

Abstract
Many patients with pancreatic cancer have metastases to distant organs at the time of initial presentation. Recent studies examining the evolution of pancreatic cancer at the genetic level have shown that clonal complexity of metastatic pancreatic cancer is already initiated within primary tumors, and organ-specific metastases are derived from different subclones. However, we do not yet understand to what extent the evolution of pancreatic cancer contributes to proteomic and signaling alterations. We hypothesized that genetic heterogeneity of metastatic pancreatic cancer results in heterogeneity at the proteome level. To address this, we employed a model system in which cells isolated from three sites of metastasis (liver, lung, and peritoneum) from a single patient were compared. We used a SILAC-based accurate quantitative proteomic strategy combined with high-resolution mass spectrometry to analyze the total proteome and tyrosine phosphoproteome of each of the distal metastases. Our data revealed distinct patterns of both overall proteome expression and tyrosine kinase activities across the three different metastatic lesions. This heterogeneity was significant because it led to differential sensitivity of the neoplastic cells to small molecule inhibitors targeting various kinases and other pathways. For example, R428, a tyrosine kinase inhibitor that targets Axl receptor tyrosine kinase, was able to inhibit cells derived from lung and liver metastases much more effectively than cells from the peritoneal metastasis. Finally, we confirmed that administration of R428 in mice bearing xenografts of cells derived from the three different metastatic sites significantly diminished tumors formed from liver- and lung-metastasis-derived cell lines as compared with tumors derived from the peritoneal metastasis cell line. Overall, our data provide proof-of-principle support that personalized therapy of multiple organ metastases in a single patient should involve the administration of a combination of agents, with each agent targeted to the features of different subclones.
Approximately half of the patients with pancreatic cancer are initially diagnosed with metastases to distal sites, with the commonest sites being the liver, lung, and peritoneum (1). Therapeutic strategies against metastases could help reduce the high mortality rates associated with this cancer (2). Understanding the nature of metastatic pancreatic cancer at a systems level can enable the discovery of potential targets for the development of targeted therapies.
Pancreatic cancer has been shown to be a genetically evolving and heterogeneous disease (3–5). Clonal diversity and evolution of cancer genomes have also been demonstrated based on the isolation of distinct clonal populations purified directly from patient biopsies by means of flow cytometry followed by genomic characterization (6). A number of reports have documented the adoption of a proteomic approach for the discovery of potential biomarkers in pancreatic cancer (7, 8). However, these studies generally assume pancreatic cancers to be homogeneous, and the emphasis is placed on identifying molecules that are common across a broad array of tumors. There is a lack of studies systematically examining the proteomic changes or signaling pathways across pancreatic cancers to dissect the nature of the heterogeneity of each clone. An excellent setting in which the heterogeneity of tumors can be studied systematically is in a patient harboring metastases to several distant sites. To this end, we chose cells isolated from three metastatic pancreatic lesions of a single patient. The exomes of each tumor site were previously sequenced to study the progression of pancreatic cancer, and the results showed that all cell lines were identical for the genetic status of driver mutations (e.g. KRAS, TP53, and SMAD4) (9). Our hypothesis was that a better understanding of the proteomic consequences of the heterogeneity derived from genetic changes, and possibly other types of alterations, might provide additional opportunities to identify therapeutic targets.
In order to precisely quantify differences across the proteomes of multiple metastatic pancreatic cancer lesions, we employed a SILAC-based1 quantitative proteomics strategy combined with high-resolution mass spectrometry (10, 11). Based on changes observed at the whole-proteome level, we found that a class of cell surface receptors showed significant enrichment with the highest alteration of their expression among the three metastatic pancreatic cancer cell lines examined (i.e. peritoneum, lung, and liver). Because the total protein levels provide information about the static levels of proteins and not their activity per se, we decided to examine the activation of phosphorylation-driven pathways, many of which are activated by cell surface receptors. To globally examine tyrosine phosphorylation-based signaling pathways, we carried out mass spectrometric analysis of purified tyrosine phosphorylated peptides enriched using anti-phosphotyrosine antibodies. As a result, we observed differential activation of tyrosine kinases in the three different sites of metastases. For example, Axl receptor tyrosine kinase was found to be hyperphosphorylated in lung and liver metastases relative to peritoneal metastasis. Expression of Axl receptor tyrosine kinase in primary and matched pancreatic cancers on tissue microarrays was validated by immunohistochemistry. Given such unique patterns of activation of pathways, it was possible that tumors derived from different sites could show differences in their sensitivity to pathway inhibitors. To test this, we performed experiments in which we screened cell lines derived from each metastatic site against a panel of small molecule inhibitors. We observed that the three metastatic pancreatic cancers had differential sensitivities to different inhibitors. For example, cells derived from the peritoneal metastasis were highly sensitive to lapatinib, whereas greater sensitivity to the Axl inhibitor R428 was observed in the lung metastasis cell line. Finally, we showed that treatment of mice bearing xenografts from these different pancreatic cancer cell lines with R428, an inhibitor of Axl receptor tyrosine kinase, led to reduction of tumors with evidence of activation of Axl.
RESULTS
Our goal was to study the differences in the proteomes of metastases derived from various sites in the same individual. For this purpose, we created low-passage cell lines isolated from three different metastatic sites of pancreatic cancer from a single patient (liver, lung, and peritoneum) at autopsy and carried out both total proteome and tyrosine phosphoproteome analyses as described below.
Significant Alterations in the Proteome of Metastatic Lesions Revealed by Quantitative Proteomics
To systematically explore the similarities and subtle differences of expressed proteomes, we employed an unbiased SILAC-based quantitative proteomics in which cells from the peritoneal metastasis were unlabeled (i.e. “light”) and cells from lung metastasis and liver metastasis were labeled with “medium” lysine (2H4) and arginine (13C6) or “heavy” lysine (13C615N2) and arginine (13C615N4) (Fig. 1A). After mixing of an equal amount of protein lysates, the samples were separated via SDS-PAGE, and the gel was excised into 24 slices and processed for LC-MS/MS experiments. High-quality tandem mass spectrometry data with high accuracy and high resolving power (resolution = 120,000 at 400 m/z) were acquired on an LTQ-Orbitrap Elite mass spectrometer. An average and a median of mass errors were estimated to 0.69 ppm and to 0.45 ppm, respectively (Fig. 1B). A Mascot search was carried out, and a median Mascot score was found to be 59 at a 1% false discovery rate, as shown in Fig. 1C. We obtained substantial coverage of the proteome with precise quantitative measurements of >4,200 proteins. Remarkably, although the large majority of proteins were unchanged, 28% and 18% of the proteome of cells derived from liver metastasis were differentially expressed >2-fold relative to cells from the peritoneal and lung metastases, respectively, as shown in Fig. 1D. We also observed a similar result in pairwise comparison. Overall an average of 25% of the proteome was differentially expressed between different metastatic pancreatic cancer cell lines. As described earlier (9), pancreatic cancer metastases to the peritoneum may be genetically divergent from distant metastases. When looking at the correlation of differential proteome expression among cells from three metastatic lesions (i.e. peritoneum, lung, and liver), we observed a similar pattern in that cells from the lung metastasis were indeed closer in their proteomic profile to cells from the liver metastasis than the peritoneal metastasis, as shown in Fig. 2A.
Fig. 1.

A SILAC strategy combined with high-resolution mass spectrometry for differential proteome analysis. A, schematic of a SILAC-based quantitative proteomics for differential proteome analysis of cells isolated from metastatic pancreatic cancer in distal organs. B, highly accurate mass measurements were carried out with sub-ppm error on the LTQ-Orbitrap Elite mass spectrometer. C, distribution of Mascot scores of all peptide-spectrum matches at 1% false discovery rates shows a median Mascot score of ∼59 derived from high-quality tandem mass spectrometry data acquired in this study. D, over 4,200 proteins were observed in this experiment. When two metastatic pancreatic cancer proteomes (i.e. peritoneal metastasis versus liver metastasis) were compared, ∼28% of proteins were found to vary by >2-fold among metastases, although most of the proteins remained unchanged.
Fig. 2.

Receptor tyrosine kinases as a highly regulated class of proteins in metastatic pancreatic cancer revealed by quantitative proteomics analysis. A, proteomes of liver metastasis and lung metastasis were found to be more similar to each other than to that of peritoneum metastasis. This result is consistent with genetic data from in our previous study showing that lung pancreatic metastasis is evolutionarily closer to liver pancreatic metastasis. This indicates that a pattern of proteome expression among genetically close clones is closely related. In other words, a genetically different clone may have a different proteome expression. B, the pie chart shows that ∼42% of proteomes in cells derived from distal metastatic pancreatic cancers were categorized in proteins that were highly variable when pairwise comparison of three metastases was carried out. C, bioinformatics analysis showed that receptor/signal transducer activities were enriched in altered proteome.
Receptors and Signaling Molecules Are Among the Most Differentially Expressed Proteins Across Metastases
As shown in Fig. 1D, given that proteomes were differentially expressed across the three metastatic lesions, we wished to determine whether these changes occurred across all categories of proteins or were somehow biased toward certain classes of molecules. To do so, we first carried out a pairwise comparison of all three proteomes and classified proteins as “less variable” (less than 2-fold change in abundance across any two samples) or “highly variable” (more than 2-fold change in abundance across any two samples). As shown in Fig. 2B, 42% of the proteome was found to be highly variable when compared across any two sites of metastatic pancreatic cancer in the individual. Based on a Gene Ontology functional analysis using molecules found to be highly variable in their expression at the protein level, we observed an enrichment of receptor activity and signal transducer activity among the molecular functions (Fig. 2C). In the case of cellular components, we also found an enrichment of cell surface proteins and extracellular proteins. We were then interested in determining whether any classes of proteins were resistant to alterations (i.e. were less variable in their expression levels) across the three types of metastatic lesions. As somewhat expected, proteins involved in critical functions such as RNA/DNA/protein binding were found to vary less across cells derived from different metastases (Fig. 2C). In addition, ribosomal components were highly resistant to change in their expression. Taken together, these data suggest that receptors and other signaling molecules that show differential expression might lead to activation of different signaling pathways in the various metastatic lesions.
Metastatic Lesions Show Unique Patterns of Activation of Tyrosine Kinase Signaling Pathways
Based on our analysis of the total proteome, we sought to directly test whether tyrosine-phosphorylation-based signaling pathways that are downstream of many cell surface receptors are differentially activated in liver-, lung-, or peritoneum-derived metastases. To obtain an idea of the global expression of phosphotyrosine levels among the three metastatic pancreatic cancer cell lines, we performed a Western blot experiment using anti-tyrosine phosphorylation antibody (clone 4G10) as shown in Fig. 3A. Western blotting indicated modest changes in the regions of ∼125 kDa and ∼65 kDa. To better clarify changes in phosphotyrosine peptides, we therefore performed immunoaffinity purification of tyrosine phosphorylated peptides using anti-phosphotyrosine antibodies followed by LC-MS/MS as depicted in Fig. 3B. Phosphotyrosine peptides were purified using phosphotyrosine-specific antibodies, and tyrosine phosphorylated peptides were analyzed on an LTQ-Orbitrap mass spectrometer equipped with a nanoflow liquid chromatography system. A total of 6,033 tandem mass spectra were searched using MASCOT, assigning to 128 phosphopeptide-spectrum matches (phosphoPSMs). To confirm phosphoPSMs, we carried out a manual validation of all phosphoPSMs for all identified peptides. Localization of phosphorylation was statistically assessed by the PhosphoRS algorithm (12), and sites of phosphorylation for 13 phosphoPSMs were re-localized. Of those 128 phosphoPSMs, site probabilities for 119 were found to be greater than 90%, whereas phosphorylation for 9 phosphoPSMs was found to be ambiguously localized. In all, a total of 68 phosphorylated peptides representing 69 phosphorylation sites including 67 tyrosine residues, which suggests a highly selective affinity purification of phosphotyrosines, were identified and mapped to proteins encoded by 70 genes.
Fig. 3.
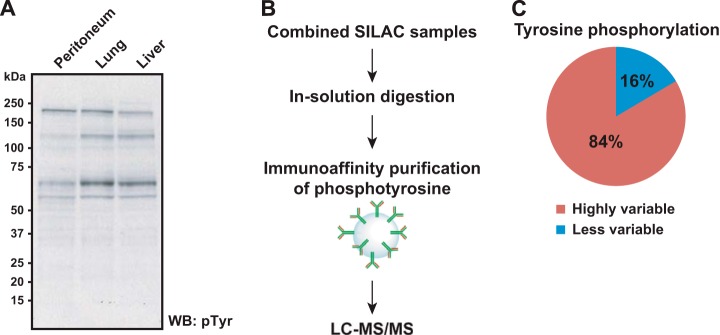
Unique activation of different signaling pathways in metastatic pancreatic cancer revealed by quantitative tyrosine phosphoproteomics analysis. A, a pattern of tyrosine phosphorylation levels among pancreatic cancer metastases was briefly screened via Western blot experiment using anti-phosphotyrosine antibody. B, schematics of SILAC-based quantitative tyrosine phosphoproteomics of multiple pancreatic cancer metastases. Tyrosine phosphorylated peptides were enriched via immunoaffinity purification using anti-phosphotyrosine antibodies. C, the pie chart shows that a greater portion (∼89%) of identified tyrosine phosphorylation sites was found to be altered.
DAVID (13) analysis showed that several pathways based on KEGG (14) were significantly activated, including axon guidance (p value = 4.4e-13), focal adhesion (p value = 2.0e-10), ErbB signaling pathway (p value = 4.7e-6), and VEGF signaling pathway (p value = 2.5e-5). The supplemental material shows the KEGG pathways with molecules found to be phosphorylated. As listed in supplemental Table S1, there were genes whose protein products were observed to be phosphorylated along with sites of phosphorylation, as well as potential upstream kinases that could phosphorylate them. For example, a peptide (VLEDDPEATpYTTSGGK) derived from EphA2 receptor tyrosine kinase was found to be phosphorylated at a position of Tyr772. Based on the PhosphoSitePlus database (15), the site can be autophosphorylated by itself. The phosphorylation levels among the three cell lines were found not to be significantly different. As another instance, a peptide (LIEDNEpYTAR) shared among proteins encoded by multiple genes such as YES1, LCK, FYN, and SRC was found to be highly phosphorylated in the peritoneum-derived metastatic cell line relative to the liver-metastasis-derived line. This phosphorylation can be mediated by multiple kinases such as LCK, FYN, SRC, PDGFRB, and PRKACA. Overall, tyrosine phosphoproteomics analysis resulted in the identification of a number of molecules activated in metastatic pancreatic cancer. Fig. 3C further shows that ∼84% of tyrosine phosphorylations quantitated were found to be highly variable (i.e. >2-fold changes in any two comparisons). These results support our hypothesis that the activity of molecules involving signal transduction (i.e. tyrosine kinases) is largely altered among metastatic pancreatic cancer clones derived from a single patient.
Differential Sensitivity of Metastases Revealed by in Vitro Screening of Small Molecule Inhibitors
Our data from total proteome and tyrosine phosphoproteome analyses clearly reveal that there are profound differences in metastases derived from different target organ sites from the same patient. In light of these findings, we hypothesized that cells derived from these different metastases might also exhibit differential sensitivity to drugs. To test this, we used a panel of small molecule inhibitors to systematically screen drug sensitivity across each cell line (16, 17). The panel used in our experiment included inhibitors targeting receptor tyrosine kinases such as EGFR, ErbB2, Axl, Met, and VEGFR, among others. As shown in Fig. 4A, we observed that among the small molecule inhibitors that were relatively potent in inhibiting proliferation, the effect was not uniform across the three metastatic cell types. For example, GSK-1838705A (18), which targets anaplastic lymphoma kinase, insulin-like growth factor-I receptor, and insulin receptor, was more potent against cells derived from lung metastasis, whereas lapatinib (19), which targets EGFR and ErbB2, was found to be more potent against cells derived from peritoneal metastases (Fig. 4A).
Fig. 4.

Axl receptor tyrosine kinase as a potential therapeutic target revealed by inhibitor screening assay and in vivo mouse study. A, the heat map shows the differential sensitivity of metastatic pancreatic cancer cells derived from peritoneum, lung, and liver to small molecule inhibitors. B, mice xenografted with metastatic pancreatic cancer cells derived from lung and liver were more sensitive to R428, a small molecule inhibitor to Axl receptor tyrosine kinase in vivo mouse model, than mice xenografted with cells derived from peritoneum. C, five representative immunohistochemistry images of Axl staining on matched primary and metastatic pancreatic cancers from five different patients are depicted (A31, A29, A55, A89, and A92). A31 is an example of low expression in the primary and matched metastasis. The arrow in A31 primary indicates strong positive labeling of endothelium within a small capillary, as compared with negative labeling of the neoplastic cells. The negative labeled cancer cells are circled with a dashed line in A31 metastasis. A29 is an example of strong positive labeling in the primary carcinoma and weak labeling in the metastasis. By contrast, A55 is an example of weak labeling in the primary and strong labeling in the metastasis. A89 and A92 are examples of strong positive labeling in both the primary and the matched metastasis. The arrow in the A89 metastasis indicates a negative labeled normal bile duct.
Validation of Axl as a Potential Therapeutic Target in Pancreatic Cancer Using a Preclinical Mouse Model
One of the molecules of interest was Axl receptor tyrosine kinase. Axl belongs to a less well-characterized receptor tyrosine kinase subfamily (which includes Tyro3 and Mer) of the 20 known families of receptor tyrosine kinases in human (20). Axl is known to be overexpressed in tumor types such as acute leukemia, breast cancer, gastric cancer, lung cancer, and pancreatic cancer (21). It has recently been reported that resistance to EGFR-targeted therapy in patients with lung cancer can be driven by the activation of Axl receptor tyrosine kinase (22). We found hyperphosphorylation of Axl on Tyr634, Tyr702, and Tyr703 in the lung- and liver-metastasis-derived cell lines but not the peritoneal metastasis cell line. To assess the function of hyperphosphorylation on Axl, we performed validation experiments using a preclinical mouse model system. In this experiment, each metastatic pancreatic cancer cell line was subcutaneously injected into nude mice. Subsequently, mice were administered R428, a small molecule inhibitor of Axl receptor tyrosine kinase. As shown in Fig. 4B, there was a significant reduction in tumor mass observed in the lung and liver pancreatic metastasis cell lines as compared with the peritoneum metastasis cell line upon treatment with the Axl inhibitor.
Finally, we performed immunohistochemical labeling to detect Axl expression in primary and metastatic pancreatic cancer tissues from patients with known patterns of failure and metastatic burden at autopsy. Strong positive labeling for Axl in the primary carcinoma was positively correlated with similar levels of labeling in the patients' matched metastases (Table I). Even more striking was the observation that higher expression of Axl in the primary tumors, or in the metastatic tissues, correlated with a higher overall metastatic burden in patients (Table II and Fig. 4C), consistent with the observation that patients with high metastatic burden most often have liver and lung metastases.
Table I. Correlation of immunohistochemical labeling for total Axl in 29 matched primary and metastatic pancreatic cancers (p = 0.029, two-sided Fisher's exact test).
| Primary carcinoma |
|||
|---|---|---|---|
| Low Axl expression | High Axl expression | ||
| Metastasis | Low Axl expression | 13 | 4 |
| High Axl expression | 4 | 8 | |
Table II. Correlation of immunohistochemical labeling for total Axl in primary pancreatic cancer tissues to their metastatic burden at autopsy (p = 0.028, two-sided Fisher's exact test).
| Metastatic burden |
||||
|---|---|---|---|---|
| None | 1 to 10 metastases | 11 to 100 metastases | >100 metastases | |
| Low Axl expression | 2 | 7 | 4 | 6 |
| High Axl expression | 3 | 0 | 2 | 10 |
DISCUSSION
Metastatic pancreatic cancer is a deadly disease with almost no effective therapeutic options. Although it is now understood that there is a clonal evolution both in primary tumors and in metastases leading to considerable genetic heterogeneity, the differential activation of cellular signaling pathways in different subclones is poorly understood. Here, we employed high-resolution mass-spectrometry-based quantitative proteomics and tyrosine phosphoproteomics methods to understand better the effect of genetic changes on proteome and phosphotyrosine signaling in metastatic pancreatic cancer. Our discovery of activation of different profiles of tyrosine kinases in cells derived from three different sites of metastases suggests that personalized therapy for metastatic disease might have to incorporate strategies aimed at tackling this heterogeneity of signaling pathways as well.
MATERIALS AND METHODS
Reagents
Anti-phosphotyrosine antibody (HRP-conjugated, 4G10) was purchased from Millipore (Billerica, MA). Anti-phosphotyrosine mouse monoclonal antibody (PY100) conjugated to beads for immunoaffinity purification of tyrosine phosphorylated peptides was from Cell Signaling Technology (Danvers, MA). TPCK-treated trypsin was from Worthington Biochemical Corp. (Lakewood, NJ). DMEM with/without lysine/arginine, fetal bovine serum (FBS), l-glutamine, antibiotics, and phosphate-buffered saline (PBS, pH 7.4) were purchased from Invitrogen (Carlsbad, CA). SILAC amino acids, 2H4-lysine, 13C615N2-lysine, 13C6-arginine, and 13C615N4-arginine were from Cambridge Isotope Laboratories (Andover, MA). All small molecule inhibitors were from Selleck Chemicals (Houston, TX). All other reagents used in this study were from Fisher Scientific (Pittsburgh, PA).
SILAC Labeling and Peptide Preparation
Pancreatic metastatic cell lines were derived from peritoneum, lung, or liver in an individual via rapid autopsies as described previously (9) and maintained in DMEM supplemented with 20% FBS, l-glutamine, penicillin, and streptomycin at 37 °C and 5% CO2, unless mentioned. Cells derived from peritoneal metastasis were maintained in the light media, and cells isolated from lung or liver were adapted to the medium SILAC media with 2H4-lysine and 13C6-arginine or to the heavy SILAC media with 13C615N2-lysine and 13C615N4-arginine, as described earlier (10). Exponentially growing cells were washed with PBS and grown in the absence of FBS for 12 h prior to harvest.
Cells were lysed in SDS-based lysis buffer (4% SDS, 100 mm Tris, pH 7.4, 100 mm DTT), and crude protein lysates were prepared by sonication. After centrifugation at 12,000 × g, protein concentration in the supernatant was measured using the BCA method. The same amount of proteins from metastatic pancreatic cancer cells derived from peritoneum, lung, and liver were mixed, and 75 μg of protein mixture were separated on an SDS-PAGE gel. 24 bands were sliced and processed for in-gel digestion. Tryptic peptides were extracted, dried, and kept at −20 °C until analysis. For enrichment of tyrosine phosphoproteome, a mixture of peptides from three metastatic cancer cells was prepared via an in-solution tryptic digestion protocol as described earlier (17), with minor modifications. Briefly, cells were lysed in urea-based lysis buffer (20 mm HEPES, pH 8.0, 9 m urea, 1 mm sodium orthovanadate, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate) and sonicated for 30 s twice. Cell lysates were cleared by centrifugation at 20,000 × g at 15 °C for 15 min. 10 mg of proteins from each state were mixed, reduced with 4.1 mm dithiothreitol for 20 min at 60 °C, and alkylated with 8.3 mm iodoacetamide for 20 min in the dark. Proteins were diluted in 20 mm HEPES, pH 8.0, to a final concentration of 2 m urea and incubated with TPCK-treated trypsin at 25 °C overnight. Protein digests were acidified by the addition of 20% trifluoroacetic acid (TFA) to a final concentration of 1% TFA and subjected to centrifugation at 2,000 × g at room temperature for 15 min. The supernatant of protein digests (30 mg total) was loaded onto a Sep-Pak C18 cartridge (Waters, Milford, MA, C# WAT051910) equilibrated with 0.1% TFA. Peptides were eluted with 40% acetonitrile with 0.1% TFA. Eluted peptides were lyophilized and subjected to immunoaffinity purification of tyrosine phosphorylated peptides.
Immunoaffinity Purification of Tyrosine Phosphopeptides
Immunoaffinity purification (IAP) of tyrosine phosphorylated peptides was carried out as previously described (17, 23). Briefly, after lyophilization, about 30 mg of tryptic peptides was dissolved in 1.4 ml of IAP buffer (50 mm MOPS, pH 7.2, 10 mm sodium phosphate, 50 mm NaCl) and subjected to centrifugation at 2,000 × g at room temperature for 5 min. Before IAP, P-Tyr-100 beads were washed with IAP buffer twice at 4 °C, and the pH of the supernatant containing peptides was adjusted to ∼7.2 by the addition of 1 m Tris Base. For IAP, the supernatant was incubated with P-Tyr-100 beads at 4 °C for 30 min, and the beads were washed three times with IAP buffer and twice with ultrahigh pure water. Peptides were eluted twice from beads by incubating the beads with 0.15% TFA at room temperature for 15 min. Eluted peptides were cleaned via StageTip (24), and dried peptides were kept at −20 °C prior to LC-MS/MS experiment.
Liquid Chromatography–Tandem Mass Spectrometry
In-gel tryptic peptides from 24 slices were analyzed on an LTQ-Orbitrap Elite mass spectrometer with a running time of 100 min as described previously. Peptides were loaded onto a trap column (2 cm long, 75 μm inner diameter) packed in-house with the C18 material (5-μm size, 100-Å pore, Michrom Bioresources, Inc., Auburn, CA) and separated by an analytical column (20 cm long, 75 μm inner diameter) packed in-house with the C18 material (3-μm size, 100-Å pore, Michrom Bioresources, Inc.). Both precursor and higher-energy C-trap dissociation (HCD)-induced fragment ion species were measured on the Orbitrap with resolutions of 120,000 and 30,000, respectively.
LC-MS/MS analysis of tyrosine phosphorylated peptides enriched via IAP was carried out using a reversed phase liquid chromatography system connected online to an LTQ-Orbitrap XL ETD mass spectrometer (25). The peptides were loaded onto a trap column (2 cm × 75 μm, Magic C18AQ 5 μm, 100 Å) and separated by an analytical column (15 cm × 75 μm, Magic C18AQ 5 μm, 100 Å) packed in-house. Precursor mass spectra were acquired in an Orbitrap mass analyzer (Thermo Scientific) from 350–1,800 m/z at a resolution of 60,000, and tandem mass spectra produced via the collision-induced dissociation method were measured in a linear ion trap mass analyzer. The most intense eight ions (isolation width, 2.5 m/z; normalized collision energy, 35%; activation Q, 0.25; activation time, 30 ms) were subjected to sequencing in a data-dependent manner.
Mass Spectrometry Data Analysis
The acquired tandem mass spectra were searched by MASCOT (version 2.2.0) (26) and SEQUEST (27) search algorithms against a human RefSeq protein database (version 50 containing 33,833 entries along with common contaminants) using the Proteome Discoverer platform (version 1.3, Thermo Scientific). For both algorithms, the search parameters included a maximum of two missed cleavages and carbamidomethylation at cysteine as a fixed modification. N-terminal acetylation; deamidation at asparagine and glutamine; oxidation at methionine; phosphorylation at serine, threonine, and tyrosine; and SILAC labeling at lysine (13C615N2 or 2H4) and arginine (13C6 or 13C615N4) were allowed as variable modifications. For the MS data acquired on the LTQ-Orbitrap Elite mass spectrometer, the monoisotopic peptide tolerance was set at 10 ppm, and the MS/MS tolerance at 0.05 Da. The required false discovery rate was 1% at the peptide level. For the MS data acquired on the LTQ-Orbitrap XL mass spectrometer, the monoisotopic peptide tolerance was set at 10 ppm, and the MS/MS tolerance at 0.4 Da. The ratio of heavy/light for each phosphoPSM was calculated based on the quantitation node, and the probability of phosphorylation for each Ser/Thr/Tyr site on each peptide was calculated based on the PhosphoRS node (12) in Proteome Discoverer (version 1.3, Thermo Scientific). After normalizing SILAC ratios based on the median, we chose a cutoff of 2-fold alteration in any comparison. All phosphoPSMs were subjected to manual verification. A list of PSM/peptide/protein identifications is provided in the supplemental material.
In Vitro Drug Sensitivity Screening
The assay was performed as described previously (16). In brief, kinase inhibitors stored at 10 to 100 mmol/l in dimethyl sulfoxide were used in a setting of 96-well plates. The inhibitor concentrations used were in the range from ∼10 nm to 10 μm. Cells suspended in DMEM supplemented with 20% FBS were incubated for 3 days at 37 °C, 5% CO2 and subjected to a CellTiter 96 AQueous One solution cell proliferation assay (Promega, Madison, WI). An algorithm was designed and implemented using Excel and Visual Basic to provide automated IC50 calculation and therapeutic target identification. IC50 values were calculated using second-degree polynomial regression curve fitting, and the result of inhibitor sensitivity was depicted as a heat map.
In Vivo R428 Treatment
To examine the effects of R428 on the growth of three metastatic pancreatic cancer cells derived from peritoneum, lung, and liver in vivo, 5 × 106 cells were subcutaneously injected into nude mice (six mice of each line), and tumor growth was allowed. Two weeks post-injection, the mice were divided into control and experiment groups. The experiment group was treated with R428 twice daily (oral administration) at a dose of 25 mg/kg, whereas the control group was treated with media for R428. Following treatment with R428, we assessed tumor growth by measuring tumor volume every three days for a month. Data were presented as the ratio of the tumor size in the treated group to that in the untreated group.
Immunohistochemistry
Primary and metastatic cancer tissues from patients who underwent a rapid autopsy in association with the Gastrointestinal Cancer Rapid Medical Donation Program at Johns Hopkins (28) were used to construct a tissue microarray containing 208 cores (2 to 4 primary and 2 metastatic tissue cores per patient). This program was approved by the Johns Hopkins Institutional Review Board. One unstained 5-μm section was cut from this tissue microarray, and the slide was deparaffinized via routine techniques before being steamed for 20 min at 80 °C in 1× sodium citrate buffer (diluted from 10× heat-induced epitope retrieval buffer, Ventana-Bio Tek Solutions, Tucson, AZ). Slides were cooled for 5 min and incubated with anti-human total Axl antibody (AF154, R&D Systems, Minneapolis, MN) at a 1:500 dilution. Immunolabeling was detected using the IVIEW Detection Kit (Ventana Medical Systems, Inc., catalog no. 760091, Tucson, AZ) per the kit instructions, and the slide was counterstained in hematoxylin before being coverslipped. Labeling of each tissue core was independently scored by two pathologists (C.I.D. and J.P.) as negative (0), weak (1), moderate (2), or strong (3). In the event of discrepant scores (n = 2 cases), the case was re-reviewed by both pathologists at a two-headed microscope to achieve a consensus. Final scores for all cores for each primary tumor and each metastasis were averaged and categorized as low expression (scores from 0–1.5) and high expression (average score > 1.5).
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (29) with the dataset identifier PXD000750.
Supplementary Material
Footnotes
Author contributions: M.K. and A.P. designed research; M.K., Y.Z., S.Y., N.R., M.L.A., A. Marimuthu, K.M., and C.A.I. performed research; M.K., Y.Z., R.H.H., J.S.P., J.W.T., A. Maitra, C.A.I., and A.P. analyzed data; M.K., C.A.I., and A.P. wrote the paper.
* This study was supported in part by an NCI's Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036); Sol Goldman Pancreatic Cancer Research Center; a Department of Defense Era of Hope Scholar Award (W81XWH-06-1-0428); an NIH Roadmap Grant for Technology Centers of Networks and Pathways (U54GM103520); a contract (HHSN268201000032C) from the National Heart Lung and Blood Institute; a Stand Up To Cancer Dream Team Translational Cancer Research Grant, a Program of the Entertainment Industry Foundation, SU2C-AACR-DT0509 (N. R.ports and A. Maitra); NIH Grant Nos. CA140599 (C.I.D.), CA101955 (C.I.D.), and P50CA62924 (A.P., C.I.D., and A. Maitra); and the Uehara Memorial Foundation (S.Y.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SILAC
- stable isotope labeling by amino acids in cell culture
- phosphoPSM
- phosphopeptide-spectrum match
- TPCK
- L-1-tosylamido-2-phenylethyl chloromethyl ketone
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- IAP
- immunoaffinity purification.
REFERENCES
- 1. Yachida S., Iacobuzio-Donahue C. A. (2009) The pathology and genetics of metastatic pancreatic cancer. Arch. Pathol. Lab. Med. 133, 413–422 [DOI] [PubMed] [Google Scholar]
- 2. Wolfgang C. L., Herman J. M., Laheru D. A., Klein A. P., Erdek M. A., Fishman E. K., Hruban R. H. (2013) Recent progress in pancreatic cancer. CA Cancer J. Clin. 63, 318–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hruban R. H., Goggins M., Parsons J., Kern S. E. (2000) Progression model for pancreatic cancer. Clin. Cancer Res. 6, 2969–2972 [PubMed] [Google Scholar]
- 4. Iacobuzio-Donahue C. A. (2012) Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 61, 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Penchev V. R., Rasheed Z. A., Maitra A., Matsui W. (2012) Heterogeneity and targeting of pancreatic cancer stem cells. Clin. Cancer Res. 18, 4277–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruiz C., Lenkiewicz E., Evers L., Holley T., Robeson A., Kiefer J., Demeure M. J., Hollingsworth M. A., Shen M., Prunkard D., Rabinovitch P. S., Zellweger T., Mousses S., Trent J. M., Carpten J. D., Bubendorf L., Hoff D. V., Barrett M. T. (2011) Advancing a clinically relevant perspective of the clonal nature of cancer. Proc. Natl. Acad. Sci. 108, 12054–12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grønborg M., Kristiansen T. Z., Iwahori A., Chang R., Reddy R., Sato N., Molina H., Jensen O. N., Hruban R. H., Goggins M. G., Maitra A., Pandey A. (2006) Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol. Cell. Proteomics 5, 157–171 [DOI] [PubMed] [Google Scholar]
- 8. Makridakis M., Vlahou A. (2010) Secretome proteomics for discovery of cancer biomarkers. J. Proteomics 73, 2291–2305 [DOI] [PubMed] [Google Scholar]
- 9. Yachida S., Jones S., Bozic I., Antal T., Leary R., Fu B., Kamiyama M., Hruban R. H., Eshleman J. R., Nowak M. A., Velculescu V. E., Kinzler K. W., Vogelstein B., Iacobuzio-Donahue C. A. (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harsha H. C., Molina H., Pandey A. (2008) Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat. Protoc. 3, 505–516 [DOI] [PubMed] [Google Scholar]
- 11. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 12. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 13. Huang D. W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tyner J. W., Yang W. F., Bankhead A., Fan G., Fletcher L. B., Bryant J., Glover J. M., Chang B. H., Spurgeon S. E., Fleming W. H., Kovacsovics T., Gotlib J. R., Oh S. T., Deininger M. W., Zwaan C. M., Boer M. L. D., van den Heuvel-Eibrink M. M., O'Hare T., Druker B. J., Loriaux M. M. (2013) Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 73, 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhong J., Kim M.-S., Chaerkady R., Wu X., Huang T.-C., Getnet D., Mitchell C. J., Palapetta S. M., Sharma J., O'Meally R. N., Cole R. N., Yoda A., Moritz A., Loriaux M. M., Rush J., Weinstock D. M., Tyner J. W., Pandey A. (2012) TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol. Cell. Proteomics 11, M112.017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sabbatini P., Korenchuk S., Rowand J. L., Groy A., Liu Q., Leperi D., Atkins C., Dumble M., Yang J., Anderson K., Kruger R. G., Gontarek R. R., Maksimchuk K. R., Suravajjala S., Lapierre R. R., Shotwell J. B., Wilson J. W., Chamberlain S. D., Rabindran S. K., Kumar R. (2009) GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 8, 2811–2820 [DOI] [PubMed] [Google Scholar]
- 19. Rusnak D. W., Lackey K., Affleck K., Wood E. R., Alligood K. J., Rhodes N., Keith B. R., Murray D. M., Knight W. B., Mullin R. J., Gilmer T. M. (2001) The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 1, 85–94 [PubMed] [Google Scholar]
- 20. Bose R., Molina H., Patterson A. S., Bitok J. K., Periaswamy B., Bader J. S., Pandey A., Cole P. A. (2006) Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. U.S.A. 103, 9773–9778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Verma A., Warner S. L., Vankayalapati H., Bearss D. J., Sharma S. (2011) Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 10, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Z., Lee J. C., Lin L., Olivas V., Au V., LaFramboise T., Abdel-Rahman M., Wang X., Levine A. D., Rho J. K., Choi Y. J., Choi C.-M., Kim S.-W., Jang S. J., Park Y. S., Kim W. S., Lee D. H., Lee J.-S., Miller V. A., Arcila M., Ladanyi M., Moonsamy P., Sawyers C., Boggon T. J., Ma P. C., Costa C., Taron M., Rosell R., Halmos B., Bivona T. G. (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hansen A.-M., Chaerkady R., Sharma J., Díaz-Mejía J. J., Tyagi N., Renuse S., Jacob H. K. C., Pinto S. M., Sahasrabuddhe N. A., Kim M.-S., Delanghe B., Srinivasan N., Emili A., Kaper J. B., Pandey A. (2013) The Escherichia coli phosphotyrosine proteome relates to core pathways and virulence. PLoS Pathog. 9, e1003403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 25. Kim M.-S., Kandasamy K., Chaerkady R., Pandey A. (2010) Assessment of resolution parameters for CID-based shotgun proteomic experiments on the LTQ-Orbitrap mass spectrometer. J. Am. Soc. Mass Spectrom. 21, 1606–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 27. Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 28. Embuscado E. E., Laheru D., Ricci F., Yun K. J., Witzel S. de B., Seigel A., Flickinger K., Hidalgo M., Bova G. S., Iacobuzio-Donahue C. A. (2005) Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol. Ther. 4, 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (29) with the dataset identifier PXD000750.