

Abstract
Peach flesh color (white or yellow) is among the most popular commercial criteria for peach classification, and has implications for consumer acceptance and fruit nutritional quality. Despite the increasing interest in improving cultivars of both flesh types, little is known about the genetic basis for the carotenoid content diversity in peach. Here we describe the association between genotypes at a locus encoding the carotenoid cleavage dioxygenase 4 (PpCCD4), localized in pseudomolecule 1 of the Prunus persica reference genome sequence, and the flesh color for 37 peach varieties, including two somatic revertants, and three ancestral relatives of peach, providing definitive evidence that this locus is responsible for flesh color phenotype. We show that yellow peach alleles have arisen from various ancestral haplotypes by at least three independent mutational events involving nucleotide substitutions, small insertions and transposable element insertions, and that these mutations, despite being located within the transcribed portion of the gene, also result in marked differences in transcript levels, presumably as a consequence of differential transcript stability involving nonsense-mediated mRNA decay. The PpCCD4 gene provides a unique example of a gene for which humans, in their quest to diversify phenotypic appearance and qualitative characteristics of a fruit, have been able to select and exploit multiple mutations resulting from a variety of mechanisms.
Keywords: Prunus persica L. Batsch, carotenoid cleavage dioxygenase, allelic variants, transposable element, somatic revertants, nonsense-mediated mRNA decay
Introduction
Peach, the third most important temperate tree fruit species, was domesticated in China, from where it was dispersed to Europe, Africa and America (Byrne et al., 2012). Ample phenotypic diversity exists within the cultivated peach germplasm for various characteristics, including color, and consequently many cultivars of peaches are grown successfully in various climatic and geographic regions. Flesh color, one of the most popular commercial criteria for peach classification, has implications for consumer acceptance and nutritional quality (Gil et al., 2002), and improved cultivars of both flesh types are actively sought (Williamson et al., 2006). Certainly, the total carotenoid content is much higher in yellow-fleshed cultivars than in white-fleshed ones, with yellow-fleshed peaches exhibiting higher quantities of β–cryptoxanthin and β–carotene at harvest (Gil et al., 2002; Vizzotto et al., 2007; Brandi et al., 2011).
Carotenoids are a class of ubiquitous pigments involved in plant photoprotection. Moreover, they are important in the pigmentation of flowers and fruits to attract animals for pollination and seed dispersion (Moise et al., 2005), and play an important role in human appeal and health, providing a significant contribution to dietary intake of antioxidants (de la Rosa et al., 2009). Thus, the events that lead to pigment formation and degradation into compounds affecting the color, nutritional value and aroma of fruits and vegetables have important economic implications (Lewinsohn et al., 2005). Moreover, knowledge of the genetics and variability of characters related to fruit quality determines our ability to manipulate them to obtain more attractive and healthier fruits for the consumer (Illa et al., 2011). The genetic basis of variation in fruit color has been widely studied in several species of fleshy fruits, such as tomato (Solanum lycopersicum; Ballester et al., 2010; Kachanovsky et al., 2012), pepper (Capsicum annuum; Brand et al., 2012; Rodriguez-Uribe et al., 2012), orange (Citrus sinensis; Butelli et al., 2012) and grape (Vitis vinifera; Kobayashi et al., 2004; Shimazaki et al., 2011).
Various regulatory mechanisms affecting pigment content have been recognized in plant organs that accumulate carotenoids (Cazzonelli and Pogson, 2010). In many cases, their steady-state levels are determined by the rate of biosynthesis, and various steps have been identified that control the biosynthetic pathway in this manner. In tomato fruit, accumulation of lycopene is highly correlated with the regulation of genes involved in lycopene production (Bramley, 2002). In Arabidopsis and maize (Zea mays), the phytoene synthase enzyme appears to be responsible for the regulation of carotenoid biosynthesis (Li et al., 2008; Rodriguez-Villalon et al., 2009). In addition, a recent study suggested that expression of lycopene ε–cyclase and carotene isomerase is significant in predicting final carotenoid accumulation in mature apple fruit (Malus domestica; Ampomah-Dwamena et al., 2012). On the other hand, studies in strawberry (Fragaria × ananassa), grape and Citrus fruits, as well as Chrysanthemum petals and potato tubers (Solanum tuberosum), have all demonstrated that the pool of carotenoids is determined by the rate of degradation by carotenoid cleavage dioxygenases (Giuliano et al., 2003; Mathieu et al., 2005; Kato et al., 2006; Ohmiya et al., 2006; García-Limones et al., 2008; Huang et al., 2009; Campbell et al., 2010) (Figure 1).
Figure 1.
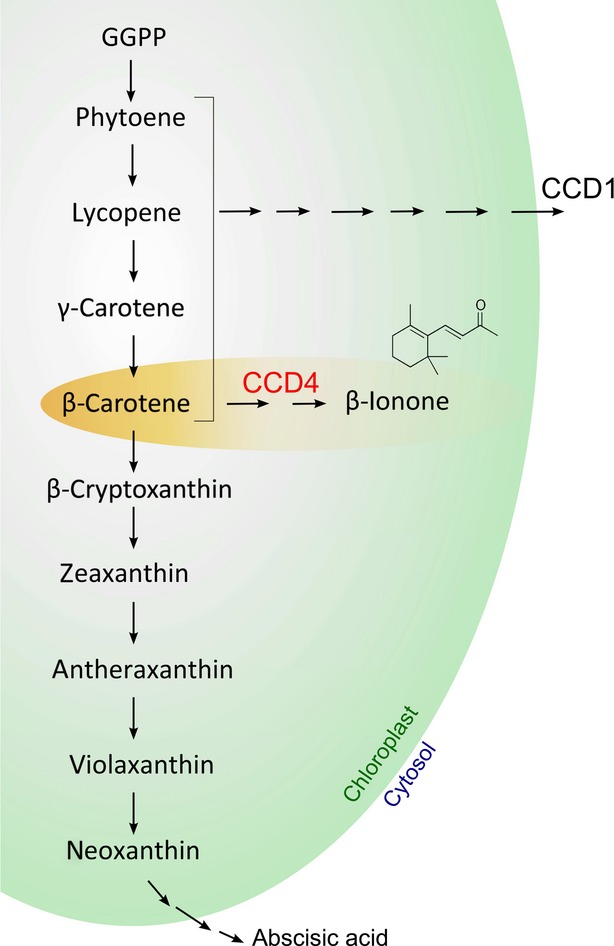
Schematic representation of the carotenoid biosynthetic pathway in plants.
The activities of the carotenoid cleavage dioxygenases 1 and 4 (CCD1 and CCD4) according to Huang et al. (2009) are shown. Although CCD1 and CCD4 enzymes cleave carotenoids at the same positions (9,10 and 9′,10’), CCD4 enzymes are more substrate-specific than CCD1. Moreover, the CCD1 enzymes are located in the cytoplasm, while CCD4 enzymes are located in plastids.
It is well-known that peach flesh color is controlled by a single locus (Y) mapping to linkage group 1 (Bliss et al., 2002), with white flesh dominant over yellow flesh (Connors, 1920; Bailey and French, 1949). Recently, the locus was fine-mapped in a high-density SNP linkage map (Martinez-Garcia et al., 2013), and located in an interval of approximately 500 kb (scaffold_1: 25 584 537–26 004 830) in the Peach v1.0 assembly (Verde et al., 2013). Remarkably, a recent study demonstrated no significant differences in the expression levels of carotenoid biosynthetic genes between white- and yellow-fleshed peach cultivars, and a gene encoding a carotenoid cleavage dioxygenase (CCD) was proposed to be the major factor responsible for carotenoid degradation in white peaches (Brandi et al., 2011). However, the Y gene has not yet been identified, and its function remains to be elucidated. The goal of the present study was to investigate the genetic basis for the carotenoid content diversity in yellow- and white-fleshed peach cultivars. Taking advantage of the availability of the peach genome sequence produced by the International Peach Genome Initiative (Verde et al., 2013) (http://www.rosaceae.org/species/prunus_persica/genome_v1.0, http://www.phytozome.net/peach), we performed in silico and in vivo analysis on several peach varieties that differ with respect to flesh color.
Results
Analysis of a carotenoid cleavage dioxygenase gene (PpCCD4) on linkage group 1
Our research focused on identification of a gene encoding a carotenoid cleavage dioxygenase enzyme, as a good candidate for contributing to color development in flesh tissues (Bliss et al., 2002; Brandi et al., 2011; Martinez-Garcia et al., 2013). Homology-based searches allowed identification of a predicted gene (ppa006109), located in pseudomolecule 1 in the peach genome sequence, encoding a protein showing 86% identity with CCD4 of Malus domestica. We named this gene PpCCD4.
Comparison between the PpCCD4 deduced amino acid sequence and CCD4s from other species indicated that the predicted peach protein was probably incomplete, lacking the N–terminus (Figure 2). Therefore, a manual annotation was performed in the region, enabling identification of a putative start codon 512 bp upstream of the predicted one. Interestingly, the gene sequence in the yellow-fleshed dihaploid ‘Lovell’ PLov2–2N, used for production of the reference sequence, contained a hypervariable (TC)8 microsatellite 47 nucleotides downstream of the start codon, where a frameshift mutation may be responsible for the incomplete protein prediction. The microsatellite within the CCD4 gene sequence (EPPISF25) was previously isolated (Vendramin et al., 2007) in an EST obtained from peach mesocarp of ‘Yumyeong’ (accession number DN677210).
Figure 2.

UPGMA consensus tree and amino acid sequence comparison of CCD4 proteins of various plant species.
Left: amino acid sequence alignment of species closely related to peach. Gray-shaded amino acids represent the N–terminus of the manually predicted protein that is not present in the automated prediction of the peach genome sequence v1.0. The conserved four iron-ligating histidine (H) residues, and the glutamates (E) or aspartates (D) giving stability to the complex, are indicated (blue and red, respectively).
Right: phylogenetic tree showing bootstrap support at critical nodes as percentages. PpCCD4, Prunus persica (ppa006109); MdCCD4, Malus domestica (ABY47995); RdCCD4, Rosa damascena (ABY60886); CcCCD4a, Citrus clementina (ABC26011); AtCCD4, Arabidopsis thaliana (NP_193652); PsCCD4, Pisum sativum (BAC10552); VvCCD4a, Vitis vinifera (XP_002268404); OsCCD4, Osmanthus fragrans (ABY60887); CmCCD4a, Chrysanthemum morifolium (ABY60885); CmCCD4b, Chrysanthemum morifolium (BAF36656); LsCCD4, Lactuca sativa (BAE72094); VvCCD4b, Vitis vinifera (XP_002270161); OsCCD4b, Oryza sativa (ABA97976); HvCCD4, Hordeum vulgare (AK248229); OsCCD4a, Oryza sativa (NP_001047858); CcCCD4b, Citrus clementina (ABC26012); VvCCD4c, Vitis vinifera (XP_002269538); AtCCD1, Arabidopsis thaliana (NP_191911).
After removing two base pairs from the microsatellite region, the PpCCD4 gene sequence consisted of 2003 bp, including two exons of 696 and 1098 bp in length and an intron 209 bp long, resulting in a protein of 597 amino acids. Comparison of PpCCD4 with CCD4s from 11 other species revealed that the peach predicted protein exhibits strong similarity to many other CCD4 proteins, grouping with MdCCD4 and RdCCD4, and showing highest similarity with the apple protein (Figure 2). As for other CCD4 proteins, PpCCD4 contains four highly conserved histidine residues, typical ligands of a non-heme iron co-factor required for (di)oxygenase activity, and conserved glutamates or aspartates that are used to stabilize iron-ligating histidines (Huang et al., 2009) (Figure 2). In addition, PpCCD4 displayed a predicted chloroplast transient peptide in its N–terminal region (Table S1), supporting a plastid localization characteristic of CCD4 enzymes (Rubio et al., 2008).
Association between PpCCD4 genotypes and flesh color in peach varieties
Based on previous observations (Connors, 1920; Bliss et al., 2002; Brandi et al., 2011; Martinez-Garcia et al., 2013), we undertook a detailed analysis of the PpCCD4 locus variation under the assumption that white-fleshed fruits possess at least one copy of a properly functioning dominant allele, and that yellow-fleshed fruits are homozygous for recessive loss-of-function mutations.
We initially analyzed 35 peach genotypes (21 yellow-fleshed and 14 white-fleshed) (Table 1) for sequence variation in the locus using a combination of microsatellite genotyping, cloning or direct Sanger sequencing of PCR products, and whole-genome Illumina resequencing (Verde et al., 2012, 2013), focusing on the above-described hypervariable microsatellite region. Two allelic variants were observed (Table 1), with fragment lengths compatible with the presence of seven and eight dinucleotide repeats, respectively. Remarkably, in silico translation of the eight repeat-containing allele resulted in a truncated protein, due to the presence of an early stop codon (Figure 3a,b), whereas the (TC)7 repeat produced a putatively functional form of the gene. We therefore expected all varieties carrying a (TC)7 allele to be white-fleshed, but exceptions to this prediction were observed in some varieties.
Table 1.
Allelic variants of PpCCD4 as related to phenotype in various white- and yellow-fleshed peach genotypes
| PpCCD4 locus | |||||
|---|---|---|---|---|---|
| Genotype | Phenotype | SSR | SNP | RE | Geographical origin |
| Armking | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| Babygold8 | Yellow | TC7/TC7 | A/A | +/+ | USA (B) |
| Big Top | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| *Bolinha | Yellow | TC7/TC7 | T/T | −/− | Brazil (B) |
| Circe | Yellow | TC8/TC8 | A/A | −/− | Italy (B) |
| Earligold | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| Elberta | Yellow | TC7/TC8 | A/A | +/− | USA (B) |
| *P. ferganensis | Yellow | TC7/TC7 | A/A | +/+ | Fergana Valley (L) |
| Fidelia | White | TC7/TC8 | A/A | −/− | USA (B) |
| Flordastar | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| *GF305 | White | TC7/TC7 | A/A | −/− | France (L) |
| IF7310828 | Yellow | TC8/TC8 | A/A | −/− | Italy (B) |
| Imera | White | TC7/TC7 | A/A | −/− | Italy – Sicily (L) |
| Kamarat | White | TC7/TC7 | A/A | −/− | Italy – Sicily (L) |
| Kurakata Wase | White | TC7/TC8 | A/A | −/− | Japan (B) |
| Leonforte | Yellow | TC7/TC8 | T/A | −/− | Italy – Sicily (L) |
| Leonforte1 | Yellow | TC7/TC7 | T/T | −/− | Italy – Sicily (L) |
| Lovell | Yellow | TC8/TC8 | A/A | −/− | USA (L) |
| Maruja | Yellow | TC7/TC8 | T/A | −/− | Spain (L) |
| Maycrest | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| Michelini | White | TC7/TC8 | A/A | −/− | Italy (L) |
| *Oro A | Yellow | TC7/TC8 | T/A | −/− | Brazil (B) |
| Percoca di Romagna 7 | Yellow | TC8/TC8 | A/A | −/− | Italy (L) |
| Pillar | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| *Quetta | White | TC7/TC8 | A/A | −/− | Pakistan (L) |
| Redhaven | Yellow | TC7/TC8 | A/A | +/− | USA (B) |
| *Shaua Hong Pantao | White | TC7/TC7 | A/A | −/− | South China (L) |
| *Shenzhou Mi Tao | White | TC7/TC7 | A/A | −/− | North China (L) |
| Silver Rome | White | TC7/TC8 | A/A | −/− | Italy (B) |
| Stark Red Gold | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| Stark Saturn | White | TC7/TC8 | A/A | −/− | USA (B) |
| Tabacchiera | White | TC7/TC7 | A/A | +/− | Italy – Sicily (L) |
| Weinberger | Yellow | TC8/TC8 | A/A | −/− | USA (B) |
| Whitecrest | White | TC7/TC8 | A/A | −/− | USA (B) |
| *Yumyeong | White | TC7/TC7 | A/A | −/− | Korea (B) |
Sequence variation in the locus was analyzed by means of a combination of microsatellite genotyping, cloning or direct Sanger sequencing of PCR products, and whole-genome Illumina resequencing for varieties indicated by an asterisk. P. ferganensis, previously reported as a different species, may be considered a P. persica genotype (Verde et al., 2013). B, breeding material; L, landraces; W, wild.
Figure 3.

Schema of the PpCCD4 allele structures and related encoded proteins.
Different colors for the coding regions are reflective of distinct sequence haplotypes. The microsatellite region and the stop codon are indicated in green and red, respectively. The inverted black triangle and the red diamond indicate the positions of conserved glutamates (E)/aspartates (D) and histidines (H) residues on the protein, respectively.
(a) A putatively functional form of the gene with a (TC)7/10 repeat, encoding a complete protein.
(b) Haplotype presenting eight dinucleotide TC repeats that causes a truncated protein.
(c) Ancestral TC7 haplotype prior to occurrence of the nonsense mutation.
(d) Haplotype with (TC)7 microsatellite and an SNP at nucleotide position 1519 causing a premature stop codon, resulting in an incomplete protein lacking a histidine and glutamate residues.
(e) Haplotype with (TC)7 microsatellite and an intronic retroelement (RE) insertion. A schematic diagram of a non-autonomous Copia-like retrotransposon (5282 bp) with putative primer binding site (PBS), polypurine tract (PPT) and long terminal repeats (LTR) is shown.
We further investigated the molecular structure of the PpCCD4 locus, focusing on the varieties that displayed discrepancies between the TC repeat variation and phenotype. Illumina resequencing data available for several varieties (Verde et al., 2012, 2013) identified a SNP occurring at position 1519, with an A→T transversion (T1519A) within the second exon of the PpCCD4 gene, leading to a premature stop codon replacing the one encoding lysine. This mutation was found heterozygously in the Oro A (Brazil), Leonforte (Italy/Sicily) and Maruja (Spain) varieties (Figure 3d) and homozygously in the Leonforte1 (Italy/Sicily) and Bolinha (Brazil) varieties. The homozygous genotypes allowed us to establish that the SNP is in-phase with the functional (TC)7 allele at the microsatellite locus, and that it is found within a rare and diverged haplotype carrying additional unique variants. The heterozygous genotypes demonstrated the presence of two independent loss-of-function mutations at the PpCCD4 locus. Interestingly, resequencing of Imera, a Sicilian white variety, showed that it is homozygous for the same rare haplotype, but does not carry the stop codon, suggesting that it contains the ancestral haplotype prior to occurrence of the nonsense mutation (Figure 3c).
However, the two mutations identified so far did not account for the entirety of the phenotypic variation observed. Four yellow-fleshed varieties (‘Babygold8’, ‘Elberta’, ‘Redhaven’ and Prunus ferganensis), carrying at least one functional (TC)7 allele at the microsatellite locus and no premature stop codon in position 1519, were further analyzed. Unsuccessful attempts to PCR amplify and/or clone the entire (TC)7 allele in these varieties led us to hypothesize that an insertion event may have occurred within the PpCCD4 gene. A strategy combining long-range PCR and next-generation sequencing of the PCR product was adopted, revealing a heterozygous insertion of 6254 bp in the intron of the PpCCD4 gene. The inserted sequence showed similarity to the long terminal repeats (LTRs) of a large family of Copia-like retrotransposons, and included a 5 bp target site duplication (CATAT), typical of LTR retroelement (RE) insertion sites (Kim et al., 1998). A putative primer binding site at position 495, complementary to tRNA-Ala(AGC), and a polypurine tract at position 5744 were also detected, but no coding region was identified in the internal region of the retrotranposon (5282 bp in length), making it a putative non-autonomous element (Figure 3e). The two LTRs of the inserted sequence, 486 bp long with 10 bp canonical inverted terminal repeats, starting with TG and ending with CA, are identical, as expected for a recent insertion (Kijima and Innan, 2010). Non-autonomous LTR retroelements that do not encode the proteins necessary for transposition and are mobilized in trans by proteins provided by functional (autonomous) elements have been described in many eukaryotic genomes (Havecker et al., 2004). In the peach reference genome sequence (Verde et al., 2013), we identified several instances of autonomous elements sharing LTR sequences with the inserted sequence in PpCCD4, and estimate the copy number of intact elements of this family to be 57 in the reference sequence.
A PCR assay, using specific primers to detect the insertion, identified the presence of a heterozygous or homozygous insertion in all varieties where the flesh color phenotype was not accounted for by the other two mutations. Sequencing of a PCR-amplified DNA fragment including the microsatellite region and part of the inserted retroelement confirmed the presence of insertions in the haplotype containing the (TC)7 allele at the microsatellite in ‘Elberta’, ‘Redhaven’ and ‘Tabacchiera’. Interestingly, ‘Babygold8’ and P. ferganensis were homozygous for the presence of the retrotransposon, in agreement with their homozygous (TC)7/(TC)7 condition in the microsatellite region and their yellow flesh color, supporting the hypothesis that the retroelement is responsible for PpCCD4 locus inactivation and flesh color determination.
Somatic revertants provide evidence that PpCCD4 is causative for the flesh color phenotype
Evidence that the white allele represents the ancestral condition derives from the observation that resequencing of cherry (Prunus avium), apricot (Prunus armeniaca) and almond (Prunus dulcis) (data not shown) and three ancestral relatives of peach (Prunus mira, Prunus davidiana and Prunus kansuensis) showed the presence of putatively functional haplotypes carrying a (TC)7 allele and no premature stop codon in all cases (Table 2). In order to conclusively demonstrate the causal relationship between variation in the gene and the phenotype, we focused on two varieties (‘Silver King’ derived from ‘Armking’ and ‘Redhaven Bianca’ derived from ‘Redhaven’) that represent natural sport mutations causing reversion of the flesh color phenotype. Simple Sequence Repeat (SSR) analysis with 16 primer pairs (data not shown) using long-living accessions in two Italian locations (Rome and Udine) confirmed the isogenicity between the revertants and the ancestral varieties. In both cases, the presence of yellow pigmentation of the suture in the white variety fruit indicates that they are chimeric mutants, with the mutation having occurred in the L–II apical cell layer, and not in the L–I layer that produces the epidermis and the cells in the suture. In ‘Redhaven Bianca’, the L–II origin of the mutation is also demonstrated by meiotic transmission of the white phenotype to its progeny (Brandi et al., 2011). ‘Armking’ and ‘Redhaven’ have different genotypes at the PpCCD4 locus, with ‘Armking’ being homozygous for the frameshift mutation in the microsatellite region [(TC)8/(TC)8] and ‘Redhaven’ being heterozygous, with one haplotype carrying the microsatellite frameshift mutation (TC)8 and the other carrying the retroelement intronic insertion. Sequence analysis of PpCCD4 in ‘Silver King’ revealed the presence of a (TC)10 allele in addition to the (TC)8 present in ‘Armking’ (Figure 3a): addition of two repeat units in the microsatellite region restores the correct reading frame present in the (TC)7 allele by adding two amino acids to the predicted PpCCD4 protein (Figure S1), and substantiates the causal relationship between PpCCD4 and flesh color in peach. The analysis of PpCCD4 in ‘Redhaven Bianca’ revealed no variation in the microsatellite region, but absence of the intronic retrotransposon insertion. This was confirmed by both a PCR assay for presence/absence of the insertion as well as sequencing of the haplotype containing the (TC)7 microsatellite allele that carries the retrolement insertion in ‘Redhaven’. Independent analysis of DNA from the fruit flesh (L–II alone) and the leaf (L–I and L–II) confirmed the periclinal chimeric nature of the ‘Redhaven’ somatic mutant, as the fruit flesh is homozygous for the absence of the insertion in the (TC)7 haplotype, while the leaf tissue is heterozygous (Table 3). Surprisingly, we were unable to detect any evidence of the previous presence of the retroelement in the (TC)7 haplotype in ‘Redhaven Bianca’.
Table 2.
Allelic variants of PpCCD4 as related to phenotype in three ancestral relatives of peach
| PpCCD4 locus | |||||
|---|---|---|---|---|---|
| Species | Phenotype | SSR | SNP | RE | Geographical origin |
| P. davidiana | White | TC7/TC7 | A/A | −/− | China (W) |
| P. mira | White | TC7/TC7 | A/A | −/− | China (W) |
| P. kansuensis | White | TC7/TC7 | A/A | −/− | China (W) |
See Table 1 for description of experiments.
Table 3.
Allelic variants of PpCCD4 as related to phenotype in two revertant genotypes
| PpCCD4 locus | |||||
|---|---|---|---|---|---|
| Revertant genotype | Phenotype | SSR | SNP | RE | Geographical origin |
| Redhaven Bianca | White | TC7/TC8 | A/A | −/− | USA |
| Silver King | White | TC10/TC8 | A/A | −/− | USA |
See Table 1 for description of experiments.
Transcriptional analysis of PpCCD4 haplotypes/alleles
None of the three mutations identified as being causative for the yellow flesh phenotype appear to have a direct effect on transcriptional regulation of the gene. Haplotype analysis in multiple resequenced varieties indicated that the only additional SNP variants in the gene and in the 5′ and 3′ flanking regions are observed in the haplotype carrying the SNP causing the nonsense mutation. However, Brandi et al., 2011) observed dramatic differences in PpCCD4 transcript levels when comparing ‘Redhaven’ and ‘Redhaven Bianca’.
Quantitative real-time PCR was used to estimate the transcript levels of PpCCD4 in the flesh of several peach varieties differing for flesh pigmentation. These results showed that, in general, considerably lower steady-state levels of PpCCD4 transcripts were observed in yellow-fleshed fruits than in white ones (Figure 4). In addition, we performed quantitative assays to estimate allele-specific transcript abundance by both PCR amplification from cDNA (Salvi et al., 2007) and from RNA-Seq data. The allele-specific analysis of transcript levels in heterozygous individuals minimizes environmental as well as trans-acting effects on transcript abundance. We used the TC microsatellite as the polymorphism to distinguish transcripts derived from the various haplotypes carrying the functionally relevant mutations.
Figure 4.
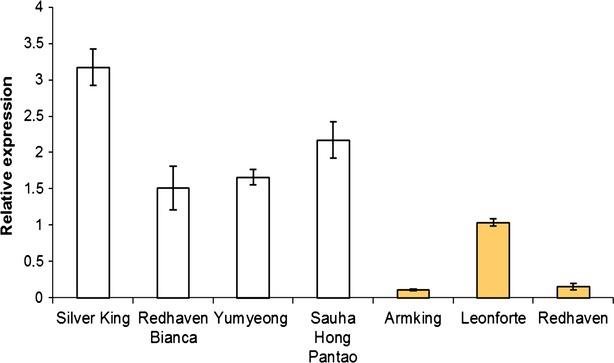
Relative expression levels of the PpCCD4 gene in various white- and yellow-fleshed genotypes.
Expression was determined by quantitative real-time PCR. Values are means ± standard deviations of three replicates.
Genomic DNA and cDNA from fruit of varieties representative of all the functional mutations identified were used as template for microsatellite analysis. Peak height was used to estimate the relative amount of DNA and mRNA (as cDNA) for the two alleles (Table 4). When the yellow (TC)8 haplotype was compared against two white [(TC)7 and (TC)10] and two yellow haplotypes [(TC)7 + RE insertion and (TC)7 + T/A nonsense SNP], the relative transcript levels for the yellow haplotypes were considerably lower than those for the white ones (from 7–13-fold), and similar to one another (Table 5). The somatic mutations resulting in a frameshift from (TC)8 to (TC)10 (‘Armking’ → ‘Silver King’) and absence of the RE insertion (‘Redhaven’ → ‘Redhaven Bianca’) corresponded to major and very similar increases in transcript levels (13.5- and 13.4-fold, respectively). Taken together, these data convincingly indicate that the various mutations causing the yellow flesh phenotype result in large differences in steady-state levels of the transcript, even though they do not appear to affect transcriptional regulation of the gene. Additional support for this hypothesis comes from the observation that whole-genome resequencing analysis of a number of genotypes (Verde et al., 2013; M. Morgante, unpublished results) reveals no additional variant between the (TC)8 and (TC)7 haplotypes (not carrying the nonsense SNP), either in the UTRs or in the promoter regions, when searching for both SNPs as well as small and large insertion/deletion polymorphisms. RNA-Seq data from ‘Silver King’ and ‘Armking’, in addition to confirming these results, allowed us to observe that a much higher proportion of spliced versus unspliced transcripts are present in ‘Silver King’ than ‘Armking’ (15.3 versus 1.5 ratio). Assuming that the unspliced transcripts correspond largely to contaminating pre-mRNA, this evidence suggests that the mutation plays a role in a mechanism affecting transcript stability through degradation rather than transcriptional regulation. Additionally, an analysis of the yellow ‘Redhaven’ transcriptome was performed in order to determine whether mature functional mRNA deriving from the retrotransposon-mutated allele may be detected. Microsatellite analysis performed on total cDNA, obtained from yellow ‘Redhaven’ flesh, revealed the presence of two peaks (Figure S2a) corresponding to the (TC)7 and (TC)8 alleles. This led us to further investigate the structure of the molecules containing the (TC)7 repeat. To this end, both a traditional and a long-range PCR approach were adopted, using cDNA as the template and specific primers for start and stop codon regions. No fragments of aberrant length, but only products of approximately 1800 bp that are compatible with normal PpCCD4 transcription, were detected. Therefore, a microsatellite amplification was performed on these products, and the presence of a single peak, corresponding to the (TC)8 allele, was observed (Figure S2b). These results support the absence of mature and functional transcripts of the (TC)7 allele in the yellow ‘Redhaven’ genotype, even though a small percentage of PpCCD4 mRNA presumably lacking the 5′ and/or 3′ terminal regions may contain the (TC)7 repeat.
Table 4.
Peak heights obtained by detection of (TC)n repeats during microsatellite analysis of genomic DNA and cDNA from varieties representative of all functionally relevant mutations identified
| Sample | Sequence haplotype | Peak height | Peak height ratio |
|---|---|---|---|
| Redhaven DNA | TC7 + RE | 9699 | 1.17 |
| TC8 | 8258 | ||
| Redhaven Bianca DNA | TC7 | 10 005 | 1.16 |
| TC8 | 8596 | ||
| Redhaven cDNA | TC7 + RE | 10 693 | 0.63 |
| TC8 | 16 980 | ||
| Redhaven Bianca cDNA | TC7 | 23 265 | 8.36 |
| TC8 | 2784 | ||
| Silver King DNA | TC10 | 6813 | 0.62 |
| TC8 | 10 902 | ||
| Silver King cDNA | TC10 | 21 712 | 8.44 |
| TC8 | 2574 | ||
| Leonforte DNA | TC7 + SNP | 17 039 | 1.20 |
| TC8 | 14 238 | ||
| Leonforte cDNA | TC7 + SNP | 12 613 | 1.28 |
| TC8 | 9833 |
The peak height ratio column shows the relative amount of the two alleles in all heterozygous genotypes.
Table 5.
Relative allelic expression levels for heterozygous varieties representative of all functionally relevant mutations. The ratio of the two alleles in the transcript pool of each heterozygous variety was derived by normalizing cDNA ratios on the basis of the peak height ratios obtained from genomic DNA indicating a perfect proportion (1:1) of the two alleles
| Variety | Haplotypes | Locus alleles | Normalized relative allelic expression levels |
|---|---|---|---|
| Redhaven | TC7+RE/TC8 | Yellow/yellow | 0.536 |
| Redhaven Bianca | TC7/TC8 | White/yellow | 7.180 |
| Silver King | TC10/TC8 | White/yellow | 13.498 |
| Leonforte | TC7+SNP/TC8 | Yellow/yellow | 1.072 |
Discussion
The flesh color of peach fruit has important implications for nutritional quality, particularly in terms of carotenoid levels (Gil et al., 2002; Cazzonelli and Pogson, 2010), and the genetic control of this trait by a single locus (Y) has been known for a long time (Connors, 1920; Bailey and French, 1949).
Carotenoid accumulation in various tissues and organs is the final result of biosynthesis, degradation and stable storage of synthesized products (Cazzonelli and Pogson, 2010). In many cases, the transcriptional regulation of carotenogenic gene expression has been shown to be essential in controlling specific carotenoid accumulation (Fray and Grierson, 1993; Harjes et al., 2008; Blas et al., 2010; Kachanovsky et al., 2012). However, other studies have demonstrated that the pool of carotenoids is at least partly determined by the rate of degradation by CCDs, which appear to have various substrate preferences (Auldridge et al., 2006; Campbell et al. 2008, Kato et al., 2006; Mathieu et al., 2005; Ohmiya et al., 2006).
The goal of identification of the gene controlling peach fruit flesh color has been pursued for a long time by various research groups. As reported in Williamson et al. (2006), the presence of a dominant allele results in lack of orange pigmentation, suggesting that the Y gene controls a step in carotenoid biochemical pathways, or degradation of one or more specific carotenoids. Indeed, the CCD4 gene has recently been proposed to be responsible for carotenoid degradation in a white peach variety (Brandi et al., 2011), and the results of our study are consistent with this hypothesis. We identified a transcript (ppa006109) encoding a CCD protein highly similar to isoform 4 of other species on chromosome 1 of the peach genome (http://www.rosaceae.org/species/prunus_persica/genome_v1.0). The availability of several peach accessions enabled us to substantiate the hypothesis that this candidate gene (named PpCCD4) is solely responsible for controlling fruit flesh color.
Examination of 37 peach genotypes and three ancestral relatives of peach (P. mira, P. davidiana and P. kansuensis) allowed identification of four variants at the locus: (i) the dominant functional allele, encoding a 597 amino acid polypeptide present in the homozygous or heterozygous state in white-fleshed fruit, (ii) a crucial frameshift mutation causing a premature stop codon in a hypervariable (TC)n microsatellite region located 47 nucleotides downstream of the start codon, (iii) an SNP occurring at position 1519 with a T/A transversion leading to a premature stop codon embedded within a diverged haplotype, and (iv) an intronic LTR retrotransposon insertion affecting PpCCD4 transcript stability. Remarkably, two putatively inactive forms of the gene are always recognized in yellow-fleshed genotypes in various arrangements.
The haplotypes of yellow peaches appear to have arisen from various white ones by independent mutation events, and the analysis of ancestral relatives of peach, with white flesh and functional haplotypes, lends support to this hypothesis. All three main mutational mechanisms generating diversity in plants, i.e. point mutations, replication slippage and transposable element movement, appear to have contributed to the diversification of flesh color in peach.
We have identified a retrotransposon insertion that accounts for a proportion of the variation in flesh color, similar to what has been identified in orange and grape (Kobayashi et al., 2004; Butelli et al., 2012). In this study, an enzyme directly involved in pigment degradation is involved in the mutational event, rather than a transcription factor.
The correlation between phenotypic variation and molecular analyses provides evidence for a causal relationship between the PpCCD4 mutation and flesh color in peach. Somatic revertants offer a conclusive confirmation of this model. As was somewhat to be expected on the basis of the known mutation rates, revertants were observed only for two of the three yellow haplotypes, i.e. those involving replication slippage and transposable element insertion, and not for the one involving a nucleotide substitution. ‘Silver King’ and ‘Redhaven Bianca’ are white-fleshed peaches arising as bud sport mutants of the original yellow varieties ‘Armking’ and ‘Redhaven’, respectively. In both cases, the presence of yellow pigmentation at the suture in the white variety fruit indicates that they are chimeric mutants, with the mutation having occurred in the L–II apical cell layer and not in the L–I layer producing the epidermis and the cells in the suture. However, whereas the change in the number of TC repeats in a microsatellite region originating in ‘Silver King’ was fully expected, the mechanism by which ‘Redhaven Bianca’ originates appears to be more complex. The yellow ancestral genotype represents the first example in peach of a natural retroelement-mediated gene inactivation as the origin of variation, and it is thought to be isogenic with ‘Redhaven Bianca’ except for the mutation causing the change in flesh color. The presence of the retroelement insertion in the leaf DNA (derived from all histogenic layers) and its absence in fruit flesh DNA in ‘Redhaven Bianca’ supports the hypothesis that a mutation involving only L–II has occurred in ‘Redhaven’ and is causative for white flesh.
The vast majority of LTR retrotransposons do not excise and may cause reversion events through unequal homologous recombination between the two LTRs, leaving a solo LTR that is often no longer sufficient to cause gene inactivation [see Kobayashi et al. (2004) for an example related to fruit color]. These insertions are irreversible, rarely undergoing precise excision (Huang et al., 2008), but the occurrence of precise excision of a Drosophila retrotransposon (Kuzin et al., 1994) suggests that a similar process is conceivable, even if it is almost impossible to provide direct proof of this event. An alternative hypothesis is that rare cells present in ‘Redhaven’, representing the ancestral status prior to the retroelement insertion, may have substituted, via displacement, the histogen involved in fruit and gamete development. Chimerism in peach was first studied as variability in the level of ploidy (cytochimeras), and histogenetic factors determining peach sports have been known for decades. The difference in the pattern of tissues developed from the L–II and L–III layers in chimeric fruits is due to a variable rate of mitotic activity in different portions of the tissues derived from the two layers (Dermen, 1956; Yeager and Meader, 1956). However, invasion of a cell layer by mutated underlying cells has been well documented in grape (Walker et al., 2006). In this respect, the L–III layer may represent a ‘reserve’ of ancestral cells, but further studies on cell layer-specific genotyping are required to provide new information concerning this hypothesis.
Taken together, our data demonstrate that yellow pigmentation results in two of three haplotypes from loss of function of the PpCCD4-encoded protein and in all three cases from mutations that occur within the transcribed region of the gene However, interestingly, the quantitative real time–PCR analysis revealed that the mutations determining yellow flesh also result in significantly lower levels of PpCCD4 transcripts. As flesh color appears to be controlled by a single gene, the reason for this differential expression must be found at the locus itself. Support for this hypothesis is also provided by the observation that phenotypic reversion from yellow to white resulting from mutations that restore the correct reading frame is accompanied by restoration of the steady-state transcript levels observed in the ancestral white haplotype. The marked imbalance in the allelic expression of heterozygous white genotypes indicates that the various mutations causing yellow flesh result in large differences in steady-state levels of the transcript, probably affecting its stability, as also supported by RNASeq data obtained from Armking and Silver King. This may be a result of nonsense-mediated mRNA decay, which detects premature stop codons to target the transcripts for degradation (van Hoof and Green, 2006). It is likely that alleles affected by both the microsatellite mutation and the SNP, occurring in exons 1 and 2, respectively, may be subject to nonsense-mediated mRNA decay (Figure S3), as this mechanism in plants may be independent of the exon–exon junction position (van Hoof and Green, 2006). On the other hand, the absence of mature and functional transcripts of the allele containing the retroelement has also been established in this work, and may originate from improper intron splicing determined by the presence of a transposable element. It is therefore evident that, in addition to providing examples of a variety of mutational mechanisms that result in phenotypic diversity, the PpCCD4 gene also provides an example of a gene for which a variety of mechanisms result in wide differences in transcript levels without involving control of transcription.
Unlike other traits in both plants and animals [e.g. white berries in wine grape varieties (Kobayashi et al., 2004), yellow endosperm in maize (Palaisa et al., 2003), alternate gaits in horses (Andersson et al., 2012), and short legs in dogs (Parker et al., 2009)], where a single mutation on a single background haplotype has been selected by humans in their attempt to improve plant or animal breeds, the PpCCD4 gene provides a unique example of a gene for which humans have been able to exploit a variety of mutations resulting from a variety of mechanisms. Association mapping relying on linkage disequilibrium of such a gene would be particularly challenging as a consequence of two independent factors: allelic heterogeneity on one hand, and the fact that two of the mutations causing the yellow phenotype occurred very recently in the same haplotype background as the ancestral white variant. The analysis of somatic mutants showing revertant phenotypes proved extremely valuable in determining the causal relationship between the PpCCD4 gene and yellow flesh. Somatic mutants are widely available in vegetatively propagated fruit trees, and represent a very important resource for future attempts to link genotype to phenotype.
Experimental procedures
Plant material
Twenty-one genotypes of yellow-fleshed peach (‘Armking’, ‘Babygold8’, ‘BigTop’, ‘Bolinha’, ‘Circe’, ‘Earligold’, ‘Elberta’, P. ferganensis, ‘Flordastar’, IF7310828, ‘Leonforte, ‘Leonforte1’, ‘Maruja’, ‘Maycrest’, ‘Oro A’, ‘Percoca di Romagna 7’, ‘Pillar’, PLov2–2N, ‘Redhaven’, ‘Stark Red Gold’ and ‘Weinberger’) and 16 accessions of white-fleshed genotypes (‘Fidelia’, ‘GF305’, ‘Imera’, ‘Kamarat’, ‘Kurakata Wase’, ‘Michelini’, ‘Quetta’, ‘Redhaven Bianca’, ‘Shaua Hong Pantao’, ‘Shenzhou Mitao’, ‘Silver Rome’, ‘Silver King’, ‘Stark Saturn’, ‘Tabacchiera’, ‘Whitecrest’ and ‘Yumyeong’), plus three wild peach relatives (P. mira, P. davidiana and P. kansuensis) were analyzed in this study. ‘Redhaven Bianca’ and ‘Silver King’ are two sport mutation of the cultivars ‘Redhaven’ and ‘Armking’, respectively. ‘Bolinha’, P. mira and P. davidiana were grown at the Institut National de la Recherche Agronomique in Avignon (France) and ‘Leonforte’ was grown at the University of Palermo, Italy. ‘Redhaven’ peach trees (yellow and white) were grown at the experimental farm of Udine University (north-eastern Italy). All other varieties were grown at the Consiglio per la Ricerca e la Sperimentazione in Agricoltura – Centro di Ricerca per la Frutticoltura experimental farm (Rome, Italy). From all accessions, young leaves or fruit were collected and stored at −80°C for DNA or RNA extraction.
DNA and RNA extraction
Peach leaf tissues were ground in liquid nitrogen and DNA was extracted as described by Zhang et al. (1995) with minor modifications.
Total RNA was obtained from the mesocarp of peach fruit at harvest as described by Falchi et al. (2010). The final RNA pellet was resuspended in RNase-free water and checked for integrity on a 1% agarose gel. RNA samples were stored at −80°C.
cDNA synthesis and quantitative real-time PCR analysis
A 10 μg aliquot of total RNA was treated with DNase (Promega, www.promega.com) to remove contamination by genomic DNA. The reaction mix was incubated at 37°C for 30 min, and the RNA was purified and concentrated using an RNeasy MinElute clean-up kit (Qiagen, www.qiagen.com), according to the manufacturer’s instructions. An aliquot of RNA was quantified using a NanoDrop 1000 spectrophotometer (www.nanodrop.com), and electrophoretically separated on a 1% agarose gel to check integrity. Reverse transcription on purified RNA was performed using the Superscript VILO cDNA synthesis kit (Invitrogen, www.invitrogen.com) according to the manufacturer’s instructions.
Specific primers, amplifying a 111 bp region, were designed on the ppa006109 transcript (forward 5′-GGTTTGATGTGCCTGGTTTT-3′; reverse 5′-AGCAGAGCACACAATGGAGA-3′), and used to perform quantitative RT–PCR reactions with SYBR® Green PCR Master Mix (5PRIME, www.5prime.com) in an MJ Opticon 2 system (BIO-RAD, www.bio-rad.com). All experiments were performed in triplicate under the same conditions: first step at 50°C for 2 min, denaturation step at 95°C for 3 min, followed by 41 cycles of 94°C for 15 sec, 56°C for 20 sec and 72°C for 30 sec.
All quantifications were normalized to ubiquitin-conjugating enzyme (accession number BF717254) amplified under the same conditions with primers 5′-CCCACCTGATTACCCTTTCA-3′ (forward) and 5′-GATCTGTCAGCAGTGAGCA-3′ (reverse). Differences in PpCCD4 gene expression among fruits of different varieties were calculated according to the ΔΔCt method (Pfaffl, 2001).
Identification of the candidate gene controlling peach fruit flesh color
The coding sequence from apple MdCCD4 (ABY47995) was used as query sequence to perform a tblastx search of the peach genome (http://services.appliedgenomics.org/blast/public/). The best match (E–value 0.0, identity 86%) corresponded to the second exon (scaffold_1: 25 640 331–25 641 440) of a predicted transcript (ppa006109) in the peach genome (http://services.appliedgenomics.org/fgb2/iga/prunus_public/gbrowse/prunus_public/). The localization of the ppa006109 transcript in scaffold_1 made this gene a good candidate for flesh color determination.
CCD4 sequence retrieval and alignment, and phylogenetic tree construction
Protein sequences of CCD4 in various species were identified by searching public databases available at NCBI (http://www.ncbi.nlm.nih.gov), and multiple alignments of amino acid sequences were produced using the web-based version of ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The multiple sequence alignment obtained was used to create a bootstrap consensus tree inferred from 1000 replicates. The tree topology was generated by the neighbor-joining method of MEGA version 4 software (Tamura et al., 2007), with bootstrap support at critical nodes indicated as percentages.
Detection of microsatellite variation and allele-specific expression assay
Genomic DNA samples from various accessions, extracted as previously described, were used as PCR templates for microsatellite (TC)n region amplification. The reaction included specific primers (5′-GCAGTGAAGGGCAATACCAG-3′ and 5′-TGTGGAGGTGGGTTTTGAAG-3′), and the forward oligonucleotide was labeled with a fluorescent dye (6-fluorescein amidite, 6–FAM). PCR reactions were performed using HotMaster Taq DNA polymerase (5PRIME) according to the manufacturer’s instruction. PCR products were diluted 1:100, and 2 μl were added to 0.2 μl of a GS500 LIZ size standard and 78 μl of Hi-Di formamide (Applied Biosystems, www.appliedbiosystems.com) and separated by capillary electrophoresis using an ABIPrism 3730xl DNA analyzer (Applied Biosystems). Alleles were called and sized using GeneMapper software (Applied Biosystems).
The polymorphisms determined by the length of the microsatellite were utilized for a quantitative allele-specific expression assay. In detail, the amount of each of the two different-sized alleles represented in the PCR product was estimated by measuring the corresponding peak height. The same analysis was performed in both genomic DNA (as a control) and cDNA. As expected, the allele ratios in genomic DNA did not substantially deviate from unity in all genotypes analyzed, and they were used for cDNA peak ratio normalization. Normalized allelic cDNA ratios deviating from unity indicate differential expression of the two alleles.
Cloning and sequencing of PpCCD4
A targeted sequencing approach was adopted in order to examine the polymorphism of PpCCD4 alleles from non-resequenced varieties. To this purpose, PCR reactions were performed using HotMaster Taq DNA polymerase (5PRIME) according to the manufacturer’s instructions with specific primers. The primers 5′-GGGTGATCCAATGCCTAAGA-3′ (forward) and 5′-GGCTCTCTAGCCACGAAAAA-3′ (reverse) were used for SNP detection. The primers 5′-AGAATGTGGTCCCCTCCTCT-3′ (forward) and 5′-TGGTCAGATTTGCACTCACC-3′ (reverse), designed on UTR regions, were used for amplification of the complete gene. PCR products were purified using Agencourt magnetic beads (Beckman Coulter, www.beckmancoulter.com), and subjected to direct Sanger sequencing on an ABIPrism 3730xl DNA analyzer (Applied Biosystems) using Big Dye Terminator chemistry. When an accurate distinction between two alleles in heterozygous genotypes was necessary, a cloning step was included before sequencing the PCR products. Cloning was performed using a TOPO TA cloning kit with Top 10 F′ cells (Invitrogen), and samples were purified using the Wizard Plus Minipreps kit (Promega).
Isolation and de novo assembly of LTR retrotransposons from the Redhaven genotype
A long-range PCR reaction was optimized in order to test the hypothesis of a large insertion at the PpCCD4 locus in the yellow-fleshed Redhaven genotype. In detail, two high-Tm primers flanking the gene were designed (forward 5′-TCCCATTTTGCAGTGAAGGGCAAT-3′; reverse 5′- CGGGGCAGCCTCACATCTGC-3′) and used to perform the PCR reaction with AccuTaq LA DNA polymerase (Sigma-Aldrich, www.sigmaaldrich.com) according to the manufacturer’s instructions. The following protocol was used: 30 sec at 98°C, 25 cycles of 15 sec at 94°C, 20 sec at 65°C and 23 min at 68°C, and a 10 min final elongation time. As expected, the PCR reaction generated a double product (corresponding to the two alleles with and without the insertion). The longest product (approximately 8000 bp) was extracted and purified from the agarose gel using an E.Z.N.A.® gel extraction kit (Omega Bio–tek, www.omegabiotek.com), suitable for large PCR product purification. An additional step of precipitation with ethanol and sodium acetate was added in order to avoid possible interference of kit reagents with subsequent downstream applications. The result of purification was checked by gel electrophoresis, and the product was quantified using a Qubit™ fluorometer (Invitrogen). Finally, a library was prepared for Illumina sequencing using Illumina Nextera library preparation kits (Epicentre Biotechnologies, www.epibio.com), and sequenced using an Illumina MiSeq reagent kit v1 (300 cycles), producing 196 186 paired end reads of 150 bp long for a total of 58.9 Mb, corresponding to more than 7000 x coverage of the region. Reads were trimmed for low-quality regions using rNA (Vezzi et al., 2012) and assembled de novo using CLC Genomics Workbench 5.1 (CLC Bio, www.clcbio.com) with default parameters except k-mer size, which was set to 51.
PCR screening for LTR retrotransposon insertion
PCR-based screening was performed in order to determine the absence/presence of the LTR retrotransposon in various genotypes, and its homozygous/heterozygous status in the genome. Four primers were used (Table S2): forward primer RE1 and reverse primer RE4, which are specific to the gene of interest, and primers RE2 and RE3, which are complementary to the LTR sequences. For each genotype, three PCR reactions were performed: one using primers RE1 and RE2, a second with primers RE3 and RE4, and a third using primers RE1 and RE4. Products of the PCR reactions were detected by agarose gel electrophoresis. The third reaction was expected to be successful only in absence of the retrotransposon insertion; both reactions involving primers based on LTR regions were associated with retrotransposon presence. The occurrence of all three products in the PCR indicated heterozygous retrotransposon insertion.
Acknowledgments
This work was financially supported by the Italian Ministry of Agricultural, Food and Forestry Politics (MiPAAF, http://www.politicheagricole.it), Projects ‘Drupomics’ (grant number DM14999/7303/08), ‘Agronanotech’ (grant number DM686/7303/08) and the European Research Council under the European Union’s Seventh Framework Program (FP/2007–2013)/ERC grant agreement number 294780 (http://erc.europa.eu). We are grateful to I. Jurman (Istituto di Genomica Applicata, via J.Linussio 51, 33100 Udine, Italy) for technical assistance in the sequencing of PCR products. We acknowledge T. Pascal and B. Quilot (Génétique et Amélioration des Fruits et Légumes, Institut National de la Recherche Agronomique, Avignon, France) for kindly providing leaf material and phenotypic information on some accessions, and T. Caruso (Dipartimento Scienze Agrarie e Forestali, University of Palermo, Italy) for the Leonforte variety. We are grateful to Amy Iezzoni (Michigan State University, Department of Horticulture, East Lansing. MI) for critical reading of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Comparison between nucleotide and deduced amino acid sequences of the (TC)8 and (TC)10 repeat alleles in ‘Armking’ and ‘Silver King’ revertants.
Microsatellite analysis performed on cDNA from Redhaven fruit flesh.
Premature stop codons.
Prediction of subcellular localization of the PpCCD4 protein.
Primers used for PCR screening of LTR retrotransposon insertion.
References
- Ampomah-Dwamena C, Dejnoprat S, Lewis D, Sutherland P, Volz RK, Allan AC. Metabolic and gene expression analysis of apple (Malus × domestica) carotenogenesis. J. Exp. Bot. 2012;63:4497–4511. doi: 10.1093/jxb/ers134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson LS, Larhammar M, Memic F, et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature. 2012;488:642–646. doi: 10.1038/nature11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auldridge ME, Block A, Vogel JT, Dabney-Smith C, Mila I, Bouzayen M, Magallanes-Lundback M, DellaPenna D, McCarty DR, Klee HJ. Characterization of three members of the Arabidopsis carotenoid cleavage dioxygenase family demonstrates the divergent roles of this multifunctional enzyme family. Plant J. 2006;45:982–993. doi: 10.1111/j.1365-313X.2006.02666.x. [DOI] [PubMed] [Google Scholar]
- Bailey JS, French AP. The Inheritance of Certain Fruit and Foliage Characters in the Peach. Amherst, MA: University of Massachusetts Press; 1949. [Google Scholar]
- Ballester AR, Molthoff J, de Vos R, et al. Biochemical and molecular analysis of pink tomatoes: deregulated expression of the gene encoding transcription factor SlMYB12 leads to pink tomato fruit color. Plant Physiol. 2010;152:71–84. doi: 10.1104/pp.109.147322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blas AL, Ming R, Liu Z, Veatch OJ, Paull RE, Moore PH, Yu Q. Cloning of the papaya chromoplast-specific lycopene β–cyclase, CpCYC–b, controlling fruit flesh color reveals conserved microsynteny and a recombination hot spot. Plant Physiol. 2010;152:2013–2022. doi: 10.1104/pp.109.152298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss FA, Arulsekar S, Foolad MR, Becerra V, Gillen AM, Warburton ML, Dandekar AM, Kocsisne GM, Mydin KK. An expanded genetic linkage map of Prunus based on an interspecific cross between almond and peach. Genome. 2002;45:520–529. doi: 10.1139/g02-011. [DOI] [PubMed] [Google Scholar]
- Bramley PM. Regulation of carotenoid formation during tomato fruit ripening and development. J. Exp. Bot. 2002;53:2107–2113. doi: 10.1093/jxb/erf059. [DOI] [PubMed] [Google Scholar]
- Brand A, Borovsky Y, Meir S, Rogachev I, Aharoni A, Paran I. pc8.1, a major QTL for pigment content in pepper fruit, is associated with variation in plastid compartment size. Planta. 2012;235:579–588. doi: 10.1007/s00425-011-1530-9. [DOI] [PubMed] [Google Scholar]
- Brandi F, Bar E, Mourgues F, Horváth G, Turcsi E, Giuliano G, Liverani A, Tartarini S, Lewinsohn E, Rosati C. Study of ‘Redhaven’ peach and its white-fleshed mutant suggests a key role of CCD4 carotenoid dioxygenase in carotenoid and norisoprenoid volatile metabolism. BMC Plant Biol. 2011;11:24. doi: 10.1186/1471-2229-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli E, Licciardello C, Zhang Y, Liu J, Mackay S, Bailey P, Reforgiato-Recupero G, Martin C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012;24:1242–1255. doi: 10.1105/tpc.111.095232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne D, Raseira M, Bassi D, Piagnani M, Gasic K, Reighard G, Moreno M, Pérez S. Peach. In: Badenes ML, Byrne DH, editors. Fruit Breeding. New York: Springer Verlag; 2012. pp. 505–569. [Google Scholar]
- Campbell R, Ducreux LJM, Morris WL, Morris JA, Suttle JC, Ramsay G, Bryan GJ, Hedley PE, Taylor MA. The metabolic and developmental roles of carotenoid cleavage dioxygenase4 from potato. Plant Physiol. 2010;154:656–664. doi: 10.1104/pp.110.158733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzonelli CI, Pogson BJ. Source to sink: regulation of carotenoid biosynthesis in plants. Trends Plant Sci. 2010;15:266–274. doi: 10.1016/j.tplants.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Connors CH. Some notes on the inheritance of unit characters in the peach. Proc. Am. Soc. Hortic. Sci. 1920;16:24–36. [Google Scholar]
- Dermen H. Histogenetic factors in color and fuzzless peach sports. J. Hered. 1956;47:64–76. [Google Scholar]
- Falchi R, Cipriani G, Marrazzo T, Nonis A, Vizzotto G, Ruperti B. Identification and differential expression dynamics of peach small GTPases encoding genes during fruit development and ripening. J. Exp. Bot. 2010;61:2829–2842. doi: 10.1093/jxb/erq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fray RG, Grierson D. Identification and genetic analysis of normal and mutant phytoene synthase genes of tomato by sequencing, complementation and co-suppression. Plant Mol. Biol. 1993;22:589–602. doi: 10.1007/BF00047400. [DOI] [PubMed] [Google Scholar]
- García-Limones C, Schnäbele K, Blanco-Portales R, Luz Bellido M, Caballero JL, Schwab W, Muñoz-Blanco J. Functional characterization of FaCCD1: a carotenoid cleavage dioxygenase from strawberry involved in lutein degradation during fruit ripening. J. Agric. Food Chem. 2008;56:9277–9285. doi: 10.1021/jf801096t. [DOI] [PubMed] [Google Scholar]
- Gil MI, Tomás-Barberán FA, Hess-Pierce B, Kader AA. Antioxidant capacities, phenolic compounds, carotenoids, and vitamin C contents of nectarine, peach, and plum cultivars from California. J. Agric. Food Chem. 2002;50:4976–4982. doi: 10.1021/jf020136b. [DOI] [PubMed] [Google Scholar]
- Giuliano G, Al–Babili S, von Lintig J. Carotenoid oxygenases: cleave it or leave it. Trends Plant Sci. 2003;8:145–149. doi: 10.1016/S1360-1385(03)00053-0. [DOI] [PubMed] [Google Scholar]
- Harjes CE, Rocheford TR, Bai L, et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science. 2008;319:330–333. doi: 10.1126/science.1150255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havecker ER, Gao X, Voytas DF. The diversity of LTR retrotransposons. Genome Biol. 2004;5:225. doi: 10.1186/gb-2004-5-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Green PJ. NMD in plants. In: Maquat LE, editor. Nonsense-Mediated mRNA Decay. New York: Landes Bioscience; 2006. pp. 167–172. [Google Scholar]
- Huang X, Lu G, Zhao Q, Liu X, Han B. Genome-wide analysis of transposon insertion polymorphisms reveals intraspecific variation in cultivated rice. Plant Physiol. 2008;148:25–40. doi: 10.1104/pp.108.121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FC, Molnár P, Schwab W. Cloning and functional characterization of carotenoid cleavage dioxygenase 4 genes. J. Exp. Bot. 2009;60:3011–3022. doi: 10.1093/jxb/erp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illa E, Eduardo I, Audergon J, et al. Saturating the Prunus (stone fruits) genome with candidate genes for fruit quality. Mol. Breed. 2011;28:667–682. [Google Scholar]
- Kachanovsky DE, Filler S, Isaacson T, Hirschberg J. Epistasis in tomato color mutations involves regulation of phytoene synthase 1 expression by cis-carotenoids. Proc. Natl Acad. Sci. USA. 2012;109:19021–19026. doi: 10.1073/pnas.1214808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Matsumoto H, Ikoma Y, Okuda H, Yano M. The role of carotenoid cleavage dioxygenases in the regulation of carotenoid profiles during maturation in citrus fruit. J. Exp. Bot. 2006;57:2153–2164. doi: 10.1093/jxb/erj172. [DOI] [PubMed] [Google Scholar]
- Kijima TE, Innan H. On the estimation of the insertion time of LTR retrotransposable elements. Mol. Biol. Evol. 2010;27:896–904. doi: 10.1093/molbev/msp295. [DOI] [PubMed] [Google Scholar]
- Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;8:464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Goto-Yamamoto N, Hirochika H. Retrotransposon-induced mutations in grape skin color. Science. 2004;304:982. doi: 10.1126/science.1095011. [DOI] [PubMed] [Google Scholar]
- Kuzin A, Lyubomirskaya N, Khudaibergenova B, Ilyin Y, Kim A. Precise excision of the retrotransposon gypsy from the forked and cut loci in a genetically unstable Drosophila melanogaster strain. Nucleic Acids Res. 1994;22:4641–4645. doi: 10.1093/nar/22.22.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinsohn E, Sitrit Y, Bar E, Azulay Y, Ibdah M, Meir A, Yosef E, Zamir D, Tadmor Y. Not just colors – carotenoid degradation as a link between pigmentation and aroma in tomato and watermelon fruit. Trends Food Sci. Technol. 2005;16:407–415. [Google Scholar]
- Li F, Vallabhaneni R, Yu J, Rocheford T, Wurtzel ET. The maize phytoene synthase gene family: overlapping roles for carotenogenesis in endosperm, photomorphogenesis, and thermal stress tolerance. Plant Physiol. 2008;147:1334–1346. doi: 10.1104/pp.108.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garcia P, Parfitt D, Ogundiwin E, Fass J, Chan H, Ahmad R, Lurie S, Dandekar A, Gradziel T, Crisosto C. High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.) Tree Genet. Genomes. 2013;9:19–36. [Google Scholar]
- Mathieu S, Terrier N, Procureur J, Bigey F, Günata Z. A carotenoid cleavage dioxygenase from Vitis vinifera L.: functional characterization and expression during grape berry development in relation to C13-norisoprenoid accumulation. J. Exp. Bot. 2005;56:2721–2731. doi: 10.1093/jxb/eri265. [DOI] [PubMed] [Google Scholar]
- Moise AR, von Lintig J, Palczewski K. Related enzymes solve evolutionarily recurrent problems in the metabolism of carotenoids. Trends Plant Sci. 2005;10:178–186. doi: 10.1016/j.tplants.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Ohmiya A, Kishimoto S, Aida R, Yoshioka S, Sumitomo K. Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in Chrysanthemum petals. Plant Physiol. 2006;142:1193–1201. doi: 10.1104/pp.106.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaisa KA, Morgante M, Williams M, Rafalski A. Contrasting effects of selection on sequence diversity and linkage disequilibrium at two phytoene synthase loci. Plant Cell. 2003;15:1795–1806. doi: 10.1105/tpc.012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker HG, VonHoldt BM, Quignon P, et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science. 2009;325:995–998. doi: 10.1126/science.1173275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Uribe L, Guzman I, Rajapakse W, Richins RD, O’Connell MA. Carotenoid accumulation in orange-pigmented Capsicum annuum fruit, regulated at multiple levels. J. Exp. Bot. 2012;63:517–526. doi: 10.1093/jxb/err302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Villalon A, Gas E, Rodriguez-Concepcion M. Phytoene synthase activity controls the biosynthesis of carotenoids and the supply of their metabolic precursors in dark-grown Arabidopsis seedlings. Plant J. 2009;60:424–435. doi: 10.1111/j.1365-313X.2009.03966.x. [DOI] [PubMed] [Google Scholar]
- de la Rosa LA, Alvarez-Parrilla E, Gonzalez-Aguilar GA. Fruit and Vegetable Phytochemicals: Chemistry, Nutritional Value and Stability. Ames, IA: John Wiley & Sons; 2009. [Google Scholar]
- Rubio A, Rambla JL, Santaella M, Gómez MD, Orzaez D, Granell A, Gómez-Gómez L. Cytosolic and plastoglobule-targeted carotenoid dioxygenases from Crocus sativus are both involved in beta-ionone release. J. Biol. Chem. 2008;283:24816–24825. doi: 10.1074/jbc.M804000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi S, Sponza G, Morgante M, et al. Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl Acad. Sci. USA. 2007;104:11376–11381. doi: 10.1073/pnas.0704145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki M, Fujita K, Kobayashi H, Suzuki S. Pink-colored grape berry is the result of short insertion in intron of color regulatory gene. PLoS ONE. 2011;6:8. doi: 10.1371/journal.pone.0021308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Vendramin E, Dettori MT, Giovinazzi J, Micali S, Quarta R, Verde I. A set of EST-SSRs isolated from peach fruit transcriptome and their transportability across Prunus species. Mol. Ecol. Notes. 2007;7:307–310. [Google Scholar]
- Verde I, Bassil N, Scalabrin S, et al. Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS ONE. 2012;7:e35668. doi: 10.1371/journal.pone.0035668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I, Abbott AG, Scalabrin S, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013;45:487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- Vezzi F, Del Fabbro C, Tomescu AI, Policriti A. rNA: a fast and accurate short reads numerical aligner. Bioinformatics. 2012;28:123–124. doi: 10.1093/bioinformatics/btr617. [DOI] [PubMed] [Google Scholar]
- Vizzotto M, Cisneros-Zevallos L, Byrne DH, Ramming DW, Okie WR. Large variation found in the phytochemical and antioxidant activity of peach and plum germplasm. J. Am. Soc. Hortic. Sci. 2007;132:334–340. [Google Scholar]
- Walker AR, Lee E, Robinson SP. Two new grape cultivars, bud sports of Cabernet Sauvignon bearing pale-coloured berries, are the result of deletion of two regulatory genes of the berry colour locus. Plant Mol. Biol. 2006;62:623–635. doi: 10.1007/s11103-006-9043-9. [DOI] [PubMed] [Google Scholar]
- Williamson J, Peace C, Bliss F, Garner D, Crisosto C. Evidence for a single locus controlling flesh color, senescent leaf color, and hypanthium color in peach. J. Am. Soc. Hortic. Sci. 2006;131:256–260. [Google Scholar]
- Yeager AF, Meader EM. A flesh-color chimera in the peach. J. Hered. 1956;47:77–78. [Google Scholar]
- Zhang H-B, Zhao X, Ding X, Paterson AH, Wing RA. Preparation of megabase-size DNA from plant nuclei. Plant J. 1995;7:175–184. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison between nucleotide and deduced amino acid sequences of the (TC)8 and (TC)10 repeat alleles in ‘Armking’ and ‘Silver King’ revertants.
Microsatellite analysis performed on cDNA from Redhaven fruit flesh.
Premature stop codons.
Prediction of subcellular localization of the PpCCD4 protein.
Primers used for PCR screening of LTR retrotransposon insertion.