, La Jolla, CA) to construct the genes of the Q2, Q3, and Q4 mutant TcLDHs, and with a KOD Plus mutagenesis kit (Toyobo, Osaka, Japan) to construct the genes of the Q4/P1 and Q4/P2 mutant TcLDHs, using expression plasmid p8T4 (29). The mutant TcLDHs were expressed in E. coli MV1184 cells and purified essentially according to the procedure for the wild-type enzyme. The purity of the enzyme preparations was examined by SDS-PAGE according to Laemmli (38).
Enzyme Assays and Protein Concentration Determination
It is known that TcLDH shows sigmoidal saturation curves for substrates in the absence of FBP (12, 21–25), but no significant cooperativity on NADH binding, giving dissociation constants of 0.9–1.2 μm for NADH independently of FBP or the P mutations (25). The enzyme assay for TcLDH was performed at 30 °C in 50 mm sodium MOPS buffer (pH 7.0) containing 0.1 mm NADH, which is a saturating concentration for TcLDH, and sodium pyruvate (varied between 0.01 and 50 mm). The decrease in absorbance at 340 nm caused by the consumption of NADH was monitored. One unit was defined as the catalytic rate of the conversion of 1 μmol of substrate per min. Protein concentrations were determined by the Bradford method (39) with Bio-Rad protein assay protein reagent (Bio-Rad), using bovine serum albumin as a standard protein. The Hill equation (Equation 1) (40) was used for sigmoidal curve fitting to obtain kinetic parameters such as the Hill coefficient (nH) and half-saturating concentrations (S0.5) of pyruvate by curve fitting of the data with KaleidaGraph.
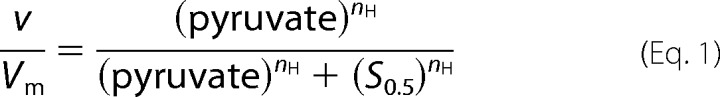 |
RESULTS
Changes in Quaternary and Tertiary Structures during Allosteric Transition
The unliganded and liganded (with NADH, oxamate, and FBP) structures of TcLDH were determined at 1.8 and 2.0 Å, respectively (Table 1). The two structures obviously differ from each other in their quaternary structures, the P-axis-related dimers taking on open and closed conformations (Fig. 3A), as in the case of the T and R states of BlLDH (9) and LcLDH (16), respectively. This indicates that TcLDH also has two major conformational states, the T and R states, in the allosteric transition, although the enzyme undergoes a more markedly smaller quaternary structural change (average RMSD, 2.69 Å) than BlLDH (3.69 Å) and LcLDH (4.89 Å) do. Although the structure of the unliganded (T state) TcLDH tetramer is virtually the same as that of the apo TtLDH, the liganded (R state) structure shows better agreement with that of the holo TtLDH except for the active site loop structure (positions 98–109). This loop is closed (subunits A and C) or disordered (subunits B and D) in the holo TtLDH, but open in all the four subunits of the R state TcLDH, although the two enzymes consistently bind NADH and oxamate molecules in their active sites (Fig. 3A). When the active site loop region was omitted from the calculation, the average RMSD between the two structures is only 0.48 Å, indicating that the holo TtLDH structure forms the R state structure, although it contains no bound FBP molecule. This is not surprising because the two enzymes consistently undergo the MWC type of allosteric transition like LcLDH and BlLDH.
TABLE 1.
Data collection and refinement statistics
| Data sets | Unliganded | Liganded with NADH, oxamate, and FBP |
|---|---|---|
| Data collection statistics | ||
| Protein Data Bank entry | 3VPG | 3VPH |
| Space group | P21 | C2 |
| Unit cell parameters | ||
| a (Å) | 54.24 | 146.74 |
| b (Å) | 135.07 | 67.84 |
| c (Å) | 85.88 | 147.34 |
| β (°) | 94.44 | 94.02 |
| Resolution (Outer shell) ( Å) | 29.2-1.80 (1.90-1.80) | 29.4-2.00 (2.11-2.00) |
| Unique reflections | 111,821 (16,008) | 96,717 (13,750) |
| Completeness (%) | 98.4 (96.6) | 98.9 (96.9) |
| Rmerge (%) | 6.3 (27.7) | 4.9 (27.0) |
| Mean <I/σ(I)> | 7.1 (2.7) | 16.9 (4.1) |
| Refinement statistics | ||
| Rfactor (%) | 19.1 | 20.9 |
| Rfree (%) | 22.7 | 24.6 |
TcLDH has virtually an identical secondary structural element composition to those of other representative LDHs, except for the region between βJ and α1/2G (positions 210a-220) (Fig. 2). The βJ-α1/2G loop region is called the “flexible surface area,” where both the primary and three-dimensional structures are poorly conserved in known LDHs. TcLDH has a small helix (the αT helix) at positions 210a-213 in this region like LcLDH (16), LpLDH (19), and Thermotoga maritima LDH (TmLDH) (41), whereas this helix is partially unwound in G. stearothermophilus LDH (8) and completely unwound in BlLDH (9) and dogfish muscle LDH (6). Like LcLDH (16), TcLDH does not show a significant change in the secondary structural elements between the two state structures, and the α1/2G helix is constitutively kinked at position 234, whereas in BlLDH the α1/2G helix is kinked only in the R state structure (9). TcLDH and TtLDH exhibit essentially the same structural changes between their two state structures except for in the active site loop area (Fig. 3B). In the tertiary structure, TcLDH also shows an apparently smaller structural change (0.89 Å) than BlLDH (1.26 Å) and LcLDH (0.97 Å), the latter of which consistently has open active site loops in the two states like TcLDH (16).
TcLDH exhibits a marked structural change in the catalytic domain (positions 166–331) like BlLDH (9) and LcLDH (16) and particularly great movements in two regions (mobile regions (MRs)), MR1 (positions 172–185) and MR2 (positions 211–221) (Figs. 2 and 3B). A marked structural change is also observed in the N-terminal half of the α1/2G helix (α1G), which is divided into α1G and α2G by the conserved kink at position 234, but no significant change is observed in the following α2G helix, α1/2G-α3G loop, or α3G helix, unlike in the case of LcLDH or BlLDH. Superimpositioning of the T and R state structures clearly indicates the different allosteric motions of the three LDHs (Fig. 3C). In TcLDH, the C-terminal area of the α2F helix (MR1) and the flexible surface area (MR2) are mostly moved, which greatly shift in position in the same direction. In contrast, in BlLDH, these regions do not markedly move, instead the α1/2G-α3G loop area greatly moves together with the active site loop, which forms the closed conformation in the R state as in the case of TtLDH. LcLDH shows an apparently intermediate type of structural change between those of TcLDH and BlLDH, both the MR1-MR2 and α1/2G-α3G areas moving equivalently.
Structural Change in the Active Site
The R state TcLDH has essentially an active site structure identical to that of the R state TtLDH except for the conformation of the active site loop (Fig. 4A) and binds NADH and oxamate essentially at the same positions and orientations as in TtLDH. It is known that conserved His195, Arg109, Arg171, and Thr246 (Fig. 2) play important roles in the catalytic function of LDH through direct interaction with the bound pyruvate molecule (6, 42–45). The oxamate molecule in TcLDH forms proper hydrogen bonds with all these residues except Arg109, which is located in the active site loop. In the R state TcLDH, in addition, the imidazole of His195, the acid/base catalyst of LDH, forms the proper catalytic pair with the carboxyl group of Asp168, which stabilizes both the ground and transition states of the enzyme-NADH-pyruvate ternary complex during the catalytic process of LDH (46). Hence, the R state (ternary complex) structure of TcLDH is unique in the lack of the Arg109-oxamate interaction, but thus may not be surprising because Arg109 is less important for the substrate binding than for the hydrogen transfer step (46).
FIGURE 4.

Structural changes in the active sites (A and B) and FBP binding sites (C and D). One subunit (subunit A) is colored salmon, and the Q-axis and P-axis-related subunits are colored cyan and green, respectively. Red dotted lines in A and B indicate hydrogen bonds. oxm indicates the oxamate molecule. A, left, the active sites of the R state structures of TcLDH and TtLDH (semitransparent). Right, the T and R (semitransparent) state structures of TcLDH. B, the active sites of the T and R (semitransparent) state structures of LcLDH (left panel) and BlLDH (right panel). C, the FBP binding sites of the R state structures of TcLDH and TtLDH (semitransparent) (left panel) and T and R (semitransparent) state structures of TcLDH (right panel). The glycerol molecule in the R state TcLDH is indicated as Gol. D, the FBP binding sites of the T and R (semitransparent) state structures of LcLDH (left panel) and BlLDH (right panel). Nitrate and sulfate ions from the solvent in the T and R state LcLDH structures are denoted as NO3 and SO4, respectively. The unliganded T state structure of BlLDH is essentially the same as the liganded T state structure, except for the configuration of the Arg173 side chain (9, 48); therefore only Arg173 (Arg173(U)) is shown for the unliganded BlLDH structure.
Like in BlLDH (9) and LcLDH (16), in TcLDH the side chain of Arg171 is orientated outside the active site in the T state, and the Arg171 orientation is switched through a change in the contact between the α2F helix and the αC helix on the Q-axis-related subunit (αC(Q)) (Fig. 4, A and B), where His68 (Q) blocks the alternative orientation of Arg171 through steric hindrance. However, TcLDH obviously differs from BLLDH or LcLDH in the motions of the two helices. In TcLDH, the α2F helix greatly rotates by 10° around its N-terminal area, but the αC(Q) helix (3°) only slightly rotates around its C-terminal area. On the other hand, in LcLDH, both the αC(Q) helix (10°) and the α2F helix (6°) greatly rotate, and in BlLDH the αC(Q) helix (12°) rotates much more than the α2F helix (3°). As in the case of LcLDH, in TcLDH, the Asp168 carboxyl group is allowed to be slightly (0.8 Å) more separate from the His195 imidazole through the α2F helix rotation in the T state structure. In TcLDH and LcLDH, constitutive intersubunit hydrogen bonds are consistently formed between the αC(Q) helix and the α1/2G-α3G loop areas. In LcLDH, therefore, the α1/2G-α3G loop area moves together with the αC(Q) helix between the two states, which allows Thr246 of the loop to be more separate from the active site in the T state structure, whereas in TcLDH this loop area or Thr246 does not markedly move. On the other hand, BlLDH exhibits drastic structural changes in the αC(Q) helix and α1/2G-α 3G areas, which form intersubunit hydrogen bonds only in the R state structure. In the T state structure of BlLDH, consequently, His68 (Q) is deeply intercalated into the active site to form hydrogen bonds with Asp168, which allows Thr246 to completely point away from the active site and instead allows Ile240 to occupy the position in which the substrate molecule would be accommodated in the R state. TcLDH thus apparently undergoes the smallest and simplest structural change in the active site area among the three enzymes.
Structural Change in the FBP Binding Site
As in the cases of G. stearothermophilus LDH (8) and BlLDH (9), the R state TcLDH binds two FBP molecules per tetramer at the P-axis subunit interface (Fig. 3A) essentially in the same manner (Fig. 4, C and D), where Arg173 and His188 of two juxtaposed subunits form salt bridges with the two phosphate groups of FBP. In particular, the two His188 of TcLDH form multiple hydrogen bonds with two phosphate groups of FBP, whereas each of the two Arg173 forms monodentate or bidentate hydrogen bonds with each of the two phosphates. The FBP binding site of TcLDH also contains two glycerol molecules, but these molecules appear to be artifacts caused by the crystallization solution, because glycerol shows no significant effects on the catalytic properties of TcLDH (data not shown). The two imidazolium rings of His188 are positioned in parallel at ∼4 Å distance in the R state TcLDH structure and greatly point away from the binding site in the T state structure (Fig. 4C). It is notable that the R state TtLDH has essentially the same His188 conformation, although it contains no FBP molecule in the binding site (28), this being consistent with the MWC pre-existing theory. In the TcLDH structure, the FBP phosphate also forms a hydrogen bond with Tyr190 (except for in subunit B), which is highly conserved in allosteric LDHs (Fig. 2). Because TcLDH and TtLDH agree less in the orientation of Tyr190 than that of His188, the motion of Tyr190 may depend on the FBP binding according to the induced fit theory, although the exact function of Tyr190 remains uncertain.
Unlike in the case of TcLDH or TtLDH, the structure of BlLDH or LcLDH is not evidential as to the distinct FBP affinity between their two states, because these enzymes have similar conformations as to His188 to that of the R state TcLDH in both the two states (Fig. 4D). Although the unliganded T state structure of BlLDH was also determined (47), it differs from the liganded one only in the orientation of the Arg173 side chain. For TtLDH, the 1-Mut (Protein Data Bank code 2XXJ) and 5-Mut (Protein Data Bank code 4A73) enzymes also form an R state-like conformation as to His188 in their crystal structures (28), although they consistently have the T state overall structures, suggesting that the switching of His188 is quite critically controlled in TcLDH and TtLDH. It was previously suggested that electrostatic repulsion of Arg173 and His188 destabilizes the R states of TcLDH and BlLDH, because the two sets of Arg173 and His188 closely face each other at the P-axis subunit interface (9). During the T to R transition, the two His188 actually approach each other by 1.3 Å (at Cα), but the two Arg173 are rather separated by 0.3 Å. The switching of His188 appears to arise from the critical changes in the relative positions of His188 and Arg173.
The Core of Allosteric Motion
MR1 and MR2 exhibit many van der Waals and hydrogen bond contacts between them in the two state structures and thereby constitute a rigid mobile body for the allosteric motion in TcLDH (Fig. 5). His179, Leu180, and Arg181 are located at the center of MR1 and form four constitutive hydrogen bonds with Arg216 and the main chains of Arg218 and Ala219, which are located at the center of MR2 (Fig. 5C). The Arg181 main chain forms an additional inter-region hydrogen bond with the Arg216 side chain in the R state structure, and the Arg218 side chain forms an additional inter-region hydrogen bond with the His179 main chain in the T state structure. In the cases of BlLDH and LcLDH, the corresponding regions also exhibit many van der Waals contacts, but no inter-region hydrogen bond. The MR1-MR2 area rotates around the area that comprises the N-terminal region of the α2F helix, and the β-sheet of the βG (the C-terminal part) and βH strands, which form a rigid immovable support for the rotating motion through the hydrogen bonds between them (Fig. 5A). The discontinuous βG strand also functions as a hinge for the rotating motions of the following βG-βI-βJ sheet and MR2, and the main chain of Ala189 likely mediates the coordinated motions of the α2F helix and the βG-βI-βJ sheet through constitutive hydrogen bonds with the Arg173 side chain of the α2F helix. The motion of MR1-MR2 is further linked to the motions of the N-terminal part of the α1/2G helix (α1G helix), the βG-βH loop, and the αH helix (Fig. 5A). The α1G helix has many van der Waals contacts with the α2F helix in the vicinity of the kink in the two state structures and also hydrogen bond contacts with the βG-βH turn between Arg233 and Asp197. On the other hand, the N-terminal area of the αH helix moves together with the βG-βI-βJ sheet through constitutive hydrogen bond contacts with a small helix between the βI and βJ strands. The MR1-MR2 area, like the α1G and αH helices, is highly exposed to the solvent on the surface of the protein and therefore can flexibly move during the allosteric transition of TcLDH.
FIGURE 5.

The core of allosteric motion in TcLDH. A, allosteric motion and hydrogen bond network of MR1, MR2, and related regions. The subunit A structures of the R and T (semitransparent) states were superimposed by means of least squares deviation for Cα atoms. The region of α2F to MR2 is shown in purple, and the α1/2G and αH helices are in green and cyan, respectively. The red broken lines indicate hydrogen bonds in the R state structure except for the intrahelical hydrogen bonds. B, ribbon diagram superimposing the R and T states of TcLDH as to MR1 and MR2 in the P-axis-related dimer. One subunit (subunit A in Fig. 3A) is colored magenta or salmon for the R state or T state, respectively, and the P-axis-related subunit is colored green or yellow for the R state or T state, respectively. The P-axis-related dimers of the two state structures were superimposed by means of least squares deviation for only one subunit (subunit A) of a dimer. Red and blue dotted lines indicate consistent intersubunit hydrogen bonds in the R and T states, respectively. C, structures of the MR1-MR2 area in the R (left panel) and T (right panel) states of TcLDH (left panel) and TtLDH (right panel). MR1 and MR2 are colored red and purple, respectively, and the other regions of the same subunit are colored salmon. The P-axis-related subunit is colored green. The red dotted lines indicate the hydrogen bonds between MR1 and MR2, and the blue dotted lines indicate the intersubunit hydrogen bonds that MR1 and MR2 form with the P-axis-related subunits.
The α2F-βG loop of MR1 has intersubunit contacts with the α3G-βK loop of the P-axis-related subunit, and the Ala183 main chain forms a constitutive hydrogen bond with the Lys266(P) main chain in the two state structures (Fig. 5B). In the case of LcLDH, the α2F-βG loop forms an intersubunit linkage for allosteric motion to the α1/2G-α3G(P) loop of the P-axis-related subunit via the α3G-βK(P) loop and α3G(P) helix, and therefore the α2F and α1/2G-α3G cooperatively move in the active site of this enzyme (16). On the other hand, TcLDH exhibits no marked motion in the α3G-βK loop or α3G helix (Figs. 3B and 4A), indicating that it does not have such a specific intersubunit linkage. When the P-axis-related dimers of the two state structures are superimposed by means of least squares deviation for only one subunit of a dimer, the MR1 of the P-related subunits is located at virtually the same position in the two state structures (Fig. 5B). In TcLDH, thus, the motion of the α2F-βG loop is linked only to the whole P-axis-related subunit, leading to the open and closed conformational changes of the P-axis-related dimer (Fig. 3A). The αT-α1/2G loop of MR2 also forms constitutive salt bridges with the P-axis-related intersubunit between Arg216 and Glu299(P) and moves together with the whole P-axis-related subunit. In the NAD-binding domain (positions 22–162) of TcLDH, the αB (RMSD = 0.7–1.2 Å) and αC (0.8–1.4 Å) helices slightly but significantly move between the two states, whereas the core β-sheet (βA to βF strands) of the Rossmann fold does not significantly move (Fig. 3B, left). These two helices form constitutive hydrogen bond contacts with the α3G(Q) helix and α1/2G-α3G(Q) loop at the Q-axis subunit interface and therefore move together with the whole Q-axis-related subunit through the quaternary structural change. The motion of the MR1-MR2 area is thus linked with all the structural changes during the allosteric transition.
Kinetic Properties of the Wild-type and Mutant TcLDHs
The catalytic properties of TcLDH in the pyruvate reduction at pH 7.0 at 30 °C were evaluated (Fig. 6A and Table 2), as in the case of TtLDH (28). In the absence of FBP, the wild-type TcLDH showed a slightly sigmoidal saturation curve (Hill coefficient, nH = 1.4) for pyruvate and exhibited an apparent S0.5 value of 36 mm, which is ∼102-fold greater than that of TtLDH. In the presence of 0.1 mm FBP, on the other hand, TcLDH showed an apparently hyperbolic saturation curve for pyruvate, and exhibited an ∼4-fold improved Vmax value and a 103-fold reduced pyruvate Km value (40 μm), which is equivalent to that of TtLDH (∼60 μm), These results indicate that TcLDH actually has a more T state-sided allosteric equilibrium than TtLDH.
FIGURE 6.

Catalytic profiles of the wild-type and mutant TcLDHs. A, pyruvate saturation profiles for the wild-type and mutant TcLDHs at pH 7.0. Left panel, pyruvate saturation curves for the Q mutant enzymes in the absence of FBP. The reaction velocities for the wild-type enzyme (gray), and the Q2 (L67E/H68D) (blue), Q3 (L67E/H68D/E178K) (green), Q4(K) (L67E/H68D/E178K/A235K) (purple), and Q4(R) (L67E/H68D/E178K/A235R) (red) mutant enzymes were measured with the indicated concentrations of pyruvate. Center panel, pyruvate saturation curves without FBP (circles) and with 0.1 mm FBP (diamonds). The symbols for the Q4(K) and Q4(R) enzymes are purple and red, respectively, according to the left panel. The concentrations of pyruvate are plotted on a logarithmic scale. Right panel, pyruvate saturation curves showing the additive activation effects of the P1 (R173Q) and P2 (R173Q/R216L) mutations on the Q4 mutant enzymes. The colors of the symbols indicate the difference in the Q4 mutation of the enzymes, i.e. the enzymes containing no Q mutation (gray) or containing the Q4(K) (purple) or Q4(R) (red) mutation, and the shapes of the symbols indicate the differences in the P mutations for the enzymes with no P mutation (circles) and with the P1 (squares) or P2 (triangles) mutations. The lines indicate the calculated saturation curves obtained with the kinetic parameters shown in Table 2. B, pH profiles of the kinetic parameters during pyruvate reduction by the wild-type and Q4(R)/P2 mutant enzymes without FBP. The pH dependence of the Vmax (left panel), S0.5 (center panel), and Vmax/S0.5 (right panel) values for the wild-type (gray) and Q4(R)/P2 mutant (red) enzymes are logarithmically plotted versus pH. The buffers used for the assay were 50 mm sodium acetate (triangles), MES (squares), MOPS (circles), and Tris-HCl (diamonds). The inset in the left panel shows the pH dependence of the Hill coefficient (nH) in the reaction of the wild-type enzyme. The short lines above and below the symbols in the left and center panels indicate standard deviations in the curve fittings. The broken lines in the center and right panels were drawn without a slope or with a slope of 1.0.
TABLE 2.
Kinetic parameters of the wild-type and P and Q mutant TcLDHs for substrate pyruvate
The parameters were determined as described under “Experimental Procedures,” and standard deviations are shown in parentheses.
| S0.5 | Vmax | Vmax/S0.5 | nH | |
|---|---|---|---|---|
| mm | units/mg | units/mg/mm | ||
| − FBP | ||||
| Wild type | 36 (7) | 11 (1) | 0.29 | 1.4 (0.1) |
| I67E/H68D (Q2) | 24 (1) | 43 (1) | 2.81 | 2.4 (0.1) |
| I67E/H68D/E178K (Q3) | 20 (1) | 31 (0.3) | 1.40 | 1.8 (0.1) |
| I67E/H68D/E178K/A235K (Q4(K)) | 9.3 (0.4) | 47 (1) | 5.1 | 1.6 (0.1) |
| I67E/H68D/E178K/A235R (Q4(R)) | 7.3 (0.8) | 40 (1) | 5.5 | 1.6 (0.1) |
| R173Q (P1) | 3.2 (0.2) | 10.6 (0.4) | 3.3 | 1.3 (0.1) |
| R173Q/R216L (P2) | 0.33 (0.10) | 9.6 (1.2) | 29 | 0.8 (0.1) |
| Q4(K)/P1 | 0.93 (0.02) | 27.1 (0.3) | 29 | 1.4 (0.1) |
| Q4(R)/P1 | 0.76 (0.04) | 40.0 (0.9) | 53 | 1.3 (0.1) |
| Q4(K)/P2 | 0.26 (0.02) | 43.4 (1.3) | 167 | 1.1 (0.1) |
| Q4(R)/P2 | 0.079 (0.03) | 41.0 (0.5) | 520 | 1.0 (0.1) |
| + FBP | ||||
| Wild type | 0.040 (0.002) | 40.1 (1.3) | 1000 | 1.2 (0.1) |
| I67E/H68D (Q2) | 0.13 (0.01) | 41.9 (0.8) | 322 | 1.1 (0.1) |
| I67E/H68D/E178K (Q3) | 0.50 (0.08) | 75.7 (5.5) | 151 | 0.9 (0.1) |
| I67E/H68D/E178K/A235K (Q4(K)) | 0.075 (0.003) | 45.1 (0.8) | 601 | 1.2 (0.1) |
| I67E/H68D/E178K/A235R (Q4(R)) | 0.041 (0.002) | 51.4 (0.9) | 1250 | 1.1 (0.1) |
We also characterized four Q mutant TcLDHs, the Q2 (L67E/H68D), Q3 (L67E/H68D/E178K), Q4(K) (L67E/H68D/E178K/A235K), and Q4(R) (L67E/H68D/E178K/A235R) enzymes. The A235K and A235R replacements for the Q4(K) and Q4(R) enzymes mimic the Q-axis intersubunit area of nonallosteric LpLDH (Figs. 1A and 2) and TtMDH (Fig. 1B), respectively. In the absence of FBP, all the Q mutant enzymes consistently exhibited ∼4-fold increased Vmax values, but the Q4(K) and Q4(R) enzymes exhibited only 3.9- and 4.9-fold smaller S0.5 values than the wild-type enzyme and still showed marked positive cooperativity in the pyruvate binding (nH = 1.6). Notably, each of the Q mutations did not show an additive effect on the pyruvate saturation. The Q2 enzyme exhibited a 1.5-fold smaller pyruvate S0.5 and markedly greater homotropic cooperativity (nH = 2.4) than the wild-type enzyme. Nevertheless, the Q3 enzyme exhibited an only 1.2-fold smaller S0.5 than the Q2 enzyme and lower homotropic cooperativity (nH = 1.8) again. In the presence of FBP, on the other hand, the Q mutant enzymes consistently exhibited virtually the hyperbolic shapes of pyruvate saturation curves and ∼102-fold smaller S0.5 values, although they exhibited somewhat larger S0.5 values than the wild-type enzyme except for the Q4(R) enzyme. Hence, the Q4 mutations did not greatly change the allosteric equilibrium, unlike in the case of LcLDH (20).
In contrast, the P mutations, the P1 (R173Q) and P2 (R173Q/R216L) replacements, did not markedly increase the Vmax values, but 10- and 102-fold reduced the pyruvate S0.5 values, respectively (Fig. 6A and Table 2). The P mutations also significantly reduced the positive homotropic cooperativity, and the P2 enzyme exhibited a virtually hyperbolic pyruvate saturation curve. The P mutations additively reduced both the pyruvate S0.5 and nH values of the Q4 enzymes without a reduction in the Vmax values. Consequently, the Q4(K)/P2 and Q4(R)/P2 enzymes exhibited only 6.5- and 2-fold greater pyruvate Km values than the wild-type enzyme that was activated with FBP. It is known that a high concentration of FBP rather inhibits the catalytic reaction of TcLDH (16), reducing both Vmax and Km (data not shown), although the exact mechanism underlying the inhibition remains uncertain. Under the standard assay conditions (pH 7.0, 30 °C, 1.0 mm pyruvate), FBP exhibited its maximal activation effect at 0.1–0.4 mm and exhibited an apparent half-inhibition concentration (IC50) of ∼2 mm for the enzyme reaction of TcLDH (data not shown). FBP exhibited no marked activation effect on the mutant enzymes with the R173Q replacement (data not shown) but exhibited a marked inhibitory effect on the Q4(R)/P2 enzyme at high concentration, exhibiting an apparent IC50 of 4 mm, suggesting that TcLDH has another binding site for FBP than the known one.
pH Profiles of the Wild-type and Mutant TcLDHs
Vertebrate nonallosteric LDHs exhibit a typical pH profile of acid/base catalytic enzymes in the catalytic reaction (6). Because the pyruvate binding depends on the protonated form of His195, pyruvate Km of LDH is highly affected by the pH conditions, whereas Vmax is virtually independent of pH. Bacterial allosteric LDHs show more complicated pH profiles, because their allosteric properties are also affected by pH (16), and the FBP binding usually requires the protonated form of His188 (11, 13). In the case of TcLDH, the optimal pH for the enzyme reaction apparently shifts depending on the FBP concentration, because the inhibitory effect of FBP also depends on pH (24). In this study, we simply evaluated the pH profiles of the wild-type and mutant TcLDHs in the absence of FBP.
The wild-type enzyme exhibited virtually constant Vmax values, and pH-dependent S0.5 and Vmax/S0.5 values in the pH range of 5.0–7.5 (Fig. 6B). The catalytic reaction of TcLDH seemed to be affected by ion strength of buffers, particularly in the case of the wild-type enzyme (data not shown). In this case, nevertheless, the effect of ion strength was much weaker as compared with the pH effect. It also showed high pH dependence as to the homotropic cooperativity (nH) and exhibited no significant cooperativity above pH 7.5. The individual Vmax or S0.5 value could not be determined at pH 8.0 because of the weak enzyme activity. These results suggest that the protonated form of His195 promotes the T to R transition caused by pyruvate, and that the T state enzyme is more predominantly involved in the reaction at higher pH. On the other hand, the Q4(R)/P2 enzyme exhibited no significant cooperativity at pH 5.0–8.5 and showed constant Vmax and pH-dependent Km values at higher pH, as in the case of the vertebrate enzymes. Although the mutant enzyme exhibited apparently greater slopes of Km and Vmax/Km than 1.0 under alkaline conditions, it roughly had an apparent pKa of ∼6.7 in the Km and Vmax/Km profiles.
Thermal Properties of the Wild-type and Mutant TcLDHs
TtLDH shows a constant substrate Km (∼50 μm) in the presence of FBP independently of temperature but a markedly temperature-dependent Km in the absence of FBP, giving a 2–3-fold smaller Km at 80 °C (∼150 μm) than that of 30 °C (∼350 μm) (28). On the other hand, TcLDH exhibited slight cooperativity in pyruvate binding (nH = 1.3–1.6) and the temperature-dependent pyruvate S0.5 values at 30 to 80 °C in the absence of FBP, exhibiting a 10-fold smaller S0.5 at 80 °C than 30 °C (Fig. 7A). As in the case of TtLDH, TcLDH exhibited a virtually temperature-independent S0.5 (Km) of 30–40 μm in the presence of FBP (20–100 μm), although the exact Km value could not be clearly determined because of the complicated activation inhibition effects of FBP, as described above. At 80 °C, accordingly, TcLDH showed an only 102-fold reduced pyruvate S0.5 with FBP.
FIGURE 7.

Thermal profiles of the wild-type and Q4 and Q4/P2 mutant TcLDHs. A, Arrhenius plots of the S0.5 (left panel), Vmax (center panel), and Vmax/S0.5 (right panel) values for the wild-type (black) and Q4(R) (blue) and Q4(R)/P2 (red) mutant TcLDHs. The pH of the assay mixtures was adjusted to 7.0 at room temperature. B, inactivation of the wild-type (black) and Q4(K) (left, blue), Q4(K)/P2 (left, red), Q4(R) (right, blue), and Q4(R)/P2 (right, red) mutant TcLDHs on heat treatment. The enzymes were diluted to 0.3 mg/ml with 50 mm sodium MOPS and then treated for 30 min at the indicated temperatures. The circles represent the mean values for three independent experiments.
The Q4(R) enzyme exhibited significant homotropic cooperativity (nH = ∼1.6) below 50 °C, but no apparent cooperativity above 65 °C in the absence of FBP. This enzyme showed lower temperature dependence as to pyruvate S0.5 and exhibited only a 1.8-fold smaller substrate S0.5 than the wild-type enzyme at 80 °C (Fig. 7A). The Q4(R)/P2 enzyme exhibited no significant homotropic cooperativity and constantly a 102-fold smaller S0.5 value than the Q4(R) enzyme in the range of 30–80 °C. The two mutant enzymes exhibited virtually the same Vmax values, which were constantly 4–5-fold greater than those of the wild-type enzyme. These three types of TcLDH exhibited similar temperature dependence as to Vmax values, the apparent activation energies (ΔEa) being 10.8 ± 1.5, 10.6 ± 0.1, and 9.1 ± 0.6 kcal/mol for the wild-type, Q4(R) and Q4(R)/P2 enzymes, respectively.
The Q4(R) TcLDH exhibited a significantly higher apparent denaturation temperature (T½) (97.5 °C) than the wild-type enzyme (95 °C), whereas the Q4(K) enzyme showed apparently the same T½ as that of the wild-type enzyme (Fig. 7B). The inactivation of the Q4(R) and Q4(K) enzymes consistently occurred more abruptly around the T½ than that of the wild-type enzyme, suggesting that the mutations increased the activation enthalpy of heat inactivation (ΔH‡), as in the case of LcLDH (20). The P2 mutation further increased the thermal stabilities of the Q4(K) and Q4(R) mutant enzymes, and the Q4(R) and Q4(R)/P2 enzymes consistently exhibited higher thermal stabilities than the Q4(K) and Q4(K)/P2 enzymes, respectively (Fig. 7B).
Clusters of Positively Charged Residues
Inspection of the electrostatic surfaces in the TcLDH structures revealed that surface patches of positive surface potential exist around the R symmetry axis and are focused on and disperse from the axis in the R and T states, respectively (Fig. 8A). Interestingly, one of the patches is generated mostly from Arg181 and Arg218, which are located at the centers of MR1 and MR2, respectively. These two residues thus form a cluster of positive charges together with Arg173, His179, and Arg216 (named the MR1-MR2 cluster). The other patch comprises Met22 (the N-terminal amino group), Arg47, His74, and Arg92, which form another cluster of positive charges around the core β-sheet of the Rossmann fold NAD-binding domain, together with Lys23 and Arg82 (named the N domain cluster). These two clusters are unique in TcLDH, because the basic residues in them are poorly conserved in other LDHs including LcLDH and BlLDH (Fig. 2). The two clusters face each other across the Q-axis subunit interface, and the MR1-MR2 cluster moves closer to and away from the N domain cluster in the R and T states, respectively, whereas the N domain cluster is virtually immovable. This motion of the MR1-MR2 area (at Cα atoms) (Fig. 8B) corresponds to the motion in the tertiary structure (Fig. 3B), although it is slightly greater with the quaternary structural change, and the central parts of MR1 and MR2 are 4–5 Å more distant from the N domain cluster (Q) in the T state structure. Regarding the basic residues of the MR1-MR2 area, the side chains generally tend to move more than the main chains, particularly in the cases of Arg173, Arg181, His188, and Arg218 (Fig. 8C). It is noteworthy that the Arg171 side chain is exceptionally orientated toward the N domain cluster in the T state structure, where it is 6–7 Å closer to the cluster than in the R state one. It is also notable that Glu178 apparently interrupts the static repulsion between Arg171 and the N domain cluster because of its carboxyl group (Fig. 8C).
FIGURE 8.

Clusters of positive charges and allosteric motion of TcLDH. Changes in intersubunit interactions at the P- and R-axis subunit interfaces. A, surface electrostatic potential models of the R (left panel) and T (right panel) state structures of TcLDH. The models show electronegative (red), electropositive (blue), and electroneutral (white) amino acid side chains. The panels show the details of the regions indicated by yellow boxes. B, changes in the distances between the amino acid residues of the two clusters of positive charge in the allosteric motion of TcLDH. The distances were measured between the Cα atoms of the indicated positioned residues and Met22 (purple), Arg47 (red), His74 (blue), Arg80 (green), and Arg92 (orange). The line graphs indicate the differences of distances: (distance in the T state structure) − (distance in the R state structure). C, structures of the cluster areas of positively charged residues in the R (left panel) and T (right panel) states of TcLDH. The subunits are colored according to A.
The N domain cluster also faces several basic residues of the R-axis-related subunit, such as the N-terminal amino group, and the basic side chains of Lys23, Arg92, Arg256, and Arg259 (Fig. 8C). Because the motion of the MR1-MR2 cluster is coupled with that of the P-axis-related subunit (the R-axis-related subunit for the N domain cluster), these basic groups also move toward and away from the N domain cluster in the R and T state structures, respectively, through the quaternary structural changes of the enzyme.
DISCUSSION
The R state structure of TcLDH provides a new structural snapshot of the catalytic process of LDHs, indicating that the ternary complex LDH has two stable conformational states with open and closed conformations of the active site loop. Furthermore, this structure provides a snapshot of the allosteric motion of TcLDH, where tightly bound MR1-MR2 moves as the core independently of the active site loop and mediates the allosteric equilibrium mostly through electrostatic repulsion against the Q-axis-related N domain cluster (Fig. 8). This structural observation is well consistent with the results obtained on chemical modification (22–25) or amino acid replacement of the basic amino acid residues (12, 13, 25), because such modifications consistently reduce the static repulsion. In addition, the structural comparison explains the distinct allosteric properties of TcLDH and TtLDH (A154G/H179Y), because His179 is located just at the center of MR1, whereas Ala154 is located in the α1F-βF loop on the protein surface. First of all, the H179Y replacement reduces the net positive charge of the MR1-MR2 area, changing the static balance between the two clusters. Second, the replacement relaxes the MR1-MR2 linkage, because His179 plays a key role in the hydrogen bond network between MR1 and MR2 (Fig. 5C). As compared with TcLDH, actually, TtLDH has fewer inter-region hydrogen bonds in the MR1-MR2 area, which is obviously more asymmetric as to the four subunits than that of TcLDH, particularly in the R state structure. In this area, in TtLDH, the positively charged groups of Arg181, Arg216, and Arg218 are markedly rearranged in the two state structures, and in the R state structure, Arg218 is 2 Å more separate from the N domain cluster than in TcLDH. Even in TtLDH, the R218A replacement (1-Mut) further reduces the pyruvate Km value (28), suggesting that the allosteric equilibrium of the enzyme is still mediated by static repulsion between the positively charged clusters.
It is also notable that TcLDH undergoes a markedly different structural change from that of BlLDH or LcLDH between the two states (Figs. 3 and 4), indicating that the allosteric machineries of LDHs have highly diverged during evolution. In TcLDH, the MR1-MR2 area shifts without a secondary structural change, and the active site is rearranged only through rotation of the α2F helix, which comprises Asp168 and Arg171, through the MR1-MR2 motion. In contrast, in BlLDH, the active site is directly rearranged through a secondary structural change of the α2G-α3G area, which is the center of the allosteric structural change in this enzyme, and in LcLDH, both the MR1-MR2 and α2G-α3G areas shift through the unique intersubunit linkage of motion between the MR1 and α2G-α3G(P) (16). The change in Gibbs free energy (ΔG) between the two states was estimated to be 7.3 kcal/mol for TcLDH (at pH 6.75) (25), whereas the ΔG values for BlLDH (9) and LcLDH (16) were 8.0 and 3.4 kcal/mol, respectively. TcLDH nevertheless showed markedly lower cooperative substrate binding (nH = 1.4) than BlLDH (nH = 2.4) (14, 26) and LcLDH (nH = 2.0) (16) in the absence of FBP, although it exhibits a higher nH value at lower pH (Fig. 7B). This finding indicates that for TcLDH the pyruvate affinity is less strictly switched between the two states, which is consistent with the smaller structural change in its active site. Instead, in TcLDH, the structure of the FBP binding site more strictly changes, i.e. the affinity to FBP, through a great change in the orientation of His188 (Fig. 4, C and D). Actually, TcLDH appears to exhibit virtually full activation by FBP alone (25, 26), whereas BlLDH requires an additional activation effect of pyruvate for full activation (9, 14, 48).
The small structural change of TcLDH in the active site area (Figs. 1, C and D, and 4A) is also likely responsible for the small effect of Q4 mutations on the FBP-independent activity (Fig. 6A). The mutations introduce intersubunit salt bridges between the α2F and αC(Q) and the α2G and αC(Q) helices, respectively, in the vicinity of the active site. However, in TcLDH, only the α2F helix moves between the two states, whereas in LcLDH the α2F, α2G, and αC(Q) helices all move (Fig. 4, A and B). The Q4 mutations therefore likely stabilize the T and R states of TcLDH less selectively and induce a less marked shift of the allosteric equilibrium. It is also notable that the Q3 enzyme exhibits virtually the same S0.5 value as the Q2 enzyme (Fig. 6A and Table 2). Because Glu178 is located in MR1, the E178K replacement likely increases the static repulsion between the positively charged clusters, canceling out the activation effect of the intersubunit salt bridges. This emphasizes that the MR1-MR2 area is actually a pivot in the allosteric motion of TcLDH. It is also notable that the Q2 enzyme exhibits a markedly higher nH value than the wild-type enzyme, although the Q3 enzyme exhibits a nH value virtually equivalent to that of the wild-type enzyme (Fig. 7A and Table 2). In the Q2 enzyme, Asp68 likely inhibits the substrate binding through static repulsion, particularly when Arg171 is orientated outside the active site in the T state, and thereby allows the substrate affinity of the enzyme to more strictly change between the two states. In the Q3 enzyme, in contrast, the E178K replacement likely promotes the substrate affinity of the T state enzyme, because Glu178 apparently stabilizes the orientation of Arg171 in the T state structure by canceling out the repulsion between the N domain cluster and Arg171 (Fig. 8C). These findings thus suggest that Glu178 plays important roles in both the strict change of the substrate affinity and the modulation of the allosteric equilibrium between the two states. Comprehensively, the Q4 mutations appear to slightly increase the FBP-independent activity mostly through changes in the catalytic properties of each of the two state enzymes rather than a change in the allosteric equilibrium.
This study demonstrated the uniquely simple but precise allosteric machinery of TcLDH, where the MR1-MR2 area pivotally moves during the allosteric motion and mediates the allosteric equilibrium through electrostatic repulsion within the protein molecule. TcLDH is a heat-stable enzyme (21) and exhibits an ∼30 °C higher denaturation temperature than LcLDH (16) and LpLDH (18) (Fig. 7B), although the crucial thermal stability of BlLDH has not been examined. It was reported that TtLDH (27) and T. maritima LDH (41), another hyperthermostable LDH (49), have increased numbers of intersubunit hydrogen bonds or salt bridges as compared with heat-labile LDHs. Nevertheless, TcLDH has an equivalent number of intersubunit hydrogen bonds (15–16 per monomer) to that of LcLDH (15 per monomer) and an even markedly lower number than that of LpLDH (26 per monomer). This suggests that the number of intersubunit hydrogen bonds should be limited for allosteric LDHs, which require flexible changes of the quaternary structure. In TcLDH, the flexible surface area (MR2) uniquely binds tightly to MR1 to form the rigid core of allosteric motion and thus appears to compensate for its sufficient thermal stability for poor intersubunit interactions because of its rigid subunit structure and compact allosteric motion. It was reported that 5-Mut TtLDH shows markedly lower thermal stability than the wild-type enzyme (28). For MWC-type allosteric enzymes, there are two possible ways to simply shift the allosteric equilibrium to the R state: i.e. destabilization of the T state structure and stabilization of the R state one. The former way likely decreases the intrinsic stability of allosteric LDHs, which usually exhibit a T state-sided allosteric equilibrium. The 5-Mut mutation is designed to reduce unique salt bridge interactions within the TtLDH molecule and likely destabilizes the T state structure more greatly than the R state one. On the other hand, the Q4(K) LcLDH does not exhibit increased thermal stability, and there may be even slightly reduced thermal stability, despite the increased intersubunit salt bridges (20). This is also reasonable because the mutation destabilizes the intrinsically stable T state structure in return for stabilization of the labile R state structure. In the case of TcLDH, it is also reasonable that the Q4(K) and Q4(R) mutations, particularly the latter one, increase the thermal stability of the enzyme (Fig. 7B), because it can be presumed that the mutations nonselectively stabilize the two state structures, unlike in the case of LcLDH. The higher thermal stability of the Q4(R) enzyme suggests that Arg235 undergoes a stronger intersubunit salt bridge interaction than Lys235 in both the two state structures. In contrast to the Q4 mutations, the P2 mutation does not increase or even reduces intersubunit salt bridges, and selectively stabilizes the R state in allosteric equilibrium (25), but significantly increases the thermal stability of TcLDH (Fig. 7B). This unusual effect of the P2 mutation emphasizes the unique regulation mechanism of TcLDH, i.e. mediation of the allosteric equilibrium through electrostatic repulsion. Static repulsion within a protein generally destabilizes the compact structure of a globular protein. Hence, the P2 mutation does not reduce or even increases the intrinsic stability of TcLDH, although it selectively reduces the relative stability of the T state in allosteric equilibrium. This indicates that TcLDH somewhat sacrifices its protein stability in return for its allosteric machinery involving intramolecular static repulsion. Nevertheless, TcLDH likely minimizes the sacrifice of thermal stability with the alternative T state structure, which reduces the static repulsion by dispersing the charged clusters in the open quaternary structure. TcLDH thus appears to gain its allostery and sufficient thermal stability through its compact allosteric motion and intrinsically stable T state structure, which provides the minimum necessary stability for the enzyme.
This work was performed with the approval of Photon Factory Program Advisory Committee Proposals 2004G136 and 2006G160. This work was supported by Grant-in-Aid 23580120 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H. T.).
- LDH
- l-lactate dehydrogenase
- BlLDH
- B. longum LDH
- FBP
- fructose 1,6-bisphosphate
- LcLDH
- L. casei LDH
- LpLDH
- L. pentosus LDH
- MWC model
- Monod-Wyman-Changeux model
- RMSD
- root mean square deviation
- TcLDH
- T. caldophilus LDH
- TtLDH
- T. thermophilus LDH
- TtMDH
- T. thermophilus MDH
- MR
- mobile region.
REFERENCES
- 1. Monod J., Wyman J., Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 2. Koshland D. E., Jr., Némethy G., Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 [DOI] [PubMed] [Google Scholar]
- 3. Fischer E. (1894) Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dt. Chem. Ges. 27, 2985–2993 [Google Scholar]
- 4. Koshland D. E. (1958) Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 44, 98–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fetler L., Kantrowitz E. R., Vachette P. (2007) Direct observation in solution of a preexisting structural equilibrium for a mutant of the allosteric aspartate transcarbamoylase. Proc. Natl. Acad. Sci. U.S.A. 104, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holbrook J. J., Liljas A., Steindel S. J., Rossmann M. G. (1975) Lactate dehydrogenase. In The Enzymes (Boyer P. D., ed) 3rd ed., Vol. 11, pp. 191–292, Academic Press, New York [Google Scholar]
- 7. Garvie E. I. (1980) Bacterial lactate dehydrogenases. Microbiol. Rev. 44, 106–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wigley D. B., Gamblin S. J., Turkenburg J. P., Dodson E. J., Piontek K., Muirhead H., Holbrook J. J. (1992) Structure of a ternary complex of an allosteric lactate dehydrogenase from Bacillus stearothermophilus at 2.5 Å resolution. J. Mol. Biol. 223, 317–335 [DOI] [PubMed] [Google Scholar]
- 9. Iwata S., Kamata K., Yoshida S., Minowa T., Ohta T. (1994) T and R states in the crystals of bacterial l-lactate dehydrogenase reveal the mechanism for allosteric control. Nat. Struct. Biol. 1, 176–185 [DOI] [PubMed] [Google Scholar]
- 10. Hensel R., Mayr U., Woenckhaus C. (1983) Affinity labeling of the allosteric site of the l-lactate dehydrogenase of Lactobacillus casei. Eur. J. Biochem. 135, 359–365 [DOI] [PubMed] [Google Scholar]
- 11. Clarke A. R., Wigley D. B., Barstow D. A., Chia W. N., Atkinson T., Holbrook J. J. (1987) A single amino acid substitution deregulates a bacterial lactate dehydrogenase and stabilizes its tetrameric structure. Biochim. Biophys. Acta 913, 72–80 [DOI] [PubMed] [Google Scholar]
- 12. Matsuzawa H., Machida M., Kunai K., Ito Y., Ohta T. (1988) Identification of an allosteric site residue of a fructose 1,6-bisphosphate-dependemt l-lactate dehydrogenase of Thermus caldophilus GK24: production of a non-allosteric form by protein engineering. FEBS Lett. 233, 375–378 [Google Scholar]
- 13. Schroeder G., Matsuzawa H., Ohta T. (1988) Involvement of the conserved histidine-188 in the l-lactate dehydrogenase from Thermus caldophilus GK24 in allosteric regulation by fructose 1,6-bisphosphate. Biochem. Biophys. Res. Commun. 152, 1236–1241 [DOI] [PubMed] [Google Scholar]
- 14. Fushinobu S., Kamata K., Iwata S., Sakai H., Ohta T., Matsuzawa H. (1996) Allosteric activation of l-lactate dehydrogenase analyzed by hybrid enzymes with effector-sensitive and -insensitive subunits. J. Biol. Chem. 271, 25611–25616 [DOI] [PubMed] [Google Scholar]
- 15. Feldman-Salit A., Hering S., Messiha H. L., Veith N., Cojocaru V., Sieg A., Westerhoff H. V., Kreikemeyer B., Wade R. C., Fiedler T. (2013) Regulation of the activity of lactate dehydrogenases from four lactic acid bacteria. J. Biol. Chem. 288, 21295–21306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arai K., Ishimitsu T., Fushinobu S., Uchikoba H., Matsuzawa H., Taguchi H. (2010) Active and inactive state structures of unliganded Lactobacillus casei allosteric l-lactate dehydrogenase. Proteins 78, 681–694 [DOI] [PubMed] [Google Scholar]
- 17. Grau U. M., Trommer W. E., Rossmann M. G. (1981) Structure of the active ternary complex of pig heart lactate dehydrogenase with S-lac-NAD at 2.7 A resolution. J. Mol. Biol. 151, 289–307 [DOI] [PubMed] [Google Scholar]
- 18. Taguchi H., Ohta T. (1992) Unusual amino acid substitution in the anion-binding site of Lactobacillus plantarum non-allosteric l-lactate dehydrogenase. Eur. J. Biochem. 209, 993–998 [DOI] [PubMed] [Google Scholar]
- 19. Uchikoba H., Fushinobu S., Wakagi T., Konno M., Taguchi H., Matsuzawa H. (2002) Crystal structure of non-allosteric l-lactate dehydrogenase from Lactobacillus pentosus at 2.3 Å resolution: specific interactions as subunit interfaces. Proteins 46, 206–214 [DOI] [PubMed] [Google Scholar]
- 20. Arai K., Ichikawa J., Nonaka S., Miyanaga A., Uchikoba H., Fushinobu S., Taguchi H. (2011) A molecular design that stabilizes active state in bacterial allosteric l-lactate dehydrogenases. J. Biochem. 150, 579–591 [DOI] [PubMed] [Google Scholar]
- 21. Taguchi H., Yamashita M., Matsuzawa H., Ohta T. (1982) Heat-stable and fructose 1,6-bisphosphate-activated l-lactate dehydrogenase from an extremely thermophilic bacterium. J. Biochem. 91, 1343–1348 [DOI] [PubMed] [Google Scholar]
- 22. Taguchi H., Matsuzawa H., Ohta T. (1984) l-Lactate dehydrogenase from Thermus caldophilus GK24, an extremely thermophilic bacterium. Desensitization to fructose 1,6-bisphosphate in the activated state by arginine-specific chemical modification and the N-terminal amino acid sequence. Eur. J. Biochem. 145, 283–290 [DOI] [PubMed] [Google Scholar]
- 23. Koide S., Yokoyama S., Matsuzawa H., Miyazawa T., Ohta T. (1989) Conformation of NAD+ bound to allosteric l-lactate dehydrogenase activated by chemical modification. J. Biol. Chem. 264, 8676–8679 [PubMed] [Google Scholar]
- 24. Machida M., Matsuzawa H., Ohta T. (1985) Fructose 1,6-bisphosphate-dependent l-lactate dehydrogenase from Thermus aquaticuas YT-1, an extreme thermophilile: Activation by citrate and modification reagents and comparison with Thermus caldophilus GK24 l-lactate dehydrogenase. J. Biochem. 97, 899–909 [DOI] [PubMed] [Google Scholar]
- 25. Koide S., Yokoyama S., Matsuzawa H., Miyazawa T., Ohta T. (1992) Conformational equilibrium of an enzyme catalytic site in the allosteric transition. Biochemistry 31, 5362–5368 [DOI] [PubMed] [Google Scholar]
- 26. Machida M., Yokoyama S., Matsuzawa H., Miyazawa T., Ohta T. (1985) Allosteric effect of fructose 1,6-bisphosphate on the conformation of NAD+ as bound to l-lactate dehydrogenase from Thermus caldophilus GK24. J. Biol. Chem. 260, 16143–16147 [PubMed] [Google Scholar]
- 27. Coquelle N., Fioravanti E., Weik M., Vellieux F., Madern D. (2007) Activity, stability and structural studies of lactate dehydrogenases adapted to extreme thermal environments. J. Mol. Biol. 374, 547–562 [DOI] [PubMed] [Google Scholar]
- 28. Colletier J. P., Aleksandrov A., Coquelle N., Mraihi S., Mendoza-Barberá E., Field M., Madern D. (2012) Sampling the conformational energy landscape of a hyperthermophilic protein by engineering key substitutions. Mol. Biol. Evol. 29, 1683–1694 [DOI] [PubMed] [Google Scholar]
- 29. Koide S., Iwata S., Matsuzawa H., Ohta T. (1991) Crystallization of allosteric l-lactate dehydrogenase from Thermus caldophilus and preliminary crystallographic data. J. Biochem. 109, 6–7 [PubMed] [Google Scholar]
- 30. Ono M., Matsuzawa H., Ohta T. (1990) Nucleotide sequence and characteristics of the gene for l-lactate dehydrogenase of Thermus aquaticus YT-1 and the deduced amino acid sequence of the enzyme. J. Biochem. 107, 21–26 [DOI] [PubMed] [Google Scholar]
- 31. Kunai K., Machida M., Matsuzawa H., Ohta T. (1986) Nucleotide sequence and characteristics of the gene for l-lactate dehydrogenase of Thermus caldophilus GK24 and the deduced amino-acid sequence of the enzyme. Eur. J. Biochem. 160, 433–440 [DOI] [PubMed] [Google Scholar]
- 32. Powell H. R. (1999) The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 55, 1690–1695 [DOI] [PubMed] [Google Scholar]
- 33. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 34. Jones T. A., Zou J.-Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, Palo Alto, CA [Google Scholar]
- 36. Kraulis P. J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–950 [Google Scholar]
- 37. Merritt E. A., Bacon D. J. (1997) Raster3D photorealistic molecular graphics. Methods Enzymol. 277, 505–524 [DOI] [PubMed] [Google Scholar]
- 38. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 39. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 40. Dixson M., Webb E. C. (1979) Enzymes, pp. 400–402, Longman, London [Google Scholar]
- 41. Auerbach G., Ostendorp R., Prade L., Korndörfer I., Dams T., Huber R., Jaenicke R. (1998) Lactate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: the structure at 2.1 Å resolution reveal strategies for intrinsic protein stabilization. Structure 6, 769–781 [DOI] [PubMed] [Google Scholar]
- 42. Clarke A. R., Wigley D. B., Chia W. N., Barstow D., Atkinson T., Holbrook J. J. (1986) Site-directed mutagenesis reveals role of mobile arginine residue in lactate dehydrogenase catalysis. Nature 324, 699–702 [DOI] [PubMed] [Google Scholar]
- 43. Hart K. W., Clarke A. R., Wigley D. B., Waldman A. D., Chia W. N., Barstow D. A., Atkinson T., Jones J. B., Holbrook J. J. (1987) A strong carboxylate-arginine interaction is important in substrate orientation and recognition in lactate dehydrogenase. Biochim. Biophys. Acta 914, 294–298 [DOI] [PubMed] [Google Scholar]
- 44. Bur D., Clarke T., Friesen J. D., Gold M., Hart K. W., Holbrook J. J., Jones J. B., Luyten M. A., Wilks H. M. (1989) On the effect on specificity of Thr246-Gly mutation in l-lactate dehydrogenase of Bacillus sterothermophilus. Biochem. Biophys. Res. Commun. 161, 59–63 [DOI] [PubMed] [Google Scholar]
- 45. Sakowicz R., Kallwass H. K., Parris W., Kay C. M., Jones J. B., Gold M. (1993) Threonine 246 at the active site of the l-lactate dehydrogenase of Bacillus stearothermophilus is important for catalysis but not for substrate binding. Biochemistry 32, 12730–12735 [DOI] [PubMed] [Google Scholar]
- 46. Clarke A. R., Wilks H. M., Barstow D. A., Atkinson T., Chia W. N., Holbrook J. J. (1988) An investigation of the contribution made by the carboxylate group of an active site histidine-aspartate couple to binding and catalysis in lactate dehydrogenase. Biochemistry 27, 1617–1622 [DOI] [PubMed] [Google Scholar]
- 47. Iwata S., Ohta T. (1993) Molecular basis of allosteric activation of bacterial l-lactate dehydrogenase. J. Mol. Biol. 230, 21–27 [DOI] [PubMed] [Google Scholar]
- 48. Fushinobu S., Ohta T., Matsuzawa H. (1998) Homotropic activation via the subunit interaction and allosteric symmetry revealed on analysis of hybrid enzymes of l-lactate dehydrogenase. J. Biol. Chem. 273, 2971–2976 [DOI] [PubMed] [Google Scholar]
- 49. Wrba A., Jaenicke R., Huber R., Stetter K. O. (1990) Lactate dehydrogenase from the extreme thermophile Thermotoga maritima. Eur. J. Biochem. 188, 195–201 [DOI] [PubMed] [Google Scholar]
- 50. Eventoff W., Rossmann M. G., Taylor S. S., Torff H. J., Meyer H., Keil W., Kiltz H. H. (1977) Structural adaptations of lactate dehydrogenase isozymes. Proc. Natl. Acad. Sci. U.S.A. 74, 2677–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]