

Background: Immune activation through a balanced cell surface expression of human NKG2D ligands is crucial for the elimination of diseased cells.
Results: Methylselenol induces the expression of the NKG2D ligands MICA/B but specifically inhibits ULBP2 protein expression.
Conclusion: Methylselenol regulates NKG2D ligand expression on transcriptional and posttranscriptional levels.
Significance: Methylselenol is the first identified metabolite that diversely regulates NKG2D ligands, and, therefore, its implementation could improve NKG2D-based immune therapy.
Keywords: Autophagy, Molecular Cell Biology, Natural Killer Cells (NK cells), Selenium, Trafficking, Tumor Immunology
Abstract
For decades, selenium research has been focused on the identification of active metabolites, which are crucial for selenium chemoprevention of cancer. In this context, the metabolite methylselenol (CH3SeH) is known for its action to selectively kill transformed cells through mechanisms that include increased formation of reactive oxygen species, induction of DNA damage, triggering of apoptosis, and inhibition of angiogenesis. Here we reveal that CH3SeH modulates the cell surface expression of NKG2D ligands. The expression of NKG2D ligands is induced by stress-associated pathways that occur early during malignant transformation and enable the recognition and elimination of tumors by activating the lymphocyte receptor NKG2D. CH3SeH regulated NKG2D ligands both on the transcriptional and the posttranscriptional levels. CH3SeH induced the transcription of MHC class I polypeptide-related sequence MICA/B and ULBP2 mRNA. However, the induction of cell surface expression was restricted to the ligands MICA/B. Remarkably, our studies showed that CH3SeH inhibited ULBP2 surface transport through inhibition of the autophagic transport pathway. Finally, we identified extracellular calcium as being essential for CH3SeH regulation of NKG2D ligands. A balanced cell surface expression of NKG2D ligands is considered to be an innate barrier against tumor development. Therefore, our work indicates that the application of selenium compounds that are metabolized to CH3SeH could improve NKG2D-based immune therapy.
Introduction
NKG2D ligands are induced on the cell surface of a variety of stressed, transformed, and infected cells, whereas the expression on healthy human cells is low. The immune system recognizes NKG2D ligand-positive cells through the NKG2D receptor, a major activating receptor expressed on natural killer cells, NKT cells, CD8+ T cells, γδ T cells, and some activated CD4+ T cells (1–4). There are eight different human NKG2D ligands described, belonging to the MIC (MICA and MICB) and UL16-binding protein (ULBP1–6) families (5). All ligands are MHC class I-related glycoproteins (6). Different forms of cellular stress result in increased NKG2D ligand surface expression, including heat shock, virus infection, inflammatory cytokines, histone deacetylase (HDAC) inhibitors, propionic acid, retinoic acid, proteasome inhibitors, Toll-like receptor (TLR) signaling, and DNA damage response (7–17). Moreover, surface expression of NKG2D ligands on a variety of tumors derived from different origins present an attractive target for anticancer therapy (18–20).
Selenium is a fundamental nutrient in the human diet. Uptake of 50–60 μg of the trace element per day is recommended for healthy adults (21). In the body, ingested selenium is metabolized to a variety of low molecular weight compounds and selenoproteins. In the latter case, selenium is incorporated as selenocysteine. The low molecular weight compounds are divided into organic and inorganic forms, and both can be used as nutritional and supplemental sources. Inorganic selenium is mainly represented by selenate and selenite, whereas the selenoamino acids selenomethionine (SeMet)3 and selenium methylselenocysteine (MSC) are members of organic selenium forms and can be identified in vegetables such as garlic and onions (22, 23). Methylselenic acid (MSA) is a synthetic selenium compound (24, 25) used in several in vitro and animal studies as a stable stripdown variant of MSC containing no amino acid moiety and only one methyl group (26). The metabolism of these organic and inorganic selenium compounds is complex and closely regulated, with two key metabolites, selenide (H2Se) and methylselenol (CH3SeH), being important for the biological function of the selenium compounds (Scheme 1). Selenite is reduced in presence of GSH into H2Se (27). The compounds SeMet and selenocysteine (SeCys2) are also primarily converted to H2Se and incorporated into selenoproteins or selenosugars (27). In contrast, the methylated selenium compounds MSC and MSA are converted into CH3SeH via the enzyme β-lyase or reducing agents, respectively. CH3SeH is either demethylated into H2Se or further methylated to dimethylselenide (DMse) and trimethylselenonium (TMse). In vitro, CH3SeH immediately forms the volatile DMDSe. High concentrations of generated DMDSe can be converted back to CH3SeH (28). Furthermore, SeMet can also be directly converted to CH3SeH in the presence of γ-lyase activity (29). Both metabolites, H2Se and CH3SeH, are highly reactive and volatile, and the equilibrium between H2Se and CH3SeH depends on methylation and demethylation activities as well as the removal of selenium from the derived products. The methylation of H2Se into CH3SeH is a rate-limiting step, and CH3SeH is only produced from H2Se when this compound is available in high excess (27). In addition, the cellular thioredoxin and glutaredoxin systems, which are essential to maintain the intracellular redox balance, play a role in the reduction of various selenium compounds into H2Se or CH3SeH (30, 31). Selenium is one of the most extensively studied chemopreventive compounds but has also been suggested to have anticarcinogenic effects on many types of cancer, including bladder, prostate, lymphoma, and breast cancer (32, 33). In this regard, generated selenoproteins have been suggested to function either as antioxidants (34) or to alter the level of genes involved in cancer development (35). Moreover, selenium metabolites have anticarcinogenic activity. A prominent example is CH3SeH, which has exhibited a stronger prevention of prostate cancer compared with H2Se (36). Selenium intervenes with two hallmarks of cancer: apoptosis and angiogenesis. Studies have shown that selenium compounds, most likely CH3SeH, can induce caspase-mediated apoptosis in cancer cells (37–39). Angiogenesis is affected by CH3SeH precursors because of its inhibition of VEGF from several cancer cell lines (40). Other studies have shown that metabolites of MSC and SeMet inhibit HDAC activity (41, 42) and that CH3SeH is able to inhibit PKC activity by redox modifications of cysteines (43, 44). More recently, selenium compounds have also been applied in cancer treatment (33). An anticancer effect was restricted to selenium compounds that could generate active metabolites during in vivo metabolism. In this context, preclinical as well as clinical trials showed that SeMet (45), MSA (46), and selenite (47, 48) mediated tumor suppression. Moreover, adjuvant selenium therapy in addition to chemotherapy caused a synergistic effect regarding the induction of apoptosis and improvement of the overall clinical outcome of cancer patients (49).
SCHEME 1.
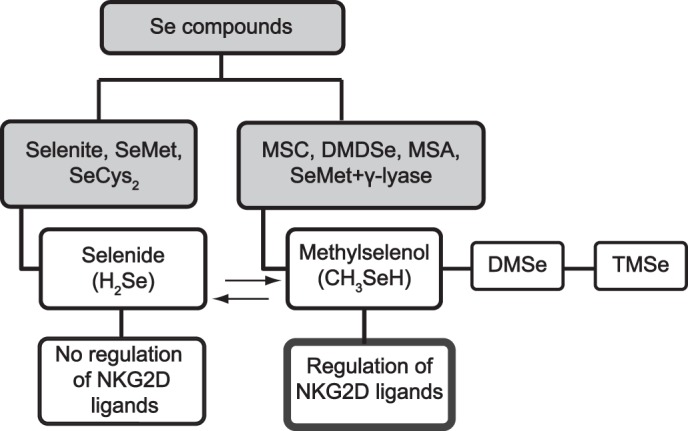
Regulation of NKG2D ligands by different selenium compounds. Selenium compounds are divided into organic and inorganic forms. The metabolism of these organic and inorganic selenium compounds is complex and closely regulated, with two key metabolites: Selenide (H2Se) and methylselenol (CH3SeH). On the basis of the current understanding of the selenium metabolism, NKG2D ligands are only regulated by CH3SeH-generating selenium compounds.
Autophagy is an evolutionary ancient pathway that ensures that cells can maintain their cell-autonomous homeostasis through the removal of intracellular material by lysosomal degradation (50, 51). Moreover, autophagy is utilized by infected cells to eliminate intracellular pathogens and likely serves as one of the earliest forms of eukaryotic defense against intracellular pathogens (52). Autophagy is characterized by the translocation of microtubule-associated protein 1 light chain 3 (LC3) from the cytoplasm to the autophagosome, where it is targeted to the lysosome for degradation (53).
We have shown previously that the synthetic selenium compound MSA modulates NKG2D ligands (54). In this study, we investigated the effect of different selenium compounds, metabolites, or intermediates with regard to expression of NKG2D ligands, and we identified CH3SeH as a key metabolite involved in the regulation of NKG2D ligands.
EXPERIMENTAL PROCEDURES
Cells
Two Jurkat T cell lines were used in this study. Jurkat E6-1 was purchased from the ATCC, and Jurkat Tag-9 was provided by Dr. Carsten Geisler (Department of International Health, Immunology, and Microbiology, University of Copenhagen, Denmark). Jurkat Tag-9 cells are stably transfected with the large T antigen from SV40, and they were used primarily for transient transfection studies. Jurkat cells were grown in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% FBS, 2 mm glutamine, and 2 mm penicillin and streptomycin. U20S cells (human osteosarcoma) were purchased from Millipore as part of the FlowCellectTM GFP-LC3 reporter autophagy assay kit (catalog no. CF200096). U20S cells were cultured in 10% DMEM + GlutaMAX with 2 mm penicillin and streptomycin, 1× non-essential amino acids (from 100× stock), 10 mm HEPES, and 250 mg/ml geneticin. All cells were incubated at 37 °C and 5% CO2.
Reagents
FR901228 was provided by the NCI, National Institutes of Health (Bethesda, MD). l-selenomethionine (SeMet) (catalog no. 561505) was from Calbiochem. Sodium selenite (selenite) and sodium selenate (selenate) (catalog no. S5261), SeCys2 (catalog no. M6680), MSC (catalog no. M6680), dimethyl diselenide (DMDSe) (catalog no. 328502), MSA (catalog no. 541281), EGTA (catalog no. E3889), wortmannin (catalog no. W1628), 3-methyladenine (catalog no. M9281), MG132 (catalog no. M7449), and propidium iodine (PI) (catalog no. P4170) were from Sigma-Aldrich. l-methionine γ-lyase (catalog no. 42616-25-1) was from Wako Pure Chemical Industries. TRIzol reagent (catalog no. 15596-026) was from Invitrogen.
Transient Transfections and Constructs
Jurkat Tag-9 cells were transiently transfected using Nucleofector technology (Lonza). In brief, Jurkat Tag-9 cells were resuspended in 100 μl of Ingenio electroporation solution (Mirus Bio LLC), mixed with 1 μg of plasmid/1 × 106 cells, and pulsed using the Nucleofector program G-010. The MICA promoter plasmid p3.2k-WT-GFP and the +2 control plasmid have been described previously (55). Promoter activity was calculated by multiplying the percentage of GFP-expressing cells with the mean fluorescence of these cells. The plasmids Ub-M-GFP (catalog no. 11938), Ub-R-GFP, catalog no. 11939), and Ub-G76-GFP (catalog no. 11941) used to study the proteasome inhibition were purchased from Addgene.
Flow Cytometry
Cells were washed twice in cold PBS and stained with the specific antibodies at a dilution of 1:100 for 30 min at 4 °C. Following antibody incubation, the cells were washed, resuspended, and analyzed in PBS. For staining of dead cells, the cells were incubated with 1 μg/ml PI for at least 5 min at room temperature prior to analysis. Staining of apoptotic cells by Annexin V was carried out by washing and staining the cells in buffer containing 10 mm Hepes (pH 7.4), 0.14 m NaCl, and 2.5 mm CaCl2. Lysosome accumulation was investigated by staining the cells with 4-nitro-7-(1-piperazinyl)-2,1,3-benzoxadiazole according to the protocol of the manufacturer (Cayman Chemical Co., catalog no. 600310). In brief, 4-nitro-7-(1-piperazinyl)-2,1,3-benzoxadiazole (1:1000)-stained cells were incubated for 10 min at 37 °C, centrifuged, and resuspended in cell-based assay buffer. The flow cytometry data acquisition was performed on a BD Accuri C6 flow cytometer and CFlow software, and the analysis of the collected data was carried out in FCSexpress 3.0/Treestar FlowJo. The following antibodies were used in this study: PE-conjugated mouse anti-hMICA/B (BD Biosciences, catalog no. 558352), APC-conjugated mouse anti-hULBP2/5/6 (R&D Systems, catalog no. FAB1298A), and FITC-conjugated Annexin V (BD Biosciences, catalog no. 556419).
Quantitative Real-time PCR
Jurkat E6-1 cells were treated for 4 h with FR901228, MSA, selenite and H2O2 (catalog no. H1009) or left untreated. RNA was isolated from 1 × 106 Jurkat E6-1 cells using TRIzol reagent and reverse-transcribed using SuperScript III reverse transcriptase enzyme according to the protocol of the manufacturer. PCR was performed using standard conditions. MICA primer sequences were as follows: MICA_523_Fwd, 5′-GCCATGAACGTCAGGAATTT-3′; MICA_760_Rev, 5′GACGCCAGCTCAGTGTGATA-3′. ULBP2 primer sequences were as follows: ULBP2_378_Fwd, 5′-CAGAGCAACTGCGTGACATT-3′; ULBP2_610_Rev, 5′-GGCCACAACCTTGTCATTCT-3′. The housekeeping gene ribosomal protein, large, P0 (RPLP0) was used as a normalization control. RPLP0 primer sequences were as follows: RPLP0_Fwd, 5′-GCTTCCTGGAGGGTGTCC-3′; RPLP0_Rev, 5′-GGACTCGTTTGTACCCGTTG-3′. All primers were purchased from Eurofins MWG Operon. Quantitative real-time PCR was performed using the Brilliant II SYBR Green QPCR Master Mix kit with low ROX (Stratagene). The real-time PCR reactions were performed on a Stratagene Mx3000P QPCR system thermocycler.
High-performance LC-Inductively Coupled Plasma MS
Jurkat E6-1 cells were treated for 4 h with 20 ng/ml FR901228, 5 μm MSA, or were left untreated. The cells were lysed with methanol (50% final concentration), and cell supernatants were injected into the HPLC system. The HPLC system was an Agilent 1100 system comprising an isocratic pump (G1310A) and an auto sampler (G1313A) and was run with Chemstation software (all from Agilent, Germany). The chromatographic column was a Gemini C18 (3 μm, 110 Å, 50 × 1 mm inner diameter) (Phenomenex, Denmark). The mobile phase was 0.1% formic acid in 40% methanol. The flow rate was 100 μl/min. Injection volumes were 10 μl. The chromatographic instrument was hyphenated to an inductively coupled plasma mass spectrometer. The inductively coupled plasma mass spectrometer was an Elan 6000 (PerkinElmer Life Sciences SCIEX) equipped with a jacketed cyclonic spray chamber (water-cooled to 4 °C) and a MicroMist microconcentric nebulizer (AR30-1-FM02E, both from Glass Expansion, West Melbourne, Australia). Cones were made of platinum. Nebulizer gas flow, lens voltage, and radio frequency power were optimized daily via microliter per minute infusion of a 100-ppb matrix-matched selenite standard solution. 76Se, 77Se and 82Se isotopes were monitored. The intensity of the 82Se isotope was used for quantitative calculations. The instrument was run by the Elan software version 3.4 (PerkinElmer Life Sciences), and the chromatographic data were handled by TotalChrom software (PerkinElmer Life Sciences).
Proteasome Sensor Vector Kit Assay
The induction of proteasome inhibition after MG132, FR901228, and MSA treatment was investigated by using the proteasome-sensitive fluorescent reporter ZsProSensor-1 according to the protocol of the manufacturer (Clontech, catalog no. 632425). In brief, 3 × 106 Jurkat Tag-9-cells were transfected as described above and incubated with 0.2 μm MG132, 20 ng/ml FR901228, and 5 μm MSA for 18 h. The cells emitted green fluorescence when there was a drop in proteasome activity, which was analyzed by flow cytometry.
GFP-LC3 Reporter Autophagy Assay
The induction of autophagy after FR901228 and MSA treatment was investigated by using a FlowCellectTM GFP-LC3 reporter autophagy assay kit (Millipore, catalog no. FCCH100181) according to the protocol of the manufacturer. In brief, 30,000 U20S cells were seeded into a 96-well plate. Duplicates of adhered cells were treated with either 20 ng/ml FR901228 or 5 μm MSA for 18 h or were left untreated. After the treatment, medium was aspirated, and cells were washed once with 1× Hanks' balanced salt solution. One copy of FR901228, MSA, or untreated cells was incubated for 2 h with 200 μl of plating medium containing autophagy reagent A (100 μm), and the second half was kept in culture medium. Post-incubation, the plating medium was aspirated, and cells were washed once with 200 μl of 1× Hanks' balanced salt solution. Cells were detached using versene, resuspended in 200 μl of fresh culture medium, and transferred to a new 96-well (U-bottom) plate. Cells were pelleted by centrifugation at 300 × g for 5 min at room temperature, and the supernatant was discarded. Next, cells were resuspended in 100 μl of 1× autophagy reagent B and pelleted immediately at 300 × g for 5 min at room temperature. To remove residual 1× autophagy reagent B, cells were washed once with 1× assay buffer. Cells were analyzed in 200 μl of 1× assay buffer by flow cytometry as described above.
RESULTS
Methylselenol-generating Selenium Compounds Induce MICA/B Surface Expression
The monomethylated, synthetic selenium compound MSA is directly reduced to CH3SeH (26) and induces the expression of the NKG2D ligands MICA/B (54). To investigate the role of CH3SeH in relation to NKG2D ligand regulation, different selenium compounds were tested for their ability to modulate surface expression of the NKG2D ligands. The tested compounds included MSC, which is converted to CH3SeH by the enzyme β-lyase; DMDSe, which is converted directly to CH3SeH; SeMet and SeCys2, which are primarily converted to H2Se or directly converted to CH3SeH by γ-lyase activity (SeMet + γ-lyase); as well as selenite and selenate, which are precursors of H2Se (Scheme 1). Jurkat E6-1 cells were incubated with the different selenium compounds for 18 h, and surface expression of MICA/B and ULBP2 was analyzed by flow cytometry. Jurkat T cells display a low basal level of MICA/B and a high basal level of ULBP2 cell surface expression. A clear trend emerged when the different selenium compounds were tested. All selenium compounds metabolized into CH3SeH. MSA, MSC, and DMDSe induced MICA/B but not ULBP2 surface expression (Fig. 1A). MSC induced maximal MICA/B expression at a concentration of 500 μm, which was ∼100 times higher compared with DMDSe and MSA (5 μm). This is most likely due to limited expression of the β-lyase needed for the conversion into CH3SeH (56). The precursors of H2Se, selenite, selenate, SeCys2, and SeMet had no effect on MICA/B or ULBP2 surface expression (Fig. 1B). In these experiments, the HDAC inhibitor FR901228 (20 ng/ml) and MSA (5 μg/ml) were used as positive controls. The different concentrations of each selenium compound were on the basis of our previous studies (57) as well as on work performed by other researchers in the field (26) Treatment of Jurkat T cells with the highest concentration of the selenium compounds MSA, MSC, selenite, and selenate induced cell death in 10–20% of the cells (Fig. 1C). Notably, all tested selenium compounds caused less cell death compared with the HDAC inhibitor FR901228, which is well known to regulate NKG2D ligands (16). To further assure that selenium regulation of NKG2D ligands occurs in living cells, we additionally show that MICA/B up-regulation after MSA treatment only occurs in viable, non-apoptotic cells, whereas apoptotic cells (Annexin V+) did not up-regulate MICA/B (Fig. 1D, left and center panels). In previous experiments we have shown that FR901228 only increases NKG2D ligand surface expression of non-apoptotic cells (58, 59). Therefore, treatment with FR901228 was used as a control (Fig. 1D, right panel). To strengthen our hypothesis that CH3SeH is required for MICA/B surface expression, we tested whether the combined treatment of SeMet and γ-lyase induced the surface expression of MICA/B in Jurkat E6-1 cells. We performed this experiment because SeMet is converted to CH3SeH in the presence of γ-lyase activity. As seen in Fig. 1E, left panel, the combined treatment of 5 μm SeMet and 0.02–0.04 U γ-lyase, but not SeMet alone, resulted in MICA/B surface expression. Again, no change in ULBP2 surface expression was observed (Fig. 1E, right panel). Our results strongly suggest that the generation of CH3SeH during the treatment with selenium compounds is required for cell surface induction of the NKG2D ligands MICA/B.
FIGURE 1.

Methylselenol-generating selenium compounds induce MICA/B surface expression. A, Jurkat E6-1 cells were treated with various concentrations of MSC and DMDSe for 18 h. FR901228 and MSA were used as positive controls for induction. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. B, Jurkat E6-1 cells were treated with the indicated concentrations of selenite, selenate, SeCys2, and SeMet for 18 h. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. C, Jurkat E6-1 cells were left untreated (UT) or treated with the highest concentrations of selenium compounds used in A and B for 18 h. For staining of dead cells, the cells were incubated with 1 μg/ml PI and analyzed by flow cytometry (FL-2 channel). D, Annexin V and anti-MICA/B staining of Jurkat E6-1 cells treated with 5 μm MSA and FR901228 for 18 h. The cutoff for MICA/B staining was set according to isotype control-treated cells (not shown for FR901228 treatment); FSC, forward scatter. E, cells were exposed to either 0.5 or 50 μm SeMet with or without the addition of 0.02 units/ml or 0.04 units/ml METase (methionine γ-lyase + PLP) for 18 h. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. All bar graphs show mean ± S.D., and all experiments are representative of at least two independent experiments.
Methylselenol Activates the Transcription of the NKG2D Ligands MICA and ULBP2
We and others have shown that NKG2D ligands can be regulated on the transcriptional level (55, 60, 61). To investigate whether CH3SeH also causes this transcriptional activation of NKG2D ligands, Jurkat Tag-9 cells were transiently transfected with a MICA promoter containing plasmid as well as a promoterless (+2) variant. Post-transfection, the cells were treated with MSC, selenite, 20 ng/ml FR901228 (control), and 5 μm MSA (control) for 18 h. The promoter activity was calculated by multiplying the percentage of GFP-expressing cells with the mean fluorescence of these cells (55). The CH3SeH-generating compound MSC directly stimulated the transiently transfected MICA promoter construct, whereas selenite had no effect (Fig. 2A). Non-transfected and selenite-treated cells showed a slight increase in fluorescence intensity compared with the other untransfected cells. This might be due to increased autofluorescence, a recognized effect of selenite treatment (62). However, this effect is likely quenched by the strong GFP signal during the transfection experiments. To elaborate these findings, we tested NKG2D ligand mRNA induction after the treatment with CH3SeH-, and non-CH3SeH-generating selenium compounds. For this experiment, Jurkat E6-1 cells were treated with (10 μm) selenite, 20 ng/ml FR901228 (control), and 5 μm MSA (control) for 4 h. RNA was isolated and reverse-transcribed into cDNA, followed by real-time quantitative PCR analysis. The level of MICA and ULBP2 mRNA was induced by the CH3SeH-generating selenium compound MSA, whereas MICA and ULBP2 mRNA was not induced by selenite (Fig. 2B). These experiments further strengthen our hypothesis that CH3SeH is the key metabolite in NKG2D ligand regulation.
FIGURE 2.

Methylselenol activates the transcription of the NKG2D ligands MICA and ULBP2. A, Jurkat Tag-9 cells were transfected with either the 3.2-kb wild-type MICA promoter construct or the +2 control construct. After 24 h, cells were treated with 20 ng/ml FR901228, 5 μm MSA, 500 μm MSC, and 10 μm selenite for 18 h. Cells were analyzed for their expression of GFP by flow cytometry. B, Jurkat E6-1 cells were left untreated or treated with 20 ng/ml FR901228, 5 μm MSA, or 10 μm selenite. After 4 h, total RNA was extracted and used for quantitative real-time PCR analysis. MICA and ULBP2 mRNA expression was normalized to the housekeeping gene RPLP0 and displayed as the fold change relative to the control. Data (mean ± S.D.) are representative of at least three separate experiments.
Methylselenol-generating Selenium Compounds Inhibit ULBP2 Surface Expression after Treatment with the HDAC Inhibitor FR901228
The results stated above indicate that CH3SeH has no effect on the surface expression of ULBP2. Interestingly, the induction of surface-expressed ULBP2 after HDAC inhibitor treatment was inhibited when treated in combination with MSA (54). Therefore, we tested whether this inhibition is caused by CH3SeH-generating selenium compounds. To this end, Jurkat E6-1 cells were treated for 18 h with MSC, DMDSe, selenite, and SeMet in combination with FR901228 (20 ng/ml). The cell surface expression of NKG2D ligands was analyzed by flow cytometry. As seen in Fig. 3C, left panel, selenite did not affect the expression of NKG2D ligands, whereas the combination of FR901228 and MSC or DMDSe strongly inhibited the cell surface expression of ULBP2 (Fig. 3B). Here it should be noted that SeMet exhibited a dose-dependent inhibition of both FR901228-induced MICA/B and ULBP2 (Fig. 3C, right panel). This is likely due to an increasing number of cells going into apoptosis. These results further imply that only CH3SeH-generating selenium compounds can regulate the expression of NKG2D ligands.
FIGURE 3.

Methylselenol-generating selenium compounds inhibit ULBP2 surface expression after treatment with the HDAC inhibitor FR901228. A, Jurkat E6-1 cells were either left untreated or treated with 20 ng/ml FR901228 in combination with the indicated concentrations of MSA. After 18 h, cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. B, Jurkat E6-1 cells were treated with 20 ng/ml FR901228 in combination with the indicated concentrations of MSC or DMDSe. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. C, Jurkat E6-1 cells were treated with 20 ng/ml FR901228 in combination with the indicated amounts of selenite or SeMet. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. Gating was performed on untreated cells. All bar graphs show mean ± S.D. and are representative of at least two independent experiments.
The Induction of the NKG2D Ligands MICA/B Is Not Caused by CH3SeH Generated from HDAC Inhibitors
CH3SeH can be generated in cells through metabolism (33). Given the similarities between the effect of both HDAC inhibitors and CH3SeH on the induction of the NKG2D ligands MICA/B, we wanted to investigate whether HDAC inhibitors in general regulate NKG2D ligands through the generation of CH3SeH. This was, however, not the case. As shown in Fig. 4A, no detection of selenium-metabolized products was observed in the FR901228-treated samples, arguing against the generation of CH3SeH during FR901228 metabolism.
FIGURE 4.

Methylselenol is not generated during FR901228 treatment, whereas compounds are stable in aqueous solution. A, LC inductively coupled plasma MS analysis was performed on Jurkat E6-1 cells pretreated for 4 h with or without 20 ng/ml FR901228 or 5 μm MSA. Each sample was mixed 1:1 with methanol, and the sample supernatant was analyzed. The presence of DMDSe indicated CH3SeH formation. Peak 1 contains hydrophilic selenium species such as S-(MeSe)-GS, and peak 2 is DMeSe. B, 20 ng/ml FR901228 or 5 μm MSA were diluted in different aqueous solutions: H2O and culture medium containing FBS (CM+). Aqueous solutions were either incubated for 4 days at room temperature or freshly prepared before the addition to Jurkat E6-1 cells. After 18 h, cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. Gating was performed on untreated cells. All bar graphs show mean ± S.D. and are representative of at least three independent experiments.
The NKG2D Ligand-regulating Effect of CH3SeH and an HDAC Inhibitor Is Stable in Aqueous Solutions
Preclinical studies in nonhuman primates as well as reports from a phase I trial in adults describe the FR901228 terminal half-life to be limited to a maximal 8 h (63, 64). Furthermore, studies suggest that infusions of FR901228 over a time period of 1–4 h are most effective in terms of cytotoxicity toward multiple tumor cell lines but at the same time are least toxic (65–67). The compounds formed after MSA treatment, CH3SeH, dimethylselenide, and DMDSe, are considered to be highly volatile and difficult to detect (68). Therefore, we examined whether MSA or FR901228 diluted in different aqueous solutions for several days would alter their regulation of NKG2D ligands compared with freshly prepared solutions. As seen in Fig. 4B, 4 days of preincubation of 20 ng/ml FR901228 or 5 μm MSA in culture medium with 10% FBS or H2O did not affect their ability to induce NKG2D ligands when compared with freshly prepared MSA or FR901228. Similar results were observed with the compounds diluted in PBS or culture medium without FBS (data not shown). Therefore, the cellular effects of MSA and FR901228 are stable for 4 days in serum-containing media, which is of significant interest for therapeutic applications. The double peaks seen in the histograms could be due to the fact that a fraction of the cells will undergo apoptosis caused by the different treatments, as shown in Fig. 1D.
Methylselenol Affects the Expression of NKG2D Ligands by Regulating Autophagy
The results stated above highlight the regulatory potential of CH3SeH on the induction as well as inhibition of NKG2D ligands. This regulatory pathway is clearly distinct from the HDAC inhibitor FR901228. We then focused our attention on the mechanism used by CH3SeH to modulate NKG2D ligands. Because the stable, monomethylated compound MSA was developed specifically to generate CH3SeH just by reduction (26), all further CH3SeH-requiring experiments were performed using MSA. CH3SeH is described to act both as an oxidant and antioxidant as well as an inducer of endoplasmic reticulum stress. Therefore, compounds such as N-acetyl-cysteine (NAC) and vitamin E (antioxidants); buthionine sulfoximine (BSO) (oxidant); and thapsigargin, aristolochic acid, arsenic trioxide, and DTT (endoplasmic reticulum stressors) were tested for their ability to regulate the cell surface expression of NKG2D ligands. None of the compounds caused an effect similar to CH3SeH (data not shown), suggesting that direct regulation of oxidative stress or endoplasmic reticulum stress in general is not essential for CH3SeH regulation of NKG2D ligands. Next we focused on posttranslational pathways important for NKG2D ligand regulation. It has been recognized that proteasomal inhibition is important for the regulation of NKG2D ligands, specifically for the induction of cell surface-expressed ULBP2 (17). Hence, we hypothesized that the different expression patterns of CH3SeH and FR901228 could be a result of differences in proteasome regulation. To examine the proteasome function, Jurkat Tag-9 cells were transfected with a proteasome sensor vector (ZsProSensor-1) followed by incubation with 0.2 μm MG132, 20 ng/ml FR901228, and 5 μm MSA for 18 h. The cells emitted green fluorescence when there was a drop in proteasome activity, which was analyzed by flow cytometry. As seen in Fig. 5A, both FR901228 and CH3SeH inhibited proteasome activity almost as potently as the proteasome inhibitor MG132. Next we used an assay developed by Dantuma et al. (69) for quantifying ubiquitin/proteasome-dependent proteolysis to investigate whether there was a selective modification of proteolysis without functional inactivation. To this end, the plasmids Ub-M-GFP, Ub-R-GFP, and Ub-G76-GFP were transfected into Jurkat Tag-9 cells before treatment with FR901228, MSA, and the proteasome inhibitor MG132. The plasmid DNAs encoding Ub-R-GP and Ub-G76-GFP contain a stabilizing arginine and a mutated, non-cleavable ubiquitin moiety, respectively, but will both be degraded by the proteasome. The control plasmid Ub-M-GFP contains no degradation signal when ubiquitin is cleaved and should accumulate inside the cell (69). The treatment of both CH3SeH and FR901228 caused increased levels of GFP accumulation similar to MG132, indicating their ability to block ubiquitin/proteasome-dependent proteolysis (Fig. 5B, left and center panels). Interestingly, all treatments led to less GFP accumulation compared with untreated cells when transfected with the control Ub-M-GFP plasmid (Fig. 5B, right panel). The results suggest that the different regulation patterns of NKG2D ligand expression caused by CH3SeH and FR901228 are not due to differences in the proteasome activation state.
FIGURE 5.

Methylselenol affects the expression of NKG2D ligands by regulating autophagy and only in the presence of extracellular calcium. A, Jurkat Tag-9 cells were transfected with proteasome sensor vector (ZsProSensor-1) and incubated with 0.2 μm MG132, 20 ng/ml FR901228, and 5 μm MSA for 18 h. Cells were analyzed for accumulation of GFP via flow cytometry. B, Jurkat Tag-9 cells were transfected with the plasmids Ub-G76V-GFP, Ub-R-GFP, and Ub-M-GFP, followed by incubation for 18 h with 0.2 μm MG132, 20 ng/ml FR901228, or 5 μm MSA. After incubation, the amount of accumulated GFP was measured by flow cytometry. C, Jurkat E6-1 cells were incubated with the indicated amount of either 3-methyladenine or wortmannin in combination with 20 ng/ml FR901228 for 18 h. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. D, U20S cells were treated with 20 ng/ml FR901228 or 5 μm MSA for 18 h. The lysosome inhibitor “Reagent A” was added 1 h prior to the end of incubation. Cells were analyzed for the amount of GFP-LC3 expression using flow cytometry. Gating was performed on untreated cells. E, Jurkat E6-1 cells were treated with 10 μm chloroquine; 1, 5, or 10 μm MSA; and 100, 200, or 500 μm MSC for 18 h. Inhibition of lysosomal activity was tested by measuring 4-nitro-7-(1-piperazinyl)-2,1,3-benzoxadiazole accumulation in lysosomes via flow cytometry. F, Jurkat E6-1 cells were treated with the indicated amounts of EGTA 0.5 h prior to the addition of 20 ng/ml FR901228 or 5 μm MSA for 18 h. Cells were stained with anti-MICA/B-PE and anti-ULBP2-APC antibody and analyzed by flow cytometry. All bar graphs show mean ± S.D., and all experiments are representative of at least three independent experiments.
Studies have shown that impairment of proteasomal activity can lead to increased autophagy (70, 71), which would also explain the decreased amount of Ub-M-GFP accumulation in Fig. 5B, right panel. Because autophagy is involved in intracellular transport, not only linked to degradation, it was interesting to examine the involvement of such autophagic transport during NKG2D ligand surface expression. Autophagy is known to be induced by HDAC inhibitors (72, 73), whereas there is conflicting data regarding whether selenium compounds activate or inhibit autophagy (74, 75).
To assess whether autophagy regulates the expression of NKG2D ligands, FR901228-treated Jurkat E6-1 cells were exposed to two different autophagy inhibitors, 3-methyladenine and wortmannin, for 18 h. Treatment of 3-methyladenine inhibits autophagy by blocking autophagosome formation via the inhibition of type III PI3K, and wortmannin, more broadly, inhibits PI3K activity. Type III PI3K activity is particular important for vesicle transport during autophagy. Interestingly, both inhibitors blocked FR901228-induced surface expression of ULBP2 but not MICA/B (Fig. 5C), indicating that autophagy specifically regulates ULBP2 surface expression. This is interesting because CH3SeH also inhibited ULBP2 surface expression (Fig. 3). To investigate whether CH3SeH regulation of ULBP2 involved the autophagic transport pathway, we used a GFP-LC3 reporter assay to monitor the autophagic flux after the treatment with FR901228 and MSA-generated CH3SeH. Monitoring of the LC3 flux through the autophagy pathway is the gold standard for measuring autophagy activity and is widely applied in the field (76). During autophagy, the protein LC3 is translocated from a soluble cytoplasmic form and bound to autophagosomes. Fixation of LC3 during autophagy can be examined in U20S cells stably expressing LC3-GFP, where vesicle-bound LC3-GFP is retained after washing out free LC3-GFP using a weak detergent solution. As seen in Fig. 5D (top row), LC3-GFP accumulated after treatment with FR901228 and also accumulated to a lower extent after CH3SeH treatment. On the basis of these experiments it was not possible to distinguish whether accumulation of LC3 occurred as a result of activation of the autophagic transport pathway (flux) or because of an inhibition downstream of LC3. The inclusion of a lysosomal inhibitor, however, made it possible to distinguish whether LC3 accumulation occurred through an increased autophagic flux or as a result of buildup of LC3 because of autophagy inhibition. LC3 accumulation after FR901228 treatment was further increased after lysosomal inhibition (Fig. 5D, bottom row)). This indicates that FR901228 enhances autophagic flux. Strikingly, CH3SeH blocked the accumulation of LC3 after lysosomal inhibition as well as FR901228-mediated accumulation of LC3 (Fig. 5D, bottom row), suggesting that CH3SeH directly inhibits the autophagic flux. Inhibition of lysosomal activity was measured by 4-nitro-7-(1-piperazinyl)-2,1,3-benzoxadiazole accumulation in lysosomes, a well described method for measuring lysosomal activity in live cells (77, 78). As expected, treatments of cells with the lysosomal inhibitor chloroquine (control) lead to a robust lysosomal accumulation. The CH3SeH-generating compounds MSC and MSA also affected lysosomal activity, although not to the extent observed with chloroquine (Fig. 5E). Because the two different autophagy inhibitors 3-methyladenine and wortmannin specifically inhibited the cell surface expression of ULBP2 upon HDAC-inhibitor treatment, as did CH3SeH, our data suggest that stimulation of the autophagic transport pathway is crucial for cell surface transport of ULBP2. This hypothesis is in line with our data reported previously showing that ULBP2 traffics over an endosomal/lysosomal pathway to the cell surface (54).
The Regulation of NKG2D Ligands by CH3SeH Is Dependent on Extracellular Calcium
The level of intracellular calcium is crucial for the regulation of MICA/B and ULBP2 cell surface expression upon HDAC inhibitor treatment (58). To examine the calcium dependence of CH3SeH-regulated MICA/B and ULBP2 surface expression, Jurkat E6-1 cells were incubated with the extracellular calcium chelator EGTA before treatment with CH3SeH generated by MSA. Cells treated in combination with EGTA and FR901228 were used as a control. As shown in Fig. 5F, treatment with EGTA decreased the CH3SeH-induced cell surface expression of MICA/B in a dose-dependent manner similar to control cells. These results suggest that extracellular calcium is involved in regulating the expression of NKG2D ligands by CH3SeH.
DISCUSSION
Selenium compounds have been highly discussed as chemopreventive agents but also as potential drugs or adjuvants in cancer therapy. Especially CH3SeH has been suggested as a key metabolite in cancer prevention. Studies have shown that selenium precursors metabolized into CH3SeH are more potent tumor inhibitors than compounds derived from H2Se (26, 79). At the same time, the induction of NKG2D ligands by traditional drugs or tolerable, chemical compounds has been investigated for many years, and more knowledge is required to further improve NKG2D-based therapy. In this study, we discovered that monomethylated selenium compounds induced surface expression of the NKG2D ligands MICA/B on viable and non-apoptotic cells. Precursors of H2Se had no effect on the expression of NKG2D ligands, implying that the generation of CH3SeH is crucial for NKG2D ligand regulation. In alignment with this hypothesis, we show that treatment with SeMet in combination with γ-lyase induced surface expression of MICA/B, similar to the treatment with monomethylated selenium compounds. As noted previously, SeMet can be metabolized into CH3SeH in the presence of γ-lyase (29). Our cell culture data further demonstrated that there was a hierarchy of efficiency in inducing the surface expression of MICA/B in the order MSA ≥ DMDSe ≥ SetMet + 0.04 units γ-lyase > MSC. In general, MSA induced the surface expression of MICA/B at 1/100 the concentration of MSC. A difference in response between the selenium compounds in regard to tumor inhibition has been described previously (26). In vivo, however, the difference disappeared, and the effects caused by MSA and MSC were found to be comparable (26). The authors suggested that the high presence of β-lyase, the enzyme required to produce CH3SeH from MSC, in vivo could be responsible for this compensation. Whether this also applies to the regulation of NKG2D ligands needs to be tested in further experiments in vivo. Additionally, a continuous generation of CH3SeH might be of importance for successful modulation of NKG2D ligands in vivo to counteract the naturally occurring metabolism of CH3SeH into dimethylselenide, trimethylselenonium, or H2Se. Notably, the tested Jurkat T cell line, but also other lymphoma cell lines, can generate the volatile selenium metabolites CH3SeH, DMDSe, and dimethylselenide in vitro because of the presence of the required and functional enzymes (26, 68, 80). When taken up by cells, MSA can also be reduced to CH3SeH via a nonenzymatic process (81). For this, an excess of thiol, e.g. GSH, is required. In our study, we did not distinguish whether CH3SeH generation by enzymatic or nonenzymatic processes regulates the expression of NKG2D ligands. We did try to inhibit the thioredoxin system by treating our cellular systems with auranofin, a potent inhibitor of the selenoenzyme thioredoxin reductase (82). The treatment did not affect the cell surface expression of NKG2D ligands, implying that this type of cell stress is not responsible for the regulation of NKG2D ligands in our current setting.
The effect of both FR901228 and MSA to regulate the expression of NKG2D ligands was stable for at least 4 days when diluted in H2O, PBS, or culture medium. Even the removal of FBS, a product discussed to help drug stability and cellular uptake (83, 84), did not lead to a change of effectivity. This is restricted to our in vitro experiments and might be different in vivo, where clearance by organs will occur. In the course of these experiments, we also observed that untreated cells, cultured in the same plates as cells treated with SeMet + γ-lyase, had induced surface expression of MICA/B compared with cells cultured in separate plates (data not shown). This implies that, during the treatment process, reactive selenium gases were developed, most likely CH3SeH, that modulated the expression of NKG2D ligands in untreated wells.
Our studies revealed that only CH3SeH-generating selenium compounds regulate the expression of NKG2D ligands. Here we recognized that CH3SeH induces gene activation and up-regulation of MICA/B surface expression. In contrast, the CH3SeH-induced ULBP2 gene activation was combined with a dominant suppression of ULBP2 surface expression. This is an important regulatory difference compared with HDAC-inhibitor activity known to induce surface expression of both ligand families (8, 16), and it strongly suggests that inhibition of ULBP2 expression occurs posttranscriptionally. Therefore, we attempted to study the parallels and differences of posttranscriptional actions upon treatment with HDAC inhibitors (in our experiments, FR901228) and monomethylated selenium compounds (MSA) to regulate the surface expression of NKG2D ligands. Interestingly, monitoring of the autophagic flux by measuring the accumulation of LC3 revealed that CH3SeH blocks the autophagic transport pathway, possibly through its potential to inhibit lysosomal activity. In addition, these data suggest that autophagic stress can facilitate ULBP2 surface transport. Here the induction of ULBP2 mRNA by CH3SeH and FR901228 implies that a common stress pathway exists but that the dominating posttranscriptional effect of CH3SeH prevents ULBP2 transport to the cell surface. This hypothesis is supported by the fact that autophagy is highly evolutionarily conserved and that autophagy has been adapted for more diverse uses than clearance of pathogens and maintenance of homeostasis, e.g. fusion of autophagosomes with endosomes or MHC class II loading compartments (85). In any case, the activity of CH3SeH described here provides mechanistic insights into the initiation of endosomal/lysosomal-dependent ULBP2 cell surface transport (54).
Summarizing our data, it is highly interesting that monomethylated selenium compounds, which are fundamental nutrients and easily accessible through our diet, can modulate the expression of NKG2D ligands in cancer cells, thereby enhancing their recognition and elimination by NKG2D-expressing immune effector cells. The fact that modulation of NKG2D ligands is restricted to CH3SeH-generating compounds is especially interesting for treatment approaches because H2Se metabolites are associated with genotoxic effects in cells (86, 87). Therefore, this so far unrecognized immune regulatory effect caused by CH3SeH-generating compounds should be added to the list of chemopreventive potential mediated by selenium compounds, and in particular considered for implementation in the treatment of NKG2D ligand-expressing tumors or adjuvant therapy in general. Certain tumors, such as melanoma and prostate and ovarian cancer (19, 20, 88), secrete large amounts of soluble ULBP2. Aberrant soluble ULBP2 is immunosuppressive because of constant immune activation and subsequent down-modulation of NKG2D (89). Therefore, CH3SeH-generating compounds are potentially ideal for the treatment of ULBP2-overexpressing cancer types because they can block the immunosuppressive soluble ULBP2 and, on the other hand, induce immune activation through the induction of MICA/B.
Acknowledgments
We thank Dr. Carsten Geisler (Department of International Health, Immunology, and Microbiology, University of Copenhagen, Denmark) for providing the JTag9 cell line, Prof. K. Helin (University of Copenhagen) for the pCMV-Myc plasmid, and Dr. M. Wills (University of Cambridge) for the GFP-MICA*018 plasmid.
This study was supported by European Marie Curie Initial Training Network EngCaBra Project PITN-GA-2010-264417 and by Danish Council for Independent Research/Medical Sciences Projects DFF-1331-00169 and 271-07-0302.
- SeMet
- selenomethionine
- MSC
- selenium methylselenocysteine
- MSA
- methylselenic acid
- SeCys2
- selenocysteine
- DMDSe
- dimethyl diselenide
- PI
- propidium iodide
- MIC
- MHC class I polypeptide-related sequence
- HDAC
- histone deacetylase
- PE
- phycoerythrin
- APC
- allophycocyanin.
REFERENCES
- 1. Bauer S. (1999) Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 285, 727–729 [DOI] [PubMed] [Google Scholar]
- 2. Groh V., Bruhl A., El-Gabalawy H., Nelson J. L., Spies T. (2003) Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 100, 9452–9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Groh V., Smythe K., Dai Z., Spies T. (2006) Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat. Immunol. 7, 755–762 [DOI] [PubMed] [Google Scholar]
- 4. Vivier E., Tomasello E., Paul P. (2002) Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr. Opin. Immunol. 14, 306–311 [DOI] [PubMed] [Google Scholar]
- 5. Champsaur M., Lanier L. L. (2010) Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 235, 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raulet D. H., Gasser S., Gowen B. G., Deng W., Jung H. (2013) Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 31, 413–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andresen L., Hansen K. A., Jensen H., Pedersen S. F., Stougaard P., Hansen H. R., Jurlander J., Skov S. (2009) Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J. Immunol. 183, 897–906 [DOI] [PubMed] [Google Scholar]
- 8. Armeanu S., Bitzer M., Lauer U. M., Venturelli S., Pathil A., Krusch M., Kaiser S., Jobst J., Smirnow I., Wagner A., Steinle A., Salih H. R. (2005) Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 65, 6321–6329 [DOI] [PubMed] [Google Scholar]
- 9. Cosman D., Müllberg J., Sutherland C. L., Chin W., Armitage R., Fanslow W., Kubin M., Chalupny N. J. (2001) ULBPs, novel MHC class I-related molecules bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133 [DOI] [PubMed] [Google Scholar]
- 10. Gasser S., Orsulic S., Brown E. J., Raulet D. H. (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Groh V., Bahram S., Bauer S., Herman A., Beauchamp M., Spies T. (1996) Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. U.S.A. 93, 12445–12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jinushi M., Takehara T., Kanto T., Tatsumi T., Groh V., Spies T., Miyagi T., Suzuki T., Sasaki Y., Hayashi N. (2003) Critical role of MHC class I-related chain A and B expression on IFN-alpha-stimulated dendritic cells in NK cell activation: impairment in chronic hepatitis C virus infection. J. Immunol. 170, 1249–1256 [DOI] [PubMed] [Google Scholar]
- 13. Jinushi M., Takehara T., Tatsumi T., Kanto T., Groh V., Spies T., Kimura R., Miyagi T., Mochizuki K., Sasaki Y., Hayashi N. (2003) Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer 104, 354–361 [DOI] [PubMed] [Google Scholar]
- 14. Jinushi M., Takehara T., Tatsumi T., Kanto T., Groh V., Spies T., Suzuki T., Miyagi T., Hayashi N. (2003) Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J. Immunol. 171, 5423–5429 [DOI] [PubMed] [Google Scholar]
- 15. Schreiner B., Voss J., Wischhusen J., Dombrowski Y., Steinle A., Lochmüller H., Dalakas M., Melms A., Wiendl H. (2006) Expression of toll-like receptors by human muscle cells in vitro and in vivo: TLR3 is highly expressed in inflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. FASEB J. 20, 118–120 [DOI] [PubMed] [Google Scholar]
- 16. Skov S., Pedersen M. T., Andresen L., Straten P. T., Woetmann A., Odum N. (2005) Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 65, 11136–11145 [DOI] [PubMed] [Google Scholar]
- 17. Valés-Gómez M., Chisholm S. E., Cassady-Cain R. L., Roda-Navarro P., Reyburn H. T. (2008) Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 68, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 18. Nückel H., Switala M., Sellmann L., Horn P. A., Dürig J., Dührsen U., Küppers R., Grosse-Wilde H., Rebmann V. (2010) The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia 24, 1152–1159 [DOI] [PubMed] [Google Scholar]
- 19. Paschen A., Sucker A., Hill B., Moll I., Zapatka M., Nguyen X. D., Sim G. C., Gutmann I., Hassel J., Becker J. C., Steinle A., Schadendorf D., Ugurel S. (2009) Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin. Cancer Res. 15, 5208–5215 [DOI] [PubMed] [Google Scholar]
- 20. Wu J. D., Higgins L. M., Steinle A., Cosman D., Haugk K., Plymate S. R. (2004) Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J. Clin. Invest. 114, 560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roman M., Jitaru P., Barbante C. (2014) Selenium biochemistry and its role for human health. Metallomics 6, 25–54 [DOI] [PubMed] [Google Scholar]
- 22. Auger J., Yang W., Arnault I., Pannier F., Potin-Gautier M. (2004) High-performance liquid chromatographic-inductively coupled plasma mass spectrometric evidence for Se-“alliins” in garlic and onion grown in Se-rich soil. J. Chromatogr. A 1032, 103–107 [DOI] [PubMed] [Google Scholar]
- 23. Gammelgaard B., Jackson M. I., Gabel-Jensen C. (2011) Surveying selenium speciation from soil to cell: forms and transformations. Anal. Bioanal. Chem. 399, 1743–1763 [DOI] [PubMed] [Google Scholar]
- 24. Yan L., DeMars L. C. (2012) Dietary supplementation with methylseleninic acid, but not selenomethionine, reduces spontaneous metastasis of Lewis lung carcinoma in mice. Int. J. Cancer 131, 1260–1266 [DOI] [PubMed] [Google Scholar]
- 25. Chen Y. C., Prabhu K. S., Das A., Mastro A. M. (2013) Dietary selenium supplementation modifies breast tumor growth and metastasis. Int. J. Cancer 133, 2054–2064 [DOI] [PubMed] [Google Scholar]
- 26. Ip C., Thompson H. J., Zhu Z., Ganther H. E. (2000) In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 60, 2882–2886 [PubMed] [Google Scholar]
- 27. Suzuki K. T., Kurasaki K., Suzuki N. (2007) Selenocysteine β-lyase and methylselenol demethylase in the metabolism of Se-methylated selenocompounds into selenide. Biochim. Biophys. Acta 1770, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 28. Spallholz J. E., Shriver B. J., Reid T. W. (2001) Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: implications for the anticarcinogenic activity of l-selenomethionine and l-Se-methylselenocysteine. Nutr. Cancer 40, 34–41 [DOI] [PubMed] [Google Scholar]
- 29. Esaki N., Tanaka H., Uemura S., Suzuki T., Soda K. (1979) Catalytic action of l-methionine γ-lyase on selenomethionine and selenols. Biochemistry 18, 407–410 [DOI] [PubMed] [Google Scholar]
- 30. Fernandes A. P., Wallenberg M., Gandin V., Misra S., Tisato F., Marzano C., Rigobello M. P., Kumar S., Björnstedt M. (2012) Methylselenol formed by spontaneous methylation of selenide is a superior selenium substrate to the thioredoxin and glutaredoxin systems. PloS ONE 7, e50727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gromer S., Gross J. H. (2002) Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase: implications for the antitumor effects of selenium. J. Biol. Chem. 277, 9701–9706 [DOI] [PubMed] [Google Scholar]
- 32. Asfour I. A., El-Tehewi M. M., Ahmed M. H., Abdel-Sattar M. A., Moustafa N. N., Hegab H. M., Fathey O. M. (2009) High-dose sodium selenite can induce apoptosis of lymphoma cells in adult patients with non-Hodgkin's lymphoma. Biol. Trace Elem. Res. 127, 200–210 [DOI] [PubMed] [Google Scholar]
- 33. Brozmanová J., Mániková D., Vlčková V., Chovanec M. (2010) Selenium: a double-edged sword for defense and offence in cancer. Arch. Toxicol. 84, 919–938 [DOI] [PubMed] [Google Scholar]
- 34. Selenius M., Rundlöf A. K., Olm E., Fernandes A. P., Björnstedt M. (2010) Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signal. 12, 867–880 [DOI] [PubMed] [Google Scholar]
- 35. El-Bayoumy K., Das A., Narayanan B., Narayanan N., Fiala E. S., Desai D., Rao C. V., Amin S., Sinha R. (2006) Molecular targets of the chemopreventive agent 1,4-phenylenebis (methylene)-selenocyanate in human non-small cell lung cancer. Carcinogenesis 27, 1369–1376 [DOI] [PubMed] [Google Scholar]
- 36. Li G. X., Lee H. J., Wang Z., Hu H., Liao J. D., Watts J. C., Combs G. F., Jr., Lü J. (2008) Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 29, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang C., Wang Z., Ganther H., Lu J. (2001) Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 61, 3062–3070 [PubMed] [Google Scholar]
- 38. Wang Z., Jiang C., Lü J. (2002) Induction of caspase-mediated apoptosis and cell-cycle G1 arrest by selenium metabolite methylselenol. Mol. Carcinog. 34, 113–120 [DOI] [PubMed] [Google Scholar]
- 39. Zeng H., Wu M., Botnen J. H. (2009) Methylselenol, a selenium metabolite, induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated kinase 1/2 pathway and other cancer signaling genes. J. Nutr. 139, 1613–1618 [DOI] [PubMed] [Google Scholar]
- 40. Jiang C., Ganther H., Lu J. (2000) Monomethyl selenium-specific inhibition of MMP-2 and VEGF expression: implications for angiogenic switch regulation. Mol. Carcinog. 29, 236–250 [DOI] [PubMed] [Google Scholar]
- 41. Lee J. I., Nian H., Cooper A. J., Sinha R., Dai J., Bisson W. H., Dashwood R. H., Pinto J. T. (2009) α-Keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev. Res. (Phila.) 2, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nian H., Bisson W. H., Dashwood W. M., Pinto J. T., Dashwood R. H. (2009) α-Keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis 30, 1416–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gopalakrishna R., Gundimeda U. (2002) Antioxidant regulation of protein kinase C in cancer prevention. J. Nutr. 132, 3819S-3823S [DOI] [PubMed] [Google Scholar]
- 44. Gundimeda U., Schiffman J. E., Chhabra D., Wong J., Wu A., Gopalakrishna R. (2008) Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: relevance to selenium-induced apoptosis in prostate cancer cells. J. Biol. Chem. 283, 34519–34531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dong Y., Ganther H. E., Stewart C., Ip C. (2002) Identification of molecular targets associated with selenium-induced growth inhibition in human breast cells using cDNA microarrays. Cancer Res. 62, 708–714 [PubMed] [Google Scholar]
- 46. Dong Y., Zhang H., Hawthorn L., Ganther H. E., Ip C. (2003) Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 63, 52–59 [PubMed] [Google Scholar]
- 47. Yan L., Spallholz J. E. (1993) Generation of reactive oxygen species from the reaction of selenium compounds with thiols and mammary tumor cells. Biochem. Pharmacol. 45, 429–437 [PubMed] [Google Scholar]
- 48. Reeves P. G., Leary P. D., Gregoire B. R., Finley J. W., Lindlauf J. E., Johnson L. K. (2005) Selenium bioavailability from buckwheat bran in rats fed a modified AIN-93G torula yeast-based diet. J. Nutr. 135, 2627–2633 [DOI] [PubMed] [Google Scholar]
- 49. Micke O., Bruns F., Mücke R., Schäfer U., Glatzel M., DeVries A. F., Schönekaes K., Kisters K., Büntzel J. (2003) Selenium in the treatment of radiation-associated secondary lymphedema. Int. J. Radiat. Oncol. Biol. Phys. 56, 40–49 [DOI] [PubMed] [Google Scholar]
- 50. Klionsky D. J., Emr S. D. (2000) Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klionsky D. J. (2013) Ancient autophagy. Autophagy 9, 445–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tal M. C., Iwasaki A. (2009) Autophagy and innate recognition systems. Curr. Top. Microbiol. Immunol. 335, 107–121 [DOI] [PubMed] [Google Scholar]
- 53. Castillo K., Valenzuela V., Matus S., Nassif M., Oñate M., Fuentealba Y., Encina G., Irrazabal T., Parsons G., Court F. A., Schneider B. L., Armentano D., Hetz C. (2013) Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis. 4, e917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Uhlenbrock F., Hagemann-Jensen M., Kehlet S., Andresen L., Pastorekova S., Skov S. (2014) The NKG2D ligand ULBP2 is specifically regulated through an invariant chain-dependent endosomal pathway. J. Immunol. 193, 1654–1665 [DOI] [PubMed] [Google Scholar]
- 55. Andresen L., Jensen H., Pedersen M. T., Hansen K. A., Skov S. (2007) Molecular regulation of MHC class I chain-related protein A expression after HDAC-inhibitor treatment of Jurkat T cells. J. Immunol. 179, 8235–8242 [DOI] [PubMed] [Google Scholar]
- 56. Andreadou I., Menge W. M., Commandeur J. N., Worthington E. A., Vermeulen N. P. (1996) Synthesis of novel Se-substituted selenocysteine derivatives as potential kidney selective prodrugs of biologically active selenol compounds: evaluation of kinetics of β-elimination reactions in rat renal cytosol. J. Med. Chem. 39, 2040–2046 [DOI] [PubMed] [Google Scholar]
- 57. Lunøe K., Gabel-Jensen C., Stürup S., Andresen L., Skov S., Gammelgaard B. (2011) Investigation of the selenium metabolism in cancer cell lines. Metallomics 3, 162–168 [DOI] [PubMed] [Google Scholar]
- 58. Jensen H., Hagemann-Jensen M., Lauridsen F., Skov S. (2013) Regulation of NKG2D-ligand cell surface expression by intracellular calcium after HDAC-inhibitor treatment. Mol. Immunol. 53, 255–264 [DOI] [PubMed] [Google Scholar]
- 59. Skov S., Rieneck K., Bovin L. F., Skak K., Tomra S., Michelsen B. K., Ødum N. (2003) Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood 101, 1430–1438 [DOI] [PubMed] [Google Scholar]
- 60. Bauman Y., Nachmani D., Vitenshtein A., Tsukerman P., Drayman N., Stern-Ginossar N., Lankry D., Gruda R., Mandelboim O. (2011) An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe 9, 93–102 [DOI] [PubMed] [Google Scholar]
- 61. Nachmani D., Lankry D., Wolf D. G., Mandelboim O. (2010) The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 11, 806–813 [DOI] [PubMed] [Google Scholar]
- 62. O'Toole D., Castle L. E., Raisbeck M. F. (1995) Comparison of histochemical autometallography (Danscher's stain) to chemical analysis for detection of selenium in tissues. J. Vet. Diagn. Invest. 7, 281–284 [DOI] [PubMed] [Google Scholar]
- 63. Sandor V., Bakke S., Robey R. W., Kang M. H., Blagosklonny M. V., Bender J., Brooks R., Piekarz R. L., Tucker E., Figg W. D., Chan K. K., Goldspiel B., Fojo A. T., Balcerzak S. P., Bates S. E. (2002) Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 8, 718–728 [PubMed] [Google Scholar]
- 64. Berg S. L., Stone J., Xiao J. J., Chan K. K., Nuchtern J., Dauser R., McGuffey L., Thompson P., Blaney S. M. (2004) Plasma and cerebrospinal fluid pharmacokinetics of depsipeptide (FR901228) in nonhuman primates. Cancer Chemother. Pharmacol. 54, 85–88 [DOI] [PubMed] [Google Scholar]
- 65. Rajgolikar G., Chan K. K., Wang H. C. (1998) Effects of a novel antitumor depsipeptide, FR901228, on human breast cancer cells. Breast Cancer Res. Treat. 51, 29–38 [DOI] [PubMed] [Google Scholar]
- 66. Byrd J. C., Shinn C., Ravi R., Willis C. R., Waselenko J. K., Flinn I. W., Dawson N. A., Grever M. R. (1999) Depsipeptide (FR901228): a novel therapeutic agent with selective, in vitro activity against human B-cell chronic lymphocytic leukemia cells. Blood 94, 1401–1408 [PubMed] [Google Scholar]
- 67. Murata M., Towatari M., Kosugi H., Tanimoto M., Ueda R., Saito H., Naoe T. (2000) Apoptotic cytotoxic effects of a histone deacetylase inhibitor, FK228, on malignant lymphoid cells. Jpn. J. Cancer Res. 91, 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gabel-Jensen C., Lunøe K., Gammelgaard B. (2010) Formation of methylselenol, dimethylselenide and dimethyldiselenide in in vitro metabolism models determined by headspace GC-MS. Metallomics 2, 167–173 [DOI] [PubMed] [Google Scholar]
- 69. Dantuma N. P., Lindsten K., Glas R., Jellne M., Masucci M. G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543 [DOI] [PubMed] [Google Scholar]
- 70. Jiang H., Sun J., Xu Q., Liu Y., Wei J., Young C. Y., Yuan H., Lou H. (2013) Marchantin M: a novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis. 4, e761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., Yao T. P., Baehrecke E. H., Taylor J. P. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 72. Shao Y., Gao Z., Marks P. A., Jiang X. (2004) Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 18030–18035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yamamoto S., Tanaka K., Sakimura R., Okada T., Nakamura T., Li Y., Takasaki M., Nakabeppu Y., Iwamoto Y. (2008) Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 28, 1585–1591 [PubMed] [Google Scholar]
- 74. Ren Y., Huang F., Liu Y., Yang Y., Jiang Q., Xu C. (2009) Autophagy inhibition through PI3K/Akt increases apoptosis by sodium selenite in NB4 cells. BMB Rep. 42, 599–604 [DOI] [PubMed] [Google Scholar]
- 75. Park S. H., Kim J. H., Chi G. Y., Kim G. Y., Chang Y. C., Moon S. K., Nam S. W., Kim W. J., Yoo Y. H., Choi Y. H. (2012) Induction of apoptosis and autophagy by sodium selenite in A549 human lung carcinoma cells through generation of reactive oxygen species. Toxicol. Lett. 212, 252–261 [DOI] [PubMed] [Google Scholar]
- 76. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., Al-Younes H. M., Al-Zeer M. A., Albert M. L., Albin R. L., Alegre-Abarrategui J., Aleo M. F., Alirezaei M., Almasan A., Almonte-Becerril M., Amano A., Amaravadi R., Amarnath S., Amer A. O., Andrieu-Abadie N., Anantharam V., Ann D. K., Anoopkumar-Dukie S., Aoki H., Apostolova N., Arancia G., Aris J. P., Asanuma K., Asare N. Y., Ashida H., Askanas V., Askew D. S., Auberger P., Baba M., Backues S. K., Baehrecke E. H., Bahr B. A., Bai X. Y., Bailly Y., Baiocchi R., Baldini G., Balduini W., Ballabio A., Bamber B. A., Bampton E. T., Banhegyi G., Bartholomew C. R., Bassham D. C., Bast R. C., Jr., Batoko H., Bay B. H., Beau I., Bechet D. M., Begley T. J., Behl C., Behrends C., Bekri S., Bellaire B., Bendall L. J., Benetti L., Berliocchi L., Bernardi H., Bernassola F., Besteiro S., Bhatia-Kissova I., Bi X., Biard-Piechaczyk M., Blum J. S., Boise L. H., Bonaldo P., Boone D. L., Bornhauser B. C., Bortoluci K. R., Bossis I., Bost F., Bourquin J. P., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N. R., Brancolini C., Brech A., Brenman J. E., Brennand A., Bresnick E. H., Brest P., Bridges D., Bristol M. L., Brookes P. S., Brown E. J., Brumell J. H., Brunetti-Pierri N., Brunk U. T., Bulman D. E., Bultman S. J., Bultynck G., Burbulla L. F., Bursch W., Butchar J. P., Buzgariu W., Bydlowski S. P., Cadwell K., Cahova M., Cai D., Cai J., Cai Q., Calabretta B., Calvo-Garrido J., Camougrand N., Campanella M., Campos-Salinas J., Candi E., Cao L., Caplan A. B., Carding S. R., Cardoso S. M., Carew J. S., Carlin C. R., Carmignac V., Carneiro L. A., Carra S., Caruso R. A., Casari G., Casas C., Castino R., Cebollero E., Cecconi F., Celli J., Chaachouay H., Chae H. J., Chai C. Y., Chan D. C., Chan E. Y., Chang R. C., Che C. M., Chen C. C., Chen G. C., Chen G. Q., Chen M., Chen Q., Chen S. S., Chen W., Chen X., Chen X., Chen X., Chen Y. G., Chen Y., Chen Y., Chen Y. J., Chen Z., Cheng A., Cheng C. H., Cheng Y., Cheong H., Cheong J. H., Cherry S., Chess-Williams R., Cheung Z. H., Chevet E., Chiang H. L., Chiarelli R., Chiba T., Chin L. S., Chiou S. H., Chisari F. V., Cho C. H., Cho D. H., Choi A. M., Choi D., Choi K. S., Choi M. E., Chouaib S., Choubey D., Choubey V., Chu C. T., Chuang T. H., Chueh S. H., Chun T., Chwae Y. J., Chye M. L., Ciarcia R., Ciriolo M. R., Clague M. J., Clark R. S., Clarke P. G., Clarke R., Codogno P., Coller H. A., Colombo M. I., Comincini S., Condello M., Condorelli F., Cookson M. R., Coombs G. H., Coppens I., Corbalan R., Cossart P., Costelli P., Costes S., Coto-Montes A., Couve E., Coxon F. P., Cregg J. M., Crespo J. L., Cronje M. J., Cuervo A. M., Cullen J. J., Czaja M. J., D'Amelio M., Darfeuille-Michaud A., Davids L. M., Davies F. E., De Felici M., de Groot J. F., de Haan C. A., De Martino L., De Milito A., De Tata V., Debnath J., Degterev A., Dehay B., Delbridge L. M., Demarchi F., Deng Y. Z., Dengjel J., Dent P., Denton D., Deretic V., Desai S. D., Devenish R. J., Di Gioacchino M., Di Paolo G., Di Pietro C., Diaz-Araya G., Diaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dikic I., Dinesh-Kumar S. P., Ding W. X., Distelhorst C. W., Diwan A., Djavaheri-Mergny M., Dokudovskaya S., Dong Z., Dorsey F. C., Dosenko V., Dowling J. J., Doxsey S., Dreux M., Drew M. E., Duan Q., Duchosal M. A., Duff K., Dugail I., Durbeej M., Duszenko M., Edelstein C. L., Edinger A. L., Egea G., Eichinger L., Eissa N. T., Ekmekcioglu S., El-Deiry W. S., Elazar Z., Elgendy M., Ellerby L. M., Eng K. E., Engelbrecht A. M., Engelender S., Erenpreisa J., Escalante R., Esclatine A., Eskelinen E. L., Espert L., Espina V., Fan H., Fan J., Fan Q. W., Fan Z., Fang S., Fang Y., Fanto M., Fanzani A., Farkas T., Farre J. C., Faure M., Fechheimer M., Feng C. G., Feng J., Feng Q., Feng Y., Fesus L., Feuer R., Figueiredo-Pereira M. E., Fimia G. M., Fingar D. C., Finkbeiner S., Finkel T., Finley K. D., Fiorito F., Fisher E. A., Fisher P. B., Flajolet M., Florez-McClure M. L., Florio S., Fon E. A., Fornai F., Fortunato F., Fotedar R., Fowler D. H., Fox H. S., Franco R., Frankel L. B., Fransen M., Fuentes J. M., Fueyo J., Fujii J., Fujisaki K., Fujita E., Fukuda M., Furukawa R. H., Gaestel M., Gailly P., Gajewska M., Galliot B., Galy V., Ganesh S., Ganetzky B., Ganley I. G., Gao F. B., Gao G. F., Gao J., Garcia L., Garcia-Manero G., Garcia-Marcos M., Garmyn M., Gartel A. L., Gatti E., Gautel M., Gawriluk T. R., Gegg M. E., Geng J., Germain M., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghosh P., Giammarioli A. M., Giatromanolaki A. N., Gibson S. B., Gilkerson R. W., Ginger M. L., Ginsberg H. N., Golab J., Goligorsky M. S., Golstein P., Gomez-Manzano C., Goncu E., Gongora C., Gonzalez C. D., Gonzalez R., Gonzalez-Estevez C., Gonzalez-Polo R. A., Gonzalez-Rey E., Gorbunov N. V., Gorski S., Goruppi S., Gottlieb R. A., Gozuacik D., Granato G. E., Grant G. D., Green K. N., Gregorc A., Gros F., Grose C., Grunt T. W., Gual P., Guan J. L., Guan K. L., Guichard S. M., Gukovskaya A. S., Gukovsky I., Gunst J., Gustafsson A. B., Halayko A. J., Hale A. N., Halonen S. K., Hamasaki M., Han F., Han T., Hancock M. K., Hansen M., Harada H., Harada M., Hardt S. E., Harper J. W., Harris A. L., Harris J., Harris S. D., Hashimoto M., Haspel J. A., Hayashi S., Hazelhurst L. A., He C., He Y. W., Hebert M. J., Heidenreich K. A., Helfrich M. H., Helgason G. V., Henske E. P., Herman B., Herman P. K., Hetz C., Hilfiker S., Hill J. A., Hocking L. J., Hofman P., Hofmann T. G., Hohfeld J., Holyoake T. L., Hong M. H., Hood D. A., Hotamisligil G. S., Houwerzijl E. J., Hoyer-Hansen M., Hu B., Hu C. A., Hu H. M., Hua Y., Huang C., Huang J., Huang S., Huang W. P., Huber T. B., Huh W. K., Hung T. H., Hupp T. R., Hur G. M., Hurley J. B., Hussain S. N., Hussey P. J., Hwang J. J., Hwang S., Ichihara A., Ilkhanizadeh S., Inoki K., Into T., Iovane V., Iovanna J. L., Ip N. Y., Isaka Y., Ishida H., Isidoro C., Isobe K., Iwasaki A., Izquierdo M., Izumi Y., Jaakkola P. M., Jaattela M., Jackson G. R., Jackson W. T., Janji B., Jendrach M., Jeon J. H., Jeung E. B., Jiang H., Jiang H., Jiang J. X., Jiang M., Jiang Q., Jiang X., Jiang X., Jimenez A., Jin M., Jin S., Joe C. O., Johansen T., Johnson D. E., Johnson G. V., Jones N. L., Joseph B., Joseph S. K., Joubert A. M., Juhasz G., Juillerat-Jeanneret L., Jung C. H., Jung Y. K., Kaarniranta K., Kaasik A., Kabuta T., Kadowaki M., Kagedal K., Kamada Y., Kaminskyy V. O., Kampinga H. H., Kanamori H., Kang C., Kang K. B., Kang K. I., Kang R., Kang Y. A., Kanki T., Kanneganti T. D., Kanno H., Kanthasamy A. G., Kanthasamy A., Karantza V., Kaushal G. P., Kaushik S., Kawazoe Y., Ke P. Y., Kehrl J. H., Kelekar A., Kerkhoff C., Kessel D. H., Khalil H., Kiel J. A., Kiger A. A., Kihara A., Kim D. R., Kim D. H., Kim D. H., Kim E. K., Kim H. R., Kim J. S., Kim J. H., Kim J. C., Kim J. K., Kim P. K., Kim S. W., Kim Y. S., Kim Y., Kimchi A., Kimmelman A. C., King J. S., Kinsella T. J., Kirkin V., Kirshenbaum L. A., Kitamoto K., Kitazato K., Klein L., Klimecki W. T., Klucken J., Knecht E., Ko B. C., Koch J. C., Koga H., Koh J. Y., Koh Y. H., Koike M., Komatsu M., Kominami E., Kong H. J., Kong W. J., Korolchuk V. I., Kotake Y., Koukourakis M. I., Kouri Flores J. B., Kovacs A. L., Kraft C., Krainc D., Kramer H., Kretz-Remy C., Krichevsky A. M., Kroemer G., Kruger R., Krut O., Ktistakis N. T., Kuan C. Y., Kucharczyk R., Kumar A., Kumar R., Kumar S., Kundu M., Kung H. J., Kurz T., Kwon H. J., La Spada A. R., Lafont F., Lamark T., Landry J., Lane J. D., Lapaquette P., Laporte J. F., Laszlo L., Lavandero S., Lavoie J. N., Layfield R., Lazo P. A., Le W., Le Cam L., Ledbetter D. J., Lee A. J., Lee B. W., Lee G. M., Lee J., Lee J. H., Lee M., Lee M. S., Lee S. H., Leeuwenburgh C., Legembre P., Legouis R., Lehmann M., Lei H. Y., Lei Q. Y., Leib D. A., Leiro J., Lemasters J. J., Lemoine A., Lesniak M. S., Lev D., Levenson V. V., Levine B., Levy E., Li F., Li J. L., Li L., Li S., Li W., Li X. J., Li Y. B., Li Y. P., Liang C., Liang Q., Liao Y. F., Liberski P. P., Lieberman A., Lim H. J., Lim K. L., Lim K., Lin C. F., Lin F. C., Lin J., Lin J. D., Lin K., Lin W. W., Lin W. C., Lin Y. L., Linden R., Lingor P., Lippincott-Schwartz J., Lisanti M. P., Liton P. B., Liu B., Liu C. F., Liu K., Liu L., Liu Q. A., Liu W., Liu Y. C., Liu Y., Lockshin R. A., Lok C. N., Lonial S., Loos B., Lopez-Berestein G., Lopez-Otin C., Lossi L., Lotze M. T., Low P., Lu B., Lu B., Lu B., Lu Z., Luciano F., Lukacs N. W., Lund A. H., Lynch-Day M. A., Ma Y., Macian F., MacKeigan J. P., Macleod K. F., Madeo F., Maiuri L., Maiuri M. C., Malagoli D., Malicdan M. C., Malorni W., Man N., Mandelkow E. M., Manon S., Manov I., Mao K., Mao X., Mao Z., Marambaud P., Marazziti D., Marcel Y. L., Marchbank K., Marchetti P., Marciniak S. J., Marcondes M., Mardi M., Marfe G., Marino G., Markaki M., Marten M. R., Martin S. J., Martinand-Mari C., Martinet W., Martinez-Vicente M., Masini M., Matarrese P., Matsuo S., Matteoni R., Mayer A., Mazure N. M., McConkey D. J., McConnell M. J., McDermott C., McDonald C., McInerney G. M., McKenna S. L., McLaughlin B., McLean P. J., McMaster C. R., McQuibban G. A., Meijer A. J., Meisler M. H., Melendez A., Melia T. J., Melino G., Mena M. A., Menendez J. A., Menna-Barreto R. F., Menon M. B., Menzies F. M., Mercer C. A., Merighi A., Merry D. E., Meschini S., Meyer C. G., Meyer T. F., Miao C. Y., Miao J. Y., Michels P. A., Michiels C., Mijaljica D., Milojkovic A., Minucci S., Miracco C., Miranti C. K., Mitroulis I., Miyazawa K., Mizushima N., Mograbi B., Mohseni S., Molero X., Mollereau B., Mollinedo F., Momoi T., Monastyrska I., Monick M. M., Monteiro M. J., Moore M. N., Mora R., Moreau K., Moreira P. I., Moriyasu Y., Moscat J., Mostowy S., Mottram J. C., Motyl T., Moussa C. E., Muller S., Muller S., Munger K., Munz C., Murphy L. O., Murphy M. E., Musaro A., Mysorekar I., Nagata E., Nagata K., Nahimana A., Nair U., Nakagawa T., Nakahira K., Nakano H., Nakatogawa H., Nanjundan M., Naqvi N. I., Narendra D. P., Narita M., Navarro M., Nawrocki S. T., Nazarko T. Y., Nemchenko A., Netea M. G., Neufeld T. P., Ney P. A., Nezis I. P., Nguyen H. P., Nie D., Nishino I., Nislow C., Nixon R. A., Noda T., Noegel A. A., Nogalska A., Noguchi S., Notterpek L., Novak I., Nozaki T., Nukina N., Nurnberger T., Nyfeler B., Obara K., Oberley T. D., Oddo S., Ogawa M., Ohashi T., Okamoto K., Oleinick N. L., Oliver F. J., Olsen L. J., Olsson S., Opota O., Osborne T. F., Ostrander G. K., Otsu K., Ou J. H., Ouimet M., Overholtzer M., Ozpolat B., Paganetti P., Pagnini U., Pallet N., Palmer G. E., Palumbo C., Pan T., Panaretakis T., Pandey U. B., Papackova Z., Papassideri I., Paris I., Park J., Park O. K., Parys J. B., Parzych K. R., Patschan S., Patterson C., Pattingre S., Pawelek J. M., Peng J., Perlmutter D. H., Perrotta I., Perry G., Pervaiz S., Peter M., Peters G. J., Petersen M., Petrovski G., Phang J. M., Piacentini M., Pierre P., Pierrefite-Carle V., Pierron G., Pinkas-Kramarski R., Piras A., Piri N., Platanias L. C., Poggeler S., Poirot M., Poletti A., Pous C., Pozuelo-Rubio M., Praetorius-Ibba M., Prasad A., Prescott M., Priault M., Produit-Zengaffinen N., Progulske-Fox A., Proikas-Cezanne T., Przedborski S., Przyklenk K., Puertollano R., Puyal J., Qian S. B., Qin L., Qin Z. H., Quaggin S. E., Raben N., Rabinowich H., Rabkin S. W., Rahman I., Rami A., Ramm G., Randall G., Randow F., Rao V. A., Rathmell J. C., Ravikumar B., Ray S. K., Reed B. H., Reed J. C., Reggiori F., Regnier-Vigouroux A., Reichert A. S., Reiners J. J., Jr., Reiter R. J., Ren J., Revuelta J. L., Rhodes C. J., Ritis K., Rizzo E., Robbins J., Roberge M., Roca H., Roccheri M. C., Rocchi S., Rodemann H. P., Rodriguez de Cordoba S., Rohrer B., Roninson I. B., Rosen K., Rost-Roszkowska M. M., Rouis M., Rouschop K. M., Rovetta F., Rubin B. P., Rubinsztein D. C., Ruckdeschel K., Rucker E. B., 3rd, Rudich A., Rudolf E., Ruiz-Opazo N., Russo R., Rusten T. E., Ryan K. M., Ryter S. W., Sabatini D. M., Sadoshima J., Saha T., Saitoh T., Sakagami H., Sakai Y., Salekdeh G. H., Salomoni P., Salvaterra P. M., Salvesen G., Salvioli R., Sanchez A. M., Sanchez-Alcazar J. A., Sanchez-Prieto R., Sandri M., Sankar U., Sansanwal P., Santambrogio L., Saran S., Sarkar S., Sarwal M., Sasakawa C., Sasnauskiene A., Sass M., Sato K., Sato M., Schapira A. H., Scharl M., Schatzl H. M., Scheper W., Schiaffino S., Schneider C., Schneider M. E., Schneider-Stock R., Schoenlein P. V., Schorderet D. F., Schuller C., Schwartz G. K., Scorrano L., Sealy L., Seglen P. O., Segura-Aguilar J., Seiliez I., Seleverstov O., Sell C., Seo J. B., Separovic D., Setaluri V., Setoguchi T., Settembre C., Shacka J. J., Shanmugam M., Shapiro I. M., Shaulian E., Shaw R. J., Shelhamer J. H., Shen H. M., Shen W. C., Sheng Z. H., Shi Y., Shibuya K., Shidoji Y., Shieh J. J., Shih C. M., Shimada Y., Shimizu S., Shintani T., Shirihai O. S., Shore G. C., Sibirny A. A., Sidhu S. B., Sikorska B., Silva-Zacarin E. C., Simmons A., Simon A. K., Simon H. U., Simone C., Simonsen A., Sinclair D. A., Singh R., Sinha D., Sinicrope F. A., Sirko A., Siu P. M., Sivridis E., Skop V., Skulachev V. P., Slack R. S., Smaili S. S., Smith D. R., Soengas M. S., Soldati T., Song X., Sood A. K., Soong T. W., Sotgia F., Spector S. A., Spies C. D., Springer W., Srinivasula S. M., Stefanis L., Steffan J. S., Stendel R., Stenmark H., Stephanou A., Stern S. T., Sternberg C., Stork B., Stralfors P., Subauste C. S., Sui X., Sulzer D., Sun J., Sun S. Y., Sun Z. J., Sung J. J., Suzuki K., Suzuki T., Swanson M. S., Swanton C., Sweeney S. T., Sy L. K., Szabadkai G., Tabas I., Taegtmeyer H., Tafani M., Takacs-Vellai K., Takano Y., Takegawa K., Takemura G., Takeshita F., Talbot N. J., Tan K. S., Tanaka K., Tanaka K., Tang D., Tang D., Tanida I., Tannous B. A., Tavernarakis N., Taylor G. S., Taylor G. A., Taylor J. P., Terada L. S., Terman A., Tettamanti G., Thevissen K., Thompson C. B., Thorburn A., Thumm M., Tian F., Tian Y., Tocchini-Valentini G., Tolkovsky A. M., Tomino Y., Tonges L., Tooze S. A., Tournier C., Tower J., Towns R., Trajkovic V., Travassos L. H., Tsai T. F., Tschan M. P., Tsubata T., Tsung A., Turk B., Turner L. S., Tyagi S. C., Uchiyama Y., Ueno T., Umekawa M., Umemiya-Shirafuji R., Unni V. K., Vaccaro M. I., Valente E. M., Van den Berghe G., van der Klei I. J., van Doorn W., van Dyk L. F., van Egmond M., van Grunsven L. A., Vandenabeele P., Vandenberghe W. P., Vanhorebeek I., Vaquero E. C., Velasco G., Vellai T., Vicencio J. M., Vierstra R. D., Vila M., Vindis C., Viola G., Viscomi M. T., Voitsekhovskaja O. V., von Haefen C., Votruba M., Wada K., Wade-Martins R., Walker C. L., Walsh C. M., Walter J., Wan X. B., Wang A., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H., Wang H. G., Wang H. D., Wang J., Wang K., Wang M., Wang R. C., Wang X., Wang X., Wang Y. J., Wang Y., Wang Z., Wang Z. C., Wang Z., Wansink D. G., Ward D. M., Watada H., Waters S. L., Webster P., Wei L., Weihl C. C., Weiss W. A., Welford S. M., Wen L. P., Whitehouse C. A., Whitton J. L., Whitworth A. J., Wileman T., Wiley J. W., Wilkinson S., Willbold D., Williams R. L., Williamson P. R., Wouters B. G., Wu C., Wu D. C., Wu W. K., Wyttenbach A., Xavier R. J., Xi Z., Xia P., Xiao G., Xie Z., Xie Z., Xu D. Z., Xu J., Xu L., Xu X., Yamamoto A., Yamamoto A., Yamashina S., Yamashita M., Yan X., Yanagida M., Yang D. S., Yang E., Yang J. M., Yang S. Y., Yang W., Yang W. Y., Yang Z., Yao M. C., Yao T. P., Yeganeh B., Yen W. L., Yin J. J., Yin X. M., Yoo O. J., Yoon G., Yoon S. Y., Yorimitsu T., Yoshikawa Y., Yoshimori T., Yoshimoto K., You H. J., Youle R. J., Younes A., Yu L., Yu L., Yu S. W., Yu W. H., Yuan Z. M., Yue Z., Yun C. H., Yuzaki M., Zabirnyk O., Silva-Zacarin E., Zacks D., Zacksenhaus E., Zaffaroni N., Zakeri Z., Zeh H. J., 3rd, Zeitlin S. O., Zhang H., Zhang H. L., Zhang J., Zhang J. P., Zhang L., Zhang L., Zhang M. Y., Zhang X. D., Zhao M., Zhao Y. F., Zhao Y., Zhao Z. J., Zheng X., Zhivotovsky B., Zhong Q., Zhou C. Z., Zhu C., Zhu W. G., Zhu X. F., Zhu X., Zhu Y., Zoladek T., Zong W. X., Zorzano A., Zschocke J., Zuckerbraun B. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fehrenbacher N., Jäättelä M. (2005) Lysosomes as targets for cancer therapy. Cancer Res. 65, 2993–2995 [DOI] [PubMed] [Google Scholar]
- 78. Ishiguro K., Ando T., Goto H. (2008) Novel application of 4-nitro-7-(1-piperazinyl)-2,1,3-benzoxadiazole to visualize lysosomes in live cells. BioTechniques 45, 465, 467–468 [DOI] [PubMed] [Google Scholar]
- 79. Ip C. (1998) Lessons from basic research in selenium and cancer prevention. J. Nutr. 128, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 80. Kassam S., Goenaga-Infante H., Maharaj L., Hiley C. T., Juliger S., Joel S. P. (2011) Methylseleninic acid inhibits HDAC activity in diffuse large B-cell lymphoma cell lines. Cancer Chemother. Pharmacol. 68, 815–821 [DOI] [PubMed] [Google Scholar]
- 81. Kice J. L., Lee T. W. S. (1978) Oxidation-reduction reactions of organoselenium compounds: 1: mechanism of the reaction between seleninic acids and thiols. J. Am. Chem. Soc. 100, 5094–5102 [Google Scholar]
- 82. Talbot S., Nelson R., Self W. T. (2008) Arsenic trioxide and auranofin inhibit selenoprotein synthesis: implications for chemotherapy for acute promyelocytic leukaemia. Br. J. Pharmacol. 154, 940–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koch-Weser J., Sellers E. M. (1976) Binding of drugs to serum albumin (first of two parts). N. Engl. J. Med. 294, 311–316 [DOI] [PubMed] [Google Scholar]
- 84. Koch-Weser J., Sellers E. M. (1976) Drug therapy. Binding of drugs to serum albumin (second of two parts). N. Engl. J. Med. 294, 526–531 [DOI] [PubMed] [Google Scholar]
- 85. Levine B., Mizushima N., Virgin H. W. (2011) Autophagy in immunity and inflammation. Nature 469, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kaeck M., Lu J., Strange R., Ip C., Ganther H. E., Thompson H. J. (1997) Differential induction of growth arrest inducible genes by selenium compounds. Biochem. Pharmacol. 53, 921–926 [DOI] [PubMed] [Google Scholar]
- 87. Lu J., Jiang C., Kaeck M., Ganther H., Vadhanavikit S., Ip C., Thompson H. (1995) Dissociation of the genotoxic and growth inhibitory effects of selenium. Biochem. Pharmacol. 50, 213–219 [DOI] [PubMed] [Google Scholar]
- 88. Li K., Mandai M., Hamanishi J., Matsumura N., Suzuki A., Yagi H., Yamaguchi K., Baba T., Fujii S., Konishi I.(2009) Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol. Immunother. 58, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Groh V., Wu J., Yee C., Spies T. (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T cell activation. Nature 419, 734–738 [DOI] [PubMed] [Google Scholar]